
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monodispersed sodium hyaluronate microcapsules for transdermal drug delivery systems†
Hirotada
Hirama‡
 *a,
Yuya
Ishikura‡
b,
Shinya
Kano
a,
Masanori
Hayase
b and
Harutaka
Mekaru
a
*a,
Yuya
Ishikura‡
b,
Shinya
Kano
a,
Masanori
Hayase
b and
Harutaka
Mekaru
a
aHuman Augmentation Research Center, National Institute of Advanced Industrial Science and Technology, Chiba 277-0882, Japan. E-mail: h.hirama@aist.go.jp
bFaculty of Science and Technology, Tokyo University of Science, Chiba 278-8510, Japan
First published on 13th September 2021
Abstract
This study presents an alternative to cross-linking for the preparation of monodispersed polymer microcapsules made from water-soluble and biocompatible sodium hyaluronate (SH). Water was removed from monodispersed drug-containing SH droplets by creating an osmotic pressure difference, causing the droplets to thicken and solidify into microcapsules. The fabricated microcapsules were water-soluble and could be stored in oil for at least one month without dissolution, thereby retaining the contained drug. In addition, we used SH microcapsules to prepare biodegradable microneedles (MNs), one of the materials for transdermal drug delivery systems (TDDSs), by embedding the microcapsules in a poly(lactic-co-glycolic acid) copolymer, which was molded into a needle shape. The MNs embedded with drug-containing microcapsules released the drug over time based on the amount of microcapsules contained and were capable of penetrating the stratum corneum of the skin. These findings show the application potential of these monodispersed SH microcapsules for TDDSs, while avoiding the common issues associated with cross-linking, such as toxicity and bursting. This novel material exhibits potential for use in pharmaceutics and cosmetics.
1. Introduction
Hyaluronic acid (HA) is a natural linear polysaccharide comprising alternating D-glucuronic acid and N-acetyl-D-glucosamine disaccharide units connected via (1 → 4) glycosidic bonds.1 HA is abundant in the synovial fluid and extracellular matrix and can retain large amounts of water owing to its high negative charge. Therefore, HA plays a major role in controlling tissue hydration.2 In the field of cosmetics, HA is an excellent moisturizer used in skin care and serves as a material for skin wrinkle suppression, skin condition improvement, and facial rejuvenation.3–7 The biocompatibility, biodegradability, and water-retention ability of HA and its sodium salt, sodium hyaluronate (SH), have enabled its use in various forms, such as solutions,8–10 hydrogels,11–13 and microcapsules,14,15 in the medical field for arthritis treatment,8,16 ophthalmic treatment,9,10 and drug delivery.11,17–19 HA can bind to receptors in the liver, kidney, lymphatic vessels, and tumor tissues, and is therefore used as a carrier in drug delivery systems (DDSs) [including transdermal drug delivery systems (TDDSs), which are used for administering drugs into the skin20] to deliver genes and anticancer drugs to target cells.13,21–23 HA and SH have been used for the continuous administration of peptide and protein drugs.14,24,25 Microcapsules based on HA and SH, which protect the inclusion drug from the external environment and prevent whole-dose failure, have been developed, although certain microcapsules exhibit malfunction.26,27HA and SH for DDSs can be prepared by converting water-soluble HA and SH to water-insoluble forms via chemical or physical cross-linking.14,24,25,28 Chemical cross-linking produces cross-linked HA and SH with excellent mechanical strength and degradability control properties, but the toxicity of the cross-linking agent must be considered.11,18 Physical cross-linking (such as ionic cross-linking) does not require toxic cross-linking agents, but bursting is associated with the resulting fragile cross-linked HA and SH, which can lead to drug overdose. Therefore, new methods for the fabrication of HA- and SH-based DDS materials are required to avoid these issues.
Microcapsules can be mass-produced via monodispersion [coefficient of variation (CV) = <10%] using microchannels and are widely used as drugs, compounds, and cell carriers in the form of droplets and polymer microparticles.15,29–34 A shrinkage and gelation technique, where osmotic pressure difference and diffusion are used to generate microparticles from droplets, has been previously reported.33,35–37 Previous applications of this technique generated monodispersed sodium alginate droplets in microchannels, which were transferred to a gel plate with a high concentration of calcium chloride, which served as a cross-linking agent for sodium alginate. The droplets were dehydrated due to the osmotic pressure difference between the droplets and gel plate and simultaneously solidified via the diffusion of calcium chloride from the gel plate to the droplets. The microcapsules produced using this technique were monodispersed; their size was controlled by the flow rate of the dispersed and continuous phases introduced into the microchannel as well as the osmotic pressure difference between the droplets and the gel.
Biodegradable microneedles (MNs) are an alternative to oral and subcutaneous injections and are minimally invasive DDSs that deliver drugs through the skin.38–40 MNs, biodegradable polymers that are loaded with drugs and molded into needle shapes, penetrate the stratum corneum to deliver drugs (including polymeric and water-soluble ones) directly into the skin. Various materials (such as poly(lactic-co-glycolic acid) (PLGA),41–43 polyglycolic acid,44 and polylactic acid45) are used for MNs.
The aim of the present study was to prepare water-soluble monodispersed SH microcapsules for TDDSs using the shrinkage and gelation technique.35 Monodispersed microcapsules typically offer well-controlled release properties,46 and the solidified microcapsules are capable of maintaining monodispersity without coalescence. Drug-containing monodispersed SH droplets were generated using a microchannel, where the generated droplets were concentrated via dehydration due to the osmotic pressure difference and thickening (solidified) due to the concentration. The resulting non-crosslinked SH microcapsules were characterized based on their size, water solubility, and storage stability. In addition, we fabricated MNs with embedded microcapsules to impart sustained-release properties using PLGA, which is an established material for sustained-release preparation.41,47,48
2. Experimental
2.1 Microcapsule preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was poured into the convex mold, cured in an oven at 85 °C for 2 h, and released from the mold. Inlet and outlet holes (diameter = 1.0 mm) were punched into the PDMS slab. The PDMS slab was irradiated with oxygen plasma and permanently bonded to a glass plate. The microchannels were treated with Sigmacote (Sigma, USA) to render them hydrophobic.
:1) was poured into the convex mold, cured in an oven at 85 °C for 2 h, and released from the mold. Inlet and outlet holes (diameter = 1.0 mm) were punched into the PDMS slab. The PDMS slab was irradiated with oxygen plasma and permanently bonded to a glass plate. The microchannels were treated with Sigmacote (Sigma, USA) to render them hydrophobic.
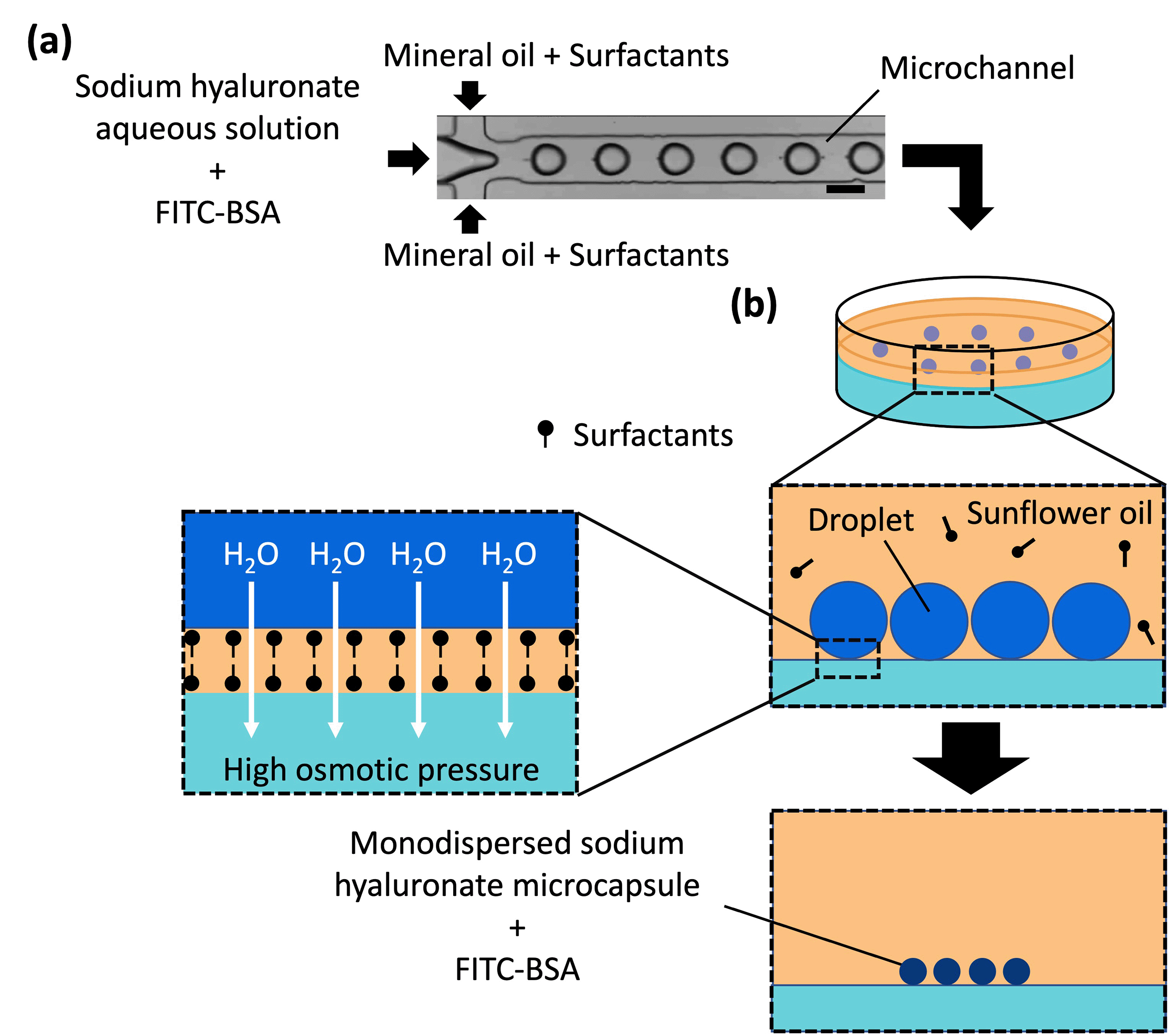 | ||
| Fig. 1 Schematic of the microcapsule fabrication procedure. (a) Droplet formation in a microchannel. The scale bar represents 100 μm. (b) Microcapsule formation from the droplets. The osmotic pressure difference between the droplet and gel plate caused migration of water from the droplet to the gel plate, thereby concentrating sodium hyaluronate to thicken and solidify the droplets. | ||
aq.; Fujifilm Wako Pure Chemical Industries, Ltd, Japan). Equal parts of dissolved agarose gel and CaCl2aq. were mixed, poured into a polystyrene dish, and cooled to room temperature (approximately 20 °C) for solidification. CaCl2 is an osmolyte and acts as a gelling agent for sodium alginate but not for SH. The resulting gel plates comprised 1 wt% agarose gel and 0.1, 0.2, 0.3, 0.4, 0.6, 0.8, 1.0, or 1.2 mol L−1 CaCl2aq. Sunflower oil (Fuji Film Wako Pure Chemical Industries, Ltd, Japan) and 1 wt% SY-Glyster CRS-75 surfactant were poured onto the gel plate, and the droplets generated using the microchannel were collected on the gel plate and incubated at room temperature for concentration and solidification. An inverted microscope (BZ-X710, KEYENCE Co., Ltd, Japan) was used to acquire the transmitted light images and fluorescence images of the droplets and the microcapsules.
2.2 Microcapsule characterization
2.3 Microneedle preparation
The suspension layer was transferred to another microtube to remove the aqueous phase, and the volume of the suspension layer was adjusted to 1 mL by adding sunflower oil with 1 wt% SY-Glyster CRS-75 surfactant. The microcapsules were counted using a disposable cell counting plate (Fukae Kasei Co., Ltd, Japan), and the desired number of microcapsules (1.2 × 106, 2.4 × 106, or 3.6 × 106) was transferred to another microtube. The oil phase was completely replaced with silicone oil by settling the microcapsules in the oil phase via centrifugation at 750 rcf for 1.5 min. The oil phase was removed, and silicone oil was added. Centrifugation was repeated to separate the microcapsules from the silicone oil. The replacement process was repeated twice to obtain a microcapsule suspension. The silicone oil used in this study (which is used as a cosmetic additive) had a lower boiling point than the heating temperature for dissolving PLGA. Because silicone oil can volatilize during the molding of MNs (in contrast, sunflower oil does not volatilize and phase separates from PLGA), we used it as a replacement for sunflower oil. Additionally, the viscosity of silicone oil was lower than the appropriate oil phase viscosity for droplet shrinkage and solidification (according to our previous study33). Therefore, sunflower oil was replaced with silicone oil after (but not before) the droplet shrinkage and solidification.
:1) was poured onto a convex metal mold (3 × 3 cm, stainless steel, base diameter = 200 μm, height = 700 μm, distance between needle centers = 600 μm, and number of needles = 256 (16 × 16); Yoneyama Mold. Co., Ltd, Japan) (Fig. 2). The mold was cured in an oven at 85 °C for 2 h and then peeled from the metal mold to obtain a concave PDMS-based mold. The microcapsule suspension was mixed with 0.2 g PLGA (Sigma Aldrich, USA) melted on a hot plate set at 250 °C. The PLGA and microcapsule mixture was applied to a concave PDMS-based mold. A polyvinyl chloride (PVC) substrate (3 cm × 3 cm × 1 mm) was pressed onto the mixture, cooled to room temperature to solidify, and peeled from the concave PDMS-based mold to form MNs.
 | ||
| Fig. 2 Microneedle (MN) fabrication procedure. (1) The polydimethylsiloxane (PDMS) mixture was poured onto a convex metal mold and solidified via oven curing. (2) PDMS was peeled from the metal mold to obtain a concave PDMS-based mold. (3) poly(lactic-co-glycolic acid) (PLGA) was melted on a hot plate. (4) Microcapsules containing the drug were added. (5) PLGA with the microcapsules was poured onto the PDMS-based mold. (6) The mold was filled with PLGA by pressing a polyvinyl chloride (PC) substrate over the applied PLGA. (7) MNs were fixed on the PC substrate after cooling at room temperature (approximately 20 °C) and peeling from the mold. | ||
2.4 Microneedle characterization
3. Results and discussion
3.1 Microcapsule formation from droplets
Droplets were generated using microchannels and served as templates for microcapsules. A preliminary study (Table S1, ESI†) indicated that a dispersed phase comprising 0.01 wt% SH aqueous solution and 0.5 mg mL−1 FITC–BSA was suitable. The flow of the dispersed aqueous phase and continuous oil phase (mineral oil with 1 wt% surfactant) into the microchannels at a constant flow rate facilitated the continuous formation of monodispersed droplets (Table 1).| Flow rate of the dispersed phase [mL h−1] | Flow rate of the continuous phase [mL h−1] | Droplet diameter [μm] | CV [%] |
|---|---|---|---|
| 1.7 | 2.0 | 102 ± 2 | 2.4 |
| 1.0 | 2.5 | 88 ± 1 | 1.6 |
| 1.7 | 5.0 | 76 ± 1 | 1.3 |
| 1.0 | 7.5 | 53 ± 1 | 1.9 |
Microcapsules were produced from the droplets by removing water to concentrate and solidify the droplets. Because no crosslinking agent was used in this method, we inferred that dehydration caused the polymer in the droplets to thicken and eventually solidify. (Note: CaCl2 did not act as a cross-linker for SH, as shown in Fig. S3, ESI†) As mentioned in our previous report,35 water was removed from the droplets by placing the droplets on a gel plate containing CaCl2 aq with high osmotic pressure. The diameters of the droplets on the gel plate were measured over time (Fig. 3 and Fig. S4, ESI†); the droplet diameter decreased and eventually became constant and monodispersed. The concentration of calcium chloride was proportional to the osmotic pressure: a higher concentration in the gel plate led to faster shrinkage and a smaller droplet diameter. The droplet diameter decreased to 14–20% of its original diameter, thereby increasing the concentration of the contents by a factor of 125–358. In addition, the shrinkage of the droplets did not lead to breakage due to mechanical stimulation during handling with a pipette and centrifugation. This can be attributed to the difference in osmotic pressure due to the generation of droplets via dehydration instead of cross-linking.
 | ||
| Fig. 3 Microcapsule formation using the original droplet generated at a flow rate of 1.0 mL h−1 in the dispersed phase and 2.5 mL h−1 in the continuous phase. (a) Shrunken droplets, which contain FITC-BSA and sodium hyaluronate. Microscopic images of the droplets on a polystyrene dish were captured. 0.1, 0.2, and 0.3 mol L−1 indicate the concentrations of CaCl2 contained in the gel plate. Scale bars measure 50 μm. Scale bars in the insets measure 20 μm. (b) Droplet shrinkage on a gel plate containing CaCl2aq. over time (N = 50). | ||
Droplet shrinkage was characterized based on the relationship between the shrinkage rate and the osmotic pressure difference between the droplet and the CaCl2aq. in the gel plate. Specifically, droplets with three different diameters were used to investigate the difference in shrinkage behavior (herein referred to as the shrinkage rate) for each original droplet size (Fig. 4). A previous study33 defined shrinkage rate as the absolute value of the slope when the droplet diameter is plotted against time. A higher osmotic pressure difference led to a faster shrinkage rate, regardless of the original droplet diameter. In addition, smaller droplets exhibited a faster shrinkage rate at a given osmotic pressure difference. A previous study on the shrinkage behavior of sodium alginate droplets based on the same experimental system revealed that contact with calcium chloride initiated cross-linking within the sodium alginate droplets to achieve gradual solidification.33 Furthermore, the shrinkage rates of sodium alginate droplets also increased with the increasing osmotic pressure difference. However, the slope of the droplet diameter–time plot for the SH droplets was 18 in the present study, which was substantially higher than that for the sodium alginate droplets (2.6) in a previous study.33 This can be attributed to the polymer network formed within the sodium alginate droplets via cross-linking, which prevented the movement of water from inside the droplet to the surface of the droplet.
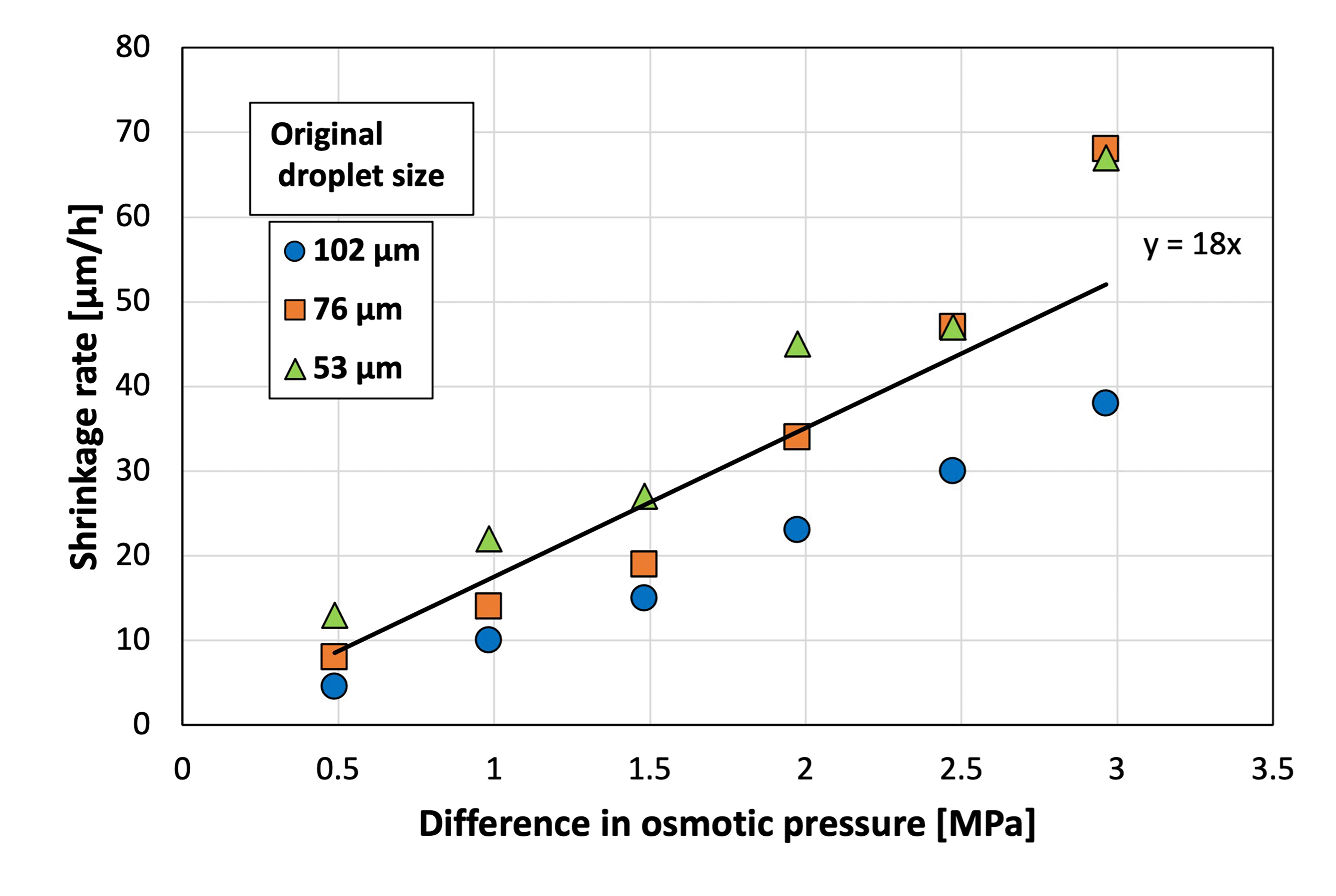 | ||
| Fig. 4 Shrinkage rate at various osmotic pressure differences between the sodium hyaluronate aqueous solution and CaCl2aq. (N = 50). | ||
To investigate the difference in shrinkage behavior (herein referred to as the shrinkage factor) for each original droplet size and to compare the behavior of cross-linked (i.e., sodium alginate) and non-cross-linked (i.e., SH) materials, the shrinkage factor was plotted against the osmotic pressure difference between the droplet and the aqueous calcium chloride solution in the gel plate (Fig. 5). A previous study33 defined the shrinkage factor as the volume change of the droplet using eqn (1):
| Shrinkage factor (%) = (original droplet volume − shrank droplet volume)/(original droplet volume) × 100 | (1) |
 | ||
| Fig. 5 Shrinkage factor at various osmotic pressure differences between the sodium hyaluronate aqueous solution and CaCl2aq. (N = 50; CV < 5.0%). | ||
The shrinkage factor increased with the increasing osmotic pressure difference, and eventually reached ∼99.9%. Therefore, the size of SH microcapsules can be controlled by adjusting the diameter of the original droplets and the osmotic pressure difference. The shrinkage factor of the sodium alginate droplets stabilized at an osmotic pressure difference of ∼10 MPa.33 In comparison, the shrinkage factor of the SH droplets stabilized at a smaller osmotic pressure difference of ∼2 MPa. This indicated that the water in the non-cross-linked SH droplets responded to a smaller osmotic pressure difference than the cross-linked sodium alginate droplets, and this can be attributed to the polymer network formed during cross-linking. In addition, theoretically, the osmotic pressure is constant at a constant concentration, temperature, and volume, regardless of the pH value, as shown in the following equation:
| Π = cRT, | (2) |
The traditional “solvent removal method” has been reported as a method for dehydrating and solidifying droplets.51 This method solidifies the droplets by evaporating the liquid present in the system. This principle is simple and well established; however, applying this method to the water-in-oil droplets in our study is difficult due to the use of oil, which has a relatively high boiling point.
FITC in the microcapsules prepared by this method was completely water-soluble without insolubilization in spite of the poor solvent environment.
In addition to the water-soluble content such as FITC–BSA, the insoluble content (e.g., inorganic particles) can be used in this method.33 In the case of microcapsule generation using insoluble content, in principle, the minimum size of the resulting microcapsules is close to the total volume of the content. This indicates that the obtained microcapsule size is affected by the osmotic pressure and the amount of content.
The phenomenon of droplet condensation driven by water removal, which has been reported in previous studies,52,53 has been quantitatively evaluated using water activities (calculated based on the solute concentration of the droplet). In these previous studies, the solute concentration of the droplet could be calculated from the droplet size because the solute does not flow out or in. Conversely, as we reported in our previous study,35 in our droplet shrinkage process, the ions contained in the gel plate flow into the droplet. The measurement of solute concentration in the droplet is limited by the complexity of measuring the ion influx. In future, when it becomes possible to measure the concentration of the ions flowing into the droplet, it will be possible to discuss the final microcapsule size in detail from the viewpoint of water activities, as has been done in previous studies.
3.2 Microcapsule characterization
The water solubility and preservation stability of the microcapsules were evaluated to demonstrate their applicability as a TDDS. The microcapsules instantly dissolved in water, but were insoluble in oil and ethanol (Fig. 6). This suggested that transdermal administration of microcapsules containing an active ingredient (i.e., a drug) leads to rapid dissolution of the microcapsules upon contact with water present in the skin, thereby releasing the drug. The preservation stability of the microcapsules was demonstrated by immersing the microcapsules in the oil phase to prevent drying during storage at 4 °C and ambient temperature for 1 month (Fig. 7). The fluorescence intensity was measured to monitor the release of the model drug (i.e., FITC–BSA) contained in the microcapsules, whereas the diameter was measured to detect the dissolution of SH in the oil phase. At both storage temperatures, the fluorescence intensity increased slightly, and no change in diameter was observed. The slight increase in fluorescence intensity was smaller for the microcapsules stored at 4 °C, suggesting that refrigeration of the microcapsules allows for more stable storage than maintaining them at ambient temperatures in terms of inclusion release. The water-soluble protein may have migrated toward the center of the microcapsule, away from the oil, over time. In addition, the fact that the intensity in the microcapsules did not decrease in this way suggests that the encapsulated model drug can be retained in the microcapsules (indicating high encapsulation efficiency). This also implies that the size of the microcapsules can be controlled in this method without considering the change in the encapsulation efficiency. | ||
| Fig. 6 Solubility of microcapsules in (a) surfactant-containing sunflower oil, (b) ethanol, and (c) water. | ||
 | ||
| Fig. 7 (a) Fluorescence intensity and (b) diameter of the microcapsules immersed in oil and stored at 4 °C and ambient temperatures (22–28 °C) (N = 50). | ||
As the microcapsule size and fabrication parameters (such as CaCl2 concentration) change, the specific surface area and physical properties of the microcapsules may change, respectively. These may affect the encapsulation efficiency and water solubility of the model drug contained in the microcapsules.
3.3 Application of microcapsules to microneedles
PLGA-based MNs containing microcapsules were prepared in a 256 (16 × 16) needle structure (Fig. 8a–c). The dimensions of the convex metal mold and fabricated MNs were measured (Table 2), revealing that the base diameters were almost the same but the height of the MNs was smaller than that of the convex metal mold. Furthermore, the tip diameter of the prepared MNs was larger than that of the convex metal mold because the tip of the needles in the concave PDMS-based mold could not be filled with high-viscosity PLGA. MNs with a sharper needle tip may be fabricated by designing a metal mold with a larger height and tip diameter or by reducing the viscosity of PLGA.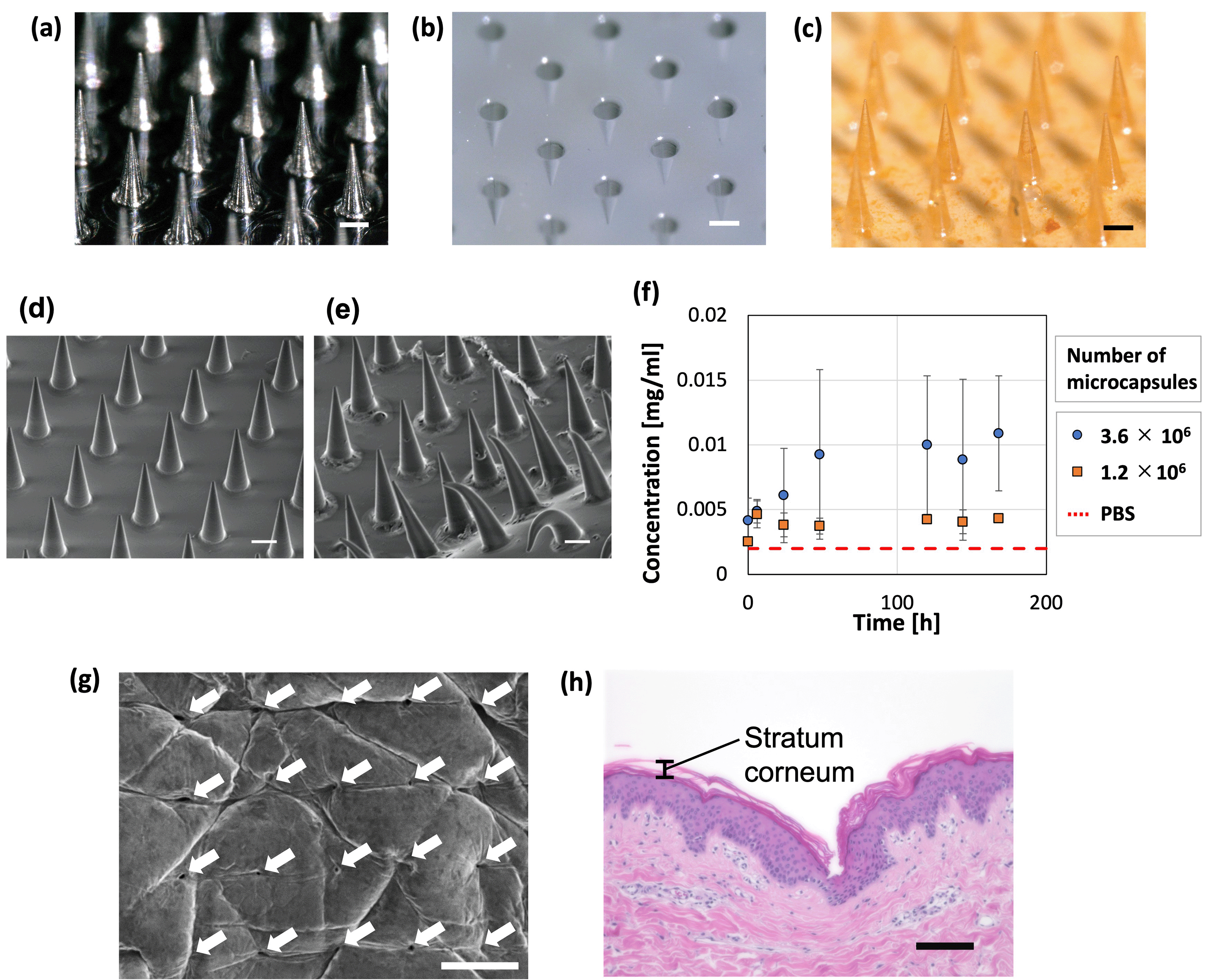 | ||
| Fig. 8 Application to microneedles. Images of the (a) convex metal mold, (b) concave polydimethylsiloxane-based mold, and (c) microneedles containing microcapsules (scale bars = 200 μm). SEM image of the MNs (d) before and (e) after immersion in PBS for 7 days (scale bars = 200 μm). (f) Concentration of FITC-BSA (released from MNs) in PBS over time, meaning the time course of cumulative release to PBS; the dashed line represents the measurement of PBS without FITC-BSA (N = 3). Skin puncture ability of the microneedles (MNs) illustrated as a (g) surface SEM image (scale bar = 500 μm) and (h) cross-sectional micrograph (showing a representative image of a histological section) of the porcine skin surface after the insertion and removal of the MNs containing 2.4 × 106 microcapsules (scale bar = 100 μm). The puncture hole was observed on the surface of the skin. | ||
| Base diameter [μm] | Height [μm] | Tip diameter [μm] | |
|---|---|---|---|
| Metal mold | 253 ± 2 | 697 ± 1 | 19 ± 1 |
| Fabricated MNs | 252 ± 3 | 681 ± 1 | 23 ± 2 |
Release of the model drug, FITC–BSA, from the fabricated MNs was evaluated; deformation of the needle and substrate was observed after inserting the MNs in PBS (Fig. 8d–f). For the fabrication of the MNs, we used microcapsules whose shrinkage behavior is described in Fig. 3. Therefore, the following conditions were used to produce microcapsules of the same size as in Fig. 3. Droplets were generated at a flow rate of 1.0 mL h−1 in the dispersed phase (comprising 0.01 wt% SH aqueous solution and 0.5 mg mL−1 FITC–BSA) and 2.5 mL h−1 in the continuous phase (mineral oil with 1 wt% surfactant). The generated droplets were contracted and solidified on a gel plate containing 0.2 mol L−1 CaCl2. To examine the trend of FITC–BSA concentration in PBS at a constant level (meaning that the release is complete), the time course was measured for 3 min in this study. Additionally, because PLGA melted by heating is highly viscous, the microcapsules added to PLGA could remain in the container and be lost when transferred to the PDMS mold. The loss of microcapsules can be avoided by mixing the molten PLGA and microcapsules on the PDMS mold. If the mixing of PLGA and microcapsules in a container is necessary, depending on the application, the loss of microcapsules may be reduced by using a container made of a material that does not easily adhere to PLGA. Consequently, the FITC–BSA concentration of the PBS increased over time. This increase was higher for the MNs with more microcapsules (3.6 × 106), indicating that MNs containing more microcapsules released more FITC–BSA. The MNs containing 3.6 × 106 microcapsules led to an increase in the drug concentration of PBS for ∼48 h, after which no significant change was observed, and drug release was complete. These findings show that MNs can be potentially used for sustained-release drug administration that may be controlled by changing the material properties of the MNs and microcapsules. In addition, the MNs with less microcapsules (1.2 × 106) showed a constant FITC–BSA concentration in PBS immediately after immersion in PBS (earlier than the MNs with more microcapsules). This suggests that the drug release was completed relatively early in the MNs with less microcapsules compared to those with more microcapsules. The amount of drug released in this study was relatively small (∼0.011 mg). For applications that required additional administration, the release (dose) rate could be further increased by increasing the number of microcapsules embedded in the MNs.
The ability of the MNs to puncture the skin was examined by pressing the MNs against porcine skin, where an array of puncture holes was formed on the surface of the skin (Fig. 8g). The MNs penetrated the stratum corneum (Fig. 8h), which is a barrier layer containing lipids. Therefore, drugs that cannot pass through the stratum corneum, such as macromolecular drugs and water-soluble drugs, would be delivered using the proposed TDDS. A section of skin that could not be penetrated by the MNs was observed (Fig. S5, ESI†). The inability to penetrate was attributed to pressure distribution caused by skin elasticity and the bed of nail effect.54 This can be improved by fabricating MNs with sharper needles and an optimized needle pitch.
4. Conclusions
Monodispersed SH microcapsules were produced by applying an approach based on the shrinkage and gelation technique instead of cross-linking. This method uses non-toxic materials that are widely used as additives in cosmetics and pharmaceuticals. The size of the microcapsules was controlled according to the original droplet size and the osmotic pressure difference between the droplets and aqueous solution in the gel plate. The microcapsules were water-soluble and could be stored stably for one month. Furthermore, sustained release of SH microcapsules was observed by producing PLGA-based MNs with embedded microcapsules. The MNs released the drug into PBS over an extended time period, which was dependent on the amount of microcapsules contained. In addition, the MNs were capable of penetrating the stratum corneum. These results demonstrate that the developed SH microcapsules can be potentially used for sustained-release preparation and avoid issues commonly associated with cross-linking, such as toxicity and bursting. While physically cross-linked gels such as ionic cross-linked gels, which form a relatively fragile structure, could burst and cause discontinuous drug release in the presence of water, the developed microcapsules (and microcapsules in MNs) could stably dissolve and release drugs in the presence of water in the skin without bursting due to their water solubility. This novel material shows excellent application potential in pharmaceutics and cosmetics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partly supported by JSPS KAKENHI (Grant Number JP18K14109).Notes and references
- T. C. Laurent, U. B. G. Laurent and J. R. E. Fraser, Ann. Rheum. Dis., 1995, 54, 429–432 CrossRef CAS PubMed.
- J. R. E. Fraser, T. C. Laurent and U. B. G. Laurent, J. Intern. Med., 1997, 242, 27–33 CrossRef CAS PubMed.
- J. E. Rayahin, J. S. Buhrman, Y. Zhang, T. J. Koh and R. A. Gemeinhart, ACS Biomater. Sci. Eng., 2015, 1, 481–493 CrossRef CAS PubMed.
- Y. Mansouri and G. Goldenberg, Cutis, 2015, 96, 85–88 Search PubMed.
- M. Oe, T. Tashiro, H. Yoshida, H. Nishiyama, Y. Masuda, K. Maruyama, T. Koikeda, R. Maruya and N. Fukui, Nutr. J., 2016, 15, 10 Search PubMed.
- M. Oe, S. Sakai, H. Yoshida, N. Okado, H. Kaneda, Y. Masuda and O. Urushibata, Clin., Cosmet. Invest. Dermatol., 2017, 10, 7 Search PubMed.
- S. N. A. Bukhari, N. L. Roswandi, M. Waqas, H. Habib, F. Hussain, S. Khan, M. Sohail, N. A. Ramli, H. E. Thu and Z. Hussain, Int. J. Biol. Macromol., 2018, 120, 1682–1695 CrossRef CAS PubMed.
- C. H. Kim, B. J. Lee, J. Yoon, K. M. Seo, J. H. Park, J. W. Lee, E. S. Cho, J. J. Hong, Y. S. Lee and J. H. Park, J. Vet. Med. Sci., 2001, 63, 1083–1089 CrossRef CAS PubMed.
- S. Shimmura, M. Ono, K. Shinozaki, I. Toda, E. Takamura, Y. Mashima and K. Tsubota, Br. J. Ophthalmol., 1995, 79, 1007–1011 CrossRef CAS PubMed.
- P. Aragona, G. Di Stefano, F. Ferreri, R. Spinella and A. Stilo, Br. J. Ophthalmol., 2002, 86, 879–884 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314, 10.1039/b506989k.
- N. El Kechai, E. Mamelle, Y. Nguyen, N. Huang, V. Nicolas, P. Chaminade, S. Yen-Nicolay, C. Gueutin, B. Granger, E. Ferrary, F. Agnely and A. Bochot, J. Controlled Release, 2016, 226, 248–257 CrossRef CAS PubMed.
- Y. G. Lei, M. Rahim, Q. Ng and T. Segura, J. Controlled Release, 2011, 153, 255–261 CrossRef CAS PubMed.
- S. K. Hahn, S. J. Kim, M. J. Kim and D. H. Kim, Pharm. Res., 2004, 21, 1374–1381 CrossRef CAS PubMed.
- M. Khanmohammadi, S. Sakai, T. Ashida and M. Taya, J. Appl. Polym. Sci., 2016, 133, 43107 CrossRef.
- V. M. Goldberg and J. A. Buckwalter, Osteoarthritis Cartilage, 2005, 13, 216–224 CrossRef CAS PubMed.
- S. Liu, M. N. Jin, Y. S. Quan, F. Kamiyama, H. Katsumi, T. Sakane and A. Yamamoto, J. Controlled Release, 2012, 161, 933–941 CrossRef CAS.
- Y.-J. Jin, T. Ubonvan and D.-D. Kim, J. Pharm. Invest., 2010, 40, 33–43 CrossRef CAS.
- G. L. Huang and H. L. Huang, J. Controlled Release, 2018, 278, 122–126 CrossRef CAS.
- K. N. How, W. H. Yap, C. L. H. Lim, B. H. Goh and Z. W. Lai, Front. Pharmacol., 2020, 11, 1105 CrossRef CAS.
- H. Lee, K. Lee and T. G. Park, Bioconjugate Chem., 2008, 19, 1319–1325 CrossRef CAS PubMed.
- X. H. Fan, X. S. Zhao, X. K. Qu and J. Fang, Int. J. Pharm., 2015, 496, 644–653 CrossRef CAS PubMed.
- K. W. Chun, J. B. Lee, S. H. Kim and T. G. Park, Biomaterials, 2005, 26, 3319–3326 CrossRef CAS PubMed.
- R. Zhang, Z. B. Huang, M. Y. Xue, J. Yang and T. W. Tan, Carbohydr. Polym., 2011, 85, 717–725 CrossRef CAS.
- S. K. Hahn, J. S. Kim and T. Shimobouji, J. Biomed. Mater. Res., Part A, 2007, 80a, 916–924 CrossRef CAS PubMed.
- M. G. Bah, H. M. Bilal and J. T. Wang, Soft Matter, 2020, 16, 570–590 RSC.
- M. Lengyel, N. Kállai-Szabó, V. Antal, A. J. Laki and I. Antal, Sci. Pharm., 2019, 87, 20 CrossRef CAS.
- Y. Luo, K. R. Kirker and G. D. Prestwich, J. Controlled Release, 2000, 69, 169–184 CrossRef CAS PubMed.
- L. R. Shang, Y. Cheng and Y. J. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS PubMed.
- T. P. Lagus and J. F. Edd, J. Phys. D: Appl. Phys., 2013, 46, 114005 CrossRef.
- Z. H. Nie, W. Li, M. Seo, S. Q. Xu and E. Kumacheva, J. Am. Chem. Soc., 2006, 128, 9408–9412 CrossRef CAS PubMed.
- J. Clausell-Tormos, D. Lieber, J. C. Baret, A. El-Harrak, O. J. Miller, L. Frenz, J. Blouwolff, K. J. Humphry, S. Koster, H. Duan, C. Holtze, D. A. Weitz, A. D. Griffiths and C. A. Merten, Chem. Biol., 2008, 15, 427–437 CrossRef CAS PubMed.
- K. Aketagawa, H. Hirama, H. Moriguchi and T. Torii, Microfluid. Nanofluid., 2013, 17, 217–224 CrossRef.
- S. Seiffert and D. A. Weitz, Polymer, 2010, 51, 5883–5889 CrossRef CAS.
- H. Hirama, T. Kambe, K. Aketagawa, T. Ota, H. Moriguchi and T. Torii, Langmuir, 2013, 29, 519–524 CrossRef CAS PubMed.
- K. Aketagawa, H. Hirama and T. Torii, J. Mater. Sci. Chem. Eng., 2013, 01, 1–5 CAS.
- H. Hirama and T. Inoue, Chem. Lett., 2017, 46, 460–462 CrossRef CAS.
- F. S. Meng, A. Hasan, M. M. N. Babadaei, P. H. Kani, A. J. Talaei, M. Sharifi, T. G. Cai, M. Falahati and Y. Cai, J. Adv. Res., 2020, 26, 137–147 CrossRef CAS PubMed.
- S. Sivasankaran and S. Jonnalagadda, J. Controlled Release, 2021, 330, 797–811 CrossRef CAS PubMed.
- N. Dabholkar, S. Gorantla, T. Waghule, V. K. Rapalli, A. Kothuru, S. Goel and G. Singhvi, Int. J. Biol. Macromol., 2021, 170, 602–621 CrossRef CAS PubMed.
- J. H. Park, M. G. Allen and M. R. Prausnitz, Pharm. Res., 2006, 23, 1008–1019 CrossRef CAS PubMed.
- L. Y. Chu and M. R. Prausnitz, J. Controlled Release, 2011, 149, 242–249 CrossRef CAS PubMed.
- M. Kim, B. Jung and J. H. Park, Biomaterials, 2012, 33, 668–678 CrossRef CAS PubMed.
- J. H. Park, M. G. Allen and M. R. Prausnitz, J. Controlled Release, 2005, 104, 51–66 CrossRef CAS PubMed.
- S. Aoyagi, H. Izumi, Y. Isono, M. Fukuda and H. Ogawa, Sens. Actuators, A, 2007, 139, 293–302 CrossRef CAS.
- Y. J. Xia and D. W. Pack, Chem. Eng. Sci., 2015, 125, 129–143 CrossRef CAS.
- L. M. Sanders, B. A. Kell, G. I. Mcrae and G. W. Whitehead, J. Pharm. Sci., 1986, 75, 356–360 CrossRef CAS PubMed.
- B. Mukherjee, K. Santra, G. Pattnaik and S. Ghosh, Int. J. Nanomed., 2008, 3, 487–496 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- A. Rotem, O. Ram, N. Shoresh, R. A. Sperling, M. Schnall-Levin, H. D. Zhang, A. Basu, B. E. Bernstein and D. A. Weitz, PLoS One, 2015, 10, e0116328 CrossRef PubMed.
- E. Mathiowitz, W. M. Saltzman, A. Domb, P. Dor and R. Langer, J. Appl. Polym. Sci., 1988, 35, 755–774 CrossRef CAS.
- J. U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. W. Jia, K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129, 8825–8835 CrossRef CAS PubMed.
- M. Fukuyama, L. Zhou, T. Okada, K. V. Simonova, M. Proskurnin and A. Hibara, Anal. Chim. Acta, 2021, 1149, 9 CrossRef PubMed.
- S. P. Davis, B. J. Landis, Z. H. Adams, M. G. Allen and M. R. Prausnitz, J. Biomech., 2004, 37, 1155–1163 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00528f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |