
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous phase control and carbon intercalation of MoS2 for electrochemical hydrogen evolution catalysis†
Jun
Xu‡
abc,
Xinjiao
Cui‡
abc,
Zhengwen
Fan
a,
Xinxin
Zhu
a,
Wei
Guo
bd,
Zhizhong
Xie
a,
Dan
Liu
 a,
Deyu
Qu
a,
Haolin
Tang
bd and
Junsheng
Li
*abd
a,
Deyu
Qu
a,
Haolin
Tang
bd and
Junsheng
Li
*abd
aSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: li_j@whut.edu.cn
bFoshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory, Xianhu hydrogen Valley, Foshan 528200, P. R. China
cResearch Center for Materials Genome Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
dHubei provincial key laboratory of fuel cell, Wuhan University of Technology, 122 Luoshi Road, Wuhan 430070, P. R. China
First published on 13th September 2021
Abstract
Molybdenum sulfide is considered to be an alternative material to commercial platinum catalysts for the electrocatalytic hydrogen evolution reaction (HER). 1T-MoS2 has higher hydrophilicity and electronic conductivity than 2H-MoS2 and is more favorable for the HER. However, 1T-MoS2 is not stable and easily transformed into a more stable 2H phase during the synthesis process. Therefore, it is crucial to find a suitable method to synthesize highly active MoS2 HER electrocatalysts. In this work, a carbon intercalated 1T-MoS2 electrocatalyst (MoS2-2C) is synthesized via a glucose-assisted hydrothermal approach. The introduction of glucose in the synthesis not only induces the formation of active 1T-MoS2 but also modifies the electronic structure of the in-plane sites of 1T-MoS2. The electronic status of MoS2-2C is studied in detail. Our results show that the free energy of HER at the optimal adsorption site of MoS2-2C is as low as 0.10 eV. Therefore, it shows promising HER catalytic activity (η = 217 mV@10 mA cm−2) and stability in 0.5 M H2SO4.
1. Introduction
MoS2 is considered a potential catalyst for the hydrogen evolution reaction (HER). The practical HER activity of MoS2 is largely determined by its phase structure. A typical MoS2 material has 5 common phase structures, namely 1H, 2H, 1T, 1T’, and 3R phases.1–4 The most thermodynamically stable MoS2 phase is the 2H phase.5 However, only the edge sites of 2H-MoS2 display good HER catalytic activity, and a large number of in-plane sites are catalytically inert.6,7 In comparison, 1T-MoS2 shows better electronic conductivity8,9 and higher hydrophilicity.10 In addition, it has a unique electronic structure that is favorable for the HER. Unfortunately, 1T-MoS2 is thermodynamically unstable and thus difficult to prepare with routine synthetic approaches.11It is of great interest to obtain stable 1T-MoS2 through a simple and controllable method for the development of high-performance MoS2-based HER catalysts. Typical methods for the synthesis of 1T-MoS2 can be divided into “top-down” and “bottom-up” methods. The “top-down method” mainly includes the ion-intercalation method.2,9,12–17 A commonly used method is lithium-intercalation,13,17 where Li+ can be used as an electron donor after intercalation, promoting the transformation of the 2H phase to the 1T phase. However, the synthetic process is complicated. Moreover, the conversion rate of 1T-MoS2 obtained by this process is generally lower than 80%.11,13 The “bottom-up method” consists of hydro-/solvothermal methods,18–25 the Chemical Vapor Deposition (CVD) method,26,27etc. Many studies have recently reported the synthesis of 1T-MoS2 by solvothermal methods, such as that by Yang et al. who grew vertically aligned 2H-1T MoS2 in DMF in a Teflon-lined stainless steel autoclave.19 This method mainly relies on the growth of MoS2 in a solvent at low temperatures.19,28,29 The HER activity of 1T-MoS2 can be further boosted by the introduction of a secondary synergistic phase that tunes the electronic status and regulates the conductive pathways of 1T-MoS2. Intercalation of MoS2 with carbon-based materials has been attempted to improve the HER performance.22,24,30–32 However, most of the reported approaches could not simultaneously achieve a high 1T phase yield, effective modulation of the electronic structure and improvement in the electronic conductivity.
In this work, carbon-intercalated 1T-MoS2 (MoS2-2C) was synthesized by hydrothermal treatment and subsequent carbonization with glucose as the carbon source. By control over the amount of glucose used in the synthesis, an almost pure 1T-MoS2 phase was formed. The glucose-derived carbon did not only stabilize the 1T phase but also suppressed the stacking of MoS2 layers, thereby increasing the exposure of catalytically active sites. Besides, the synergy of C and MoS2 effectively modifies the electronic structure of the S sites, thus further improves the activity of MoS2-2C. As a result of these favorable features, MoS2-2C exhibits a promising HER performance in the acid system.
2. Experimental section
2.1 Synthetic procedures
2.2 Characterization
Raman spectroscopy was used to characterize the chemical composition and chemical state. The measurement range was 20–3000 cm−1 (532 nm). The XRD pattern was obtained with the Empyrean X-ray diffractometer manufactured by Malvern Panalytical Company in the Netherlands. The wide-angle X-ray diffraction test was performed and the scanning range was 5–90°. The scanning electron microscope testing was conducted using a JSM-IT300 field emission scanning electron microscope manufactured by JEOL Co., Ltd and the accelerating voltage was 20.0 kV. The transmission electron microscope testing was performed using a JEM-2100F transmission electron microscope made by JEOL Ltd. The XPS spectrum was obtained from Thermo Scientific K-Alpha from ThermoFisher (Mono Al Kα). The nitrogen adsorption–desorption curves were measured by the ASAP2460 analyzer from Micromeritics, USA, and the test was performed under 77 K liquid nitrogen. The sample was degassed at 200 °C for 12 h before testing. The Elementar vario MICRO cube was used to analyze the content of C, H, N, and S in the sample.2.3 Electrochemical Measurement
5 mg sample was evenly dispersed in 900 μL isopropanol, 100 μL deionized water, and 20 μL of Nafion solution (5 wt%). 20 μL of the above ink was transferred to the rotating disk electrode (5 mm). 20 wt% commercial Pt/C was chosen to make a comparison. After naturally drying, the film was applied to test. All electrochemical tests were completed by the Correst Electrochemical Workstation in a three-electrode system in 0.5 M H2SO4. The rotating disk electrode coated with the catalyst acted as the working electrode, the saturated calomel electrode (SCE) was selected as the reference electrode, and the graphite electrode was applied as the counter electrode. The electrode was activated firstly by CV scanning during −0.3 to 0.4 V (vs. SCE) and the scan speed was set as 500 mV s−1. The polarization curve was obtained by linear sweep voltammetry (LSV), the rotating disk electrode speed was set to 1600 rpm, the potential scanning range was −1 to 0 V (vs. SCE), and the scanning speed was 2 mV s−1. To obtain the Cdl value, CV scans were performed at scan speeds of 20, 40, 60, 80, and 100 mV s−1, and the scan potential window was set to −0.25 to −0.05 V (vs. SCE). The ECSA can be calculated using eqn (1).33 The electrochemical impedance spectrum (EIS) test was measured at a potential of −0.5 V (vs. SCE). The frequency test range is 0.1 to 105 Hz and the amplitude is 5 mV. The stability was tested by the CV method, the scan range was set to −0.3–0.1 V (vs. RHE), the number of cycles was set to 2000, and the changes in the LSV curve were compared before and after 2000 cycles. The chronoamperometry test was also applied under an overpotential of 260 mV (without iR correction) to explore the stability. iR compensation (100%) was applied using the solution resistance measured by the EIS tests. The potential was converted by eqn (2), and the value was obtained by calibration test.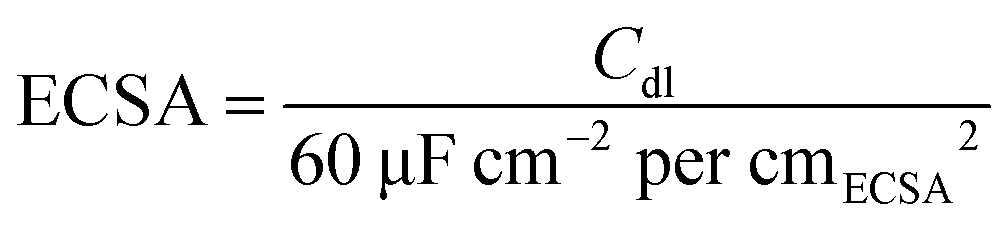 | (1) |
| ERHE = ESCE + 0.269 V | (2) |
2.4 Theoretical calculations
The 1T-MoS2 unit cell was imported from the crystal structure database, and the unit cell constant was optimized. A slab model was built toward (002), the vacuum layer was set to 15 Å, and the cell constant was 13.33 Å. The structure of VS-1T-MoS2 with sulfur defects was obtained by deleting an S atom based on the above structure. To simulate the effect of carbon (C) on the hydrogen evolution activity of 1T-MoS2, a heterojunction structure of 1T-MoS2/C was constructed in which C was simulated by the graphite structure. The hydrogen adsorption model was built by adding H atoms about 1.41 Å above the S atoms on the corresponding structure surface.All model optimization and energy calculation were completed by the DMol3 module of Materials Studio.34,35 The generalized gradient approximation method (GGA) and Perdew–Burke–Ernzerhof (PBE) function were used to deal with the exchange–correlation energy,36 and the DSPP (DFT Semi-core Pseudopots) method was used to process the core electrons.37 The basis set chosen was DNP. The convergence criteria for energy change, max force and displacement of atoms were set to 1 × 10−5 Ha, 2 × 10−3 Ha Å−1 and 5 × 10−3 Å, respectively. After a convergence test, the accuracy of orbital cutoff was set to 4.9 Å and the k points for structural optimization and calculation of electronic structure properties were set to 4 × 4 × 1 and 6 × 6 × 1, respectively. To simulate the aqueous environment, the Conductor-like Screening Model (COSMO) model38 was introduced when calculating the hydrogen adsorption energy, the solvent was water and the electrostatic constant was set to 78.54. Besides, considering the existence of van der Waals forces in two-dimensional materials, Grimme's DFT-D method was used to correct van der Waals forces in the calculation process.39 The calculation and analysis of electron density differences were completed by the CASTEP module.40 The calculation formulas were as follows.
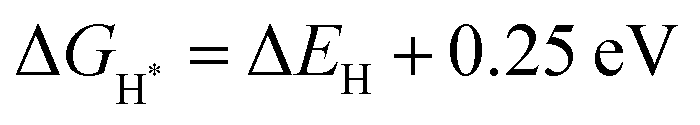 | (3) |
| ΔEH = E(MoS2 + H) − E(MoS2) − 1/2E(H2) | (4) |
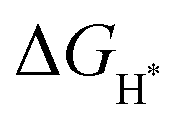 represents the free energy of the reaction and it can be calculated using eqn (3),41 ΔEH represents the energy of adsorption, E(MoS2 + H) represents the energy of the surface with an H adsorbed, E(MoS2) represents the energy of the MoS2 surface, and E(H2) represents the energy of hydrogen.
represents the free energy of the reaction and it can be calculated using eqn (3),41 ΔEH represents the energy of adsorption, E(MoS2 + H) represents the energy of the surface with an H adsorbed, E(MoS2) represents the energy of the MoS2 surface, and E(H2) represents the energy of hydrogen.
Results and discussion
The samples were prepared with a simple hydrothermal process, followed by a carbonization procedure as shown in Scheme 1. By varying the glucose content used in the hydrothermal process, we synthesized MoS2-1C, MoS2-2C, MoS2-5C, pure MoS2, and pure C (only glucose was added).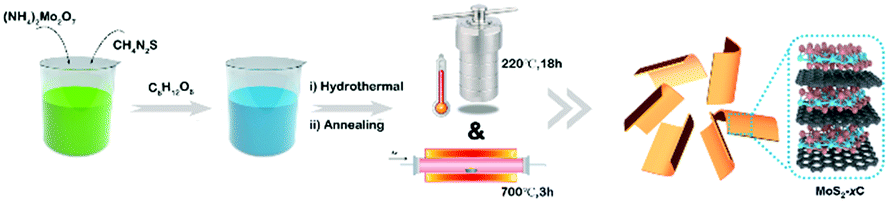 | ||
| Scheme 1 Schematic illustration for the synthesis of MoS2-xC. | ||
Then, the effect of glucose on the structure of MoS2, especially the phase structure, was explored through characterizations and theoretical calculations. As we can see from the Raman spectrum (Fig. 1(a)), the pure MoS2 sample only has the in-plane vibration E2g1 peak (378 cm−1) and the out-of-plane vibration A1g peak (404 cm−1), which are assigned to 2H-MoS2. Characteristic peaks of 1T-MoS2, J1 (142 cm−1), J2 (232 cm−1), and J3 (330 cm−1) peaks can be observed in the spectra of MoS2-1C and MoS2-2C samples. In addition, the obvious E1g peak at 278 cm−1 can also be seen that is related to the octahedral structure of 1T-MoS2.11 These results indicated glucose assisted the formation of 1T-MoS2. The Raman spectra of MoS2-1C, MoS2-2C, and MoS2-5C also showed peaks assigned to the carbon (D band at 1360 cm−1 and G band at 1580 cm−1) in Fig. 1(a).42 Particularly, only peaks from the carbon could be identified in the Raman spectra of MoS2-5C, which resulted from the excess glucose used in the synthesis.
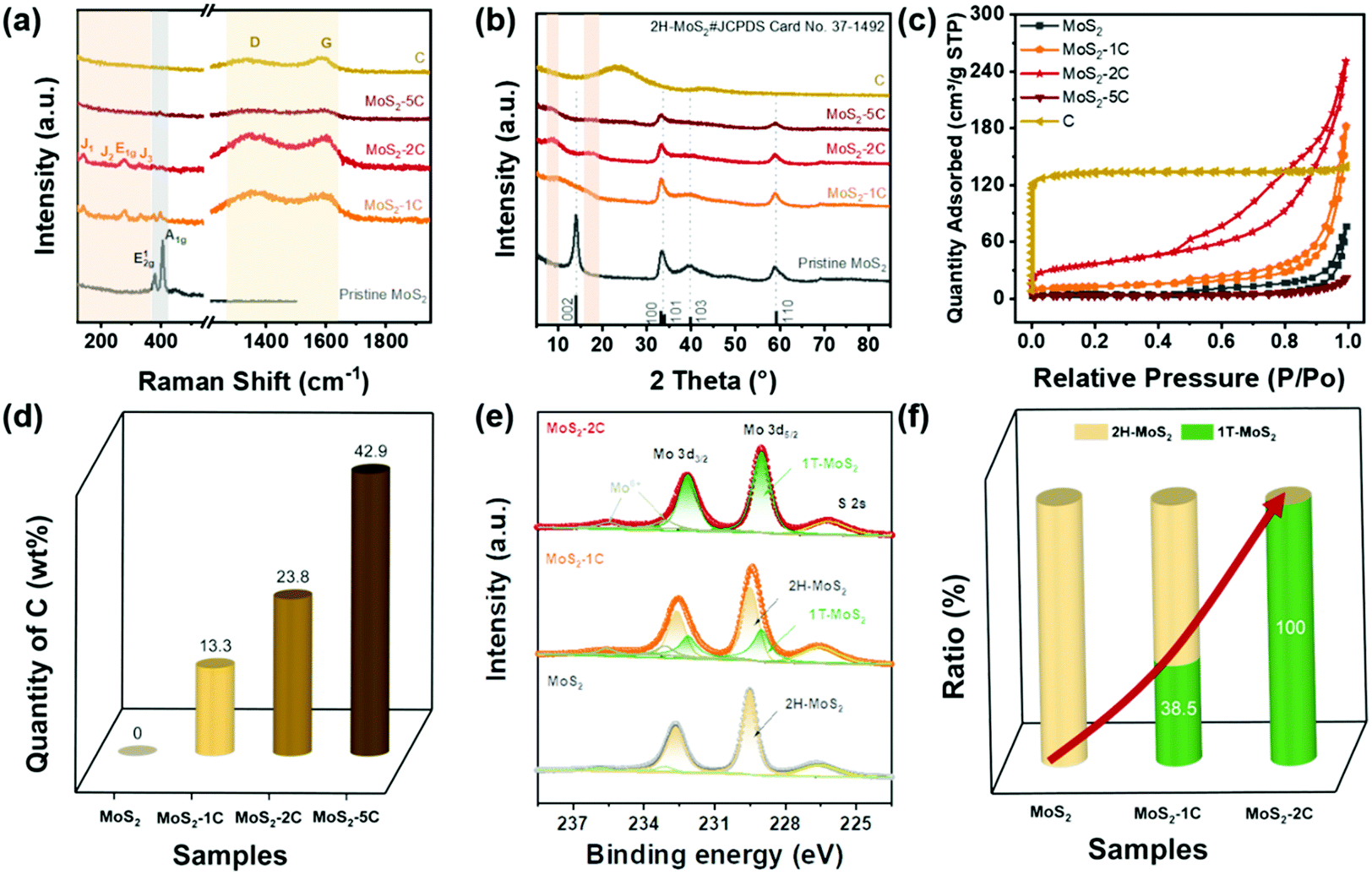 | ||
| Fig. 1 (a) Raman spectrum, (b) XRD pattern for MoS2-xC and C, (c) N2 adsorption–desorption type IV isotherms measured at 77 K of MoS2-xC and C, (d) the comparison of carbon content in MoS2-xC, and (e and f) XPS spectra comparisons among MoS2, MoS2-1C, and MoS2-2C. | ||
XRD measurements were conducted to further analyze the crystal structure of the catalysts. It's noted in Fig. 1(b) that pristine MoS2 showed peaks at 14.38°, 32.68°, 33.51°, and 39.54°, corresponding to (002), (100), (101), and (103) of 2H-MoS2 (JCPDS card number: 37-1492), respectively. While for MoS2-xC, the (002) peak shifted negatively, which appeared at 9.83°, 9.04°, and 8.85° for MoS2-1C, MoS2-2C, and MoS2-5C, respectively. These interplanar spacings were calculated to be 0.90 nm, 0.98 nm, and 1.00 nm. Besides, a second diffraction peak of the MoS2-2C was present at 2θ = 17.75°. These results further proved that 1T-MoS2 was successfully synthesized with the addition of glucose.11
The porous features of the samples were investigated with a N2 adsorption–desorption isotherm. As shown in Fig. 1(c), MoS2, MoS2-1C, MoS2-2C, and MoS2-5C exhibit typical IV isotherm characteristics. The hysteresis loop is attributed to common features of the seam structure of two-dimensional MoS2. The pore size distribution curves were obtained by the BJH method (Fig. S1, ESI†), and MoS2-1C, MoS2-2C and raw carbon display plenty of micropores and mesopores. The BET surface area and pore volume were analyzed from isotherms and are shown in Table S1 (ESI†). Among them, MoS2-2C displays the largest specific surface area (130 m2 g−1) and pore volume (0.39 cm3 g−1), which may be related to its high 1T phase ratio and less stacked structures. Since additional precursors were introduced in the synthesis of MoS2-xC, the morphology of the thus-formed carbon is distinct from the pristine carbon (synthesized using identical hydrothermal conditions). Therefore, the measured BET surface area of MoS2-xC was largely different from that of pristine carbon. Compared to MoS2-xC, pristine carbon has an abundant microporous structure, which contributed to its large specific surface area.
The relative contents of different elements in different samples were estimated through organic element analysis (Fig. 1(d) and Table S2 ESI†). The mass percentage of C in the MoS2-2C sample is about 23.8 wt%. Parts of carbon were intercalated to the interlayer of MoS2 which was proved by the increased interlayer spacing determined from XRD (Fig. 1(b)) and HRTEM (Fig. 2(b)) analysis. In addition, carbon aggregates also formed between MoS2 nanoparticles (Fig. S9(b), ESI†). The proportion of these two types of carbon is difficult to identify and will be investigated in future studies. It is worth noting that the sample may contain a small amount of O element, but no obvious oxide species were observed in the test and through characterization such as XRD, indicating that the O content is extremely low. Assuming that oxygen is absent in MoS2-xC, the atomic percentage of Mo and S can be calculated. The nS/nMo in all MoS2-xC samples is less than 2, which indicates these samples contain S vacancy defects that may result from the excess thiourea.43
 | ||
| Fig. 2 (a) SEM image of MoS2, MoS2-xC and C. (b) TEM image of MoS2-2C. (c) The enlarged HRTEM image of MoS2-2C (the white cycles represent the region of S vacancy). (d) High-Angle Annular Dark Field TEM images of the nanoflake and corresponding element mapping of MoS2-2C. | ||
The element and phase composition of MoS2-2C, MoS2-1C, and MoS2 were further analyzed by the XPS spectra (Fig. 1(e), and Fig. S2 and S3, ESI†). As shown in Fig. 1(e), the binding energies of Mo 3d5/2 and 3d3/2 peaks are located at 229.0 eV and 232.1 eV in 1T-MoS2, respectively.11 The binding energies of Mo 3d5/2 and 3d3/2 peaks are 229.5 eV and 232.6 eV in 2H-MoS2, respectively.11 In addition, there are Mo6+ peaks at binding energies of 233.1 eV and 235.6 eV, and S 2s peaks can be seen at 226.5 eV. The proportion of the 1T phase in MoS2-xC can be calculated by the ratio of integrated peak areas of the 1T phase and 2H phase (Fig. 1(f)). The proportion of the 1T phase in MoS2, MoS2-1C, and MoS2-2C was calculated to be 0%, 38.5%, and 100%, respectively. Combined with the analysis of Raman and XPS data, it can be concluded reasonably that the phase structure in MoS2-xC can be effectively adjusted by controlling the amount of glucose in the hydrothermal synthesis. The S vacancy was further verified by the XPS results (eqn (S2) and Table S3, ESI†). The values of n(s)/n(Mo) in 1T-MoS2 for MoS2-1C and MoS2-2C are both smaller than 2.0 while the values in 2H-MoS2 for MoS2-1C and MoS2 are very close to 2.0, which indicates that S vacancies preferably existed in the 1T phase of MoS2-1C and MoS2-2C. Furthermore, it was proved by the smaller formation energy of the S vacancy in 1T-MoS2 obtained by DFT calculations (Fig. S5 and eqn (S1), ESI†). The existence of S vacancies may enhance the intrinsic hydrogen evolution activity of 1T-MoS2.
The morphology of MoS2-xC was observed using a scanning electron microscope (SEM, Fig. 2(a)). Pristine MoS2 seems like nanoflowers with abundant nanoflakes on the surface. The morphology of MoS2-1C and MoS2-2C is similar to that of pristine MoS2, but their size is significantly smaller, which suggests that glucose effectively inhibits the layer stacked during the growth of MoS2. The nanostructures of MoS2-1C and MoS2-2C can provide abundant reaction sites for the hydrogen evolution reaction. With the amount of glucose increasing in the synthesis, carbon is the main component wrapping MoS2 layer by layer. The surface of the MoS2-5C nanoparticle is smooth and similar to that of pristine C (Fig. 2(a)). TEM and EDS were carried out on MoS2-2C to further explore the internal structure and composition of the sample. In the HRTEM image of MoS2-2C (Fig. 2(b)), the interlayer spacing was measured to be 0.95 nm. This value is close to the layer spacing of 0.98 nm measured by XRD (Fig. 1(b)), which also proved that the 1T phase structure was successfully obtained. The distance between atoms was also analyzed with an HRTEM image, and the relative atomic coordinate position 1T-MoS2 structure can be observed (Fig. 2(c)). The atomic signal was projected into the same plane, and the distance between the Mo atom and S atom is about 0.21 nm, while the atomic distance between S and S is about 0.17 nm, which is quite different from the overlap of the projection signals of two S atoms in the 2H phase structure. Moreover, some defects can be observed in HRTEM images (marked with white circles in Fig. 2(c)). Fig. 2(d) shows High-Angle Annular Dark Field TEM images and corresponding element mapping of MoS2-2C. As can be seen in the figure, the Mo, S, and C elements are evenly distributed in the structure, which suggests the even intercalation of carbon in the 1T-MoS2 layers.
To analyze the synergy between carbon and 1T-MoS2, a series of theoretical calculations were carried out. Different structural models, including 1T-MoS2, 1T-MoS2 with S vacancy (Vs-1T-MoS2) and carbon intercalated 1T-MoS2 with S vacancy (Vs-1T-MoS2/C) (shown in Fig. S4, ESI†). Firstly, the electron density difference of Vs-1T-MoS2/C was calculated and the result is shown in Fig. 3(a). The red region indicates the electron-rich area, and the purple region indicates the lack of electrons. This result suggests electron transfer from intercalated C to 1T-MoS2. This kind of electron transfer process may enhance the structural stability of 1T-MoS2.11 Such a modification on the electronic structure of 1T-MoS2 may enhance its electrocatalytic activity. The free energy of HER was calculated on 1T-MoS2, Vs-1T-MoS2 and Vs-1T-MoS2/C (Fig. 3(b)). For the pure 1T-MoS2, the value is determined to be −0.95 eV which means the H atom could be easily adsorbed onto the S site. However, the hydrogen generation and desorption process are kinetically difficult. After an S-vacancy is introduced in 1T-MoS2, the free energy is reduced to −0.32 eV, which is closer to the ideal value of 0 eV, while the value of Vs-1T-MoS2/C is only 0.10 eV, which suggests that a carbon-intercalated structure can modify the electron structure of 1T-MoS2 and is beneficial for the HER process.
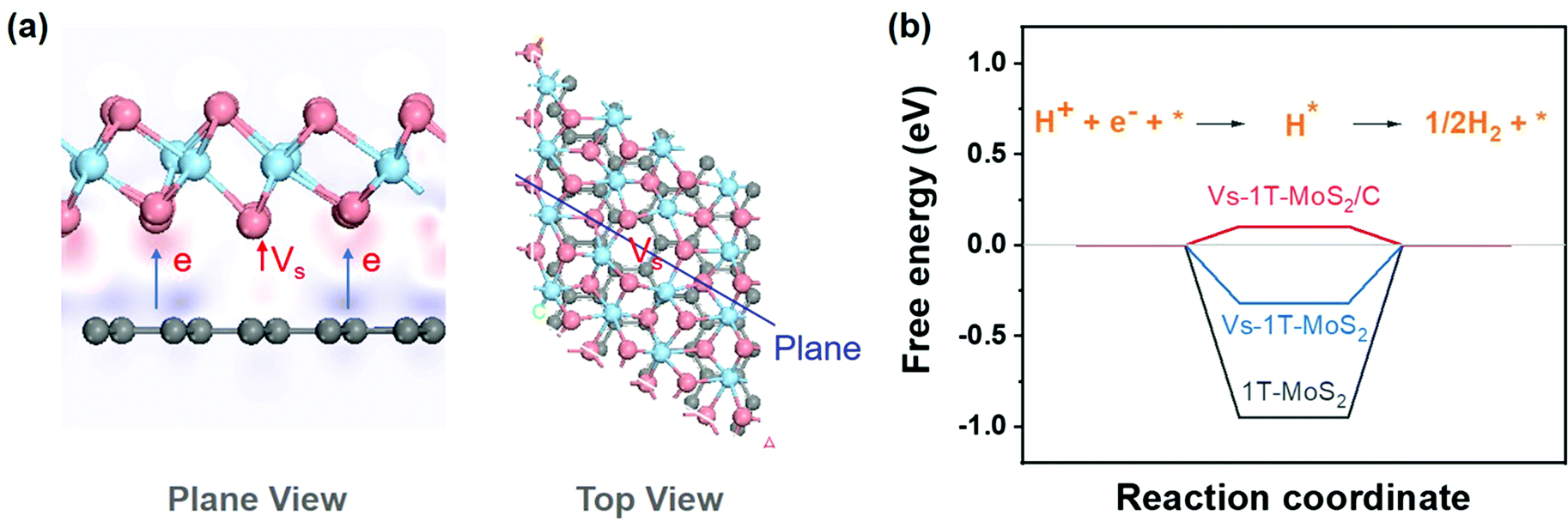 | ||
| Fig. 3 (a) Diagram of electron density difference (pink atoms represent S, blue atoms represent Mo, and gray atoms represent C). (b) The diagram of free energy change of the HER. | ||
Based on the above discussion, MoS2-2C has a high proportion of 1T phase, a large specific surface area, and favorable electronic structures. Therefore, its better HER activity can be expected. A series of electrochemical tests were performed to explore the electrochemical performance of MoS2-xC and the test device is shown in Fig. S6 (ESI†). Fig. 4(a) shows the LSV polarization curves of different samples. The onset overpotentials of MoS2, MoS2-1C, MoS2-2C, MoS2-5C, and C were 220, 203, 120, 242, and 382 mV, respectively. When the current density reached 10 mA cm−2, the overpotential values were 366, 291, 217, 461, and 620 mV, respectively. Among all the samples, MoS2-2C had the best electrochemical performance. The HER Tafel slope of MoS2-2C was only 94 mV dec−1, which indicates faster kinetics, while the Tafel slopes of MoS2, MoS2-1C, MoS2-5C, and C were 144, 98, 207, and 258 mV dec−1, respectively (Fig. 4(b)).
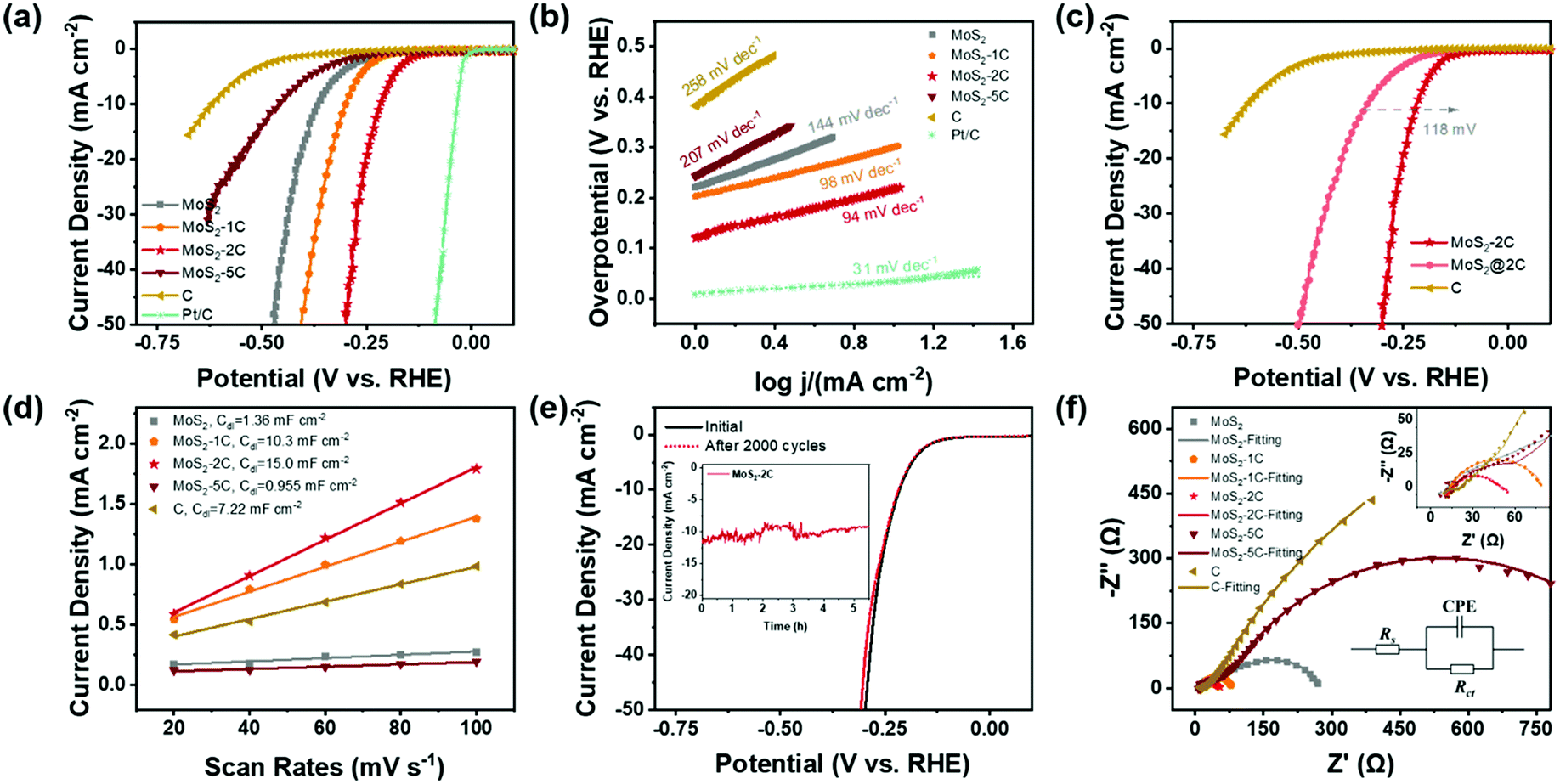 | ||
| Fig. 4 (a) LSV curves (100% iR-corrected), (b) Tafel plots (calculated from iR corrected lines), and (c) LSV curves of different samples. (d) Capacitive currents of the samples as a function of scanning rate in a non-faradaic region. (e) LSV curves of MoS2-2C before and after 2000 CV cycles (chronoamperometry test under an overpotential of 260 mV without iR correction inserted). (f) EIS spectra of MoS2-xC and C in 0.5 M H2SO4 at −0.5 V versus SCE. | ||
To exclude the influence of varying MoS2 content in different samples when assessing their electrochemical performance, the measured LSV curves were normalized to the weight of MoS2 by the results of elemental analysis (Fig. S7, ESI†). The same performance trend was observed among these samples and MoS2-2C still exhibited the highest activity. To explore the role of carbon in the HER, MoS2 synthesized under the same synthesis conditions was directly mixed with the same quantity of carbon as MoS2-2C. The electrochemical performance of this composite sample (termed MoS2@2C) is also characterized. When the current density reached 10 mA cm−2, the overpotential of MoS2@2C is 118 mV higher than MoS2-2C (Fig. 4(c)), indicating that the excellent HER performance of MoS2-2C not only stems from the improved electronic conductivity due to the incorporation of carbon but mostly originates from the higher composition of an active 1T phase.
CV measurements were conducted to discuss the electrochemical surface area of the samples. Fig. S8 (ESI†) displays the CV curves obtained for each sample at different scanning speeds in the non-faradaic current region and Fig. 4(d) shows capacitive currents as a function of scanning rate in a non-faradaic region. The electric double layer capacitances Cdl of MoS2, MoS2-1C, MoS2-2C, MoS2-5C, and carbon are respectively obtained as 1.36, 10.3, 15.0, 0.955, and 7.22 mF cm−2. The capacitance value of the electric double layer of the ideal smooth oxide electrode is 60 μF cm−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 so that the ECSA value of MoS2, MoS2-1C, MoS2-2C, MoS2-5C, and C was 22.7, 172, 250, 15.9, and 120 cmECSA2, respectively. This result agrees well with the BET surface area result and MoS2-2C had the highest electrochemically active surface area. Besides, the sample also showed little attenuation (Fig. 4(e)) after 2000 cycles of CV scanning from −0.3 V to 0.1 V (vs. RHE). The SEM and XPS tests of MoS2-2C were conducted after the chronoamperometry test was conducted under an overpotential of 260 mV (vs. RHE) for 10 hours. There was no obvious change in the morphology of MoS2-2C after 10 h stability test, which still retained the nanoflower-like shape in Fig. S9 (ESI†). We compared the high-resolution XPS spectra of Mo 3d and S 2p of MoS2-2C after and before the stability test (Fig. S10, ESI†), which did not display obvious element state and composition changes that happened after the stability test. Therefore, MoS2-2C developed in this work was proved to exhibit promising stability.
33 so that the ECSA value of MoS2, MoS2-1C, MoS2-2C, MoS2-5C, and C was 22.7, 172, 250, 15.9, and 120 cmECSA2, respectively. This result agrees well with the BET surface area result and MoS2-2C had the highest electrochemically active surface area. Besides, the sample also showed little attenuation (Fig. 4(e)) after 2000 cycles of CV scanning from −0.3 V to 0.1 V (vs. RHE). The SEM and XPS tests of MoS2-2C were conducted after the chronoamperometry test was conducted under an overpotential of 260 mV (vs. RHE) for 10 hours. There was no obvious change in the morphology of MoS2-2C after 10 h stability test, which still retained the nanoflower-like shape in Fig. S9 (ESI†). We compared the high-resolution XPS spectra of Mo 3d and S 2p of MoS2-2C after and before the stability test (Fig. S10, ESI†), which did not display obvious element state and composition changes that happened after the stability test. Therefore, MoS2-2C developed in this work was proved to exhibit promising stability.
The electrochemical impedance spectroscopy test and analysis were also performed (Fig. 4(f)) and the charge transfer resistance (Rct) was obtained by fitting. MoS2-2C also has the smallest charge transfer resistance value (only 35 Ω). The relevant electrochemical performance data are summarized in Table S4 (ESI†). Such a low Rct of MoS2-2C can be correlated to its unique electronic structure, as outlined in the theoretical calculations. A brief comparison of the electrochemical performance of MoS2-2C with representative HER catalysts reported recently is also provided (Table S5, ESI†). It can be seen from the table that MoS2-2C has promising HER catalytic activity among many similar materials reported.
Conclusions
In this paper, a carbon intercalated 1T-MoS2 electrocatalyst (MoS2-2C) was synthesized as an HER electrocatalyst. The structure and performance characterizations show that MoS2-2C has a high content of catalytically active 1T phase. Besides, it displays abundant S vacancies and a big electrochemical surface area. The intercalated carbon can not only increase the electron density of MoS2 to stabilize 1T-MoS2 but can also modify the electronic structure of the S site, leading to an optimized HER free energy (as low as 0.10 eV). Therefore, MoS2-2C shows promising HER activity (η = 217 mV@10 mA cm−2) and stability in 0.5 M H2SO4. We expect that the synthetic approach and the understanding of the performance of MoS2-2C can provide insight into the future development of advanced HER catalysts.Author contributions
Jun Xu and Xinjiao Cui contributed equally. Jun Xu, Xinjiao Cui, and Zhengwen Fan performed the conceptualization, data curation and formal analysis; Xinxin Zhu conducted the investigation; Wei Guo and Junsheng Li contributed to the funding acquisition, project administration and supervision; Dan Liu, Deyu Qu, Zhizhong Xie and Haolin Tang contributed to the methodology; Jun Xu and Junsheng Li contributed to the visualization and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 51972254), Foshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory (XHD2020-002-04) and Fundamental Research Funds for the Central Universities (WUT: 2020IB025) for the financial support.Notes and references
- M. Chhowalla, D. Voiry, J. Yang, H. S. Shin and K. P. Loh, MRS Bull., 2015, 40, 585–591 CrossRef CAS.
- Z. He and W. Que, Appl. Mater. Today, 2016, 3, 23–56 CrossRef.
- A. P. Nayak, T. Pandey, D. Voiry, J. Liu, S. T. Moran, A. Sharma, C. Tan, C.-H. Chen, L.-J. Li, M. Chhowalla, J.-F. Lin, A. K. Singh and D. Akinwande, Nano Lett., 2015, 15, 346–353 CrossRef CAS PubMed.
- T. Heine, Acc. Chem. Res., 2015, 48, 65–72 CrossRef CAS PubMed.
- W. Zhao and F. Ding, Nanoscale, 2017, 9, 2301–2309 RSC.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- S. S. Chou, Y.-K. Huang, J. Kim, B. Kaehr, B. M. Foley, P. Lu, C. Dykstra, P. E. Hopkins, C. J. Brinker, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2015, 137, 1742–1745 CrossRef CAS PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Adv. Energy Mater., 2018, 8, 1703482 CrossRef.
- R. Zhang, I. L. Tsai, J. Chapman, E. Khestanova, J. Waters and I. V. Grigorieva, Nano Lett., 2016, 16, 629–636 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- S. Yan, W. Qiao, X. He, X. Guo, L. Xi, W. Zhong and Y. Du, Appl. Phys. Lett., 2015, 106, 012408 CrossRef.
- P. Gao, L. Wang, Y. Zhang, Y. Huang and K. Liu, ACS Nano, 2015, 9, 11296–11301 CrossRef CAS PubMed.
- D. Kiriya, M. Tosun, P. Zhao, J. S. Kang and A. Javey, J. Am. Chem. Soc., 2014, 136, 7853–7856 CrossRef CAS PubMed.
- Y. Li, L. Wang, S. Zhang, X. Dong, Y. Song, T. Cai and Y. Liu, Catal. Sci. Technol., 2017, 7, 718–724 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- J. Yang, K. Wang, J. Zhu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 31702–31708 CrossRef CAS PubMed.
- Z. Liu, Z. Gao, Y. Liu, M. Xia, R. Wang and N. Li, ACS Appl. Mater. Interfaces, 2017, 9, 25291–25297 CrossRef CAS PubMed.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- S. Jayabal, G. Saranya, Y. Liu, D. Geng and X. Meng, Sustainable Energy Fuels, 2019, 3, 2100–2110 RSC.
- Z. Liu, L. Zhao, Y. Liu, Z. Gao, S. Yuan, X. Li, N. Li and S. Miao, Appl. Catal., B, 2019, 246, 296–302 CrossRef CAS.
- N. Wang, Y. Zhou, S. Yousif, T. Majima and L. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 34430–34440 CrossRef CAS PubMed.
- H. Niu, Z. Zou, Q. Wang, K. Zhu, K. Ye, G. Wang, D. Cao and J. Yan, Chem. Eng. J., 2020, 399, 125672 CrossRef CAS.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- L. Tao, X. Duan, C. Wang, X. Duan and S. Wang, Chem. Commun., 2015, 51, 7470–7473 RSC.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao, T. Hu, Z. Sun and S. Wei, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- S. Shi, D. Gao, B. Xia, P. Liu and D. Xue, J. Mater. Chem. A, 2015, 3, 24414–24421 RSC.
- A. R. P. Santiago, T. He, O. Eraso, M. A. Ahsan, A. N. Nair, V. S. N. Chava, T. Zheng, S. Pilla, O. Fernandez-Delgado, A. Du, S. T. Sreenivasan and L. Echegoyen, J. Am. Chem. Soc., 2020, 142, 17923–17927 CrossRef PubMed.
- J. He, G. Hartmann, M. Lee, G. S. Hwang, Y. Chen and A. Manthiram, Energy Environ. Sci., 2019, 12, 344–350 RSC.
- H. Deng, C. Zhang, Y. Xie, T. Tumlin, L. Giri, S. P. Karna and J. Lin, J. Mater. Chem. A, 2016, 4, 6824–6830 RSC.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- Q. Tang and D.-E. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00681a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |