
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThienyltriazine based conjugated porous organic polymers: tuning of the porosity and band gap, and CO2 capture†
Neha Rani
Kumar
a,
Prasenjit
Das
b,
Abhijeet R.
Agrawal
a,
Sanjay K.
Mandal
 *b and
Sanjio S.
Zade
*a
*b and
Sanjio S.
Zade
*a
aDepartment of Chemical Sciences and Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, India. E-mail: sanjiozade@iiserkol.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Mohali, Sector 81, Manauli PO, S. A. S. Nagar, Mohali, Punjab 140306, India. E-mail: sanjaymandal@iisermohali.ac.in
First published on 18th September 2021
Abstract
A series of four thiophene and triazine containing conjugated porous polymers (CPPs) comprising the same building block, tris(thienyl)triazine, is synthesized using the alkyne cyclotrimerization reaction for TT-CPP1, Sonogashira coupling reaction for TT-CPP2, Glaser coupling reaction for TT-CPP3 and Stille coupling reaction for TT-CPP4. By varying the spacer between tris(thienyl)triazine units, the porosity (3.6, 4.8 and 5.3 nm, respectively) and the band gap value (2.43, 2.11 and 2.03 eV, respectively) of TT-CPP1, TT-CPP2 and TT-CPP3 have been efficiently tuned. All the TT-CPPs display significant thermal stability up to 450 °C. Nitrogen and sulfur of triazine and thiophene moieties, respectively, act as electron-donating centers in these porous polymer frameworks, resulting in excellent adsorption of the Lewis acidic CO2 molecule. A maximum CO2 uptake of 11.4 wt% (2.6 mmol g−1) at 263 K under 100 kPa pressure has been observed for TT-CPP1 compared to the other two CPPs due to the small pore size and strong adsorbate–adsorbent interactions. Configurational bias Monte Carlo (CBMC) molecular simulations have shed light on the selective capture, position and binding energy of CO2 for TT-CPP1/2/3.
Introduction
Porous organic polymers (POPs), which include polymers of intrinsic microporosity (PIMs),1 hyper-cross-linked polymers (HCPs),2 covalent triazine-based frameworks (CTFs)3 and conjugated microporous or porous polymers (CMPs or CPPs),4–6 are gaining more recognition compared to their inorganic counterparts as they have inherent remarkable chemical and physical stability along with the possibility to tune their structure. Among these POPs, CPPs are generally synthesized by employing metal-catalyzed cross-coupling reactions that result in the formation of extended π-conjugated 3D networks.7 A judicious selection of the monomer and the polymerization conditions or the techniques employed can enable one to have easy control over the physical and chemical properties of CPPs. Another advantage of conjugated porous systems is that various chemical functionalities can be introduced into the pores by employing different organic reactions.7 The presence of a rigid π-conjugated structure imparts these materials with excellent thermal and chemical stability, larger surface areas and tunable porosity.8,9 CPPs have received wide attention from the research community for their potential applications in gas storage,10–16 production of hydrogen,17–19 catalysis,20–26 iodine capture,27 light-harvesting,28 and supercapacitors.29In the past couple of years, CPPs/CMPs are gaining wide recognition in the field of organic electronics.30,31 This necessitates one to have control over their electronic properties because each energy application of this class of materials demands different optimum band gap values: solar cell systems (Eg = 1.65–3.27 eV for entire visible-light absorption),32 CO2 reduction (Eg = 1.75–3.0 eV)33 and water splitting (Eg = 1.5–2.5 eV).34,35 To tune the band gap in CPPs, it is necessary to have structural control. However, efforts for such an outcome are significantly less in the literature. One significant contribution in this regard was recently made by Coskun and group, where they reported band gap tailoring via acid-dependent in situ cyclization.36
An insight into the literature reveals that the most efficient strategies5 to establish structural control in conjugated porous polymers are employed: (i) by controlling the conjugated structure and morphology, (ii) by controlling the porous structure, (iii) by tuning the length and geometry of the monomer, (iv) by employing a statistical copolymerization technique, and (v) by tuning the reaction conditions. Furthermore, it is now a well-proven fact that the presence of electron-rich aromatic rings and large heteroatom content can enhance the host–guest interaction of CPPs and boost their overall performance to act as the storehouse of gases. Thiophene, a sulfur-containing aromatic electron-rich heterocycle, is a commonly employed building block in a linear conjugated polymer. However, thiophene containing conjugated porous polymers are significantly less in the literature.37–41
Inspired by the above facts and scope, herein, we present the synthesis of a class of thienyltriazine based porous polymers TT-CPP1, TT-CPP2, TT-CPP3, and TT-CPP4 using alkyne trimerization, Sonogashira-coupling, Glaser coupling, and Stille coupling reactions, respectively. The resulting frameworks display high levels of porosity (surface areas up to 545 m2 g−1), stability and CO2 adsorption up to 11.4 wt%. This structural control established over the porous structures is done by efficiently tuning the size and geometry of the building blocks (i.e., by using a spacer) and by tuning the reaction conditions. Their band gaps have also been tuned efficiently.
Results and discussion
The synthesis of the targeted CPPs was done by using functionalized 2,4,6-tri(thiophen-2-yl)-1,3,5-triazine as the core moiety. The tris(thienyl)triazine core was synthesized starting from 2-cyanothiophene by using the procedure reported by Misra and group.42 It was then converted to its triacetylene derivative 1. Scheme 1 outlines the synthesis of TT-CPP1, TT-CPP2, TT-CPP3 and TT-CPP4. TT-CPP1 was synthesized by a Co2(CO)8 mediated alkyne trimerisation reaction of 1. TT-CPP2 was synthesized by a Sonogashira coupling reaction of 1 with 2,4,6-tris(5-bromothiophen-2-yl)-1,3,5-triazine (2), whereas TT-CPP3 was synthesized by a Glaser coupling reaction of 1. On the other hand, TT-CPP4 was synthesized by the Stille coupling of tristannyl derivative 2,4,6-tri(thiophen-2-yl)-1,3,5-triazine43 with its tribromo counterpart 2. Thus, keeping the core moiety intact, we have varied the spacer (benzene, acetylene, and diacetylene) to tune the porosity and band gap energy.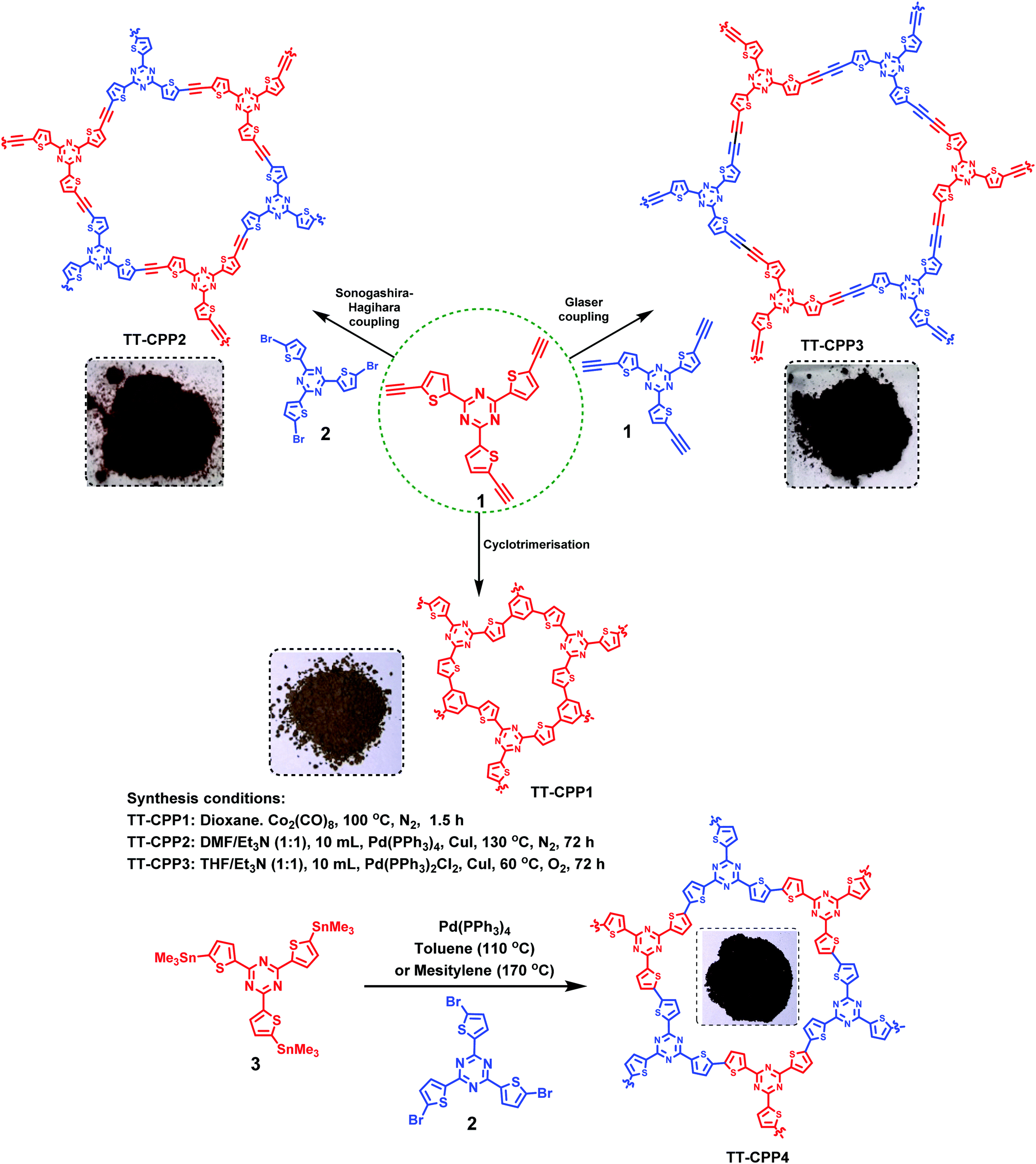 | ||
| Scheme 1 Synthesis of the trithienyltriazine based conjugated porous polymers (TT-CPP1, TT CPP2, TT-CPP3 and TT-CPP4). | ||
The chemical connectivity and structure of the networks were characterized by Fourier transform infrared (FTIR) spectroscopy (Fig. 1 and Fig. S2, ESI†), solid state 13C CP/MAS NMR spectroscopy (Fig. 2 and Fig. S1, ESI†), elemental analysis (Table S1, ESI†) and solid-state UV-visible absorption spectroscopy (Fig. 3a and Fig. S9, ESI†). The FT-IR spectra show a weak band corresponding to disubstituted C–C triple bonds at around 2175 cm−1 in TT-CPP2 and TT-CPP3 (Fig. 1, blue and red spectra). The stretching frequencies around 800, 1370 and 1500 cm−1 confirm the successful incorporation of the s-triazine moiety in the polymer frameworks, whereas the peaks at about 3080 and 1625 cm−1 are assigned to sp2 C–H stretching and C–C double bond stretching of the thiophene ring, respectively (Fig. 1, black, blue and red spectra). The IR spectra also indicate complete attenuation of the peaks at around 2100 and 3400 cm−1 in TT-CPP1-3 involving 1 as the precursor (Fig. 1 and Fig. S3, S4a, ESI†). There is also a complete disappearance of the C–Br band at 1049 cm−1 in TT-CPP2 and TT-CPP4 (Fig. 1 and Fig. S3b, S4b, ESI†). This confirms their high degree of polymerization.
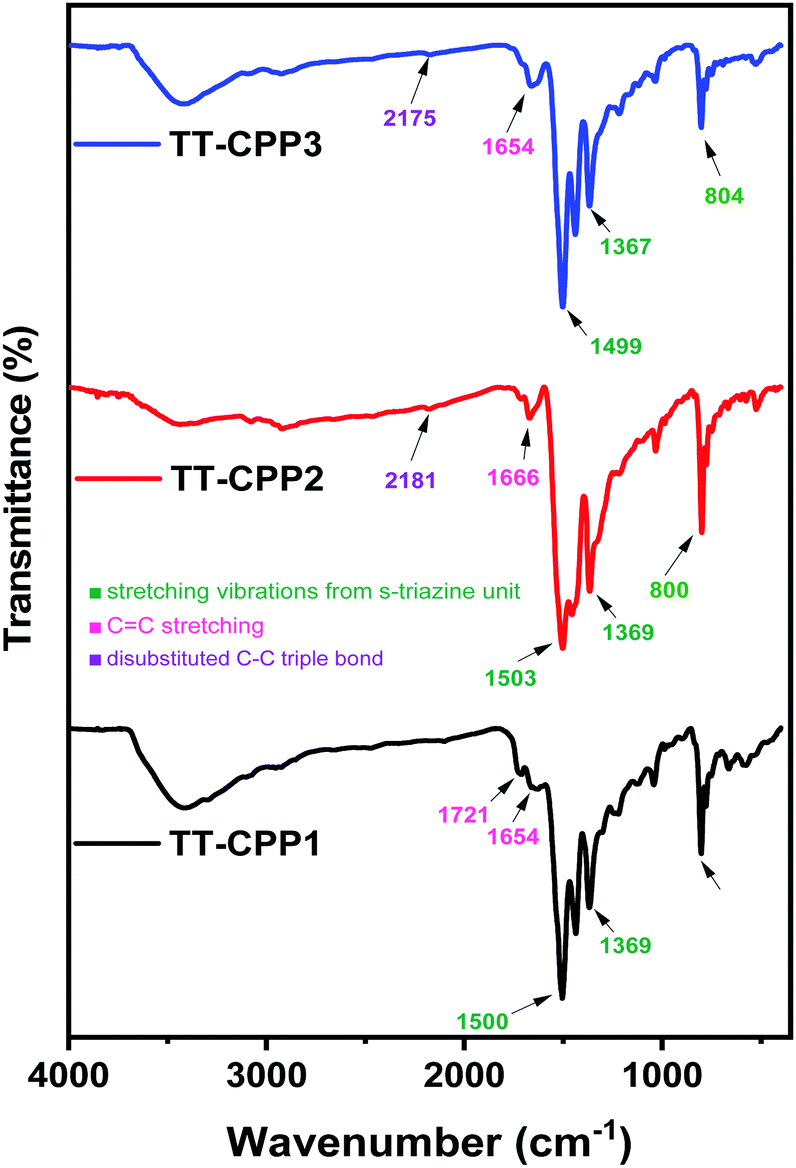 | ||
| Fig. 1 FTIR spectra of TT-CPP1, TT-CPP2 and TT-CPP3. | ||
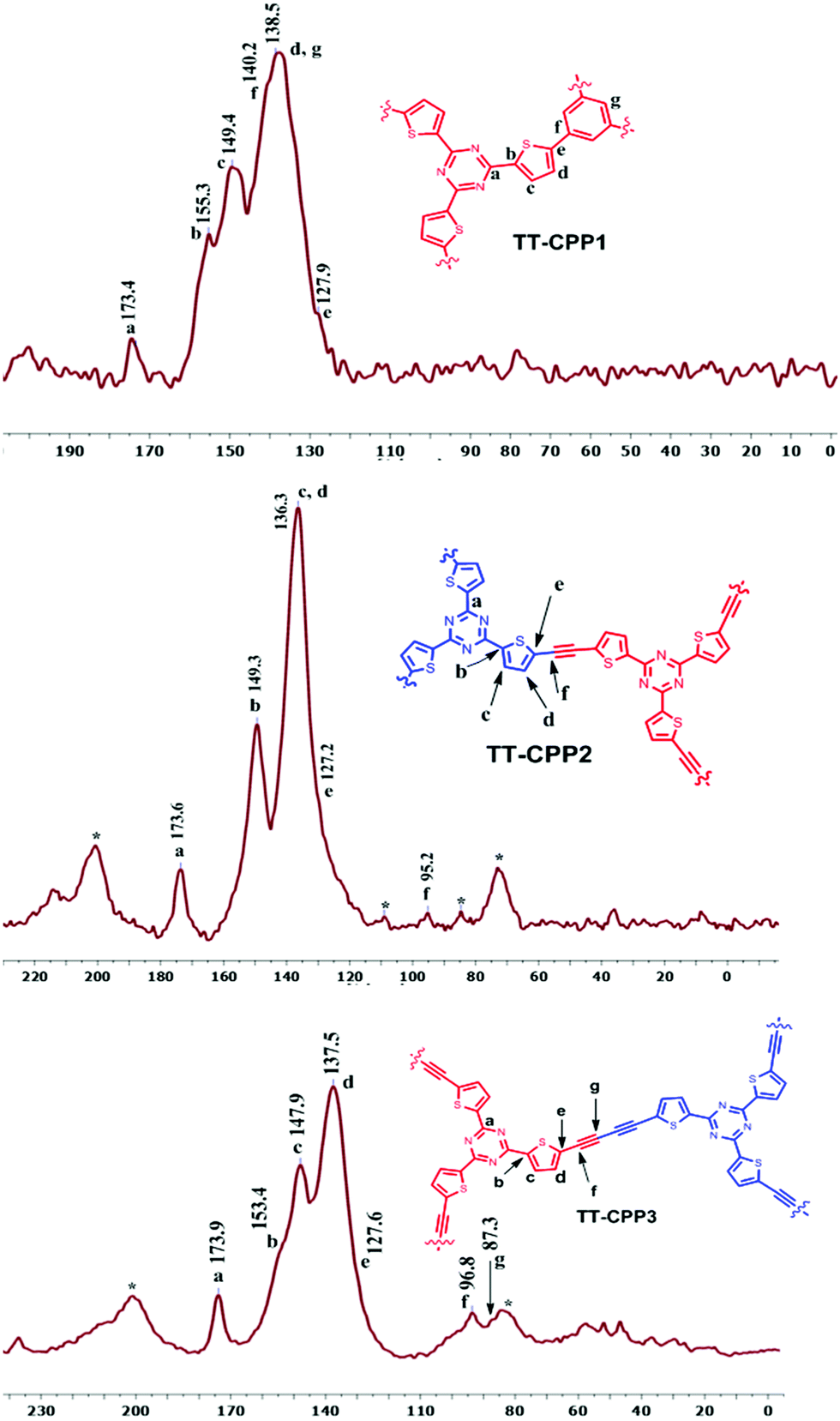 | ||
| Fig. 2 Solid-state CP-MAS 13C NMR spectra of TT-CPP1, TT-CPP2 and TT-CPP3. | ||
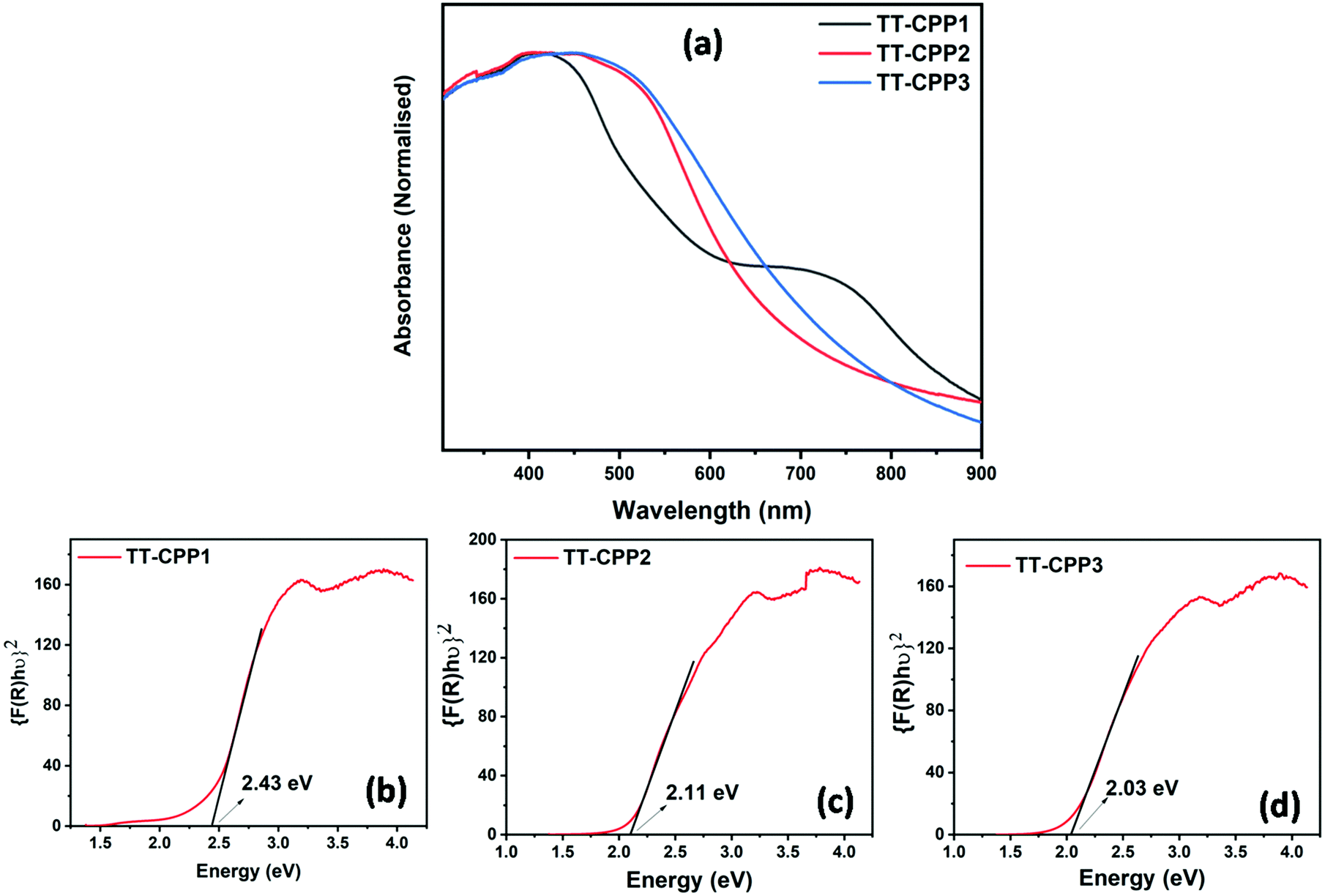 | ||
| Fig. 3 (a) Solid-state UV-visible spectra of TT-CPPs, and (b–d) Kubelka–Munk plot for the band gap calculation of TT-CPP1, TT-CPP2, and TT-CPP3, respectively. | ||
In the solid-state 13C NMR spectra (Fig. 2), TT-CPP1, TT-CPP2 and TT-CPP3 showed a peak at around 173 ppm, and TT-CPP4 showed a peak at 170 ppm, arising from the carbon atoms of the triazine core. The peaks in the range of 84–96 ppm in the spectra of TT-CPP2 and TT-CPP3 are attributed to quaternary alkyne carbons involved in polymerization. The peaks at about 153, 149, 138–135, and 127 ppm correspond to the thiophene carbons, where the first peak at 153 ppm corresponds to the thiophene carbon bonded to the triazine ring (α carbon). The peaks marked with an asterisk in the spectra of TT-CPP2 and TT-CPP3 are the spinning sidebands. By combining the results of FT-IR and solid-state 13C NMR spectroscopy, it can be concluded that the represented polymer skeletons are formed. The experimental values of the C, H, and N contents of the polymers in the elemental analysis showed some deviation from the theoretical values due to the presence of unreacted end groups and catalyst residues, as well as trapped gases and moisture, as reported earlier.44–48
The solid-state UV-visible spectra of the polymers (Fig. 3 and Fig. S9, ESI†) showed broad absorption extending from 350 nm to 800 nm for TT-CPP2, TT-CPP3 and TT-CPP4, a characteristic of the extended π-conjugation of the polymer. However, TT-CPP1 also showed an absorption peak in the low energy region extending from 650 nm to 850 nm. This can be attributed to efficient interlayer stacking between the polymer framework arising probably due to its small pore size, π–π interaction and presence of sulfur atoms. The Kubelka–Munk plot was used to calculate their band gaps (Fig. 3b–d and Fig. S10, ESI†). The Kubelka–Munk plot of TT-CPP1 (Fig. 3b) shows a very small hump in the range of 1.5 to 1.8 eV with an onset of the same at 1.5 eV which is due to the interlayer stacking in the polymer framework. Their band-gap values follow the trend TT-CPP3 < TT-CPP2 < TT-CPP4 < TT-CPP1. The observed trend is in agreement with the extent of conjugation of the polymers. On moving from polymer TT-CPP1 to TT-CPP2 and TT-CPP3, the color of the polymer powder darkens (Scheme 1), supporting the decreasing band gap and increased π-conjugation in the same order.
The extension of π-conjugation leads to a decrease in the band gap, and thus TT-CPP3 with the most extended π-conjugation has the smallest band gap of 2.03 eV, whereas TT-CPP1 with the least extension of π-conjugation has the largest band gap of 2.43 eV. This trend can also be attributed to the fact that the incorporation of a compact and rigid carbon–carbon triple bond makes the polymer framework more coplanar, and its electron-withdrawing nature enhances the donor–acceptor nature of the polymer framework, thereby lowering the band gap.
As expected, all the CPPs possess a disordered, amorphous structure, as proven by the broad diffraction peaks in the powder X-ray diffraction measurements (Fig. 4a and Fig. S7, ESI†). This also supports the fact that the polymerization reactions are kinetically controlled. The high thermal stability of the CPPs was proved by the thermal gravimetric analysis (TGA) measurement (Fig. 4b and Fig. S8, ESI†). TT-CPP2 and TT-CPP4 retained more than 95% mass at 500 °C, whereas TT-CPP1 and TT-CPP3 retained more than 85% mass at 500 °C. The impressive thermal stability of the polymers can be ascribed to the highly rigid and cross-linked polymer framework. The chemical stability of the TT-CPPs was established by dipping the samples in common organic solvents, an acid and then a base for 3 days and then recording their FTIR spectra. Compared to the FTIR spectra of the as-synthesized materials, the polymer skeleton was found to remain intact (Fig. S11, ESI†).
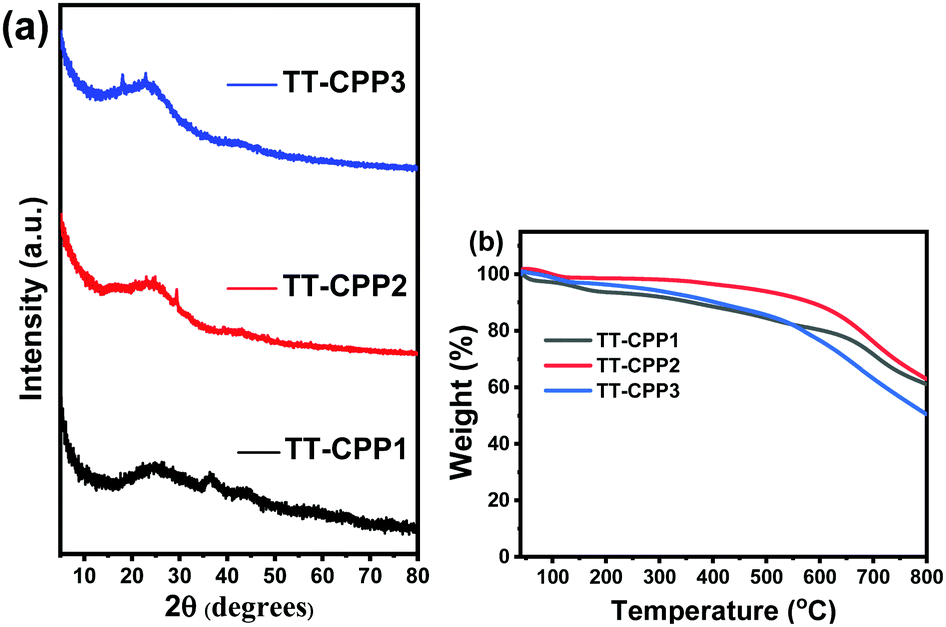 | ||
| Fig. 4 (a) Powder diffraction patterns of the TT-CPPs, and (b) TGA profiles of the TT-CPPs. | ||
Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) were used to establish the particle size and morphology of these polymers (Fig. 5 and Fig. S5, ESI†). The SEM image of TT-CPP1 (Fig. 5a and b) showed a spherical particle-like morphology, which was also confirmed by its TEM image. The SEM and TEM images of both TT-CPP2 (Fig. 5d and e) and TT-CPP3 (Fig. 5g and h) showed a fibrous morphology; the former had fibers of more ordered and uniform structure, and the latter showed a random intertwining (fibrous) porous structure. TT-CPP4 had an undefined morphology of agglomerated lumps (Fig. S5a and b, ESI†). The HRTEM images (at 20 nm scale) of all TT-CPPs showed the presence of alternate dark and bright worm-like structures, which is commonly observed in such porous structures (Fig. 5c, f and i). The permanent porosity and the pore size distributions of the polymers were studied by nitrogen sorption at 77 K. The adsorption isotherms for all the polymers were a mixture of Type II and Type IV (Fig. 6a) as per the IUPAC classification, and a sharp pore size distribution was observed (Fig. 6b). The uptake of N2 at relatively low pressures P/P°< 0.1 is very low, indicating a low level of microporosity in these CPPs. However, a steep increase in N2 uptake was observed at P/P° = 0.9–1, indicating the dominance of mesopores and macropores. The dominant meso- and macropores can arise from the interparticular voids in the porous polymer frameworks. As shown in Table 1, the BET surface area was found to be 545, 511 and 491 m2 g−1 for TT-CPP1, TT-CPP2, and TT-CPP3, respectively. TT-CPP4 showed a very low surface area of around 5 m2 g−1, indicating that the Stille coupling reaction is not an efficient synthetic strategy for the synthesis of thienyltriazine based conjugated porous polymers. This has been proven by carrying out the reaction in solvents of different boiling points like THF (reaction temperature kept at 70 °C), toluene (reaction temperature kept at 110 °C) and mesitylene (reaction temperature kept at 170 °C). All the polymers obtained were non-porous except in the case when mesitylene was used as a solvent. However, the surface area obtained was very low. Therefore, considering the inefficiency of the Stille coupling reaction in this particular case, the polymer TT-CPP4 was excluded from establishing the structure–property relationship. Guipeng Yu and group reported the template assisted synthesis of a nanotubular porous polymer having a structure similar to TT-CPP4. They synthesized the polymer by using the Yamamoto coupling reaction and the BET specific surface area was found to be 741 m2 g−1.44 The decreasing trend of the surface area from TT-CPP1 to TT-CPP3 can be attributed to the increasing size/length of the linker/spacer if the thienyltriazine system is viewed as the core. In TT-CPP1 the linker is a benzene ring, whereas in TT-CPP2 and TT-CPP3 the linkers are acetylene and diacetylene, respectively.
 | ||
| Fig. 5 SEM (a, d and g), TEM (b, e and h) and HRTEM (c, f and i) images of TT-CPP1, TT-CPP2, and TT-CPP3, respectively. | ||
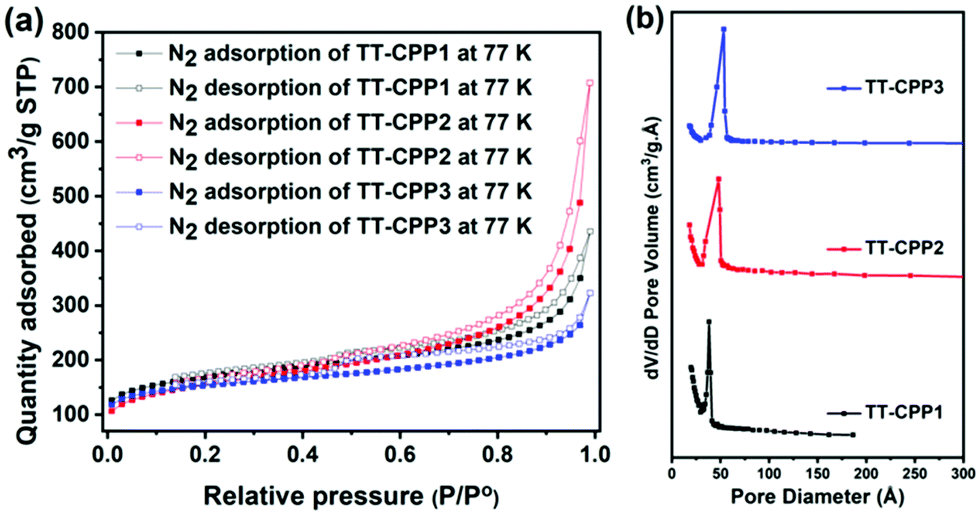 | ||
| Fig. 6 (a) N2 adsorption–desorption isotherm of the TT-CPPs at 77 K, and (b) pore-size distribution curve of the TT-CPPs. | ||
| Sample code | S BET [m2 g−1] | S Langmuir [m2 g−1] | S Micro [m2 g−1] | Average pore diameter (nm) | V Micro [cm3 g−1] | V total [cm3 g−1] | S Micro/BET (%) | V Micro/Vtotal (%) | CO2 uptake (mmol g−1) at 100 kPa | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 263 K | 273 K | 298 K | |||||||||
| a S BET is the surface area calculated from the N2 sorption isotherm. b S Micro is the microporous surface area calculated from the N2 adsorption isotherm based on the Harkins Jura method. c V Micro is the microporous volume derived from the t-plot. d V Total is the total pore volume at P/P° = 0.996. | |||||||||||
| TT-CPP1 | 545 | 964 | 252 | 3.6 | 0.132 | 0.196 | 46.23 | 67 | 2.62 | 2.18 | 1.27 |
| TT-CPP2 | 511 | 998 | 176 | 4.8 | 0.091 | 0.630 | 34.44 | 14 | 1.75 | 1.63 | 1.08 |
| TT-CPP3 | 491 | 831 | 273 | 5.3 | 0.141 | 0.383 | 55.30 | 36 | 1.42 | 1.22 | 0.76 |
The total pore volume at a relative pressure of P/P° = 0.99 was found to be 0.19, 0.63, and 0.38 cm3 g−1 for TT-CPP1, TT-CPP2, and TT-CPP3, respectively. The average pore diameter obtained using nonlocal density functional theory (NLDFT) demonstrates that the mesopores are centered in the 3 to 6 nm range for all the polymers. The expected trend for the pore size as well as the surface area of the polymers is in agreement with the experimentally observed trend.
The polymer TT-CPP1 was previously reported by Wang49 and co-workers in 2018 by direct-arylation polymerization. They reported a specific surface area of 74.9 m2 g−1 and 192.8 m2 g−1 for TT-CPP1 when the ratio of the thienyltriazine precursor and tribromo benzene precursor was 1 and 1.5, respectively. This polymer was also used for the photocatalyzed oxidative coupling of benzylamine. Its synthesis by a different reaction strategy in our case led to a 2.5 fold increase in the surface area (595 m2 g−1). This also explains the possibility of using the synthesized polymer as a photocatalyst with better efficiency. The morphology in our case was a spherical particle shape for TT-CPP1. In contrast, Wang et al. reported a fibrous morphology for the same polymer, thus depicting the effect of the reaction conditions on the morphology.
These findings show directions to efficiently tune the electronic structure as well as the band gap values (Fig. 7) in CPPs by careful selection of the monomer and varying the linker/spacer. In this case, by using an efficient heteroatom rich monomer and by varying the spacer, not only the porous characteristics such as the surface area and pore diameter but also the band gap value have been engineered, showing successful structural control (Fig. 7).
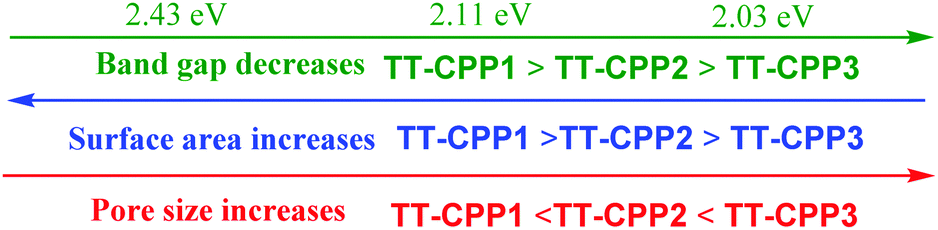 | ||
| Fig. 7 Tunable properties of the TT-CPPs. | ||
Conjugated porous polymers are finding a wide range of applications. However, they are gaining special recognition as CO2 sequestration materials.50 CO2 is one of the major greenhouse gases and is a well-known cause of the adverse effects of global warming.51 The CO2 adsorption isotherms of TT-CCP1, TT-CPP2 and TT-CPP3 at 263, 273, and at 298 K are illustrated in Fig. 8. As shown in Table 1, their CO2 adsorption capacities are calculated as 11.4 wt% (2.6 mmol g−1), 7.1 wt% (1.63 mmol g−1), and 5.3 wt% (1.22 mmol g−1) at 263 K/100 kPa for TT-CPP1, TT-CPP2, and TT-CPP3, respectively (Fig. 8a–c). The presence of N and S heteroatoms in these polymers is responsible for the high CO2 adsorption. Also, quadrupole–quadrupole and dipole–quadrupole interactions between the polymer frameworks and CO2 molecules play an important role. The CO2 adsorption is also enhanced by the presence of increased π–π interactions in the polymer skeleton due to the interaction between the quadrupole moments of CO2 and the π-clouds in the materials. The CO2 adsorption trend for the polymers is in agreement with their observed surface area calculated from the N2 sorption isotherm. The high adsorption of CO2 by TT-CPP1 may be attributed to its highest surface area amongst all the polymers, its small pore size and the high percentage of micropore volume. The reversible nature of these isotherms confirms the CO2 adsorption over TT-CPP-1/2/3 to be physisorption in nature. The CO2 isotherms measured at 263, 273 and 298 K were utilized to calculate the isosteric heat of adsorption (Qst) by employing the Clausius–Clapeyron equation. Fig. 8d shows a plot of the isosteric heats of adsorption against the CO2 uptake. These Qst values are below 40 kJ mol−1, i.e., the energy of chemical bond formation. This further supports the absence of chemisorption and the presence of strong physisorption of CO2 molecules at the polymer surface containing triazine and thiophene units. The recyclability of CO2 uptake for TT-CPP1 (taken as a representative example) is very good, as evident from the uptake after four consecutive adsorption cycles at 298 K (Fig. S12, ESI†). TT-CPP1 with a moderate surface area of 545 m2 g−1 shows decent CO2 adsorption of 2.18 mmol g−1 at 273 K and 1 bar if compared to the CO2 adsorption of some best-reported materials such as TPMTP52 (SBET = 890 m2 g−1, CO2 adsorption: 5.8 mmol g−1 at 273 K and 1 bar), P-PCz53 (SBET = 890 m2 g−1, CO2 adsorption: 5.57 mmol g−1 at 273 K and 1 bar), and HAT-CTF-450/60054 (SBET = 1090 m2 g−1, CO2 adsorption: 6.3 mmol g−1 at 273 K and 1 bar). Although TT-CPP2 and TT-CPP3 show moderate CO2 adsorption, their adsorption capacity is comparable and even higher in some cases compared to some CPPs reported in the literature with a very high surface area (Table S2, ESI†).
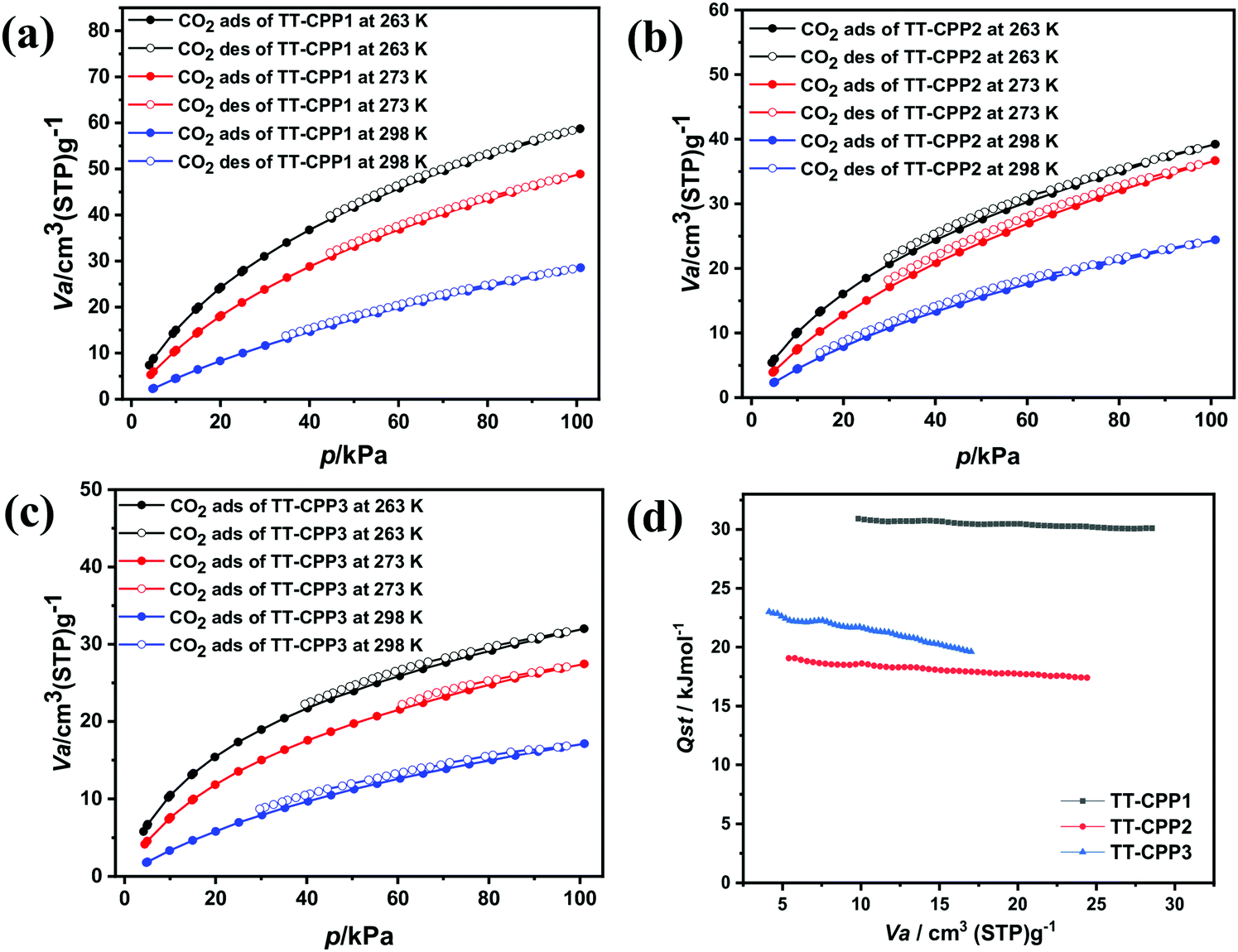 | ||
| Fig. 8 CO2 adsorption–desorption isotherms of (a) TT-CPP1, (b) TT-CPP2 and (c) TT-CPP3 at different temperatures, and (d) Qst of CO2 for the TT-CPPs. | ||
To gain a clearer understanding of the highly efficient uptake of CO2 for TT-CPP1 over TT-CPP2/3, CBMC molecular simulations in BIOVIA, Materials Studio, have been carried out. At first, TT-CPP1/2/3 containing (1 × 1 × 1) hexagonal unit cells has been optimized, and CO2 sorption was performed at 298 K up to a fixed pressure of 1 bar (Fig. 9). TT-CPP-1 displays strong quadrupole–quadrupole and dipole–quadrupole interactions with high binding energy (BE = −30.5 kJ mol−1) compared to TT-CPP2 (BE = −18.8 kJ mol−1) and TT-CPP3 (BE = −20.6 kJ mol−1) (Fig. 9). These BE values are correlated well with their pore sizes. Both the C and O atoms of CO2 interact with S of thiophene (S⋯O–C = 3.2 Å) and triazine N of TT-CPP-1 (C–O⋯N = 3.6 Å).
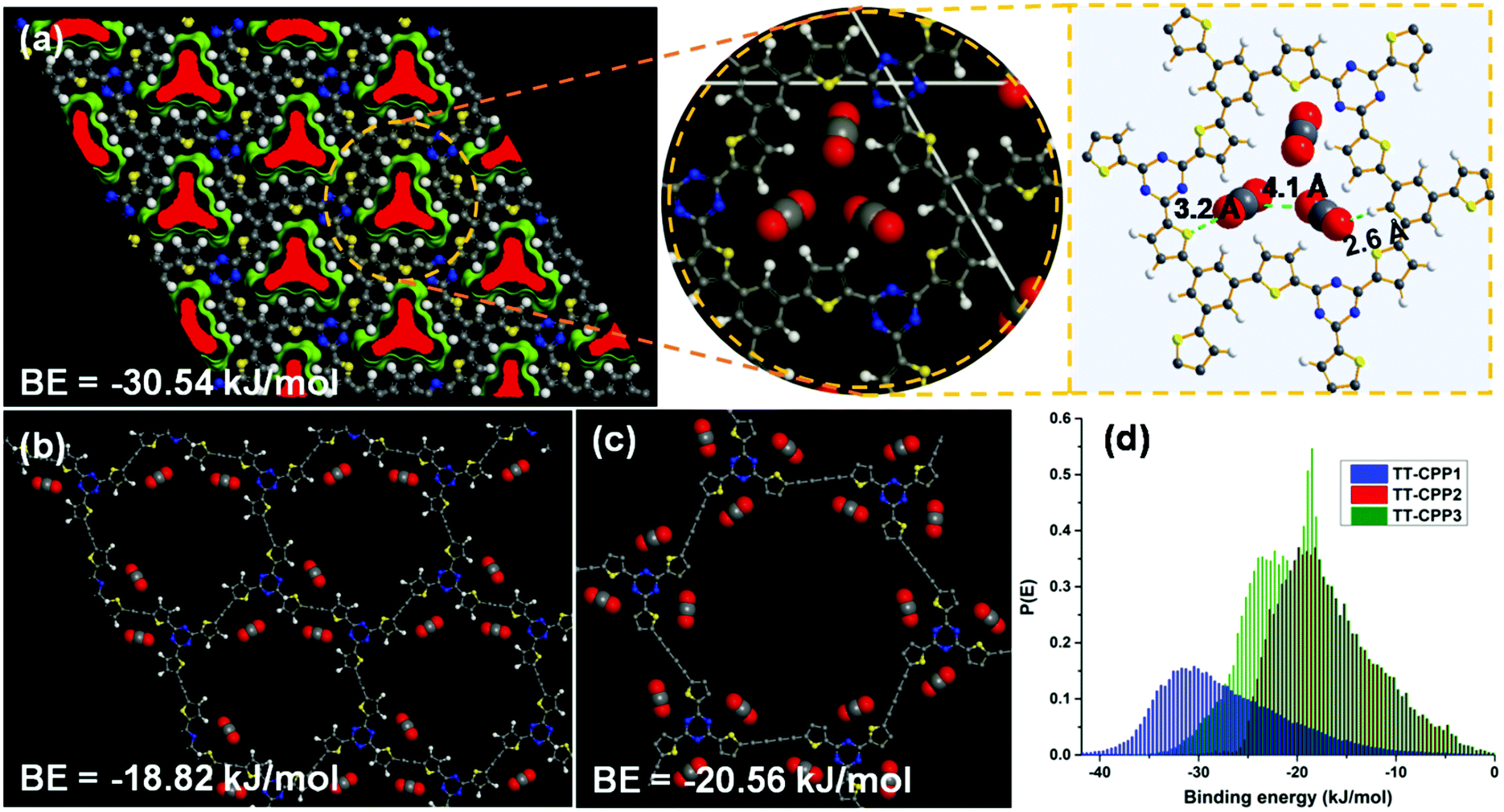 | ||
| Fig. 9 CBMC molecular simulation: sorption and location of CO2 at 298 K for (a) TT-CPP1, (b) TT-CPP2 and (c) TT-CPP-3. (d) CO2 binding energy. | ||
Conclusions
In summary, we describe the synthesis of a series of heteroatom enriched conjugated porous polymers TT-CPP (1–4) based on the thienyltriazine system. Their surface area, pore size and band gap have been tuned efficiently and hierarchically. The CPPs display high thermal and chemical stabilities and high CO2 adsorption, indicating the potential of these polymers in the CO2 sequestration process. TT-CPP1 showed the highest adsorption of about 2.6 mmol g−1 (11.4 wt%). The polymers display band gaps in the range of 2.03–2.43 eV. This study is relevant for the application of CPPs in organic electronics, optoelectronics, and photochemical water splitting, and the high sulfur content of these materials makes them a potential candidate for application in supercapacitors and lithium-ion batteries.55–60Author contributions
All authors have given their consent to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. K. M. acknowledges IISER Mohali for funding. N. R. K. thanks DST for an INSPIRE fellowship. A. R. A. thanks IISER Kolkata for a fellowship. We also acknowledge TEM, DST-FIST facility, IISER Kolkata, JEM-2100F. SERB, India is acknowledged for funding (CRG/2018/002784).Notes and references
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., 2006, 118, 1836–1839 CrossRef.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- C. Gu, D. Liu, W. Huang, J. Liu and R. Yang, Polym. Chem., 2015, 6, 7410–7417 RSC.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 12727–12731 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- M. H. Zeng, Q. X. Wang, Y. X. Tan, S. Hu, H. X. Zhao, L. S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- A. Laybourn, R. Dawson, R. Clowes, T. Hasell, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2014, 5, 6325–6333 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- J. X. Jiang, F. Su, H. Niu, C. D. Wood, N. L. Campbell, Y. Z. Khimyak and A. I. Cooper, Chem. Commun., 2008, 486–488 RSC.
- C. D. Wood, B. Tan, A. Trewin, F. Su, M. J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916–1921 CrossRef CAS.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Ghanem, K. Msayib, D. Book and A. Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802–1808 RSC.
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS.
- A. Li, R. F. Lu, Y. Wang, X. Wang, K. L. Han and W. Q. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330–3333 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Mullen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- J. X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS PubMed.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- Y. Xie, T. T. Wang, R. X. Yang, N. Y. Huang, K. Zou and W. Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- Y. B. Zhou and Z. P. Zhan, Chem. – Asian J., 2018, 13, 9–19 CrossRef CAS PubMed.
- J. Huve, A. Ryzhikov, H. Nouali, V. Lalia, G. Auge and T. J. Daou, RSC Adv., 2018, 8, 29248–29273 RSC.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- J. X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- H. Bildirir, V. G. Gregoriou, A. Avgeropoulos, U. Scherf and C. L. Chochos, Mater. Horiz., 2017, 4, 546–556 RSC.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed.
- V. S. Vyas, V. W.-h Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- H. Bohra, S. Y. Tan, J. Shao, C. Yang, A. Efrem, Y. Zhaob and M. Wang, Polym. Chem., 2016, 7, 6413–6421 RSC.
- J. Lee, O. Buyukcakir, T. Kwon and A. Coskun, J. Am. Chem. Soc., 2018, 140, 10937–10940 CrossRef CAS PubMed.
- X. Qian, Z. Q. Zhu, H. X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- H. Bohra and M. Wang, ACS Appl. Polym. Mater., 2019, 1, 1697–1706 CrossRef CAS.
- W. K. An, S. J. Zheng, Y. N. Du, S. Y. Ding, Z. J. Li, S. Jiang, Y. Qin, X. Liu, P. F. Wei, Z. Q. Cao, M. Song and Z. Pan, Catal. Sci. Technol., 2020, 10, 5171–5180 RSC.
- C. Song, B. Liu, J. Nie, C. Ma, C. Lu, F. Wang, G. Yang and Z. Chen, ACS Sustainable Chem. Eng., 2020, 8, 14377–14385 CrossRef CAS.
- T. M. Geng, C. Hu, M. Liu, C. Zhang, H. Xu and X. Wang, New J. Chem., 2020, 44, 19663–19671 RSC.
- R. Maragani and R. Misra, Tetrahedron Lett., 2013, 54, 5399–5402 CrossRef CAS.
- H. Muraoka, M. Mori and S. Ogawa, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1382–1391 CrossRef CAS.
- Z. Wang, J. Liu, Y. Fu, C. Liu, C. Pan, Z. Liu and G. Yu, Chem. Commun., 2017, 53, 4128–4131 RSC.
- Y. Xu, D. Chang, S. Feng, C. Zhang and J. X. Jiang, New J. Chem., 2016, 40, 9415–9423 RSC.
- C. Zhang, X. Yang, Y. Zhao, X. Wang, M. Yu and J. X. Jiang, Polymer, 2015, 61, 36–41 CrossRef CAS.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- M. D. W. Hussain, S. Bandyopadhyay and A. Patra, Chem. Commun., 2017, 53, 10576–10579 RSC.
- T. Jin, Y. Xiong, X. Zhu, Z. Tian, D. J. Tao, J. Hu, D. Jiang, H. Wang, H. Liu and S. Dia, Chem. Commun., 2016, 52, 4454–4457 RSC.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138(36), 11497–11500 CrossRef CAS PubMed.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS.
- H. Bohra, P. Li, C. Yang, Y. Zhao and M. Wang, Polym. Chem., 2018, 9, 1972–1982 RSC.
- K. D. Vogiatzis, A. Mavrandonakis, W. Klopper and G. E. Froudakis, Chem. Phys. Chem., 2009, 10, 374–383 CrossRef CAS PubMed.
- S. Hug, L. Stegbauer, H. Oh, M. Hirscher and B. V. Lotsch, Chem. Mater., 2015, 27, 8001–8010 CrossRef CAS.
- X. Liu, S. Wang, A. Wang, J. Chen, Z. Wang, Q. Zeng, W. Liu, Z. Li and L. Zhang, J. Phys. Chem. C, 2019, 123, 21327–21335 CrossRef CAS.
- S. K. Kundu, A. Kayet, R. Baidya, L. Satyanarayana and D. K. Maiti, ACS Omega, 2020, 5, 394–405 CrossRef CAS PubMed.
- S. K. Kundu and A. Bhaumik, ACS Sustainable Chem. Eng., 2016, 4, 3697–3703 CrossRef CAS.
- W. Huang, J. Byun, I. Rcrich, C. Ramanan, P. W. M. Blom, H. Lu, D. Wang, L. C. Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8316–8320 CrossRef CAS PubMed.
- M. E. Bhosale, R. Illathvalappil, S. Kurungot and K. Krishnamoorthy, Chem. Commun., 2016, 52, 316–318 RSC.
- Z. Wang, J. Liu, Y. Fu, C. Liu, C. Pan, Z. Liu and G. Yu, Chem. Commun., 2017, 53, 4128–4131 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for the synthesis of the monomers and polymers, 1H spectra of the precursors and solid-state 13C NMR spectra of TT-CPP4, FTIR spectra of the polymers and their precursors, complete characterization of the polymer TT-CPP4 (morphology, N2 adsorption–desorption isotherm, PXRD, TGA, solid-state UV-vis spectra, Kubelka–Munk plot for the band gap) and elemental analysis result of the polymers, FTIR spectra of the polymers after treatment with an acid and a base and 1H NMR spectra of the monomers. See DOI: 10.1039/d1ma00621e |
| This journal is © The Royal Society of Chemistry 2021 |