
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics simulations of a hydrophilic MIL-160-based membrane demonstrate pressure-dependent selective uptake of industrially relevant greenhouse gases†
Jordan
Chapman
a,
Nagasree
Garapati
a,
Vassiliki-Alexandra
Glezakou
b,
Yuhua
Duan
 c,
Jianli
Hu
a and
Cerasela Zoica
Dinu
*a
c,
Jianli
Hu
a and
Cerasela Zoica
Dinu
*a
aWest Virginia University, Morgantown, WV 26506, USA. E-mail: cerasela-zoica.dinu@mail.wvu.edu
bPacific Northwest National Laboratory, Richland, WA 99352, USA
cNational Energy Technology Laboratory, United States Department of Energy, Pittsburgh, Pennsylvania 15236, USA
First published on 7th July 2021
Abstract
Continued integration of technologies capable of achieving higher degrees of sustainability while meeting global material and energy demands is of singular importance in halting human-caused climate change. Gas separation membranes composed of metal–organic frameworks (MOFs) are considered promising candidates for such integration; owing to their modular, scalable nature and high degree of tunability they are seen essential to maintain separation functionality. However, prior to sustainable implementation, both an evaluation of MOF characteristics and an intensive examination of MOF–gas molecule interactions are necessary to fully understand the fundamental separation criteria as well as to define suitable ranges of gas separation conditions. Herein, we present our findings on the greenhouse gas separation capabilities of the hydrophilic, Al-based MIL-160 in the selective uptake of carbon dioxide (CO2) from other relevant greenhouse gases, i.e., methane (CH4), sulfur dioxide (SO2), nitrogen dioxide (NO2), and nitric oxide (NO), including gravimetric solubility, permeability, and diffusivity calculations. We found that a MIL-160 membrane has excellent applicability in the separation of gases of varying electronegativities, with a diffusivity selectivity of 72.0, 9.53, and 13.8 for CH4, NO2, and NO, respectively, relative to CO2. Further, we demonstrate that the selectivity at which gas molecules diffuse through the MIL-160 membrane varies strongly with the simulation pressure, suggesting that such membrane system is potentially an ideal candidate for the development of pressure-swing adsorption processes that achieve gas separations efficiently while mitigating the emission of greenhouse gases.
Introduction
The societal need for efficient, scalable gas separation processes has become of great significance following the enactment of international agreements that aim simultaneously to decelerate anthropogenic emissions and to implement an increased share of sustainably designed energy technologies.1–8 The separation of acid gases carbon dioxide (CO2) and hydrogen sulfide (H2S) from natural gas resources, for instance, is an important precursor to the improved economics of such processes for both producing methane (CH4) and synthesizing commodity chemicals9,10 respectively. Removal of CO2 and H2S from the feed stream prevents extensive corrosion of expensive infrastructure as well as significantly reduces downstream emissions. Complementarily, the separation of flue gas impurities such as sulfur (SOx) and nitrogen oxides (NOx), and other trace gases, offers a general platform for mitigating the hydrolytic formation of acids in the presence of water, while subsequently improving process affordability and sustainability.1,4,11 Lastly, post combustion separation of CO2 from dilute low flow rate stream aids in the industrialization and efficiency of chemical processes with specific trade-offs in capture rates.12 Whereas the storage and transportation of CO2 are facilitated by relatively mature, affordable technologies,13–15 CO2 capture—including its separation from other gases—continues to prove challenging and comprises nearly 66% of the total cost associated with carbon capture and storage (CCS) processes.5–7,16–19A number of gas adsorption and separation technologies that operate on the basis of differences in the physical properties of gases—i.e., volatility, solubility, adsorptivity, and molecular diameters of the individual gases—have been developed and implemented at the commercial scale, with varying degrees of applicability in the selective uptake of target greenhouse gases1,2,20–22 however with drawbacks. Specifically, distillation columns, absorbers, and strippers often entail considerable heat requirements and are either infeasible for the separation of incondensable gases or incur significant operating costs associated with the purchase of chemical solvents and maintenance of optimized operating conditions for instance.1,2,23 Further, such technologies require relatively large capital investment for well-defined process specifications and thus are profitable only when the scale of production is large enough with respect to both the time of operation and materials’ lifetime.4
Pressure swing adsorption (PSA) was recently exploited for the removal of vapor-phase impurities and enrichment of usable gases11,18,20,21,24,25 benefiting from low energy input and facile implementation at varied production scale.26,27 Similarly, gas separations via microporous polymer membranes have been commercially employed for the oxygen enrichment of air, recovery of hydrogen from ammonia production process streams, and the upgrade of natural gas.28 Still, challenges persist when considering gas drying, fractionation of air, scalability of off-gases and the ensuing difficulty of applicability of the bench-scale findings to consumer scale implementation.20,21 Moreover, porous polymer membranes suffer from critical tradeoffs between permeability and gas molecule selectivity, thus resulting in low efficiency of separations due to inconsistent pore size distributions and low gas selectivity.28
Metal–organic frameworks (MOFs)—porous materials with high specific surface areas, controllable nanostructures, and tunable chemical and physical properties—have recently been proposed as alternatives for CCS processes29,30 and for gas adsorption and separation.7,22,31,32 The modular nature of MOFs with aperture size distributions of angstrom-level resolution7,22,31 is envisioned to allow for high degrees of adsorption selectivity and permselectivity and improved process economics with reduced emissions. Previous studies showed that a mixed-matrix membrane (MMM) composed of a polyimide crosslinker and embedded with NH2-MIL-53, a surface-modified variant of an aluminum-based MOF, can be used for separating CH4 from CO2—exhibiting increased CO2 selectivity without significant loss of its permeability.33 The improvements were realized in part by the presence of NH2 groups that aided in regulating the MOF breathing mechanism and penetration of polyimide chains to provide configurational stability.33 Complementarily, a new humidity-resistant MOF, JNU-2, has shown promise in the separation of ethane (C2H6) from ethylene (C2H4), the single largest feedstock in the petrochemicals industry.22 The efficiency of separation was bolstered by the combination of the rational design of the JNU-2 aperture sites and the availability of high surface area.22 These authors and others showed that optimal MOF performance is often founded on the combined synergistic effects resulting from dynamic structural, chemical, mechanical, and electrical properties of resulting hybrids. However, it was also found that a significant portion of current MOFs were highly unstable in the presence of moisture in adsorption/separation processes;34 for instance, isoreticular metal–organic frameworks (IRMOFs) lose their porosity at room temperature in air, thus considerably reducing process efficiency.35 Similarly, some classes of carboxylated MOFs exhibit significant structural variations in neutral to basic media due to the deprotonation of the water molecules coordinated to their metal nodes, thus leading to a cascade of reactions with subsequent loss of native pore geometry.36–38
Herein, we hypothesized that intrinsic MOF characteristics—including selective affinity for target gas molecules, desired operability across varied temperatures and pressures, highly regular and controllable pore geometry, and structural stability in aqueous environments—will ultimately determine structure feasibility for the formation of membrane systems to be used for gas separation processes. Further, we hypothesized that the inherent chemical stability of such MOFs in solvents and caustic gases (that can otherwise cause inhibitory loss of framework native pore structure and, by extension, loss of MOF performance) will expand the applicability of such materials to a widening range of end uses.
To demonstrate our hypotheses, we used a hydrophilic MOF, MIL-160, known for its high hydrothermal stability39 and selective and reversible adsorption of water vapor.40 MIL-160 is readily synthesized under chemically mild conditions and was shown to have large potential applicability for gas sorption41 and refrigeration42 technologies. For instance, Permyakova et al. were successful in synthesizing up to 400 g per day of Al-based MIL-160 via the equimolar mixture of aluminum acetate and 2,5-furandicarboxylic acid in 2 L of water at room temperature followed by an ethanol wash and drying at 100 °C;43 the product was implemented as a water sorbent in a heat reallocation open-system reactor and it outperformed the commercial zeolite thirteen times in seasonal heat storage thus showing good promise of MIL-160-based sorbents in energy reallocation systems with a water capacity of nearly 0.36 g water g−1 MIL-160 and full regeneration of water at 80 °C.43 Solovyeva et al. reported synthesis by mixing equimolar amounts of 2,5-furandicarboxylic acid, aluminum chloride hexahydrate, and sodium hydroxide in distilled water and heating the product at 120 °C prior to washing and activating it via continuous evacuation at 150 °C for 15 h.44 Synthesized MIL-160 was implemented in an atmospheric water harvesting scheme in which the binding affinity for water and the hydrothermal stability of the material distinguished it as an ideal candidate with a specific water capacity of nearly 0.34 g water g−1 MIL-160 and a regeneration temperature of 80 °C.44 In our lab we have shown synthesis of MIL-160-alumina based membranes as supports for CO2 capture and transformation into benign byproducts upon interfacing the framework with the carbonic anhydrase enzyme.45
Brandt et al. have further confirmed the promising potential of MIL-160 in gas separation processes owing to its high selectivity for SO2 at low partial pressures, as well as its excellent stability under both humid and dry conditions.41 Lastly, Wu et al. developed a MIL-160-based membrane prepared on polydopamine-modified aluminum oxide discs via a solvothermal synthesis route that was subsequently implemented in the separation of p-xylene from its bulkier isomer o-xylene;46 the membrane was found to have a separation factor of 38.5—competitive with zeolitic metal–inorganic framework counterparts—owing to the high diffusion and adsorption selectivity of p-xylene relative to o-xylene.47 However, to our knowledge, no studies exist that directly relate the dynamic sorption behavior of different greenhouse gas species within a MIL-160 membrane or to the steady-state permeability and adsorptivity of such species. Moreover, we found no analyses that correlate the variations in chemical and physical MOF–gas molecule interactions directly to the suitability of such system for gas separation applications. Lack of such knowledge presents not only an apparent gap in performance analysis of this specific MOF but further prevents its future implementation and subsequent development of improvement strategies and applicabilities to other MOF structures as well as their integration in gas adsorption and separation processes.7,22,31,32
Methodology
General considerations for molecular dynamics simulations
Atomic-level processes critical to MOF membrane efficiency—including pressure-driven selective adsorption and selective permeability of industrially relevant gas species CO2, CH4, sulfur dioxide (SO2), nitrogen dioxide (NO2), and nitric oxide (NO)—were investigated for the selected framework aluminum (Al)-based MIL-160, constructed from the coordination of 2,5-furandicarboxylic acid with Al ions.48 All molecular dynamics (MD) simulations were performed using GROMACS software version 2016.6.49 Non-bonded force field parameters of MIL-160 including partial atomic charges derived via single point energy calculations and Mulliken analysis were taken from Cadiau et al.;50 the structure of MIL-160 was assumed to be rigid such that the atomic coordinates of MIL-160 were fixed. Transferable potentials for phase equilibria (TraPPE) parameters for CO2,51 SO2,52 NO,53 NO2,54 and CH455 were used to define both the Lenard-Jones and electrostatic interactions between the MOF membrane and gas molecules.Membrane simulations
The membrane was simulated via construction of a MOF supercell with increased numbers of unit cells in all dimensions; the process was chosen to reflect controlled scaling within the limits of computational budget. All simulations were run in the canonical NVT ensemble (constant number of molecules, volume, and temperature, respectively) and at 300 K; the simulation temperature was maintained via the V-rescale thermostat algorithm.56 van der Waals (VDW) interactions between atoms were measured at a cut-off range of 1.4 nm for sufficient sampling of gas particle–MOF interactions without burdensome computational expense, while long range electrostatics were calculated with a Particle Mesh Ewald (PME) scheme57 for the elimination of potential electrostatic artifacts. Time integration of the simulations was done following the leap-frog integration method.58 Simulation boxes with dimensions of 8.4 nm × 8.4 nm determined by the dimension of the 4 × 4 MIL-160 membrane and with varying dimensions in the z-direction depending on the target simulation pressure were constructed; a 4 × 4 × 3 MIL-160 membrane was placed in each box such that a gas bath was placed on one side of the membrane while the other side of the membrane remained a vacuum chamber, thus creating an effective pressure gradient that varied in magnitude with the number of gas molecules contained in respective gas baths. A schematic representation of the simulation scenario is shown in Fig. S.1 in the ESI.†Membrane permeation and sorption characteristics
To evaluate the membrane permeation and sorption characteristics of individual gas species, single-component gas baths—e.g., 100 mol% CO2, 100 mol% CH4, 100 mol% SO2, 100 mol% NO2, and 100 mol% NO—were inserted on one side of the MIL-160 membrane, thus emulating a physical scenario consistent with typical pressure-driven membrane processes. Gas mixtures comprised of 95 mol% CH4/5 mol% CO2, 95 mol% SO2/5 mol% CO2, 95 mol% NO2/5 mol% CO2, and 95 mol% NO/5 mol% CO2 were similarly placed on one side of the MIL-160 membrane opposite a vacuum chamber to determine the dilute CO2 separation capabilities of the MOF membrane relative to the chemical and physical properties of respective gas molecules. The pressure of each gas bath was varied from vacuum conditions to high-pressure conditions—0.2, 0.5, 1, 5, and 50 bar—by changing the number of molecules contained within the gas bath to calculate pressure-dependent sorption and permeation characteristics of the MIL-160 membrane. The simulation pressure was varied across a wide range spanning vacuum, atmospheric, and high-pressure regimes to thus survey broadly the atomic level phenomena that endow the MIL-160 membrane with its critical separation criteria even for applications related to carbon sequestration with enhanced gas recovery.59Gas bath pressures were calculated following the assumption that each gas did not deviate from ideal behaviour. Tables S.1 and S.2 in the ESI† summarize the number of gas molecules, molar composition, simulation box dimensions, and effective simulation pressure of each single-composition gas simulation and gas mixture simulation, respectively. It is noted that the simulation size of high-pressure simulations run at a gas bath pressure of 50 bar was decreased relative to all other simulations in order to avoid severely limiting computational expenses associated with a large number of atoms within the simulations.
Initial configurations were energy-minimized via the steepest descent method at a time step of 1 fs prior to simulation. MD simulations were run at a time step of 1 fs for 100 ps intervals, after which gas baths containing a depleted number of gas molecules owing to pressure-driven sorption and permeation were replaced with gas baths comprised of the same number of gas molecules and molar composition as the initial gas bath, whereas gas molecules that had permeated to the vacuum chamber were removed. Thus, temporal decreases in gas bath pressure were accounted for at each 100 ps interval and corrected via the replacement of a depleted gas bath with a gas bath containing the appropriate number of molecules pertaining to the desired gas bath pressure and molar composition. Replacement of depleted gas baths and removal of permeated gas molecules at 100 ps intervals were necessary to account for gas molecules that either were adsorbed within the membrane or permeated through the membrane into the vacuum chamber and, therefore, were needed to approximate constant pressure gradients while running sequential MD productions in the canonical NVT ensemble.60
An energy minimization in which the gas molecule atomic positions were optimized via the steepest descent method to prevent energetically prohibitive placement was run following the insertion of the gas baths at each 100 ps interval. Following energy minimization, sequential 100 ps simulations were run, and depleted gas baths were again replaced to adjust for pressure deviations. This process was extended for total data collection periods ranging from 10 ns to 120 ns simulation time as was necessary to capture both dynamic and steady-state sorption and permeation behaviors of each gas species at varied gas bath pressures. The numbers of adsorbed and permeated gas molecules were measured via a molecule-counting shell script in which gas molecules located within the MOF membranes were defined as adsorbed gas molecules and gas molecules that migrated to the vacuum side of the simulation were defined as permeated gas molecules.
The permeability Pi, gravimetric solubility Si, and diffusivity Di of each gas species were calculated from the single-component gas simulations, using eqn (1)–(3):60
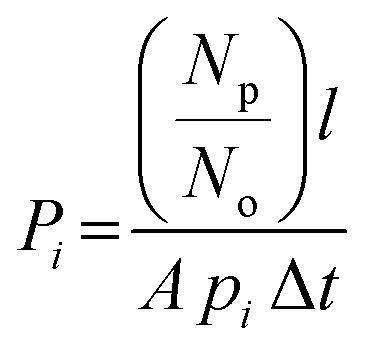 | (1) |
 | (2) |
 | (3) |
Complementarily, the permselectivity αP,i, gravimetric solubility selectivity αS,i, and diffusivity selectivity60αD,i of CH4, SO2, NO2, and NO relative to CO2 were calculated according to eqn (4)–(6):
 | (4) |
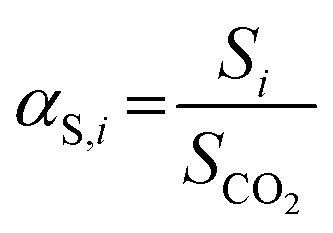 | (5) |
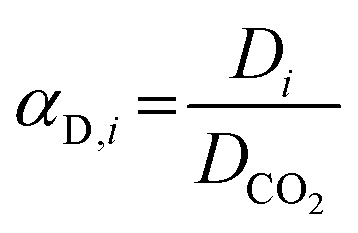 | (6) |
Statistical analysis
Statistical analysis was conducted for single-component and gas mixture selectivity calculations. The average number of adsorbed and the average number of permeated gas molecules were calculated for each single-component gas simulation in steady-state sorption and permeation regimes respectively. Further, standard deviations around the average numbers of adsorbed and permeated gas molecules were calculated for the same conditions.Similar analysis with slight adjustments in the approach was carried out for gas mixture simulations. Averages were calculated for the ratio of adsorbed gas molecules CH4, SO2, NO2, and NO to adsorbed CO2 molecules, as well as the permeated gas molecules CH4, SO2, NO2, and NO to permeated CO2 molecules, respectively, in steady-state sorption and permeation regimes. Standard deviations around the average ratios of adsorbed and average ratios of permeated gas molecules were calculated as well.
Results and discussion
Membrane permeation and sorption of single-component gases
We first conducted MD simulations emulating steady-state permeation of various gases—i.e., CO2, CH4, SO2, NO2, and NO—through a rigid MIL-160 membrane at varying pressures (i.e., 0.2, 0.5, 1, 5, and 50 bar, respectively), to probe permeability and sorption characteristics through MIL-160 defined membranes. Herein, we define steady-state conditions as those in which the number of gas molecules within the MIL-160 membrane and the rate of gas molecules permeating through the membrane approach respective constant values dependent on gas bath pressure and gas species properties. Gases were chosen based on their known and significant logistical problems within chemical refineries and power plants as associated with catalyst poisoning and evolution of corrosive species shown to damage equipment as well as contribute to the acceleration of human-caused climate change.10,61,62 For our analysis, we implemented reliable force field parameters for both gas molecules and membrane systems to thus circumvent the constraints of density functional theory (DFT) methods63 known to be computationally expensive and often prohibitive in supplementing key thermodynamic findings such as energy optimized configurations of adsorbed guest molecules with permeability resulting from the energetic preferability with which a MOF material binds target gas molecules.40,64Fig. 1 shows the sorption, permeability, and diffusivity of each single-component gas composition calculated with respect to a defined, rigid 4 × 4 × 3 MIL-160 membrane (Fig. 1(a)). The dynamic sorption and permeation profiles of simulated gas species at each pressure are presented in Fig. S.2–S.6 in the ESI.† Our analysis showed that each tested gas species followed a time-dependent trend in which the sorption of an individual molecule within the membrane reached a saturation point, i.e., the maximum loading, with the permeation rates approaching a constant rate within a simulation time depending on the employed simulation pressure. High-pressure simulations reached steady-state conditions in less time than did simulations running under vacuum and atmospheric conditions respectively. Gas permeation analysis at 5 and 50 bar elicited dynamic responses within 10 ns of simulation time, while atmospheric and vacuum conditions requiring simulation times up to 120 ns to reach steady-state sorption and permeation behaviors respectively.
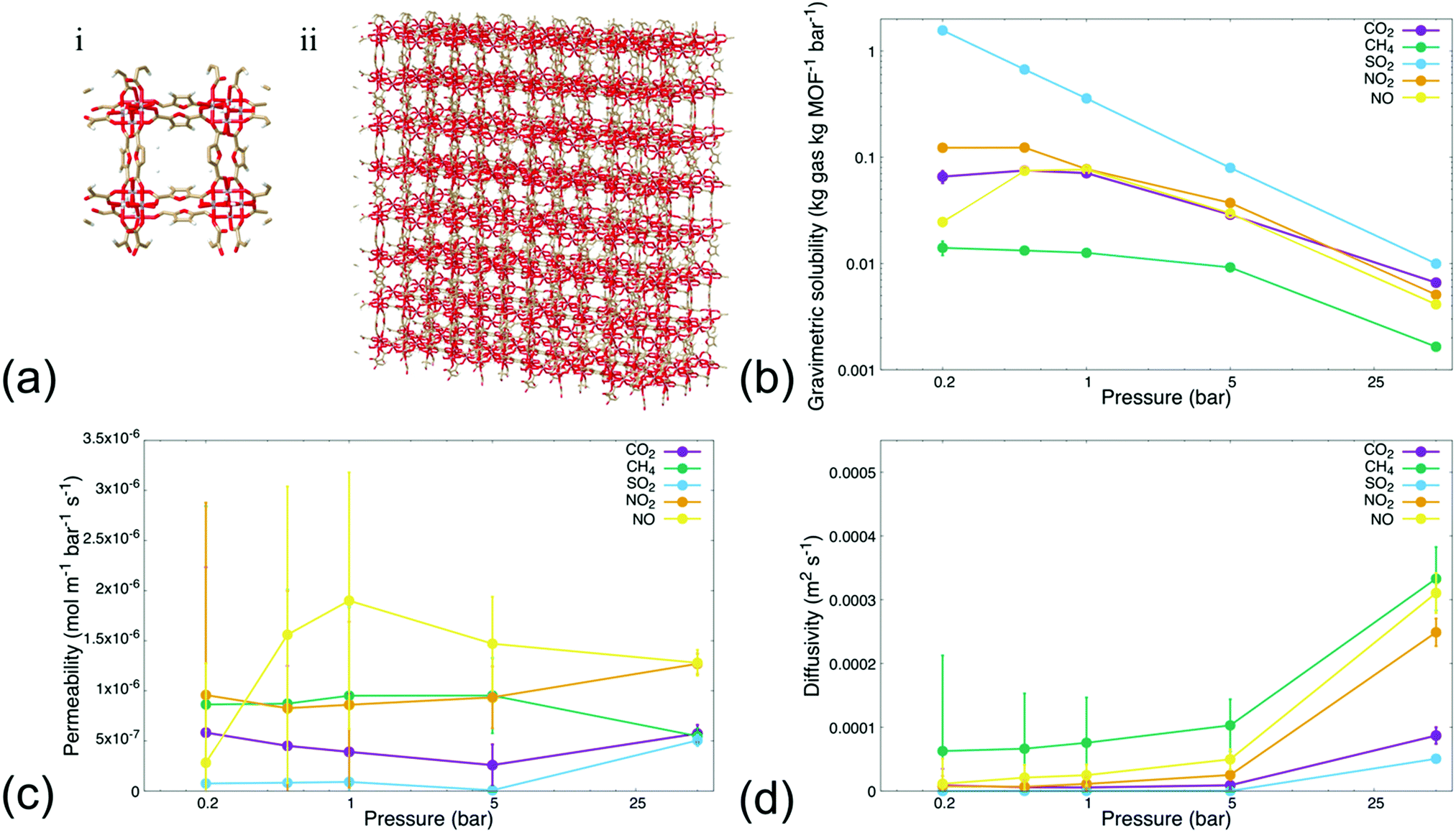 | ||
| Fig. 1 (a) MIL-160 (i) unit cell extended dimensionally to generate (ii) MOF membrane applied in gas separation simulations; (b) gravimetric solubility, (c) permeability, and (d) diffusivity of selected gas molecules sampled at varied pressures relative to a rigid 4 × 4 × 3 MIL-160 membrane. | ||
Analysis also showed that gravimetric solubility and the rate at which individual gas molecules permeated through the defined MOF membrane varied significantly with respect to the simulation pressure as well as across the simulated gas species. This was presumably due to differences in chemical and physical properties among the gas molecules being tested, with the results showing that the MIL-160 membrane possessed a higher loading capacity for oxygenated gas species relative to the non-oxygenated species, at all pressures sampled. CH4 had a lower gravimetric solubility in the MIL-160 membrane than did the oxygen-containing gas species in both high-pressure and low-pressure regimes (Fig. 1(b)). The trend in gravimetric solubility of oxygenated gas species showed a strong dependence on simulation pressure, while the solubility of CH4 varied significantly only at high pressures.
The above results are presumably due to the hydrophilic nature of MIL-160 and the electrostatic interactions of the gases with the membrane. For the first, the hydrophilic nature of the MOF can contribute to favorable interactions with the electronegative regions of the studied gas molecules to provide thermodynamically preferred configurations in which the oxygenated gas molecules can stably adsorb within the membrane structure itself.65 For the second, the electrostatic interactions are expected to play an active role in establishing critical separation criteria for potential processes employing MIL-160-based membranes.40 In particular, we found that the MIL-160 membrane has strong potential to separate to an appreciable degree gas molecules of different electronegativities, owing to stark differences in the preferability with which MIL-160 binds highly electronegative gas molecules like SO2 and CO2 over the less electronegative molecules like CH4 and NO, all in varied pressure regimes.
The general decrease in gravimetric solubility at higher pressures coincided with maximized gas molecule loadings within MIL-160, i.e., an increase in pressure did not seem to increase the number of molecules within the membrane and thus drove the gravimetric solubility downward. Our analysis also showed that both the abundance of the polar surface area of each gas molecule and the electronegativity of the oxygen atoms in the respective molecules impacted their gravimetric solubility. Moreover, the increase in oxygen atom electronegativity coincided with increased gravimetric solubility of the simulated species. Specifically, the increased electronegativity of the oxygen atoms in SO2 relative to that in CO2, NO2, and NO molecules, respectively, was expected to lead to higher molecular packing in the membrane by driving the equilibrium distance between MIL-160 and the adsorbed gas molecules downward such that a greater portion of the native MOF cage structure was preserved during the adsorption process.40 Such preservation is expected to allow for effective gas permeation in high- and low-pressure regimes, while loss of native MOF cage structure due to high gas molecule packing is seen as inhibitory to gas molecule permeation. Similarly, we found that the simulation pressure influenced the permeability of oxygenated gas species through the MIL-160 membrane to varying extents again, dependent on the interactions between gas molecules themselves and the membrane. The permeability of CH4 however did not change considerably with varied simulation pressure most likely due to the absence of strong interactions with the MIL-160 membrane.
Fig. 1(c) illustrates the variations in the permeability of each gas molecule evaluated in our study and across high- and low-pressure regimes respectively. Our analysis showed that the relationship between simulation pressure and gas molecule permeability is subject to complex tradeoffs between MOF–gas molecule interactions and associated thermodynamic barriers pertaining to the desorption process itself. In particular, the pressure at which oxygenated gas species exhibited a significant increase in permeability was influenced by the strength of the interactions between the respective gas molecule and the MIL-160 membrane. For instance, NO showed an increase in permeability starting at 0.5 bar possibly because low pressures were sufficient to partially overcome thermodynamic barriers in the desorption of the molecule from the MIL-160 membrane.41 The subsequent decrease in permeability at pressures of 5 bar and higher was likely due to diminishing increases in the number of molecules permeated. Meanwhile, SO2 had a gravimetric solubility nearly 9 times that of NO at 0.5 bar—calculated viaeqn (2) and shown in Fig. 1(b)—with an uptick in permeability at 50 bar. These results suggest that a significantly higher pressure was necessary to drive molecule permeability upward most likely due to the presence of strong interactions between SO2 and MIL-160. Such observations suggest that potential inhibition of permeation could occur due to clogging of the pore structure as well as departure from ideal gas behaviour, ultimately hindering the feasibility of implementing the MIL-160 membrane for gas separations involving relatively high concentrations of SO2.41,66
The predicted permeability of the simulated gas species was found to vary independently of the kinetic diameter of the simulated species. This finding was expected as the aperture sizes of MIL-160 are approximately 5.2 and 2.4 Å, both falling outside of the range of critical diameters of the sampled gas molecules known to be ranging from 3.17 to 3.80 Å, respectively.42
Diffusivity values were also calculated in both high- and low-pressure regimes and are shown in Fig. 1(d). Each gas molecule displayed a rise in diffusivity as the simulation pressure increased thus concurring with the increase in permeability and the decrease in gravimetric solubility, i.e., higher simulation pressures eventuated higher throughputs of gas molecules diffusing through the membrane.47 Moreover, we found that the classically described potentials,i.e., non-reactive potentials, of each gas molecule impacted trends in diffusivity that dictated individual gas separation capabilities of the MIL-160. Specifically, oxygenated gas species showed lower diffusivities in both high- and low-pressure regimes, further highlighting the inhibitory role of energetically favourable interactions between MIL-160 and more electronegative gas species such as CO2 and SO2. CH4, NO2, and NO are known to have diffusivities nearly twice those of the other gas molecules at 50 bar. CH4 showed substantially higher diffusivity—over five times higher than values calculated for CO2, NO2, and NO and three orders of magnitude higher than that calculated for SO2—even under vacuum conditions, all relative to the oxygen-containing gas molecules. This result indicates that the MIL-160 membrane has the potential to be operable across a wide range of pressures without significant degradation of its performance; stable operability in varied pressure regimes of materials implemented in the selective uptake of target greenhouse gases is regarded as a critical feature in many gas separation processes in which the separation capability of adsorbent materials relies heavily on dynamic variations in the operating pressure, i.e., PSA processes.24,67
The density of the tested gas molecules in the lateral direction of the simulation box as well as visualization of gas permeation processes provided further insights into the effect of individual gas molecule chemistry on permeability and sorption characteristics (Fig. 2). Analysis showed that gas molecule densities within the MIL-160 membrane varied significantly across pressure regimes for less electronegative gas species like CH4, NO2, and NO. This is contrary to the finding of the gas molecule density which remained relatively high at all pressures and for more electronegative gas species such as SO2 and CO2 to a lesser extent. The higher molecular packings of more electronegative gas species SO2 and CO2 are likely the result of relatively strong electrostatic interactions between gas molecules within the MOF membrane cage structures that provide energetically favorable configurations in which nearby molecules can interact.68
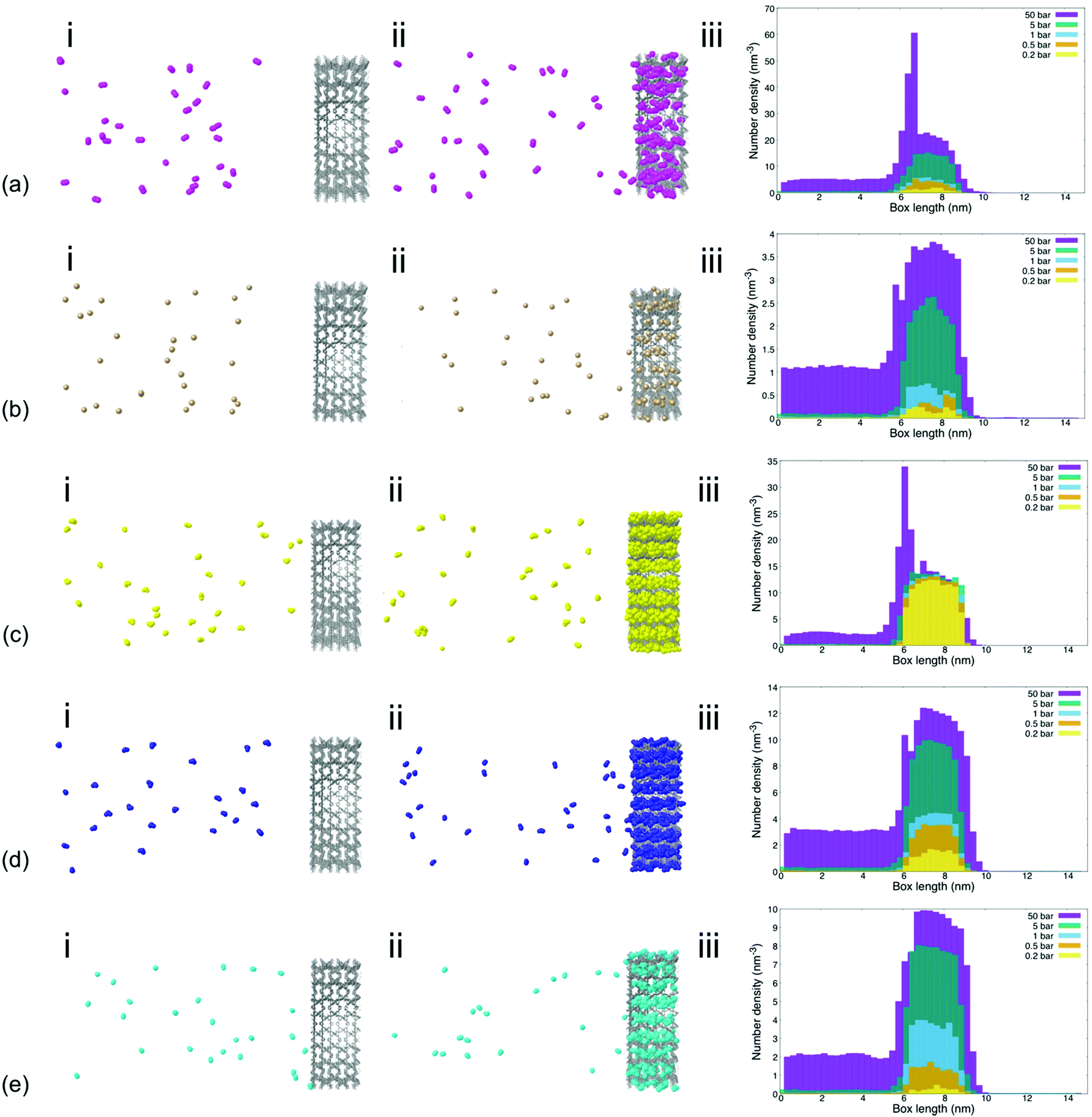 | ||
| Fig. 2 (i) Initial configuration at 1 bar, (ii) steady-state configuration at 1 bar, and (iii) number density of gas species at steady state relative to the lateral direction of the simulation box at varied simulation pressures for (a) CO2, (b) CH4, (c) SO2, (d) NO2, and (e) NO. | ||
We also found that the high binding affinities of electronegative gas molecules to the MIL-160 membrane caused high densities of SO2 within the membrane itself that are inhibitory to gas permeation even under vacuum conditions. Further, an excessively high density of gas molecules that hindered MIL-160 performance was recorded at 50 bar for CO2. The characteristic spikes in gas molecule density—i.e., a sharp increase in gas molecule density and a subsequent sharp decrease indicating a locally high molecule concentration—shown in Fig. 2 at 6 nm corresponding to the entrance of the MIL-160 membrane, were observed for both CO2 and SO2 at 50 bar corresponding to severe concentration polarization effects. This indicates significant clogging that could occur upon such gas interactions with the constructed membrane presumably due to highly favorable electrostatic interactions undergone locally at pressures where the diffusivity is not sufficient to overcome such interactions.64 This finding emphasizes that MIL-160 is not only suited for gas separation processes but further that critical separation criteria herein defined as fundamental differences in gas molecule properties could be simulated to reflect differences in gas molecule binding affinities, all at varied operating pressures. Thus, it is envisioned that the MIL-160 membrane could serve a wide range of gas separation purposes by balancing the operating pressure of the membrane system with tradeoffs in target gas molecule sorption selectivity and based on differences in gas molecule binding affinities. The tradeoffs in operating pressure and gas molecule binding affinity have been exploited in commercially viable gas separation processes such as the ones employing PSA methods.
Membrane permeation and sorption of CO2-containing gas mixtures
We further tested the extended feasibility of the MIL-160 membrane in selectively removing dilute CO2 from other industrially relevant gases. Although many definitions have previously been proposed, we define dilute CO2 as 5 mol% of each binary gas mixture thus comprising a concentration that is within the range of typical compositions studied in dilute gas separations.69 The concentrations studied herein, i.e., conditions in which CO2 is dilute compared to either NO2 or NO, lend our results applicability in the potential employment of MIL-160 in the treatment and containment of off-gas produced from the dissolution of nuclear spent fuel.70 For instance, nuclear fuel is often dissolved in nitric acid, the process leading to a significant amount of NOx contaminants as well as CO2.71 The mitigation of such contaminants reaching the atmosphere is necessary to aid alternative energy industries in attaining higher degrees of sustainability.71 The extended analyses are also meant to support emerging CCS technologies targeting uptake of CO2 at dilute concentrations and to further extend the application profiles to include large point sources of greenhouse gas emissions like power plants5–7 and chemical refineries respectively.6,47,72–74 For the analyses, the MD simulations included permeation and sorption processes of gas mixtures comprised of 5 mol% CO2 with the balance comprised of 95 mol% CH4, 95 mol% SO2, 95 mol% NO2, and 95 mol% NO respectively, with the simulations carried out at 0.2, 0.5, 1, 5, and 50 bar to corroborate single-component gas permeation results presented above. Such gas mixtures were chosen to mimic conditions in which the target molecule, i.e., CO2, corresponds to physical scenarios that are representative of current challenges in gas separation technologies.5,73The sorption selectivity, permselectivity, and diffusivity selectivity of each gas species relative to CO2 are shown in Fig. 3. Analysis of selectivity in the steady-state sorption and permeation regimes and at varied pressures including vacuum conditions and high-pressure conditions confirmed that the varied chemistries of the simulated gas molecules endowed the MOF membrane with different and very specific separation criteria. Specifically, the results showed that adsorption processes were related to the thermodynamic preferability of gas molecule binding capabilities, i.e., essential differences in MIL-160 binding affinity. This finding further supported and emphasized the selective adsorptive gas separation processes made possible through the MOF defined membranes, which is consistent with the work previously reported on MIL-160 membranes and detailing differences in the strength of MOF–gas molecule interactions as the most suitable basis for gas separations performed via MIL-160 membranes.40,42,75
 | ||
| Fig. 3 Dynamic sorption and permeation profiles of gas mixtures comprised of 5 mol% CO2 and (a) 95 mol% CH4, (b) 95 mol% SO2, (c) 95 mol% NO2, and (d) 95 mol% NO with respect to a rigid 4 × 4 × 3 MIL-160 membrane at 50 bar. | ||
Fig. 3(a) shows that the selectivity of gas molecule gravimetric solubility varied significantly across gas species and pressure regimes. The membrane exhibited high solubility selectivity for SO2 over CO2 at all pressures being simulated, congruent with our own analysis that MIL-160 possesses a higher binding affinity for SO2 than it did for CO2. The selectivity of the gravimetric solubility of SO2 remained an order of magnitude greater than the selectivity of all other gases sampled throughout the high- and low-pressure regimes, consistent with previous findings that MIL-160 is potentially applicable in the selective uptake of SO2.75 Meanwhile, the selectivity of the solubility of CH4 relative to CO2 was relatively low at all pressures being evaluated thus suggesting that differences in binding affinity for MIL-160 between CH4 and CO2 could potentially allow for the selective removal of CO2 from CH4. This finding is especially important considering that the separation of CO2 from CH4 has been identified as a particularly crucial step in the upgrade of biogas—organic gas material often derived from agricultural waste, municipal waste, and plant material—to value-added chemicals76 and hints at the potential of MIL-160 as a promising candidate in large-scale separations for biogas upgradation technologies.
The solubility selectivity values of NO2 and NO compared to CO2 were shown to vary across the pressure regimes being studied. Under vacuum and atmospheric conditions for instance, MIL-160 exhibited a slightly higher solubility for NO2 and NO than it did for CO2. However, the selectivity of NO2 and NO was found to decrease sharply at 50 bar presumably due to the increased simulation pressure overcoming thermodynamic barriers to desorption of NO2 and NO to a greater extent than those of CO2, thus resulting in CO2 binding to MIL-160 more stably. These findings are supported by our single-component gas simulations (Fig. 1(b)) in which the solubility of NO2 and NO was found to be less than that of CO2 at high pressures. The decrease in the solubility of both NO2 and NO likely coincides with the increase in permeability at pressures that are high enough to overcome the respective thermodynamic barriers associated with the desorption of NO2 and NO from the MIL-160 membrane.
Fig. 3(b) shows the calculated permselectivity values of each gas species at varied pressures relative to CO2, with analysis supporting the strong pressure and gas composition dependence on the gas separation characteristics of MIL-160. Our analysis also indicated that the permselectivity of respective gas molecules varied across orders of magnitude as the tradeoff between high MOF–gas molecule affinity and low permeability proved paramount in extending the gas separation applicability of the membrane. Less electronegative gas species like CH4, NO2, and NO all demonstrated complex pressure-dependent relationships with respect to calculated permselectivity values. In particular, the permselectivity profiles of each gas were found to correlate with the respective solubility selectivity profile. Significant changes in permselectivity occurred when pressures were sufficient in overcoming barriers associated to the desorption processes. For example, a notable change in permselectivity was calculated at a lower pressure for CH4 presumably due to the low affinity of MIL-160 for CH4 and higher pressures required to initiate desorption of the slightly more electronegative gas species. SO2 showed the lowest degree of permeability relative to less electronegative gas species in mixtures. Additionally, the permselectivity of SO2 relative to CO2 was found to be undefined at pressures 0.5 and 50 bar, i.e., no CO2 permeated through the MIL-160 membrane. Zero-approaching permeability of dilute CO2 provided further evidence that pore clogging caused by SO2 leads to logistical problems in the separability of CO2 from a highly electronegative gas when using a hydrophilic MOF support.
The kinetic diameters of the simulated gases were again predicted to have no measurable effect on the separation capabilities at the membrane interface itself. Moreover, neither the sorption selectivity nor the permselectivity of each gas species was correlated to the kinetic diameters of the greenhouse gas molecules; this was presumably due to the pore geometry of MIL-160, i.e., the aperture sizes of MIL-160 are known to be outside the range of kinetic diameters of the gas molecules sampled in our study. MIL-160 aperture sizes are approximately 5.2 and 2.4 Å, while the kinetic diameter of each gas simulated is well within that range;42 thus, our analysis demonstrated that the aperture size of MIL-160 plays a less significant role in the separation of CO2 from other greenhouse gases than do the varying strengths of MOF–gas molecule electrostatic interactions.
Our analysis of the diffusivity selectivity of each gas species relative to CO2 is shown in Fig. 3(c); it was found that the selectivity trended upward as simulation pressure increased, with the exception of SO2 which had a prohibitively high gravimetric solubility in MIL-160. On the other hand, the MIL-160 membrane showed high selectivity of CH4 relative to CO2 at all pressures tested, owing to the pronounced difference in respective gas molecule binding affinity to MIL-160. The pressure dependence of the diffusivity selectivity values of NO2 and NO was found to lie in a critical range that ultimately could determine the applicability of MIL-160 in particular gas separation applications. Specifically, the diffusivity selectivity of NO2 was found to be less than 1, i.e., in favor of CO2, at low pressures but was calculated to approach 10 at 50 bar; therefore, the operating pressure of the membrane system can potentially be altered to manipulate desired gas separation selectivity of the MIL-160 membrane. Such pressure-based functionality forms the basis of PSA processes in which MIL-160 could be implemented where the separation of CO2 from NOx is of critical importance, like flue gas separations and treatment of off-gas produced from used nuclear fuel dissolution.70,71,77
Competitive adsorption processes within the MIL-160 membrane
The dynamic sorption and permeation profiles of the simulated gas mixtures at the pressure of 50 bar are shown in Fig. 4, while the similar profiles at pressures 0.2, 0.5, 1, and 5 bar being shown in Fig. S.6–S.9 in the ESI.† Analysis indicated that the separability of dilute CO2 from other greenhouse gases at the molar compositions sampled in our study hinges on the ability of CO2 to displace already adsorbed gas species. The higher concentrations of CH4, SO2, NO2, and NO molecules in respective simulations eventuated the rapid flooding of the MIL-160 membrane with such molecules within approximately 0.5 ns of simulation followed by a slower dynamic response—about 5 ns—in which the number of CO2 molecules within the membrane increased until steady-state concentrations were attained. | ||
| Fig. 4 Dynamic sorption and permeation profiles of gas mixtures comprised of 5 mol% CO2 and (a) 95 mol% CH4, (b) 95 mol% SO2, (c) 95 mol% NO2, and (d) 95 mol% NO with respect to a rigid 4 × 4 × 3 MIL-160 membrane at 50 bar. | ||
At steady-state concentrations, the local composition of each binary mixture reflected the affinity with which MIL-160 was able to bind the respective gas molecules. While CO2 comprised only 5 mol% of the gas baths of each composition, we found that steady-state at 50 bar CO2 accounted for approximately 20 mol% of the molar composition of gas within the MIL-160 membrane in CH4/CO2 and NO/CO2 gas mixture simulations and about 16 mol% of the molar composition of gas within the MIL-160 membrane in the NO2/CO2 gas mixture simulation. Under the same conditions, CO2 accounted for only about 0.5 mol% of the molar composition of gas within the MIL-160 membrane in the SO2/CO2 gas mixture simulation, thus pointing to the clear thermodynamic preferability of MIL-160 for SO2 over CO2.
Our results are in good agreement with values calculated from both gas simulation and experimental gas sorption studies previously reported (Table 1), with the specific analysis reproducing adsorption capacities for CH4 and SO2 under similar temperature and pressure conditions when compared to those published in recent gas separation papers.40,75 The adsorption capacity of CO2 within the MIL-160 membrane at specified temperatures and pressures, however, was found to deviate from previously reported values; our analysis showed that MIL-160 possesses a lower adsorption capacity for CO2 compared to that reported in other gas simulation studies.40,75 This apparent discrepancy could be due to differences between physical scenarios defined in this work. Specifically, in our analysis, gas baths were maintained at a constant pressure without regard for controlling pressure within the MIL-160 membrane, while other authors have employed grand canonical Monte Carlo (GCMC) methods with simulation pressure controlled within the membrane.40 To our knowledge, no data exist with which to compare our adsorption capacity values of NO2 and NO, but comparison to similar analyses for different MOF materials confirms that MIL-160 has potential to perform at least as well as such MOFs.79–81
| Gas species | Calculated gas uptake (kg gas kg−1 MIL-160) @ temperature, pressure | Previously reported gas uptake (kg gas kg−1 MOF) @ temperature, pressure | MOF | Ref. |
|---|---|---|---|---|
| a Indicates values derived from empirical data. | ||||
| CO2 | 7.09 × 10−2 @ 300 K, 1 bar | 1.85 × 10−1 @ 298 K, 1 bar | MIL-160 | 75 |
| 1.67 × 10−1 @ 303 K, 1 bar | MIL-160 | 40 | ||
| 1.93 × 10−1 @ 293 K, 1 bara | MIL-160 | 78 | ||
| 3.76 × 10−2 @ 300 K, 0.5 bar | 1.41 × 10−1 @ 293 K, 0.5 bara | MIL-160 | 78 | |
| 1.31 × 10−2 @ 300 K, 0.2 bar | 7.70 × 10−2 @ 293 K, 0.2 bara | MIL-160 | 78 | |
| CH4 | 1.26 × 10−2 @ 300 K, 1 bar | 1.28 × 10−2 @ 303 K, 1 bar | MIL-160 | 40 |
| SO2 | 3.58 × 10−1 @ 300 K, 1 bar | 4.03 × 10−1 @ 298 K, 1 bar | MIL-160 | 75 |
| 4.61 × 10−1 @ 293 K, 0.97 bar | MIL-160 | 41 | ||
| 4.48 × 10−1 @ 293 K, 0.97 bara | MIL-160 | 41 | ||
| 3.35 × 10−1 @ 300 K, 0.5 bar | 4.03 × 10−1 @ 293 K, 0.5 bara | MIL-160 | 41 | |
| 3.12 × 10−1 @ 300 K, 0.2 bar | 3.78 × 10−1 @ 293 K, 0.2 bara | MIL-160 | 41 | |
| NO2 | 7.75 × 10−2 @ 300 K, 1 bar | 6.49 × 10−1 @ 298 K, 1 bar | MFM-300(Al) | 79 |
| 7.31 × 10−2 @ 298 K | UiO-66 | 80 | ||
| NO | 7.67 × 10−2 @ 300 K, 1 bar | 9.30 × 10−3 @ 298 K | Rh/MOF-177 | 81 |
Moreover, our unique approach has the advantage of allowing for complex analysis of multi-component gases which are not easily attainable through studies involving potential of mean force (PMF) analysis. Specifically, when comparing our membrane system to the membrane systems studied by Khakpay et al., we make two important distinctions.82 First, the inherent cage-like geometry of the MIL-160 membrane poses geometrical limitations on the umbrella sampling routine that would be invoked to determine the binding affinity of a particular gas molecule. It is likely that the interior cage structure of this MOF is not sufficiently large to allow for proper sampling of PMF as a gas molecule is moved away from a particular binding site. Secondly, the complexity of the chemical structure of MIL-160 compared to those of nanoporous graphene (NPG) and nanoporous graphene oxide (NPGO) sheets inundates the number of potential binding sites that could be considered of interest when undertaking a PMF analysis, with the subsequent computational load being multiplied when extended to five gas species relevant to the current study.
Our study however complements current work focusing on gas separation performance of emerging MOF materials while being insightful with respect to the expanding range of gas separations in which MIL-160 could be advantageously implemented. The identification of atomic-level phenomena that are determinant in the efficiency of a MIL-160-based membrane is further adding to the body of work previously published and highlighting hybrid supports such as polydopamine-modified aluminum oxide discs to achieve efficient separations of xylene isomers for instance.46 As such, we envision that MIL-160-based membranes prepared on a wide range of polymeric or ceramic supports can be produced to meet a number of industrially relevant gas separations. Additionally, we contend that MIL-160 materials have the potential to be particularly beneficial in the separation of target gases based on reversible, pressure- and temperature-dependent differences in gas molecule binding affinities. Similar strategies have already been employed for the efficient, low-energy uptake and full regeneration of water vapor and relatively at mild temperatures.43,44
Conclusions
Our results showed that MIL-160 has extended potential for processes designed to separate gas species with strongly differing electronegativities and different binding affinities of such gases to MIL-160 membrane itself. Specifically, we found that MIL-160 is capable of selectively binding dilute CO2 in gas mixtures comprised of less electronegative gas species—i.e., CH4, NO2, and NO, respectively—at varied pressures, which strongly altered the separation capabilities of the membrane itself as well as influenced membrane's applicability for specific uses in industrial settings like biogas upgradation and treatment of off-gas from used nuclear fuel dissolution. Further, our strategy was instrumental in exemplifying the competitive adsorption processes between greenhouse molecules within the MIL-160 membrane that were principal to the gas separation capability of the membrane itself. Our work demonstrates the significance of MD simulations in the identification of critical separation criteria, i.e., selective adsorption via MOF–gas molecule interactions that lay the foundation for scalable, sustainable gas separation processes that will meet a growing global energy demand while reducing the unwanted societal effects of anthropogenic greenhouse gas emission.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. and C. Z. D acknowledge the use of Thorny Flat super computing system at West Virginia University (WVU), which is funded in part by the National Science Foundation (NSF) Major Research Instrumentation Program (MRI) Award #1726534 and WVU.J. C. acknowledges the use of the UCSF Chimera package to generate visualizations of molecular simulations. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
V.-A. G. acknowledges support by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, project 75428 (Understanding and Control of Reactive Separations). C. Z. D acknowledges support by the National Science Foundation, 1454230.
References
- J. Kittel, E. Fleury, B. Vuillemin, S. Gonzalez, F. Ropital and R. Oltra, Mater. Corrosion, 2012, 63, 223–230 CrossRef CAS.
- S. Zhang, M. K. Taylor, L. Jiang, H. Ren and G. Zhu, Gas Separation, 2019, 25, 1–18 Search PubMed.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- W. Mazyan, A. Ahmadi, H. Ahmed and M. Hoorfar, J. Nat. Gas Sci. Eng., 2016, 30, 487–515 CrossRef CAS.
- J. K. Adewole, A. L. Ahmad, S. Ismail and C. P. Leo, Int. J. Greenhouse Gas Control, 2013, 17, 46–65 CrossRef CAS.
- C. Cao, H. Liu, Z. Hou, F. Mehmood, J. Liao and W. Feng, Energies, 2020, 13, 600–644 CrossRef CAS.
- F. Chang, J. Zhou, P. Chen, Y. Chen, H. Jia, S. M. I. Saad, Y. Gao, X. Cao and T. Zheng, Asia-Pac. J. Chem. Eng., 2013, 8, 618–626 CrossRef CAS.
- C.-F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Climate Chage, 2016, 6, 827–835 CrossRef.
- C. A. Scholes, G. W. Stevens and S. E. Kentish, Fuel, 2012, 96, 15–28 CrossRef CAS.
- B. D. Bhide, A. Voskericyan and S. A. Stern, J. Membr. Sci., 1998, 140, 27–49 CrossRef CAS.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427–2433 CrossRef CAS.
- J. Xu, Z. Wang, Z. Qiao, H. Wu, S. Dong, S. Zhao and J. Wang, J. Membr. Sci., 2019, 581, 195–213 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- D. J. Heldebrant, P. K. Koech, R. Rousseau, V.-A. Glezakou, D. Cantu, D. Malhotra, F. Zheng, G. Whyatt, C. J. Freeman and M. D. Bearden, Energy Procedia, 2017, 114, 756–763 CrossRef CAS.
- B. P. McGrail, H. T. Schaef, V.-A. Glezakou, L. X. Dang and A. T. Owen, Energy Procedia, 2009, 1, 3415–3419 CrossRef CAS.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- M. Gatti, E. Martelli, D. D. Bona, M. Gabba, R. Scaccabarozzi, M. Spinelli, F. Vigano and S. Consonni, Energies, 2020, 13, 543–574 CrossRef CAS.
- A. Streb, M. Hefti, M. Gazzani and M. Mazotti, Ind. Eng. Chem. Res., 2019, 58, 17489–17506 CrossRef CAS.
- H. T. Schaef, C. L. Davidson, A. T. Owen, Q. R. S. Miller, J. S. Loring, C. J. Thompson, D. H. Bacon, V. A. Glezakou and B. P. McGrail, Energy Procedia, 2014, 63, 7844–7851 CrossRef CAS.
- S. Sircar, Ind. Eng. Chem. Res., 2002, 41, 1389–1392 CrossRef CAS.
- C. Voss, Adsorption, 2014, 20, 295–299 CrossRef CAS.
- H. Zeng, X.-J. Xie, M. Xie, Y.-L. Huang, D. Luo, T. Wang, Y. Zhao, W. Lu and D. Li, J. Am. Chem. Soc., 2019, 141, 20390–20396 CrossRef CAS PubMed.
- D. E. Jaramillo, D. A. Reed, H. Z. H. Jiang, J. Oktawiec, M. W. Mara, A. C. Forse, D. J. Lussier, R. A. Murphy, M. Cunningham, V. Colombo, D. K. Shuh, J. A. Reimer and J. R. Long, Nat. Mater., 2020, 517–521 CrossRef CAS PubMed.
- H. Khajuria and E. N. Pistikopoulos, AIChE J., 2012, 59, 120–131 CrossRef.
- J. Zhang, I. Kutnyakov, P. K. Koech, A. Zwoster, C. Howard, F. Zheng, C. J. Freeman and D. J. Heldebrant, Energy Procedia, 2013, 37, 285–291 CrossRef CAS.
- S. Cremaschi, Comput. Chem. Eng., 2015, 81, 130–137 CrossRef CAS.
- R. Abedini and A. Nezhadmoghadam, Petroleum Coal, 2010, 52, 69–80 CAS.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- Z. Kang, L. Fan and D. Sun, J. Mater. Chem. A, 2017, 5, 10051–10724 RSC.
- S. Dong, Z. Wang, M. Sheng, Z. Qiao and J. Wang, Sep. Purif. Technol., 2020, 239 DOI:10.1016/j.seppur.2020.116580.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3435–3480 RSC.
- H. Xiang, J. H. Carter, C. C. Tang, C. A. Murray, S. Yang, X. Fan and F. R. Siperstein, Chem. Eng. Sci., 2020, 218 DOI:10.1016/j.ces.2020.115566.
- A. Kertik, L. H. Wee, K. Sentosun, J. A. R. Navarro, S. Bals, J. A. Martens and I. F. J. Vankelecom, Appl. Mater. Interfaces, 2020, 12, 2952–2961 CrossRef CAS PubMed.
- P. Guo, D. Dutta, A. G. Wong-Foy, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 2651–2657 CrossRef CAS PubMed.
- L. Bellarosa, J. M. Castillo, T. Vlugt, S. Calero and N. Lopez, Chem. – Eur. J., 2012, 18, 12260–12266 CrossRef CAS PubMed.
- N. Hanikel, M. S. Prevot and O. M. Yaghi, Nat. Nanotechnol., 2020, 15, 348–355 CrossRef CAS PubMed.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30 DOI:10.1002/adma.201704304.
- M. E. A. Safy, M. Amin, R. R. Haikal, B. Elshazly, J. Wang, Y. Wang, C. Woll and M. H. Alkordi, Chem. – Eur. J., 2020, 7109–7117 CrossRef CAS PubMed.
- D. Lenzen, P. Bendix, H. Reincsh, D. Frohlich, H. Kummer, M. Mollers, P. P. C. Hugenell, R. Glaser, S. Henninger and N. Stock, Adv. Mater., 2017, 30 DOI:10.1002/adma.201705869.
- D. D. Borges, P. Normand, A. Permiakova, R. Babarao, N. Heymans, D. S. Galvao, C. Serre, G. D. Weireld and G. Maurin, J. Phys. Chem. C, 2017, 121, 26822–26832 CrossRef.
- P. Brandt, A. Nuhnen, M. Lange, J. Mollmer, O. Weingart and C. Janiak, Appl. Mater. Interfaces, 2019, 11, 17350–17358 CrossRef CAS PubMed.
- M. Wahiduzzaman, D. Lenzen, G. Maurin, N. Stock and M. T. Wharmby, Eur. J. Inorg. Chem., 2018, 3626–3632 CrossRef CAS.
- A. Permyakova, O. Skrylynk, E. Courbon, M. Affram, S. Wang, U.-H. Lee, A. H. Valekar, F. Nouar, G. Mouchaham, T. Devic, G. D. Weireld, J.-S. Chang, N. Steunou, M. Frere and C. Serre, ChemSusChem, 2017, 10, 1419–1426 CrossRef CAS PubMed.
- M. Solovyeva, I. Krivosheeva, L. Gordeeva and Y. Aristov, Energies, 2021, 14 DOI:10.3390/en14123586.
- Q. Liu, X. Bai, H. Pham, J. Hu and C. Z. Dinu, Nanomaterials, 2021, 11 DOI:10.3390/nano11041008.
- X. Wu, W. Wei, J. Jiang, J. Caro and A. Huang, Angewandte, 2018, 57, 15354–15358 CrossRef CAS PubMed.
- Q. Qian, P. A. Asinger, M. Joo Lee, G. Han, K. M. Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- Q. Liu, J. Chapman, A. Huang, K. C. Williams, A. Wagner, N. Garapati, K. A. Sierros and C. Z. Dinu, Appl. Mater. Interfaces, 2018, 10, 41326–41337 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 19–25 CrossRef.
- A. Cadiau, J. S. Lee, D. D. Borges, P. Fabry, T. Devic, M. T. Wharmby, C. Martineau, D. Foucher, F. Taulelle, C.-H. Jun, Y. K. Hwang, N. Stock, M. F. D. Lange, F. Kapteijn, J. Gascon, G. Maurin, J.-S. Chang and C. Serre, Adv. Mater., 2015, 27, 4775–4780 CrossRef CAS PubMed.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- W. Shi and E. J. Maginn, J. Comput. Chem., 2008, 29, 2520–2530 CrossRef CAS PubMed.
- Z. Zhou, B. D. Todd, K. P. Travis and R. J. Sadus, J. Chem. Phys., 2005, 123, 054505 CrossRef PubMed.
- E. Bourasseau, V. Lachet, N. Desbiens, J.-B. Maillet, J.-M. Teuler and P. Ungerer, J. Phys. Chem. B, 2008, 112, 15783–15792 CrossRef CAS PubMed.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. G. Petersen, J. Chem. Phys., 1995, 103, 3668–3679 CrossRef CAS.
- E. Barth, K. Kyczera, B. Leimkuhler and R. D. Skeel, J. Comput. Chem., 1995, 16, 1192–1209 CrossRef CAS.
- C. M. Oldenburg, presented in part at the TOUGH Symposium 2003, 2003.
- J. Liu and J. Jiang, J. Phys. Chem. C, 2019, 123, 6607–6615 CrossRef CAS.
- S. Elhenawy, M. Khraisheh, F. AlMomani and M. Hassan, Molecules, 2020, 25 DOI:10.3390/molecules25184274.
- Y. Yang, G. Wang, S. Ge, H. Yang, M. Liu and M. Liu, Hydrogen Energy, 2020, 6329–6340 Search PubMed.
- R. M. Venable, A. Kramer and R. W. Pastor, Chem. Rev., 2019, 119, 5954–5997 CrossRef CAS PubMed.
- P. A. Monson, Microporous Mesoporous Mater., 2012, 160, 47–66 CrossRef CAS.
- B. Liu and B. Smit, J. Phys. Chem. C, 2010, 114, 8515–8522 CrossRef CAS.
- J.-Y. Zhang, J.-B. Zhang, M. Li, Z. Wu, S. Dai and K. Huang, Chem. Eng. J., 2020, 391 DOI:10.1016/j.cej.2019.123579.
- S. U. Nandanwar, D. R. Corbin and M. B. Shiflett, Ind. Eng. Chem. Res., 2020, 59, 13355–13369 CrossRef CAS.
- R. Krishna, Phys. Chem. Chem. Phys., 2015, 17, 39–59 RSC.
- S. Choi, J. H. Drese and C. W. Jones, Chem. Sustainable Energy Mater., 2009, 2, 796–854 CAS.
- Z.-M. Wang, T. Arai and M. Kumagai, Energy Fuels, 1998, 12, 1055–1060 CrossRef CAS.
- S. U. Nandanwar, K. Coldsnow, V. Utgikar, P. Sabharwall and D. E. Aston, Chem. Eng. J., 2016, 306, 369–381 CrossRef CAS.
- C. W. Jones, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 31–52 CrossRef CAS PubMed.
- W. Gao, S. Liang, R. Wang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584 RSC.
- A. Ozcan, R. Semino, G. Maurin and A. O. Yazaydin, Chem. Mater., 2020, 32, 1288–1296 CrossRef CAS PubMed.
- J.-X. Liu, J. Li, W.-Q. Tao and Z. Li, Fluid Phase Equilibria, 2021 DOI:10.1016/j.fluid.2021.112963.
- W. Zhu, F. Liu, M. Gou, R. Guo and X. Li, Green Chem. Eng., 2020, 132–143 Search PubMed.
- D. Wu, Q. Yang, C. Zhong, D. Liu, H. Huang, W. Zheng and G. Maurin, Langmuir, 2012, 18, 12094–12099 CrossRef PubMed.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schroder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- A. M. Ebrahim, B. Levasseur and T. J. Bandosz, Langmuir, 2013, 29, 168–174 CrossRef CAS PubMed.
- F. Tang, Y. Zhai, S. Li, L. Li, X. Wei, J. Hu, L. Ruan and Z. Zhang, Authorea, 2020 DOI:10.22541/au.160418532.27740129/v1.
- A. Khakpay, F. Rahmani, S. Nouranian and P. Scovazzo, J. Phys. Chem. C, 2017, 121, 12308–12320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00468a |
| This journal is © The Royal Society of Chemistry 2021 |