
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of chemical self-charging zinc ion batteries based on defect coupled nitrogen modulation of zinc manganite vertical graphene arrays†
Wenda
Qiu‡
 *ab,
Zhenchao
Lin‡
a,
Hongbing
Xiao
a,
Guoming
Zhang
a,
Hong
Gao
a,
Huajie
Feng
*c and
Xihong
Lu
b
*ab,
Zhenchao
Lin‡
a,
Hongbing
Xiao
a,
Guoming
Zhang
a,
Hong
Gao
a,
Huajie
Feng
*c and
Xihong
Lu
b
aDepartment of Environmental Engineering, Guangdong Industry Technological College, Guangzhou 510300, China. E-mail: qiuwd3@mail3.sysu.edu.cn
bMOE of the Key Laboratory of Bioinorganic and Synthetic Chemistry, KLGHEI of Environment and Energy Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China
cKey Laboratory of Electrochemical Energy Storage and Energy Conversion of Hainan Province, School of Chemistry and Chemical Engineering, Hainan Normal University, Haikou 571158, China. E-mail: Fenghuajiehk@163.com
First published on 3rd September 2021
Abstract
Self-charging power systems, which can simultaneously achieve energy harvesting and storage, play a significant role in the field of energy technology. Nevertheless, the traditional integrated systems are not only severely restricted by the energy availability, but also have a complex architecture. Herein, a novel chemical self-charging aqueous zinc ion battery (CSCAZIB) with a two-electrode structure is reported. In such a self-powered system, the discharged cathode and oxygen in air will undergo spontaneous redox reactions and self-charging. A defect coupled nitrogen modulated zinc manganite vertical graphene array (N-ZnMn2O4−x/VG) is designed as the cathode for CSCAZIBs. Benefiting from the high surface area, rich active sites, fast electron/ion diffusion, and strong structural stability, the assembled device not only shows a large specific capacity (222 mA h g−1 at 0.1 A g−1) and good rate capability (61.58% retention from 0.1 to 3 A g−1) along with admirable cycle life (92.6% retention after 3000 cycles at 1.0 A g−1), but also delivers excellent energy density (278 W h kg−1) and remarkable power density (3.62 kW kg−1). The as-assembled CSCAZIBs could be self-charged to 1.5 V after oxidation in an ambient environment for 30 h, and present a decent specific capacity of up to 176.8 mA h g−1 at 0.2 A g−1. Significantly, even in chemical or/and galvanostatic charging hybrid modes, the CSCAZIBs can still work normally. This work expands a promising prospect for rechargeable aqueous metal ion batteries, and simultaneously realizes energy harvesting, conversion and storage.
Introduction
Rechargeable batteries are important cornerstones of world economic development and human survival. Due to the high energy/power densities, superb service life and portability, rechargeable batteries are widely used in new energy vehicles, smart grids, flexible electronic equipment, distributed energy storage systems, military equipment, and urban rail transit.1,2 As far as we know, rechargeable batteries use external electrical energy to regenerate the internal active material, and can be recharged through reversible chemical reactions. Notwithstanding, there is usually no power supply system in remote areas and harsh environments, which means that rechargeable batteries cannot be used secondly and their actual value cannot be realized. To break through this bottleneck, scientists invented and created a self-charging power system, which consists of batteries and all sorts of energy harvesting technologies, for instance, photovoltaic devices,3–7 piezoelectric nanogenerators,8–10 triboelectric nanogenerators,11–13 and thermoelectrics.14–17 Regrettably, the above-mentioned integrated systems completely depend on energy resources, which cannot be obtained anytime and anywhere. Compared with conventional two-electrode batteries, the structure of these integrated systems is obviously much more complicated, which must be equipped with many additional components at the same time. Consequently, the advanced self-charging power supply system must have a simple structure and function well no matter when and where.Among the many energy sources, chemical energy is a kind of energy source stored in chemical molecules, which can be obtained anytime and anywhere, and transformed into electrical energy via oxidation–reduction reactions.18–20 As air is a rich source of oxygen, the chemical energy of oxygen has attracted widespread attention in energy conversion and storage devices, and the best known of these are metal–air batteries.21–23 Moreover, metal–air batteries could also be extended to integrated appliances, in which the metal–air batteries can provide energy for energy storage devices.19,24,25 Unfortunately, the metal–air batteries will be unable to recharge the energy loss in the energy storage device without limitation. When the energy of both parts is exhausted, it must be recharged through an external power source to recover. Quite evidently, the above-mentioned integrated devices cannot be directly charged by the continuous oxidation–reduction reactions of oxygen on the cathodes of the batteries. Recently, profiting from a good number of advantages, such as high voltage, large theoretical capacity, environmental friendliness, and source abundance, zinc manganite (ZnMn2O4) is considered as a rising star of positive materials for aqueous zinc ion batteries.26–30 For ZnMn2O4 aqueous zinc ion batteries, the intercalation of zinc ions and the reduction of manganese will take place simultaneously in the discharge process. Furthermore, manganese oxide compounds with a low valence state (such as Mn2+, Mn3+) have good redox kinetics, which should be capable of being oxidized by oxygen. As a consequence, the positive electrode ZnMn2O4 can undergo an oxidation–reduction reaction with oxygen in air after discharge, which is able to convert the chemical energy into electrical energy. Therefore, chemical self-charging aqueous zinc ion batteries (CSCAZIBs) can realize energy conversion and storage at the same time. Unfortunately, ZnMn2O4 suffers from intrinsically weak conductivity and large volume change during charging and discharging, leading to rapid capacity decay and poor rate performance that limit its further development.
In this work, we design novel nitrogen doped ZnMn2O4 with rich oxygen defects (NZnMn2O4−x) on three-dimensional vertical graphene (VG) forming NZnMn2O4−x@VG core/shell arrays as binder-free cathodes for CSCAZIBs. The specific VG skeleton not only offers an excellent conductive network for NZnMn2O4−x and improves the reaction kinetics, but also can effectively buffer volume changes during the reaction. Furthermore, the introduced symbiotic nitrogen dopant and oxygen defects can boost the intrinsic electronic conductivity and active sites of ZnMn2O4. CSCAZIB batteries have a simple two-electrode structure, and could convert the chemical energy from surrounding into electrical energy via spontaneous redox reactions and store. For this reason, the as-assembled integrated device does not need any outside power, and self-charging could be achieved by placing the NZnMn2O4−x@VG electrode in a natural environment. Most importantly, CSCAZIBs can work well in chemical or/and galvanostatic charging hybrid modes. This work is believed to bring new inspiration to the development of self-powered systems.
Experimental
Synthesis of VG arrays
We use a microwave plasma enhanced chemical vapor deposition (MPECVD) system to prepare a uniform VG array on carbon cloth. During the preparation process, the microwave source was set to 2 kW and 2.45 GHz. We first place the carbon cloth in the center of the quartz tube, and then evacuate the quartz tube to 10 mTorr. As the hydrogen flow rate reaches 90 sccm, we increased the temperature of the system to 400 °C. A microwave plasma with 550 W was used to obtain hydrogen plasma, which lasted for 2 h. The flow rate ratio of H2 to CH4 is set to be 3 : 2. Finally, we can obtain the end product after it was rinsed with deionized water several times and dried (0.5 mg cm−2).Synthesis of ZnMn2O4/VG and N-ZnMn2O4−x/VG core/shell arrays
We use an electrochemical workstation (CHI 760E) to prepare ZnMn2O4/VG through a facile anodic electrodeposition approach. In the process of sample preparation, we use a traditional three-electrode system, including a work electrode (a piece of VG array carbon cloth (2 × 3 cm2)), a counter electrode (Pt sheet), and a reference electrode (Ag/AgCl). Zinc nitrate (0.2841 g), manganese nitrate (0.5368434 g), sodium nitrate (0.147 g), and sodium dodecyl sulfate (0.28838 g) were added into deionized water (25 ml), and then stirred with a magnetic stirrer for half an hour to thoroughly dissolve it. The solution prepared above is used as the electrolyte for the anodic electrodeposition reaction. The sample prepared above was rinsed with deionized water, and then heated in air. The specific experimental parameters are as follows: the heating temperature was 300 °C, the heating time was 2 h, and the heating rate was 5 °C min−1. When the autoclave was naturally cooled to 25 °C, we obtained the ZnMn2O4 sample (5.6 mg cm−2). To prepare N-ZnMn2O4−x/VG core/shell arrays, the obtained ZnMn2O4/VG was further heat treated in NH3 at 500 °C for 4 h, and the N-ZnMn2O4−x/VG core/shell array sample is measured to be 5.8 mg cm−2.Material characterization
We used X-ray diffraction (XRD, Rigaku D-MAX 2200) to probe the crystal structure of the samples. Microscopic features were observed using a field-emission scanning electron microscope (FESEM, JSM-6330F) and a transmission electron microscope (TEM, FEI Tecnai G2 F30). X-ray photoelectron spectroscopies (XPS) were conducted and the spectral data were extracted from an ESCALab250 system. We collected the Raman spectra of the samples using a Renishaw Raman microscope, which has a laser excitation wavelength of 514 nm. A Bruker EPR spectrometer (A300-10-12) was used to collect the electron paramagnetic resonance (EPR) spectra. The synchrotron-based X-ray absorption near-edge structures (XANES) were obtained from beamlines BL10B.Electrochemical measurements
Electrochemical behaviors were investigated in CR2032 coin cells using a two-electrode system. The as-prepared ZnMn2O4/VG and N-ZnMn2O4−x/VG samples with a working area of 0.5 cm2 were directly used as the cathode, and a zinc plate was used as the anode. In the coin cells, the separator was a commercial glass fiber membrane, and the electrolyte was a Zn(CF3SO3)2 solution with a concentration of 4 mol L−1. A PARSTAT MC electrochemical workstation was adapted to measure cyclic voltammetry (CV) curves, galvanostatic charge–discharge (GCD) profiles, Mott–Schottky plots, and galvanostatic intermittent titration technique (GITT) curves as well as electrochemical impedance spectroscopy (EIS) spectra.In order to investigate the energy conversion mechanism of the CSCAZIB, which is based on the spontaneous chemical reaction of N-ZnMn2O4−x/VG and O2, the assembled N-ZnMn2O4−x/VG//Zn battery was first discharged to 0.8 V at 0.1 A g−1. Afterwards, the positive material (N-ZnMn2O4−x) after the complete discharge was rinsed with ethanol and deionized water three times, respectively. The naturally dried N-ZnMn2O4−x electrode was immersed in the electrolyte to cause the oxidation–reduction reaction with the oxygen dissolved in the electrolyte. After the reaction, the electrochemical performances of the N-Zn1−xMn2O4−x positive electrode were tested using CR2032 coin cells, which contain a zinc plate negative electrode. In order to allow oxygen in air to enter the electrolyte of the system during chemical self-charging, a hole was drilled in the positive cap of the cell in advance. Before starting the chemical self-charging process, we sealed the cell with a Kapton membrane. When the redox reaction occurs, the Kapton membrane was torn off to allow oxygen to pass through the hole. The cell would be again assembled with Kapton membrane until the chemical self-charging ends. In addition, the chemical self-charging process was executed in an environment with humidity greater than 60% during the entire operation, which can effectively avoid excessive evaporation of water in the electrolyte.
Results and discussion
The freestanding N-ZnMn2O4−x/VG shell/core arrays were prepared via an electrodeposition (ED) method and a subsequent NH3 annealing (NA) process, as schematically shown in Fig. 1a. In the first place, the freestanding VG arrays were prepared uniformly on a carbon cloth substrate by an MPECVD method. Then, these ZnMn2O4 nanoparticles were evenly deposited on the VG skeleton by a facile ED method, and the ZnMn2O4/VG shell/core arrays were formed. Finally, self-supported N-ZnMn2O4−x/VG shell/core arrays were obtained through heating ZnMn2O4/VG under an NH3 atmosphere, which was carried out at 500 °C and lasted for 4 h. The morphology variation of these samples at various stages is revealed in the SEM images (Fig. 1b–d). Thin graphene nanosheets with several nanometers are vertically grown on the carbon cloth substrate with a freestanding array structure (Fig. 1b). The VG nanosheets were connected to each other and developed into a three-dimensional porous network with an interspace of 50–500 nm, which is an excellent conductive skeleton and conducive to the rapid transmission of ions/electrons. When the ED growth is finished, the ZnMn2O4 nanoparticles are perfectly fused with the VG skeleton (Fig. 1c), in which the former is evenly covered on the latter to form a self-supporting core/shell array structure. Evidently, the ZnMn2O4/VG core/shell arrays are much thicker than the original VG arrays. After the NH3 annealing process, the microstructure of the final N-ZnMn2O4−x/VG has no obvious change compared with that of the original sample (Fig. 1d). It should be noted that the three-dimensional porous architecture was well retained throughout the process, which would help in maintaining a full contact between the electrode and electrolyte, speeding up the transmission of electrons/ions, and improving the conductivity. The microstructures and crystalline features of the three samples are further monitored with TEM measurements. It can be seen from Fig. 1e that the graphene nanosheet has a smooth corrugated texture and a very thin thickness. The distance between the graphene interlayer is gauged to be 0.37 nm, and it is in good agreement with the (002) plane of graphitic carbon (inset in Fig. 1e). The TEM image of the ZnMn2O4/VG arrays is depicted in Fig. 1f, in which the ZnMn2O4 nanoparticles can firmly adhere to the VG framework, and form a core–shell structure. As revealed in the inset of Fig. 1f, the ZnMn2O4 shell has an interlayer distance of 0.30 nm, and could be well matched with the (112) plane from the ZnMn2O4 phase (JCPDS No. 24-1133), further attesting the successful formation of the ZnMn2O4 on the VG arrays. After ammonia heat treatment, the N-ZnMn2O4−x/VG array in Fig. 1g still retains the microstructure of the ZnMn2O4/VG array, and the corresponding interlayer spacing is calculated to be 0.47 nm (inset of Fig. 1g) that belongs to the (101) plane of ZnMn2O4 (JCPDS No. 24-1133). What is more, in the HRTEM image of the N-ZnMn2O4−x/VG arrays, we can clearly see the zigzag lattice fringes, which indicate that the sample has stacking faults causing by oxygen defects. Additionally, the energy dispersive X-ray (EDS) mapping images confirm that Zn, Mn, O, C, and N elements can be homogeneously distributed on the N-ZnMn2O4−x/VG core/shell arrays (Fig. 1h–l), verifying the successful fabrication of N-ZnMn2O4−x/VG and uniform deposition on the VG array skeleton. | ||
| Fig. 1 (a) Synthesis diagram of N-ZnMn2O4−x/VG. SEM images of (b) VG, (c) ZnMn2O4−x/VG, and (d) N-ZnMn2O4−x/VG. TEM and HRTEM images of (e) VG, (f) ZnMn2O4−x/VG, and (g) N-ZnMn2O4−x/VG. (h–l) EDS elemental mapping images of C, Zn, Mn, O, and N of N-ZnMn2O4−x/VG. | ||
To further explore the crystal structure and phase evolution, XRD and Raman measurements were carried out. The diffraction peaks of the ZnMn2O4/VG and N-ZnMn2O4−x/VG samples at about 26° in Fig. S1 (ESI†) could be associated with the VG skeleton (JCPDS 75-1621). In addition to the VG (002) peak, the other diffraction peaks from both samples can correspond well to the spinel ZnMn2O4 (JCPDS No. 24-1133), indicating that they do not contain other impurities. Compared with ZnMn2O4/VG, the diffraction peaks of N-ZnMn2O4−x/VG are less intense, which is probably due to the decrease in crystallinity. The lower crystallinity added further effective diffusion channels to the electrolyte and ions, which would greatly accelerate the rate of electrochemical reactions.31 Fig. S2 (ESI†) displays the Raman spectra of the ZnMn2O4/VG and N-ZnMn2O4−x/VG. The Zn–O bond (321 and 378 cm−1) and Mn–O bond (673 cm−1) are observed in the ZnMn2O4/VG sample,28,32 and these Raman peaks manifest a distinct red-shift with decreased intensities after ammonia heat treatment, probably caused by the introduced oxygen defects and nitrogen doping.33,34 More information about the oxygen defects and nitrogen doping could be obtained from the XPS, XANES, and EPR spectra. As evidenced in Fig. S3 (ESI†), the Zn, Mn, O, C and N elements are demonstrated to be present in N-ZnMn2O4−x/VG, which is consistent with the above EDS analysis results. In the Mn 2p corelevel spectra of ZnMn2O4/VG, the Mn 2p3/2 and Mn 2p1/2 peaks are located at 642.2 and 653.9 eV, respectively (Fig. 2a). Noticeably, the two peaks shift negatively to a lower position (about 0.4 eV) for the N-ZnMn2O4−x/VG. Meanwhile, similar red-shifts are also observed in the Mn Ledge spectra (0.94 eV, Fig. 2b) and Mn 3s spectra (0.44 eV, Fig. S4, ESI†). The clear red-shift in Mn 2p and Mn Ledge as well as the Mn 3s spectra of N-ZnMn2O4−x/VG indicates that manganese ions are reduced from the trivalent to bivalent state due to the oxygen defects.33,35 The O 1s spectra of the ZnMn2O4/VG and N-ZnMn2O4−x/VG are illustrated in Fig. 2c. The characteristic peak belonging to the Mn–O bond (529.9 eV) is inspected in both samples. Yet, the N-ZnMn2O4−x/VG sample has two new strong peaks at 531.2 and 532.8 eV. Specifically, the peak at 531.2 eV is associated with the Mn–OH bond, which may result from oxygen defect sites in the N-ZnMn2O4−x/VG sample. Meanwhile, the peak at 532.8 eV is related to the O–H bond, and its strength has been greatly improved after heat treatment, indicating that a lot of surface adsorbed water molecules adhere to the surface of the N-ZnMn2O4−x/VG. These results prove that the N-ZnMn2O4−x/VG has more surface active sites and is easier to adsorb the electrolyte, leading to a smooth electrochemical reaction.36 The O K-edge spectrum is a powerful technique for characterizing the changes in the electronic valence state, and it is often used to investigate the changes between the O 1s core state and the unoccupied O 2p derived state involving hybridization with the 3d and 4sp bands in metal ions.37 The peak of N-ZnMn2O4−x/VG at 534.5 eV is much broader than that of the original ZnMn2O4/VG (Fig. 2d), and it is probably the oxygen defects in N-ZnMn2O4−x/VG that give rise to this phenomenon.26 Interestingly, no relative displacement is observed in the Zn 2p for the two samples, which reveals that there is no change in the valence state of zinc (Fig. S5, ESI†). As shown in the N 1s spectra in Fig. 2e, the N-ZnMn2O4−x/VG sample exhibits evident N–Mn bonds compared to the native ZnMn2O4/VG, which proves that the nitrogen atoms are strongly combined with the manganese atoms, which leads to the red-shift phenomenon in XPS 2p and L-edge spectra of Mn atoms.38 Moreover, the Mn K-edge XPS spectra of N-ZnMn2O4−x/VG move to a low binding energy (Fig. S6, ESI†), suggesting a higher average electron density, which results in the red shift in binding energy and photon energy of Mn element in XPS and XANES measurements.39 More evidence about the oxygen vacancies can also be obtained from the EPR spectra. The N-ZnMn2O4−x/VG shows a much stronger peak at a g value of 1.998 in Fig. 2f, indicating that Mn2+ ions are generated and oxygen vacancies are formed during the annealing process. Besides this, the Mott–Schottky plot of N-ZnMn2O4−x/VG in Fig. S7 (ESI†) has an absolute value of slope of about 2.06, which is much smaller than that of the ZnMn2O4/VG (9.26), confirming that the oxygen vacancies are successfully introduced into N-ZnMn2O4−x/VG and greatly enhanced the carrier density.
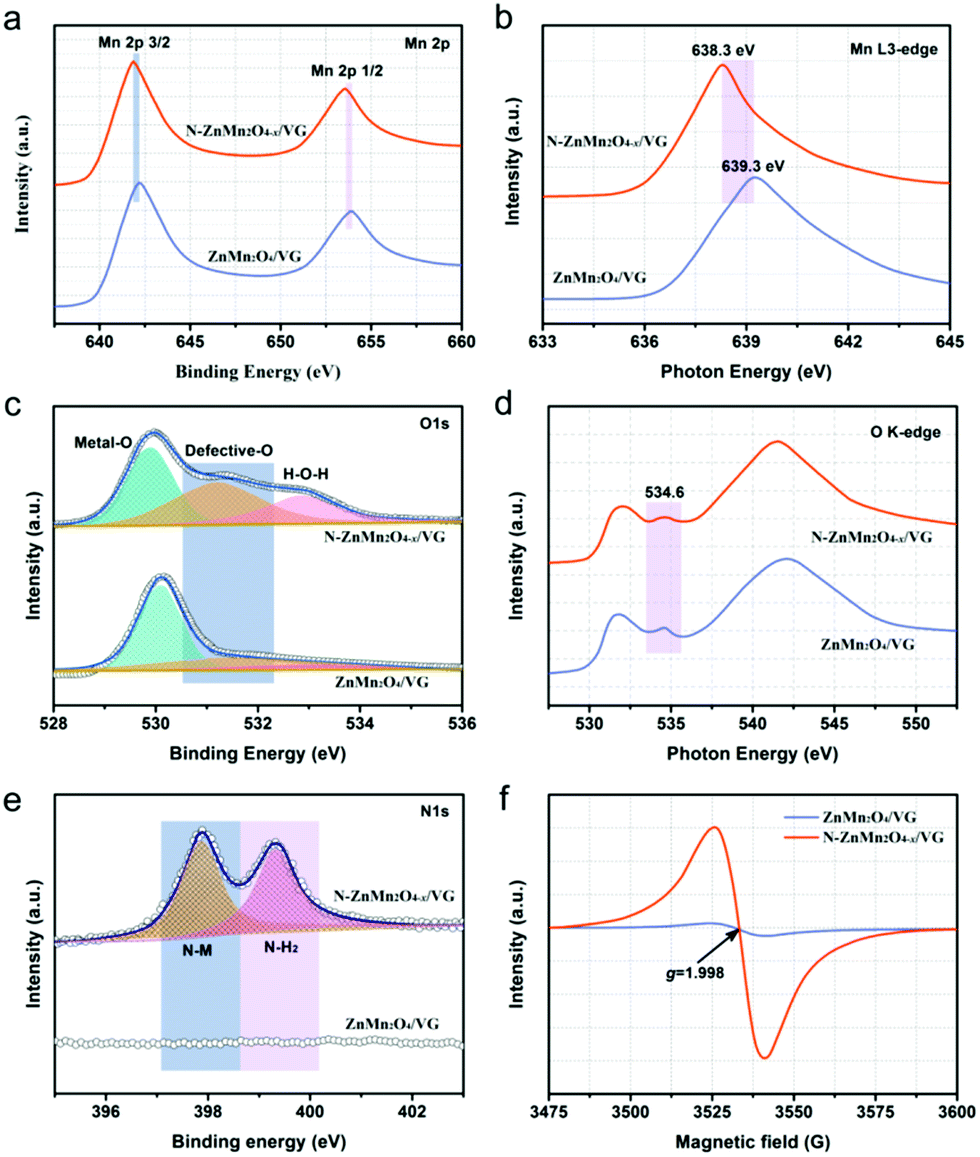 | ||
| Fig. 2 Corelevel spectra of (a) Mn 2p, (c) O 1s, and (e) N 1s. NEXAFS spectra of (b) Mn L3-edge and (d) O K-edge. (f) EPR spectra of ZnMn2O4/VG and N-ZnMn2O4−x/VG. | ||
The zinc storage performances of the N-ZnMn2O4−x/VG electrode were explored by employing it as the positive electrode for zinc ion batteries in a 4 M Zn(CF3SO3)2 electrolyte. Fig. 3a depicts the CV curves of the ZnMn2O4/VG and N-ZnMn2O4−x/VG electrodes, which were measured with 0.5 mV s−1 in the range of 0.8 to 1.8 V. Note that both ZnMn2O4/VG and N-ZnMn2O4−x/VG exhibit oxidation peaks during anodic sweeping and reduction peaks in the cathodic scan. The two well separated reduction peaks are due to the intercalation of H+ and Zn2+ step by step, and Mn4+ would be reduced to the low valence state during the process.40 However, the two oxidation peaks should be connected with the oxidation of Mn2+/Mn3+ back to the Mn4+ state upon H+ and Zn2+ extraction.41 The current density of the CV curve in N-ZnMn2O4−x/VG is obviously larger when measured against the corresponding ZnMn2O4/VG, proving the superior reactivity and utilization. In addition, the N-ZnMn2O4−x/VG electrode displays smaller polarization than the ZnMn2O4/VG counterpart at 0.1 A g−1, which can be mutually confirmed from the GCD curves shown in Fig. 3b. The first and second discharge platforms are attributed to the intercalation of H+ and Zn2+, respectively. Notably, the N-ZnMn2O4−x/VG sample has a much better charging/discharging platform and charge–discharge capacity, representing smaller polarization and higher power/energy densities that benefit from the abundant oxygen defects, nitrogen doping, and VG skeleton. These GCD curves of both samples in Fig. S8 (ESI†) that were measured at various current densities could help in achieving a similar conclusion as well. Rapid charge–discharge performance is an important index for the application of zinc ion batteries. The N-ZnMn2O4−x/VG electrode could deliver capacities of 222, 212.3, 195.6, 173.2, 153.4, and 136.7 mA h g−1, when cycled at 0.1, 0.2, 0.5, 1.0, 2.0, and 3.0 A g−1, respectively (based on N-ZnMn2O4−x/VG). Additionally, the capacity retention of 61.58% is also achieved while the rate changes from 0.1 to 3.0 A g−1. It was imperative that the capacity of the N-ZnMn2O4−x/VG electrode could be restored to 219 mA h g−1 when tested with 0.1 A g−1 again. Such a rate performance surpasses those of the ZnMn2O4/VG and other ZnMn2O4-based electrodes (Table S1, ESI†). Besides, the N-ZnMn2O4−x/VG electrode manifests a much smaller semicircle and a steeper straight line compared with the original ZnMn2O4/VG electrode (Fig. 3d), suggesting superior electron and ion diffusion rates. These results are further substantiated by the GITT measurements in Fig. 3e, from which the zinc ion diffusion coefficient (DZn) may be evaluated for both electrodes. The average DZn of the N-ZnMn2O4−x/VG electrode is in the range of 10−9 to 10−11 cm2 s−1, which is superior to that of the ZnMn2O4/VG electrode, discovering the boosted kinetics for the N-ZnMn2O4−x/VG electrode after annealing treatment, ensuring favorable rate capability. These b-values in Fig. S9 (ESI†) suggest that the zinc ion storage of the two electrodes is controlled by the surface capacitive effect. The N-ZnMn2O4−x/VG electrode has a better capacitive contribution in contrast to the primitive ZnMn2O4/VG, which signifies more advanced surface reactivity in N-ZnMn2O4−x/VG, facilitating zinc ion diffusion, high rate enhancement, and cycling stability.42,43 As revealed in Fig. 3f, the N-ZnMn2O4−x/VG electrode has a great cycle life at 1 A g−1, which delivers a reversible capacity of up to 173.2 mA h g−1 (92.6% retention) after 3000 cycles, outperforming the values of the ZnMn2O4/VG sample and the recently reported ZnMn2O4-based cathodes, as shown in Table S1 (ESI†). Additionally, the cycle lives that were tested at 0.1, 0.5, and 3.0 A g−1 are also presented in Fig. S10 (ESI†) for a more in-depth insight into its cycle stability. The Ragone plots in Fig. 3g once again prove the good electrochemical performance of the N-ZnMn2O4−x/VG//Zn battery. The N-ZnMn2O4−x/VG//Zn battery exhibits a peak specific energy density reaching 278 W h kg−1, outperforming those of Ni3S4//AC batteries (18.63 W h kg−1),44 Al//graphite batteries (40 W h kg−1),45 LiMn2O4//MoO3 batteries (45 W h kg−1),46 Ni//Bi batteries (85.8 W h kg−1),47 Ni//Fe batteries (100.7 W h kg−1),48 VS2//Zn batteries (123 W h kg−1),49 MoS2//Zn batteries (148.2 W h kg−1),50 NNA@Zn batteries (148.5 W h kg−1),51 H2V3O8/G//Zn batteries (168 W h kg−1),52 V2O5//Zn batteries (237.2 W h kg−1),53 and V2O5@PEDOT/CC//Zn batteries (243.3 W h kg−1).54 More strikingly, the top power density as high as 3.62 kW kg−1 remained ahead of those of the other reported aqueous batteries.27,55–59 For illustration purposes, two N-ZnMn2O4−x/VG//Zn batteries are connected up at 1 A g−1 in series or parallel modes, from which a twofold voltage window or capacity can be achieved (Fig. 3h). This shows that we can change the number of devices and the connection mode to attain the goal of different voltage windows and capacities. Additionally, it can be observed from Fig. 5i that N-ZnMn2O4−x/VG//Zn batteries could power a digital watch and light emitting diode (LED) lights, revealing their great application potential. The superior high rate properties and long cycle life of the N-ZnMn2O4−x/VG electrode are because of the favorable factors as mentioned below: (1) oxygen vacancies and doped nitrogen may be able to increase the active sites and boost the conductivity of the N-ZnMn2O4−x. (2) The N-ZnMn2O4−x nanoparticles are uniformly deposited on the VG skeleton with good conductivity, coupled with the three-dimensional porous network, which not only offers a large surface area and more active sites, but also improves the electron/ion transporting efficiency and diminishes polarization. (3) The three-dimensional porous skeleton and the intimate interfacial adhesion between N-ZnMn2O4−x and VG can effectively relieve the stress and maintain the structural integrity during the charging and discharging cycle. (4) The electrode without any binders and additives can make the electron and ion diffusion faster and smoother, which will greatly give impetus to the surface reaction and kinetic process. All these strong features contribute to the exceptional electrochemical properties of N-ZnMn2O4−x/VG//Zn batteries.
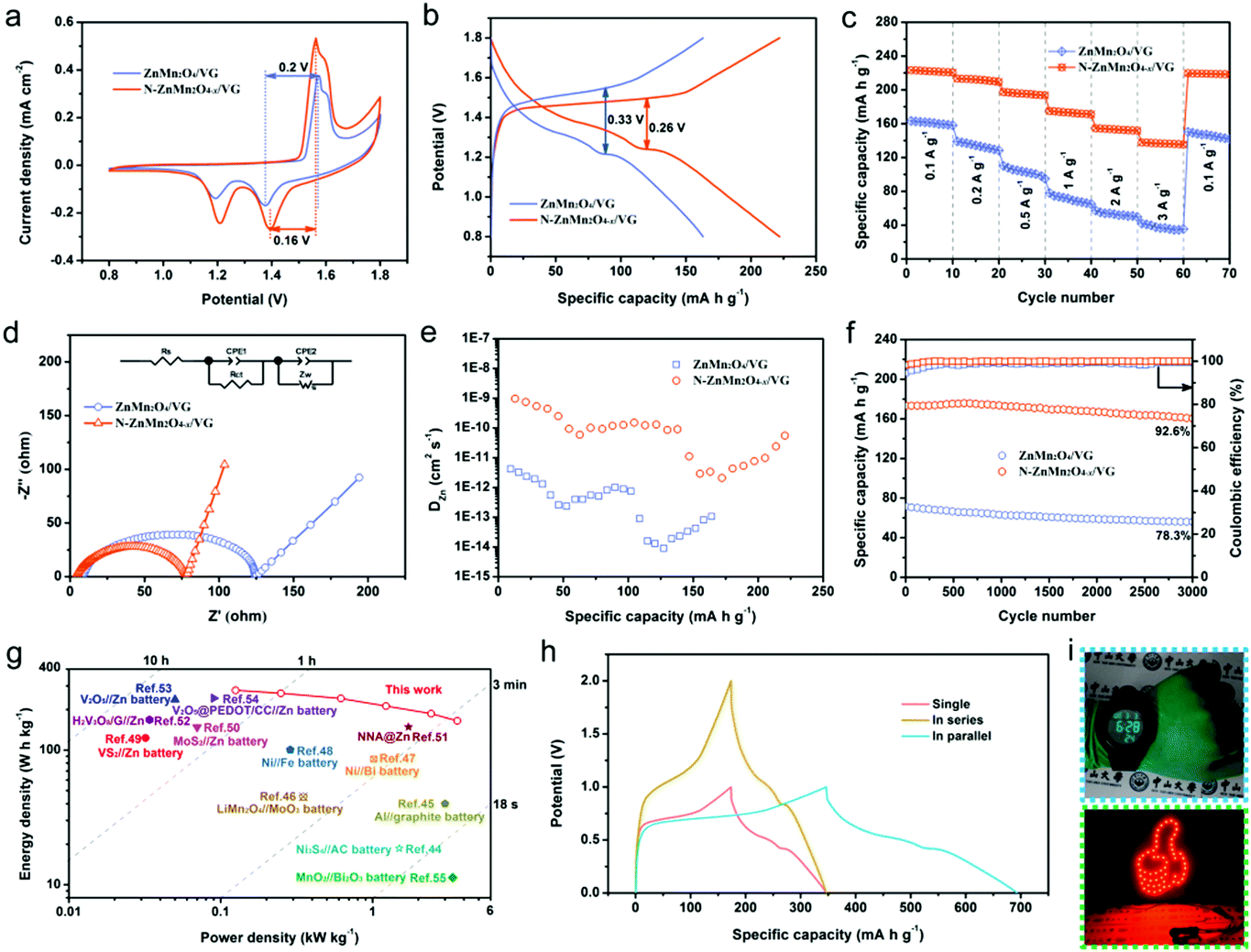 | ||
| Fig. 3 (a) CV curves, (b) GCD curves, (c) rate performance, (d) Nyquist plots, (e) calculated DZn, and (f) cycling performance of the ZnMn2O4/VG//Zn and N-ZnMn2O4−x/VG//Zn batteries. (g) Ragone plots comparing the performance of N-ZnMn2O4−x/VG//Zn battery to those of other reported aqueous rechargeable batteries.44–55 (h) GCD curves of single N-ZnMn2O4−x/VG//Zn battery and two devices in series and parallel. (i) Images of a digital watch and red LED light powered by N-ZnMn2O4−x/VG//Zn batteries. | ||
The redox potential difference (ΔE) between the reactants can give impetus to the redox reaction, which is the most direct and effective way to achieve electronic transfer.60,61 Because there is a ΔE between N-ZnMn2O4−x/VG and O2, the oxidation of manganese ions and removal of zinc ions in N-ZnMn2O4−x/VG will be able to happen spontaneously in the system, which includes oxygen dissolved in the 4 M Zn(CF3SO3)2 electrolyte. As a result, the completely discharged N-ZnMn2O4−x/VG state may restore to the charged N-Zn1−xMn2O4−x/VG state again without external power, and this can be thought of as a chemical self-charging process. When the charged N-Zn1−xMn2O4−x/VG is directly used as the cathode for zinc ion batteries, the ΔE between the cathode and anode will drive the discharge reaction. In this process, the Zn electrode loses two electrons and becomes Zn2+ inserted into N-Zn1−xMn2O4−x/VG, and the electrons move from the negative electrode to the positive electrode through the external circuit to maintain the charge balance. This system can collect chemical energy from the surrounding environment through chemical self-charging, and then convert the chemical energy into electrical energy for storage, which realizes energy collection, conversion, and storage at the same time. For the sake of exploring self-charging performances, the galvanostatic discharge curves of N-Zn1−xMn2O4−x/VG//Zn batteries with variable oxidation times were first collected. With the increase of oxidation duration, the open-circuit voltage (OCV) of the N-Zn1−xMn2O4−x/VG//Zn batteries continues to increase (Fig. 4a and b). This is because with the extension of oxidation time, manganese ions will be oxidized more and more thoroughly, and more and more zinc ions would be extracted from N-ZnMn2O4−x/VG, leading to a higher and higher cathode potential. The OCV of N-Zn1−xMn2O4−x/VG//Zn batteries can be 1.5 V and 1.52 V respectively as the oxidation time lasts for 30 h and 35 h. Apart from the OCV, the discharge capacity of the N-Zn1−xMn2O4−x/VG//Zn batteries has a great relationship with the oxidation time of N-ZnMn2O4−x/VG. The discharge capacity improves steadily with the extension of oxidation time from 0 to 30 h, and reaches a top capacity of 176.8 mA h g−1 at 30 h. These values are much better than those from self-charging in a deionized water electrolyte (Fig. S11, ESI†). In addition, even the N-Zn1−xMn2O4−x/VG//Zn batteries in different states can also show good reversibility during the chemical charging/galvanostatic discharging process. As depicted in Fig. 4c and Fig. S12 (ESI†), the N-Zn1−xMn2O4−x/VG//Zn batteries with the optimized oxidation time can release 176.8, 162.6, 149.6, 137.6, and 126.6 mA h g−1 at current densities of 0.1, 0.2, 0.3, 0.4, and 0.5 A g−1, respectively. To our delight, a capacity of 170.2 mA h g−1 could be retained when the current density was back to 0.1 A g−1. Moreover, after 100 cycles, the capacity retention rate of the N-Zn1−xMn2O4−x/VG//Zn batteries is 87.7% for the 4 M Zn(CF3SO3)2 electrolyte at 0.1 A g−1 (the N-ZnMn2O4−x/VG electrode is oxidized for 3 h), which is superior to that (87.7%) of the deionized water electrolyte (Fig. 4d and Fig. S13, ESI†), suggesting the superior self-recharge ability and good cyclic stability. Most importantly, these CSCAZIBs can freely switch between the charge and discharge modes and work well (Fig. 4e). The OCV of the N-Zn1−xMn2O4−x/VG//Zn batteries can steadily reach 1.28 V after 10 h of chemical self-charging. When the energy in the CSCAZIBs is used up, it can be self-charged again by chemical oxidation. What is even more impressive is that the N-Zn1−xMn2O4−x/VG//Zn batteries could be recharged by an external power supply, which can complete the transition from a self-charged (1.28 V) state or fully discharged (0.8 V) state to the fully charged state (1.8 V).
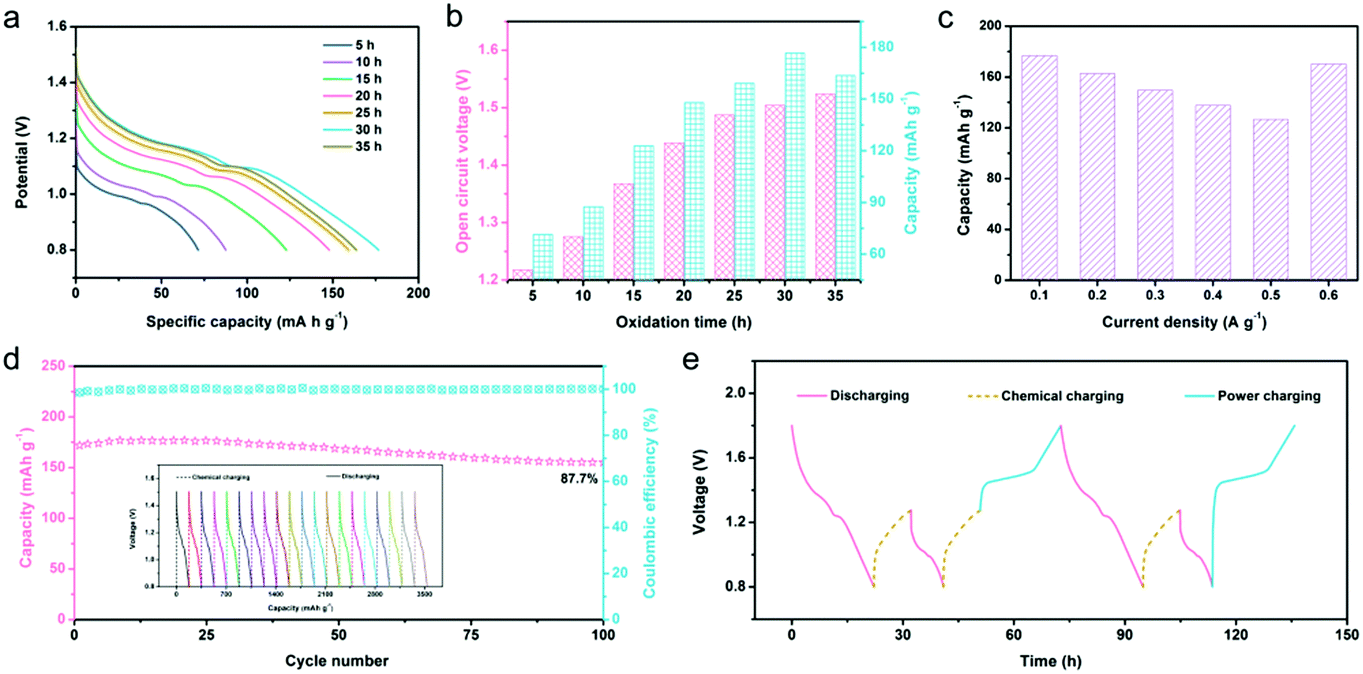 | ||
| Fig. 4 Electrochemical properties of N-Zn1−xMn2O4−x/VG//Zn batteries: (a) galvanostatic discharge curves, (b) OCV and discharge capacity at various oxidation times, (c) rate performance, (d) cycle stability (inset shows the 20th to 40th cycles), and (e) charging/discharging behavior in hybrid modes. | ||
In order to deeply investigate the reaction mechanism of the chemical self-charging process, the structure and composition evolution of the N-ZnMn2O4−x/VG electrode were studied by using all kinds of ex situ characterization techniques, such as XPS, XRD, and Raman. The Mn 2p3/2 peak in Fig. 5a has an obvious blue-shift with the prolongation of the oxidation time from 0 to 35 h. Particularly, a main Mn4+ peak is detected, while the Mn2+ peak disappears and the Mn3+ peak is obviously weakened in comparison with those of initial N-ZnMn2O4−x/VG (Fig. 5b), suggesting the oxidation of manganese in N-ZnMn2O4−x/VG in this process. In the meantime, the Zn2+ ions are extracted from the N-ZnMn2O4−x/VG framework, as confirmed by the Raman spectra in Fig. 5c. As the oxidation reaction continues, the intensity of the characteristic peaks (301 and 346 cm−1) that relate to the Zn–O bond stretching vibrations gradually decreases. Furthermore, the extraction of Zn2+ from ZnMn2O4 would result in the shrinking of the crystal lattice (Fig. 5d). When the oxidation time increases, the (103) and (211) reflections of oxidized N-ZnMn2O4−x/VG become obviously blue-shifted. Based on the Bragg equation, the distance between the (103) plane decreases from 0.274 to 0.272 nm after oxidation for 40 h, and the value for the (211) plane is changed from 0.25 to 0.248 nm (Fig. S14, ESI†). It is to be noted that the blue-shift of the XPS and XRD peaks is not observed even after 35 h (Fig. 5a and d), suggesting that the oxidation of manganese and the deintercalation of Zn2+ ions in N-ZnMn2O4−x/VG can be completed within 35 h. In this CSCAZIB system, the redox reaction between N-ZnMn2O4−x/VG and O2 can occur spontaneously. During this course, O2 accepts the electrons from N-ZnMn2O4−x/VG, and then reacts with H2O to generate OH−. The produced OH− and Zn2+ would combine with the dissociated CF3SO3− of the electrolyte to form Znx+y(CF3SO3)2y(OH)2x.43,62–64 For these analyses, the reaction mechanism between N-ZnMn2O4−x/VG and O2 can be summarized as below:
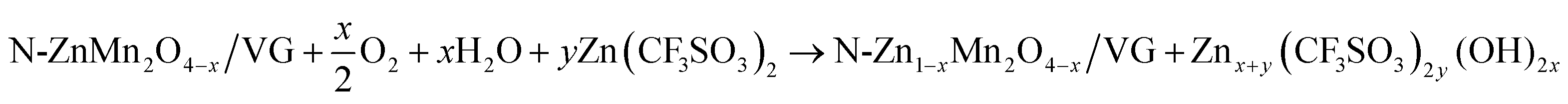 | (1) |
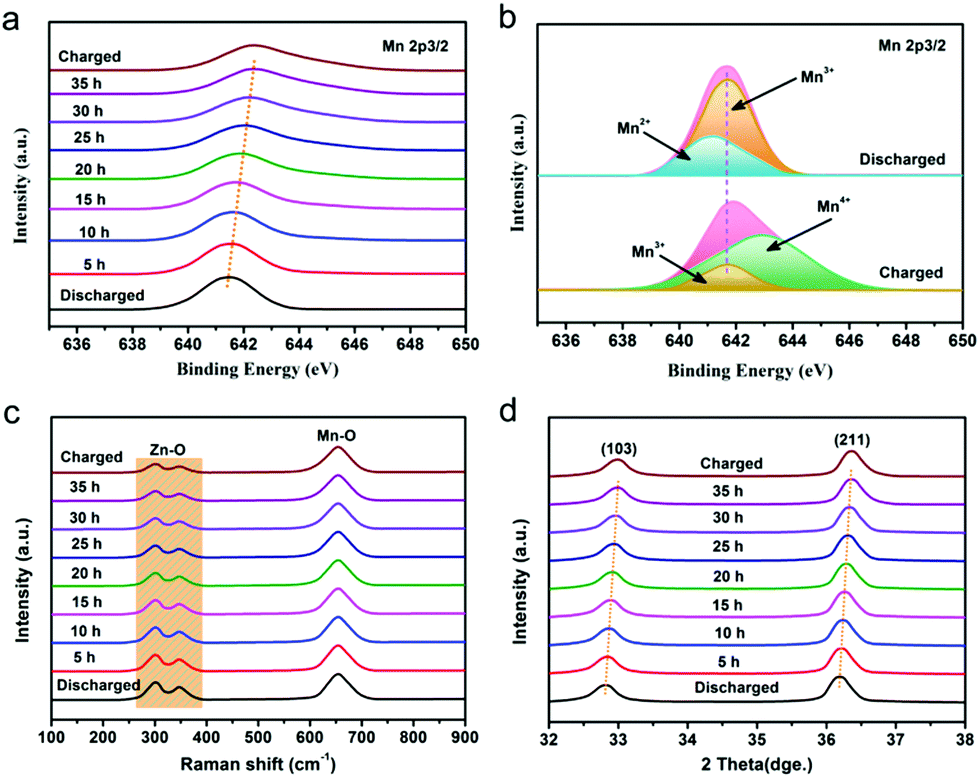 | ||
| Fig. 5 (a) Mn 2p3/2 XPS spectra, (c) Raman and (d) XRD patterns of the N-ZnMn2O4−x/VG electrodes with various oxidation times. (b) Mn 2p3/2 spectra for the charged and discharged states. | ||
Conclusions
To sum up, a novel CSCAZIB with a dual electrode has been successfully built by using an elaborate N-ZnMn2O4−x/VG cathode. The oxygen defects and nitrogen doping can make the ion/electron transport fast, create more active sites, and decrease the band-gap. Moreover, the interconnected VG arrays provide an omnibearing conductive network, high surface area, large porosity, exceptional electrolyte accessibility, and strong mechanical stability. As a consequence, the aqueous zinc ion device, which was assembled with the well-designed N-ZnMn2O4−x/VG cathode and zinc plate anode, offers superior zinc ion storage properties with a remarkable specific capacity (222 mA h g−1 at 0.1 A g−1) and superior rate capability (61.58% retention from 0.1 to 3 A g−1) along with ultralong durability (92.6% retention after 3000 cycles). Additionally, this aqueous zinc-ion battery also shows a prominent energy density (91.8 W h kg−1) and power density (27.6 kW kg−1), outperforming those of the currently existing aqueous zinc-ion batteries. Importantly, this device can realize self-charging from the spontaneous reaction between the discharged N-ZnMn2O4−x/VG electrode and O2, and be provided with the functions of energy harvesting, conversion and storage. The as-assembled CSCAZIBs could self-charge up to 1.5 V by entirely dependent on a chemical self-charging process, and achieve a specific capacity as high as 176.8 mA h g−1. Even more significantly, various chemical or/and galvanostatic charging hybrid modes can be well compatible and work properly in these CSCAZIBs. This work proves the feasibility of constructing CSCAZIBs with a dual electrode structure, and opens new opportunities for future self-charging integrated devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Basic and Applied Basic Research Project of Guangdong Province (2019A1515110827), the Science and Technology Planning Project of Guangzhou (201804010196, 202102080169), the Education Commission of Guangdong Province (2019GKTSCX015), the Advanced Functional Materials Scientific Research and Technical Service Team (X20190197), and Guangdong Training Programs of Scientific and Technological Innovation for Undergraduates (pdjh2021a0715).Notes and references
- A. Van der Ven, Z. Deng, S. Banerjee and S. P. Ong, Chem. Rev., 2020, 120, 6977 CrossRef CAS PubMed.
- Y. J. Fang, D. Y. Luan, S. Y. Gao and X. W. Lou, Angew. Chem., 2021, 133, 2 CrossRef.
- B. D. Boruah, A. Mathieson, B. Wen, S. Feldmann, W. M. Dose and M. De Volder, Energy Environ. Sci., 2020, 13, 2414 RSC.
- A. Paolella, C. Faure, G. Bertoni, S. Marras, A. Guerfi, A. Darwiche, P. Hovington, B. Commarieu, Z. Wang and M. Prato, Nat. Commun., 2017, 8, 1 CrossRef PubMed.
- B. D. Boruah, A. Mathieson, B. Wen, C. Jo, F. Deschler and M. De Volder, Nano Lett., 2020, 20, 5967 CrossRef CAS PubMed.
- B.-M. Kim, M.-H. Lee, V. S. Dilimon, J. S. Kim, J. S. Nam, Y.-G. Cho, H. K. Noh, D.-H. Roh, T.-H. Kwon and H.-K. Song, Energy Environ. Sci., 2020, 13, 1473 RSC.
- K. Xiao, L. Chen, L. Jiang and M. Antonietti, Nano Energy, 2020, 67, 104230 CrossRef CAS.
- H. Khan, N. Mahmood, A. Zavabeti, A. Elbourne, M. A. Rahman, B. Y. Zhang, V. Krishnamurthi, P. Atkin, M. B. Ghasemian and J. Yang, Nat. Commun., 2020, 11, 1 Search PubMed.
- T. Huang, Y. Zhang, P. He, G. Wang, X. Xia, G. Ding and T. H. Tao, Adv. Mater., 2020, 32, 1907336 CrossRef CAS PubMed.
- X. Guan, B. Xu and J. Gong, Nano Energy, 2020, 70, 104516 CrossRef CAS.
- L. Jin, X. Xiao, W. Deng, A. Nashalian, D. He, V. Raveendran, C. Yan, H. Su, X. Chu and T. Yang, Nano Lett., 2020, 20, 6404 CrossRef CAS PubMed.
- Y. Liu, W. Liu, Z. Wang, W. He, Q. Tang, Y. Xi, X. Wang, H. Guo and C. Hu, Nat. Commun., 2020, 11, 1 CAS.
- Y. Zou, V. Raveendran and J. Chen, Nano Energy, 2020, 77, 105303 CrossRef CAS.
- X. L. Shi, W. Y. Chen, T. Zhang, J. Zou and Z. G. Chen, Energy Environ. Sci., 2021, 14, 729 RSC.
- Y. Xiao and L. D. Zhao, Science, 2020, 367, 1196 CrossRef CAS PubMed.
- X. Zhang, Z. Bu, S. Lin, Z. Chen, W. Li and Y. Pei, Joule, 2020, 4, 986 CrossRef CAS.
- C. Y. Yang, Y. F. Ding, D. Huang, J. Wang, Z. F. Yao, C. X. Huang, Y. Lu, H. I. Un, F. D. Zhuang and J. H. Dou, Nat. Commun., 2020, 11, 1 Search PubMed.
- S. B. Wang, Y. J. Fang, X. Wang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2019, 58, 760 CrossRef CAS PubMed.
- X. F. Lu, S. L. Zhang, E. B. Shangguan, P. Zhang, S. Y. Gao and X. W. Lou, Adv. Sci., 2020, 7, 2001178 CrossRef CAS PubMed.
- Z. Luo, Y. Wang, B. Kou, C. Liu, W. Zhang and L. Chen, Energy Storage Mater., 2021, 38, 9 CrossRef.
- Q. Liu, Z. Pan, E. Wang, L. An and G. Sun, Energy Storage Mater., 2020, 27, 478 CrossRef.
- X. F. Lu, Y. J. Fang, D. Y. Luan and X. W. Lou, Nano Lett., 2021, 21, 1555 CrossRef CAS PubMed.
- C. Zhang and K. Huang, Adv. Energy Mater., 2021, 11, 2000630 CrossRef CAS.
- S. Zeng, X. Tong, S. Zhou, B. Lv, J. Qiao, Y. Song, M. Chen, J. Di and Q. Li, Small, 2018, 14, 1803409 CrossRef PubMed.
- Z. Luo, C. Liu, S. Fan and E. Liu, J. Mater. Chem. A, 2019, 7, 15131 RSC.
- H. Zhang, J. Wang, Q. Liu, W. He, Z. Lai, X. Zhang, M. Yu, Y. Tong and X. Lu, Energy Storage Mater., 2019, 21, 154 CrossRef.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894 CrossRef CAS PubMed.
- D. P. Cai, D. D. Wang, H. Huang, X. C. Duan, B. Liu, L. L. Wang, Y. Liu, Q. H. Li and T. H. Wang, J. Mater. Chem. A, 2015, 3, 11430 RSC.
- L. L. Chen, Z. H. Yang, H. G. Qin, X. Zeng and J. L. Meng, J. Power Sources, 2019, 425, 162 CrossRef CAS.
- S. Islam, M. H. Alfaruqi, D. Y. Putro, S. Park, S. Kim, S. Lee, M. S. Ahmed, V. Mathew, Y. K. Sun and J. Y. Hwang, Adv. Sci., 2021, 8, 2002636 CrossRef CAS PubMed.
- X. Wang, J. Hu, W. Liu, G. Wang, J. An and J. Lian, J. Mater. Chem. A, 2015, 3, 23333 RSC.
- G. Q. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609 CrossRef CAS PubMed.
- Y. Zhang, S. Deng, M. Luo, G. Pan, Y. Zeng, X. Lu, C. Ai, Q. Liu, Q. Xiong, X. Wang, X. Xia and J. Tu, Small, 2019, 15, 1905452 CrossRef CAS PubMed.
- Y. Zhang, S. Deng, G. Pan, H. Zhang, B. Liu, X. L. Wang, X. Zheng, Q. Liu, X. Wang, X. Xia and J. Tu, Small Methods, 2020, 4, 1900828 CrossRef CAS.
- M. Jiang, C. Fu, J. Yang, Q. Liu, J. Zhang and B. Sun, Energy Storage Mater., 2019, 18, 34 CrossRef.
- Y. Zeng, Z. Lai, Y. Han, H. Zhang, S. Xie and X. Lu, Adv. Mater., 2018, 30, 1802396 CrossRef PubMed.
- Y. J. Fang, D. Y. Luan, Y. Chen, S. Y. Gao and X. W. (David) Lou, Angew. Chem., Int. Ed., 2020, 59, 7178 CrossRef CAS PubMed.
- Y. Wu, X. Liu, D. Han, X. Song, L. Shi, Y. Song, S. Niu, Y. Xie, J. Cai, S. Wu, J. Kang, J. Zhou, Z. Chen, X. Zheng, X. Xiao and G. Wang, Nat. Commun., 2018, 9, 1425 CrossRef PubMed.
- V. Soundharrajan, B. Sambandam, S. Kim, S. Islam, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, Energy Storage Mater., 2020, 28, 407 CrossRef.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121 CrossRef CAS.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 1 Search PubMed.
- H. Wang, M. Liang, D. Duan, W. Shi, Y. Song and Z. Sun, Chem. Eng. J., 2018, 350, 523 CrossRef CAS.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang and B. J. Hwang, Nature, 2015, 520, 324 CrossRef CAS PubMed.
- W. Tang, L. Liu, Y. Zhu, H. Sun, Y. Wu and K. Zhu, Energy Environ. Sci., 2012, 5, 6909 RSC.
- Y. Zeng, Z. Lin, Y. Meng, Y. Wang, M. Yu, X. Lu and Y. Tong, Adv. Mater., 2016, 28, 9188 CrossRef CAS PubMed.
- J. Liu, M. Chen, L. Zhang, J. Jiang, J. Yan, Y. Huang, J. Lin, H. J. Fan and Z. X. Shen, Nano Lett., 2014, 14, 7180 CrossRef CAS PubMed.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- H. Li, Q. Yang, F. Mo, G. Liang, Z. Liu, Z. Tang, L. Ma, J. Liu, Z. Shi and C. Zhi, Energy Storage Mater., 2019, 19, 94 CrossRef.
- C. Xu, J. Liao, C. Yang, R. Wang, D. Wu, P. Zou, Z. Lin, B. Li, F. Kang and C. P. Wong, Nano Energy, 2016, 30, 900 CrossRef CAS.
- Q. Pang, C. Sun, Y. Yu, K. Zhao, Z. Zhang, P. M. Voyles, G. Chen, Y. Wei and X. Wang, Adv. Energy Mater., 2018, 8, 1800144 CrossRef.
- P. Hu, M. Yan, T. Zhu, X. Wang, X. Wei, J. Li, L. Zhou, Z. Li, L. Chen and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 42717 CrossRef CAS PubMed.
- D. Xu, H. Wang, F. Li, Z. Guan, R. Wang, B. He, Y. Gong and X. Hu, Adv. Mater. Interfaces, 2019, 6, 1801506 CrossRef.
- H. Xu, X. Hu, H. Yang, Y. Sun, C. Hu and Y. Huang, Adv. Energy Mater., 2015, 5, 1401882 CrossRef.
- Y. X. Zeng, X. F. Lu, S. L. Zhang, D. Y. Luan, S. Li and X. W. (David) Lou, Angew. Chem., Int. Ed., 2021, 60, 1–7, DOI:10.1002/anie.202107697.
- Y. N. Gao, H. Y. Yang, Y. Bai and C. Wu, J. Mater. Chem. A, 2021, 9, 11472 RSC.
- W. Li, K. Wang, S. Cheng and K. Jiang, Energy Storage Mater., 2018, 15, 14 CrossRef.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang and Y. Shao, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- J. Gao, S. Q. Shi and H. Li, Chin. Phys. B, 2015, 25, 018210 CrossRef.
- H. Li, L. McRae, C. J. Firby and A. Y. Elezzabi, Adv. Mater., 2019, 31, 1807065 CrossRef PubMed.
- K. Zhu, T. Wu and K. Huang, Adv. Energy Mater., 2019, 9, 1901968 CrossRef CAS.
- L. Wang, K. W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- Y. Liu, Q. Li, K. Ma, G. Yang and C. Wang, ACS Nano, 2019, 13, 12081 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD patterns, Raman spectra, XPS survey spectra, Mn 3s, Zn 2p, Mn K-edge spectra, Mott–Schottky plots, GCD curves, and surface capacitive contribution of ZnMn2O4/VG and N-ZnMn2O4−x/VG. The cycling performance of N-ZnMn2O4−x/VG//Zn batteries at 0.1, 0.5 and 3.0 A g−1. Electrochemical performance of N-Zn1−xMn2O4−x/VG//Zn batteries in deionized water. Lattice spacing of N-ZnMn2O4−x/VG with different oxidation times. Comparison of zinc storage performance of ZnMn2O4-based cathodes. See DOI: 10.1039/d1ma00727k |
| ‡ Wenda Qiu and Zhenchao Lin contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |