
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA XANES study of lithium polysulfide solids: a first-principles study†
Qing
Guo
 *ab,
Kah Chun
Lau
*b and
Ravindra
Pandey
a
*ab,
Kah Chun
Lau
*b and
Ravindra
Pandey
a
aDepartment of Physics, Michigan Technological University, Houghton, MI 49931, USA. E-mail: qinguo@mtu.edu
bDepartment of Physics and Astronomy, California State University, Northridge, CA 91330, USA. E-mail: kahchun.lau@csun.edu
First published on 23rd August 2021
Abstract
X-ray absorption spectroscopy (XAS) is used for capturing the reaction mechanisms at the molecular level via the determination of local electronic configurations inside Li–S batteries. In this paper, a comprehensive investigation of the S K-edge X-ray absorption near-edge structure (XANES) of a series of stoichiometric lithium polysulfide solids was performed, assuming that these polysulfide solids are formed during the battery cycling process. The results based on density functional theory show that the pre-edge peak in the S K-edge XANES spectra is not a common feature in the solid phase in contrast to the case of pristine polysulfide molecules where the pre-edge peak constitutes a unique signature when dissolved in liquid electrolytes. A general redshift in the S K-edge XANES peak is found when a Li vacancy is introduced in these lithium polysulfide solids. Furthermore, the first peak positions of the S K-edge XANES spectra of the lithium polysulfide solids appear within a small range (2471–2473 eV) overlapping with the white line of the α-S8 bulk (∼2472 eV). Therefore, the results suggest that the use of additional characterization methods might be necessary for accurately identifying the elusive solid phase lithium polysulfides in Li–S batteries instead of relying only on S K-edge XANES spectroscopy.
1. Introduction
The need for renewable energy sources alternative to fossil fuels has become urgent due to the increasing environmental pressure. To reduce greenhouse gas emissions, full electric vehicles (EVs) powered by Li-ion batteries (LIBs) are becoming an important alternative to the traditional internal combustion engine vehicles. However, a higher energy density battery is still needed to extend the cruising range of EVs at a lower cost. The Li–S battery system is an attractive candidate to substitute LIBs because of its ultra-high theoretical specific energy (2500 W h kg−1), which is a few times higher than that of the current LIBs.1–4 A major obstacle that prevents the commercialization of Li–S batteries is the “shuttle effect”, a phenomenon associated with the high solubility of intermediate discharge products (polysulfides) in liquid electrolytes which will reduce the active sulfur and corrode the lithium metal.5–7 Several attempts have been made to suppress the shuttle effect. One effective way is trapping intermediate polysulfides to confine them within the electrodes with which only Li+ can diffuse through the boundary, such as microporous structures8–11 and polymer coatings.7,12 The spatial confinement may lead to the formation of solid phase discharge products, and various stoichiometric ratios of Li and S (LixSy) will vary successively from elemental sulfur to intermediate polysulfides and subsequently to the final product Li2S when the battery is discharged continuously. A recent experimental study demonstrated a quasi-solid state sulfur conversion during the Li–S battery discharging process, which also indicates the presence of solid phase lithium polysulfide intermediates.13To understand these elusive intermediate species, sulfur K-edge X-ray absorption near edge structure (XANES) is a promising characterization technique to probe the sulfur element's local chemical environment/oxidation state, which has been widely used for performing a comprehensive study of the reaction mechanism inside Li–S batteries.7,10,14–17 However, interpreting the S K-edge XANES results is quite problematic due to a great variety of metastable polysulfide intermediates and the lack of XANES spectral reference fingerprints, especially in solid phase lithium polysulfides. In contrast to the lithium polysulfide molecules dissolved in liquid electrolytes, single crystalline phase Li2Sx are barely synthesized experimentally due to the thermodynamically meta-stable18 and complex properties of these lithium–sulfur compounds. Experimentally, mixtures with a specific stoichiometric ratio of lithium sulfide and sulfur have been used to establish the standards of Li2Sx.10,15 The pre-edge peak around 2470 eV of the S K-edge XANES spectrum is considered as the signal of linear Sx2− dianion formation.10,16,19 To aid the XANES spectra in experiments, first-principles-based calculations have been performed for a single polysulfide (Li2Sx) molecule dissolved in tetraglyme (TEDME) electrolyte by Pascal et al.20 The pre-edge and main peaks are assigned to the terminal and internal S atoms of the linear polysulfide dianions, respectively. Compared to polysulfide molecules in liquid electrolytes, fewer studies have focused on the solid phase of lithium polysulfides due to their low stability under ambient conditions. Recently, an experimental XANES study suggested that the solvation of polysulfides can be well suppressed by the combination of an alucone-coated C–S cathode and a carbonate-based electrolyte.7 A solid phase transition mechanism from sulfur directly to Li2S was proposed as evidenced by the absence of the pre-edge peak of polysulfides in the spectra.7 Since there is a lack of a systematic study of XANES spectra for a wide range of solid phase stoichiometric lithium polysulfides, the above interpretation appears to be somewhat ambiguous.
In this paper, we have investigated the XANES spectra of lithium polysulfide crystalline structures ranging from Li2S2 to Li2S8. The predicted structures are thermodynamically stable and are metallic or semiconducting with a small band gap compared to that of the insulating Li2S.18 Besides, the unique molecular signatures of Sx chains in the solid phase polysulfides are well preserved, analogous to conventional molecular solids. In contrast to the XANES spectra of pristine polysulfide (Sx2−) molecules in solution, the iconic pre-edge peak in the S K-edge XANES spectra of Sx2− is predicted to be absent in solids. Due to the significant effect of a Li vacancy on the XANES spectra, the results show that the local coordination of sulfur species is an important factor in dictating the XANES characteristics of lithium polysulfides.
2. Computational method
The search for low-energy crystalline structures of lithium polysulfides (Li2Sx, x = 3–8) was performed with the CALYPSO code using the particle swarm optimization methodology.21,22 The validity of this methodology for finding stable or metastable structures has been confirmed in a variety of systems, including the Li–S system.23–25 The structures totaling around ≈900 consisting of two Li2Sx formula units were searched separately for each Li2Sx stoichiometry. The underlying geometry optimization and electronic property calculations were carried out in the framework of density functional theory (DFT) using the VASP code.26,27 The projector augmented wave (PAW) method28 was applied using generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.29 Grimme's D2 method30 was applied to represent van der Waals interactions.31,32 Throughout this work, an energy cutoff of 500 eV and the Monkhorst–Pack K-point meshes with the spacing values of 0.5 Å−1 and 0.2 Å−1 in the reciprocal space were used for geometry optimization and static calculation, respectively. During the geometry optimization, the residual force on each atom was less than 0.01 eV Å−1, while the convergence criterion of total energy for the static calculation was set to be 10−7 eV.Theoretical calculations for X-ray absorption spectra (XAS) were performed within the excited-state core hole (XCH) approach33 with the modified sulfur pseudopotential with a core hole at the 1s level using the Quantum-ESPRESSO (QE) package.34,35 To obtain the wave function of the excited system, a self-consistent field (SCF) calculation on lithium polysulfides was carried out using the PWscf code34 within the QE package. Subsequently, the XANES spectra were computed via a continued fraction approach using the XSpectra code.36,37 The structural configurations of lithium polysulfides were obtained at the same level of theory using VASP. Furthermore, the energy alignment scheme proposed by Prendergast et al. was adopted,20,38,39 which allows the comparison of the DFT results with the experimental XAS results. Following this scheme, we apply the energy shift that was obtained by matching the calculated and experimental XAS spectra of an isolated S2 molecule40 to the S K-edge XANES spectra of lithium polysulfides. The validity of this approach has previously been confirmed,14–16 including examples of elemental sulfur and crystalline Li2S.41
3. Results and discussion
In the following, three low-energy structural configurations consisting of diverse sulfur chain (clusters) species for each Li2Sx (x = 2–8) (labeled as str-1, str-2, and str-3) in a crystal cell are chosen in the discussion instead of an exhaustive study of all possible configurations. It is to be noted that these polysulfides’ solid phases were found to be metastable based on the formation energy convex hull relative to that of Li2S under ambient conditions.18 For each low-energy crystalline structure, we found that the Li2Sx stoichiometry, in general, does not guarantee the presence of equivalent sulfur chains (Sx) or polysulfide (Li2Sx) species in the predicted structural configuration. In other words, various sulfur chains or polysulfide species can be present in any pre-determined chemical stoichiometry of the crystalline solid as a local minimum during the search for low-energy crystalline structures of lithium polysulfides in DFT calculations. This unique trend is found to be drastically different compared to pristine stoichiometric lithium polysulfide molecules. For example, the lowest energy Li2S4-1 structural configuration prefers the presence of S3 and S5 chains over the uniform S4 chains in the lattice. A similar trend was also observed in the molecular dynamics simulations of Li–S nanoparticles.42 From our recent findings,18 the studied lithium polysulfides are only 22–127 meV per atom unfavorable compared to the stable α-sulfur and lithium sulfide. This energy range is comparable to those of many metastable solids in Inorganic Crystal Structure Database (ICSD),43 indicating that the lithium polysulfide solids may possibly be synthesized experimentally. However, the similarity in cohesive energy values also indicates that these lithium polysulfide solid phases may co-exist or in an amorphous phase13 in experiments.The projected electronic density of states (PDOS) (Fig. S1, ESI†) shows that the electronic states are solely attributed to sulfur 3p orbitals near the Fermi level. On the other hand, the analysis of Bader's charges found that the terminal sulfur (ST) atoms possess higher charges than the internal sulfur bridge (SB) atoms in a linear sulfur chain (e.g., S3/S5 in Li2S4-1, S4/S6 in Li2S5-3 and S8 in Li2S8-1), and therefore they are likely to form a Sx2− dianion, which is analogous to the recently reported lithium polysulfide molecule that is dissolved in a liquid electrolyte20 (see Tables S1 and S2, ESI†).
To highlight the unique features of sulfur chains in the lithium polysulfide solids, we now plot the projected charge density associated with the valence and conduction bands and compare it with the molecular orbitals of the corresponding isolated polysulfide dianions (Sx2−).44 For the sake of brevity, only structures Li2S4-1 and Li2S5-3 are discussed as they consist of S3/S5 and S4/S6 chains. The unique charge distributions of Li2S4-1 and Li2S5-3 can be clearly observed in Fig. 1. We found that the iso-surface of Li2S4-1 exhibits very similar features to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the isolated molecular dianion (Sx2−) counterpart. For example, the projected charge density at the highest occupied valence band (Fig. 1a-top) of Li2S4-1 is similar to those at the HOMOs of both pristine S32− and S52− molecules, whereas the value at the lowest unoccupied conduction band (Fig. 1a-bottom) is similar to that at the LUMO of the pristine S52− molecule. For the Li2S4-1 solid, this similarity suggests that the HOMOs of the constituent S32− and S52− species are associated with the same energy level near the Fermi level. On the other hand, the LUMO of S32− is higher than that of S52− in orbital energy. Later, we show that this unique feature is reflected in its XANES spectrum. Meanwhile, for the Li2S5-3 structure (Fig. 1b), the non-degenerate HOMO and degenerate LUMO levels are associated with the S4 and S6 chains. In both cases (i.e., Li2S4-1 and Li2S5-3), the similarity in the electronic charge density of crystalline polysulfides to that of the polysulfide dianions suggests that the unique orbital characteristics of molecular polysulfides are generally well-preserved in the solid phase. This intriguing finding implies that the lithium polysulfide solids are more like molecular crystals, rather than ionic crystals, and therefore the van der Waals interaction terms governing the stability of these compounds have also been highlighted recently.31,32,41
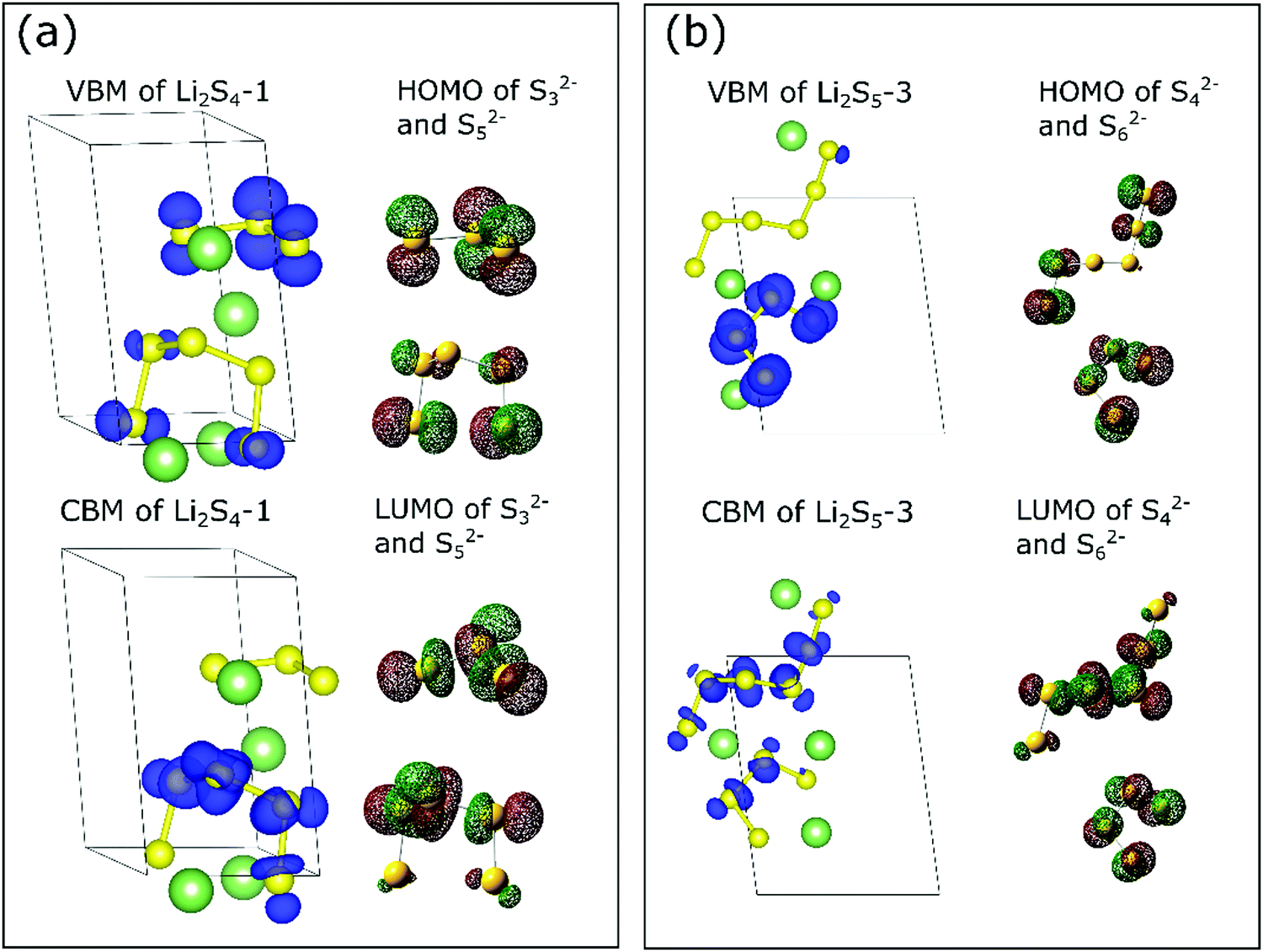 | ||
| Fig. 1 Projected charge distribution of the valence band maximum (VBM) and the conduction band minimum (CBM) of (a) Li2S4-1 and (b) Li2S5-3 solids and the HOMO and LUMO of the pristine sulfur chain dianions ((a) S32− & S52− and (b) S42− & S62−). | ||
As highlighted in the previous discussion, the individual molecular features of Sx2− anions are generally well preserved in the lithium polysulfide solids. Thus, a comparative study of the XANES spectra relative to the dissolved lithium polysulfide molecules is important. In the case of the dissolved polysulfide molecules, the pre-edge XANES peak of Sx2− has been attributed to different XANES peak positions from the terminal and internal S atoms in a polysulfide chain.20 In the present case of lithium polysulfide solids, despite having similar charge features in both terminal and internal S atoms, no distinguishable pre-edge XANES peak around 2470 eV (i.e., S K-edge) is predicted, except the case of Li2S6-1 (Fig. 2). For this crystalline structure (Li2S6-1), this configuration is unique and only S2 dimers are present in crystalline lattices, in which the nearest two parallel S2 dimers are merely separated by 2.67 Å. This small distance induces the molecular orbital split between π3px* and π3py* of a sulfur dimer which represents the final states for the S 1s electron excitation. From our analysis, the pre-edge feature of S K-edge in XANES spectra for Li2S6-1 (Fig. 2f) is attributed to this molecular orbital split as shown in Fig. S2 in the ESI.†
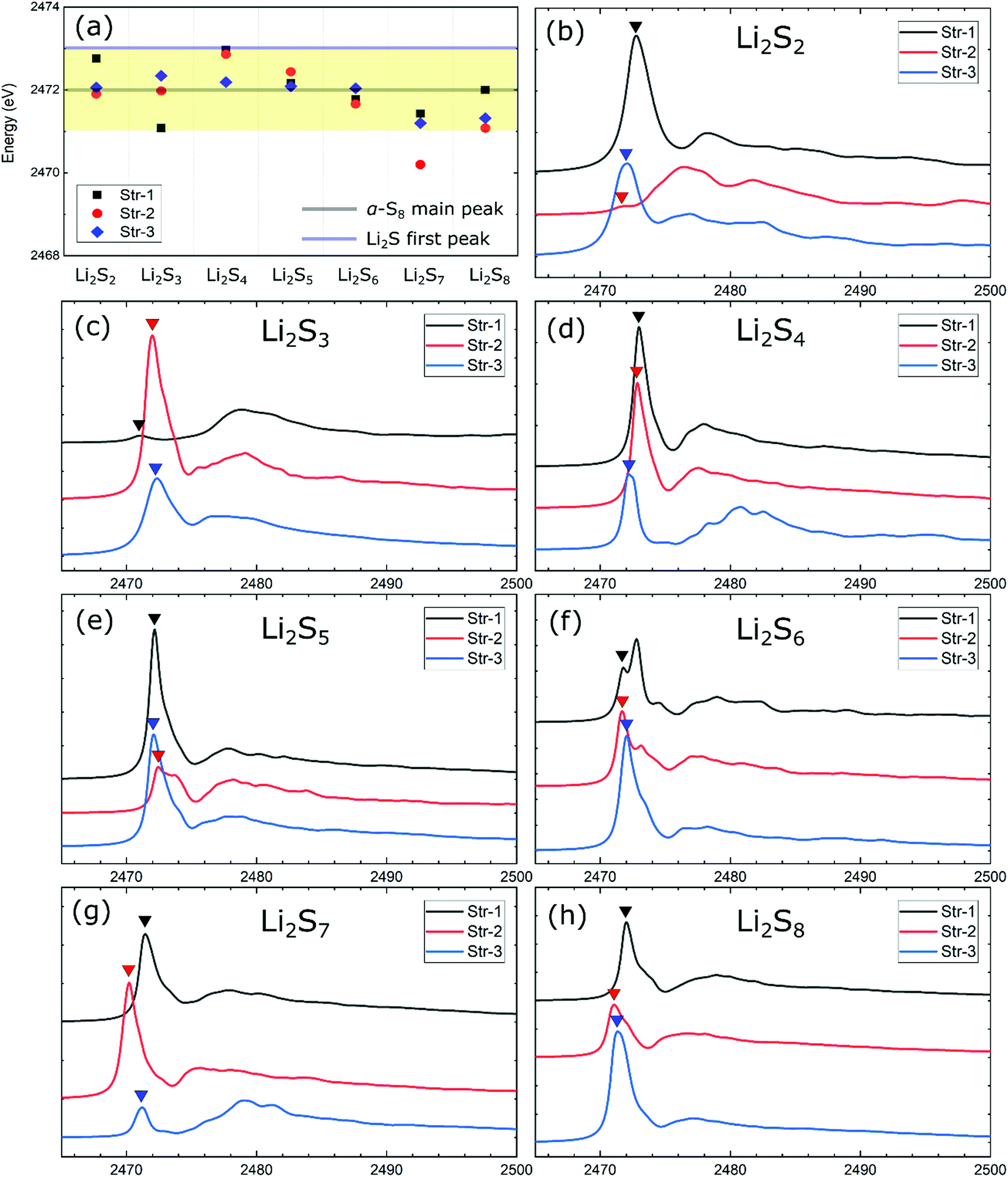 | ||
| Fig. 2 Calculated XANES spectra of Li2Sx (x = 2–8) solids. (a) The first peak positions for all the Li2Sx structures’ XANES spectra. The yellow region represents the energy range of 2471–2473 eV. The XANES spectrum for each structure with a stoichiometry of (b) Li2S2, (c) Li2S3, (d) Li2S4, (e) Li2S5, (f) Li2S6, (g) Li2S7, and (h) Li2S8. The solid triangles indicate the first peak positions for each spectrum. | ||
We note that the absence of the pre-edge XANES peak was reported in carbonate-based electrolyte Li–S batteries and was attributed to the insolubility or low solubility of polysulfides in carbonate-based electrolytes.7,45 In our opinion, the lack of distinguishable pre-edge XANES characteristics of polysulfide chains is due to the distinctive bonding environment of the polysulfide molecules in the solid phase. Instead of being solvated by organic molecules in liquid electrolytes, polysulfides in the solid phase are exposed to more substantive intermolecular interactions with the surrounding sulfur atoms including Li-ions due to the periodicity of the crystalline solid.
Considering that there exist several inequivalent S atoms in most of the lithium polysulfide structures, an exhaustive discussion on their XAS spectra could be made easier by focusing on the first adsorption peak position of the S K-edge in XANES spectra. Fig. 2 displays the predicted XANES spectra for each stoichiometric lithium polysulfide solids from Li2S2 to Li2S8. Unlike the pristine polysulfide molecules that dissolve in ether-based electrolytes,20 we found that the signature feature of the pre-edge peak is missing in the polysulfide solids. The analysis of the XANES spectra of individual atoms in the same chains found no specific differences between the terminal and the bridge S atoms (Fig. S3, ESI†). The first S K-edge peaks of these Li2Sx (x = 2–8) solids are located within a proximity of ∼2471 eV–2473 eV (Fig. 2a), and overlaps with the first peak of α-S8 at 2472 eV, but are clearly distinguishable from those of the S2− ionic species of Li2S solids that are located around 2473 eV and 2475 eV.46 Therefore, in contrast to the individual polysulfide molecules which dissolved in ether-based electrolytes,10,47 the results for polysulfide solids suggest that a distinction between polysulfide solids and the sulfur bulk solely based on the first adsorption peak position of the S K-edge in XANES spectra can be ambiguous or less conclusive.
We now focus on the analysis of XANES spectra obtained for the representative Li2S4-1 and Li2S5-3 configurations consisting of a range of sulfur chains from S3 to S6 in the crystal lattice. For Li2S4-1, the XANES main peak located at ∼2473.0 eV is mainly dominated by the contribution of the S5 chain. For the S3 component, the associated peak is located slightly higher at ∼2473.2 eV with a much lower intensity (Fig. 3a). Interestingly, this is consistent with the projected electronic density of states, which highlights the dominant contribution of S5 to forming the conduction bands (Fig. S6, ESI†). For Li2S5-3, the XANES main peak is located at ∼2472.1 eV. However, in contrast to Li2S4-1, the XANES main peak of Li2S5-3 is found to be equally contributed by S4 and S6 chains (Fig. 3b). This equivalency is reflected in the degenerate lowest unoccupied bands that are attributed to both S4 and S6 chains (Fig. 1b), providing the S 3p electronic states for the S 1s electron excitation. As expected, the electronic density of states affirms the contributions of both S4 and S6 chains in composing the lower conduction bands (Fig. S6, ESI†).
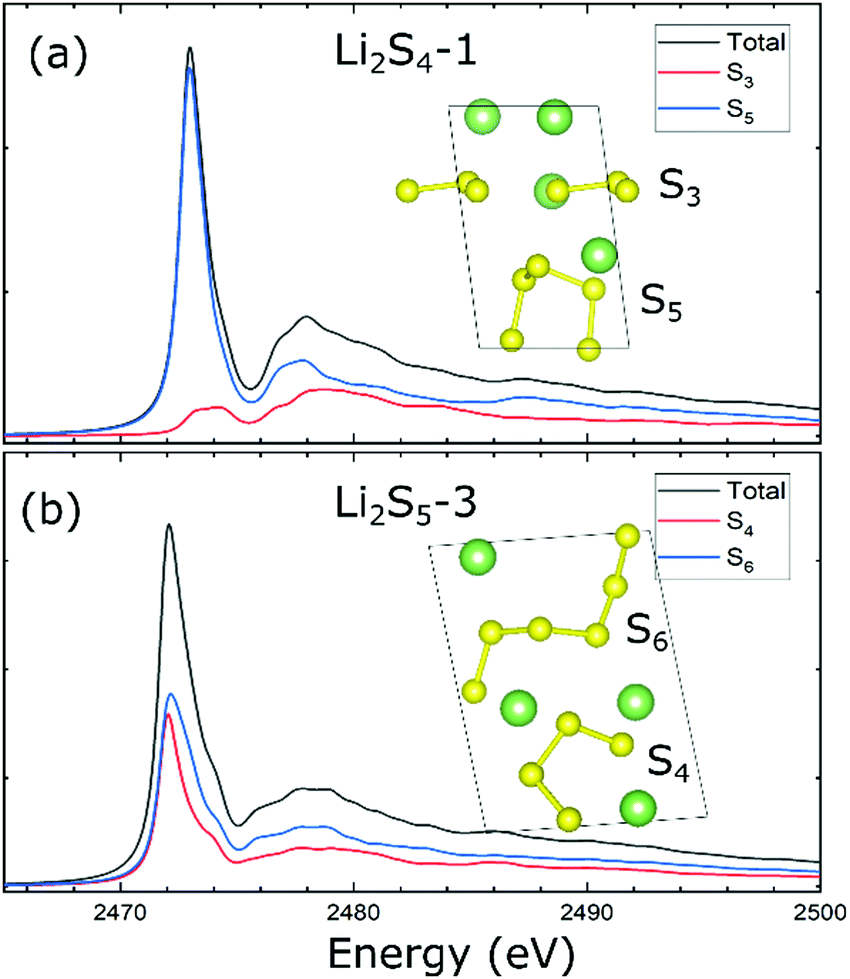 | ||
| Fig. 3 Calculated XANES spectra of (a) Li2S4-1 and (b) Li2S5-3 configurations. | ||
Next, we determine the characteristic features of XANES spectra of non-stoichiometric Li2−ySx (y = 0.5, 1, and 1.5) solids which are particularly relevant to the delithiation process during the charging process of the Li–S battery. Considering the representative Li2S4-1 and Li2S8-1 structural configurations, one, two, or three Li vacancies were created in the crystal lattice which leads to Li1.5S4, Li1S4, Li0.5S4, and Li1S8 structural configurations as shown in Fig. 4a and c. Subsequently, the optimized non-stoichiometric configurations were used to determine their XANES spectra as shown in Fig. 4b and d.
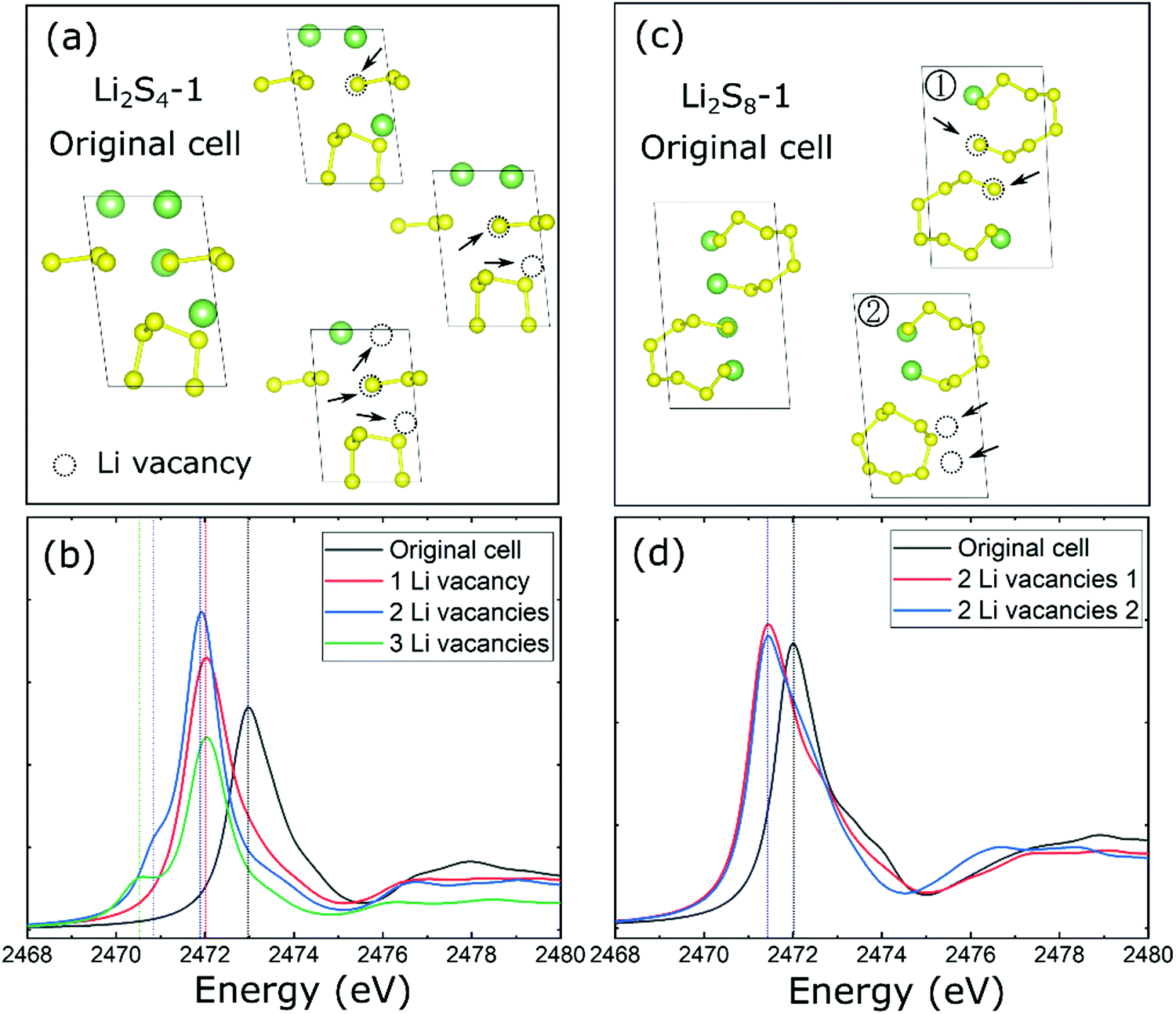 | ||
| Fig. 4 (a) The structures of Li2S4-1 with/without vacancies and (b) their calculated XANES spectra. (c) The structures of Li2S8-1 with/without vacancies and (d) their calculated XANES spectra. | ||
As shown in Fig. 4 (Fig. S4, ESI†), a Li vacancy (i.e., Li1.5S4) in the stoichiometric Li2S4 induces an energy redshift of 1.0 eV in the XANES K-edge peak which can be attributed to the lowering of the Fermi level through a missing Li atom in the unit cell. This is followed by the second Li vacancy (i.e., Li1S4) and induction of a pre-edge-like feature in the S K-edge XANES spectra. For the non-stoichiometric Li1S4, the XANES peak is associated with the total charge imbalance between S3 (0.44 e−) and S5 (1.29 e−) chains in the crystalline lattice due to the presence of Li vacancies. As shown in Fig. S5 (ESI†), the S3 unit is lowered ∼1.2 eV relative to that associated with an S5 unit, and this induces a pre-edge-like feature (Fig. 4b). More Li vacancies (i.e., 3 Li vacancies, Li0.5S4) further lowered the energy level of the pre-edge peak and caused a redshift (green line) as shown in Fig. 4b. Therefore, one can conclude that the pre-edge feature in the S K-edge spectra can be associated with the peak divergence for different S chains in the stoichiometric Li2Sx solids which is induced by the presence of Li vacancies (e.g., Li2S4-1) during the delithiation process. As noted, the origin of this pre-edge feature in polysulfide solids is quite different from that attributed to the S-terminal species found in the polysulfide molecules20 dissolved in liquid electrolytes.
In contrast to Li2S4-1, the effect of Li vacancy-induced changes in the S K-edge spectra of Li2S8-1 is found to be less significant during the delithiation process, which may be due to a smaller Li/S ratio in its unit cell. For example, the redshift of the XANES peak is only ∼0.75 eV and is found to be independent or insensitive to the position of the Li vacancy (Fig. 4c and d) in the non-stoichiometric Li1S8. Interestingly, the removal of two Li atoms from a Li2S8 chain (Fig. 4c) facilitates the formation of an S8 ring in an equilibrium configuration, thereby confirming the fact that opening/closing of the S8 ring is a reversal reaction48 depending on the lithiation/delithiation process in the Li–S battery system.
4. Summary
In this paper, the XANES spectra of a series of lithium polysulfide solids ranging from Li2S2 to Li2S8 were calculated. The results based on DFT calculations show distinctive molecular features suggesting the lithium polysulfide solids to be more like molecular solids. Analyses of the molecular orbitals, DOS near the Fermi level, and Bader charge distribution facilitate the assignment of the first peaks of the S K-edge spectra in polysulfide solids. In contrast to the pristine lithium polysulfide molecules dissolved in liquid electrolytes, the pre-edge peak is not found to be the common feature among the stoichiometric polysulfide solids, although the non-stoichiometric solids may exhibit the pre-edge feature in the S K-edge XANES spectra through the presence of the Li vacancy. Compared to stoichiometric lithium polysulfide solids, the redshift of the XANES peaks is commonly found in non-stochiometric solids when a Li vacancy is introduced. For these polysulfide solids, the first peak positions of the S K-edge XANES spectra are located within a small range of 2471–2473 eV which strongly overlaps with that of the α-S8 bulk at ∼2472 eV but it was found to be different from that of the S2− species in Li2S which is located at ∼2473–2475 eV. Overall, it is, therefore, essential to use complementary materials characterization techniques such as IR spectroscopy, Raman spectroscopy, or a “multi-modal” approach,49 in addition to XANES spectroscopy, to fully characterize the presence of polysulfides in the Li–S battery system.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The superior, high-performance computing cluster at Michigan Technological University was used to perform electronic structure calculations in this paper. Helpful support from Dr S Gowtham is gratefully acknowledged. K.C. Lau acknowledges the faculty start-up funding provided by California State University, Northridge.References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Li–O2 and Li–S Batteries with High Energy Storage, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- D. Peramunage and S. Licht, A Solid Sulfur Cathode for Aqueous Batteries, Science, 1993, 261, 1029–1032 CrossRef CAS PubMed.
- A. Fotouhi, D. J. Auger, L. O’Neill, T. Cleaver and S. Walus, Lithium-Sulfur Battery Technology Readiness and Applications—a Review, Energies, 2017, 10, 1937 CrossRef.
- S. Evers and L. F. Nazar, New Approaches for High Energy Density Lithium–Sulfur Battery Cathodes, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- Y. V. Mikhaylik and J. R. Akridge, Polysulfide Shuttle Study in the Li/S Battery System, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Lithium–Sulfur Batteries: Electrochemistry, Materials, and Prospects, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- X. Li, M. Banis, A. Lushington, X. Yang, Q. Sun, Y. Zhao, C. Liu, Q. Li, B. Wang and W. Xiao, A High-Energy Sulfur Cathode in Carbonate Electrolyte by Eliminating Polysulfides Via Solid-Phase Lithium-Sulfur Transformation, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- B. Zhang, X. Qin, G. Li and X. Gao, Enhancement of Long Stability of Sulfur Cathode by Encapsulating Sulfur into Micropores of Carbon Spheres, Energy Environ. Sci., 2010, 3, 1531–1537 RSC.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. c. D. Abruña, Yolk–Shell Structure of Polyaniline-Coated Sulfur for Lithium–Sulfur Batteries, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- M. Cuisinier, P.-E. Cabelguen, S. Evers, G. He, M. Kolbeck, A. Garsuch, T. Bolin, M. Balasubramanian and L. F. Nazar, Sulfur Speciation in Li–S Batteries Determined by Operando X-Ray Absorption Spectroscopy, J. Phys. Chem. Lett., 2013, 4, 3227–3232 CrossRef CAS.
- S. Ma, L. Wang, Y. Wang, P. Zuo, M. He, H. Zhang, L. Ma, T. Wu and G. Yin, Palladium Nanocrystals-Imbedded Mesoporous Hollow Carbon Spheres with Enhanced Electrochemical Kinetics for High Performance Lithium Sulfur Batteries, Carbon, 2019, 143, 878–889 CrossRef CAS.
- F. Wu, J. Chen, R. Chen, S. Wu, L. Li, S. Chen and T. Zhao, Sulfur/Polythiophene with a Core/Shell Structure: Synthesis and Electrochemical Properties of the Cathode for Rechargeable Lithium Batteries, J. Phys. Chem. C, 2011, 115, 6057–6063 CrossRef CAS.
- Q. Pang, A. Shyamsunder, B. Narayanan, C. Y. Kwok, L. A. Curtiss and L. F. Nazar, Tuning the Electrolyte Network Structure to Invoke Quasi-Solid State Sulfur Conversion and Suppress Lithium Dendrite Formation in Li–S Batteries, Nat. Energy, 2018, 3, 783–791 CrossRef CAS.
- R. Dominko, A. Vizintin, G. Aquilanti, L. Stievano, M. J. Helen, A. R. Munnangi, M. Fichtner and I. Arcon, Polysulfides Formation in Different Electrolytes from the Perspective of X-Ray Absorption Spectroscopy, J. Electrochem. Soc., 2018, 165, A5014–A5019 CrossRef CAS.
- K. H. Wujcik, J. Velasco-Velez, C. H. Wu, T. Pascal, A. A. Teran, M. A. Marcus, J. Cabana, J. Guo, D. Prendergast and M. Salmeron, Fingerprinting Lithium-Sulfur Battery Reaction Products by X-Ray Absorption Spectroscopy, J. Electrochem. Soc., 2014, 161, A1100–A1106 CrossRef CAS.
- M. U. Patel, I. Arčon, G. Aquilanti, L. Stievano, G. Mali and R. Dominko, X-Ray Absorption near-Edge Structure and Nuclear Magnetic Resonance Study of the Lithium–Sulfur Battery and Its Components, ChemPhysChem, 2014, 15, 894–904 CrossRef CAS PubMed.
- G. Tan, R. Xu, Z. Xing, Y. Yuan, J. Lu, J. Wen, C. Liu, L. Ma, C. Zhan and Q. Liu, Burning Lithium in Cs2 for High-Performing Compact Li2S–Graphene Nanocapsules for Li–S Batteries, Nat. Energy, 2017, 2, 17090 CrossRef CAS.
- Q. Guo, K. C. Lau and R. Pandey, Implication of Mechanical Properties of Li-S Binary Compounds Obtained from the First-Principles Study, J. Phys. Chem. C, 2021, 125, 290–294 CrossRef CAS.
- Z. Lin, C. Nan, Y. Ye, J. Guo, J. Zhu and E. J. Cairns, High-Performance Lithium/Sulfur Cells with a Bi-Functionally Immobilized Sulfur Cathode, Nano Energy, 2014, 9, 408–416 CrossRef CAS.
- T. A. Pascal, K. H. Wujcik, J. Velasco-Velez, C. Wu, A. A. Teran, M. Kapilashrami, J. Cabana, J. Guo, M. Salmeron and N. Balsara, X-Ray Absorption Spectra of Dissolved Polysulfides in Lithium–Sulfur Batteries from First-Principles, J. Phys. Chem. Lett., 2014, 5, 1547–1551 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Calypso: A Method for Crystal Structure Prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal Structure Prediction Via Particle-Swarm Optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- G. Wang, R. Pandey and S. P. Karna, Carbon Phosphide Monolayers with Superior Carrier Mobility, Nanoscale, 2016, 8, 8819–8825 RSC.
- G. Yang, S. Shi, J. Yang and Y. Ma, Insight into the Role of Li2S2in Li–S Batteries: A First-Principles Study, J. Mater. Chem. A, 2015, 3, 8865–8869 RSC.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Predicted Novel High-Pressure Phases of Lithium, Phys. Rev. Lett., 2011, 106, 015503 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, Semiempirical Gga-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. Park, H. S. Koh and D. J. Siegel, First-Principles Study of Redox End Members in Lithium–Sulfur Batteries, J. Phys. Chem. C, 2015, 119, 4675–4683 CrossRef CAS.
- Q. Guo, K. C. Lau and R. Pandey, Thermodynamic and Mechanical Stability of Crystalline Phases of Li2S2, J. Phys. Chem. C, 2019, 123, 4674–4681 CrossRef CAS.
- D. Prendergast and G. Galli, X-Ray Absorption Spectra of Water from First Principles Calculations, Phys. Rev. Lett., 2006, 96, 215502 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, Advanced Capabilities for Materials Modelling with Quantum Espresso, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- C. Gougoussis, M. Calandra, A. P. Seitsonen and F. Mauri, First-Principles Calculations of X-Ray Absorption in a Scheme Based on Ultrasoft Pseudopotentials: From A-Quartz to High-TC Compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075102 CrossRef.
- M. Taillefumier, D. Cabaret, A.-M. Flank and F. Mauri, X-Ray Absorption near-Edge Structure Calculations with the Pseudopotentials: Application to the K Edge in Diamond and A-Quartz, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 195107 CrossRef.
- A. H. England, A. M. Duffin, C. P. Schwartz, J. S. Uejio, D. Prendergast and R. J. Saykally, On the Hydration and Hydrolysis of Carbon Dioxide, Chem. Phys. Lett., 2011, 514, 187–195 CrossRef CAS.
- T. A. Pascal, C. Pemmaraju and D. Prendergast, X-Ray Spectroscopy as a Probe for Lithium Polysulfide Radicals, Phys. Chem. Chem. Phys., 2015, 17, 7743–7753 RSC.
- E. Rühl, R. Flesch, W. Tappe, D. Novikov and N. Kosugi, Sulfur 1s Excitation of S2 and S8: Core–Valence-and Valence–Valence–Exchange Interaction and Geometry-Specific Transitions, J. Chem. Phys., 2002, 116, 3316–3322 CrossRef.
- Q. Guo, K. C. Lau and R. Pandey, Novel Metallic Crystalline Phase of Li2S3, J. Phys. Chem. C, 2019, 123, 28027–28034 CrossRef CAS.
- Y. Li, N. A. Romero and K. C. Lau, Structure–Property of Lithium–Sulfur Nanoparticles Via Molecular Dynamics Simulation, ACS Appl. Mater. Interfaces, 2018, 10, 37575–37585 CrossRef CAS PubMed.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, The Thermodynamic Scale of Inorganic Crystalline Metastability, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruna, Effects of Liquid Electrolytes on the Charge–Discharge Performance of Rechargeable Lithium/Sulfur Batteries: Electrochemical and in-Situ X-Ray Absorption Spectroscopic Studies, J. Phys. Chem. C, 2011, 115, 25132–25137 CrossRef CAS.
- Y. Gorlin, M. U. Patel, A. Freiberg, Q. He, M. Piana, M. Tromp and H. A. Gasteiger, Understanding the Charging Mechanism of Lithium-Sulfur Batteries Using Spatially Resolved Operando X-Ray Absorption Spectroscopy, J. Electrochem. Soc., 2016, 163, A930 CrossRef CAS.
- K. H. Wujcik, T. A. Pascal, C. Pemmaraju, D. Devaux, W. C. Stolte, N. P. Balsara and D. Prendergast, Characterization of Polysulfide Radicals Present in an Ether-Based Electrolyte of a Lithium–Sulfur Battery During Initial Discharge Using in Situ X-Ray Absorption Spectroscopy Experiments and First-Principles Calculations, Adv. Energy Mater., 2015, 5, 1500285 CrossRef.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Recent Advances in the Polymerization of Elemental Sulphur, Inverse Vulcanization and Methods to Obtain Functional Chalcogenide Hybrid Inorganic/Organic Polymers (Chips), Polym. Chem., 2019, 10, 4078–4105 RSC.
- K. Sun, C. Zhao, C.-H. Lin, E. Stavitski, G. J. Williams, J. Bai, E. Dooryhee, K. Attenkofer, J. Thieme and Y.-c. K. Chen-Wiegart, Operando Multi-Modal Synchrotron Investigation for Structural and Chemical Evolution of Cupric Sulfide (Cus) Additive in Li-S Battery, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00450f |
| This journal is © The Royal Society of Chemistry 2021 |