
An in situ and rapid self-healing strategy enabling a stretchable nanocomposite with extremely durable and highly sensitive sensing features†
Yang
Liu
a,
Xiangqian
Fan
a,
Weimin
Feng
a,
Xinlei
Shi
a,
Fengchao
Li
a,
Jinhua
Wu
a,
Xinyi
Ji
a and
Jiajie
Liang
 *abc
*abc
aSchool of Materials Science and Engineering, National Institute for Advanced Materials, Nankai University, Tianjin 300350, P. R. China. E-mail: liang0909@nankai.edu.cn
bKey Laboratory of Functional Polymer Materials of Ministry of Education, College of Chemistry, Nankai University, Tianjin 300350, P. R. China
cTianjin Key Laboratory of Metal and Molecule-Based Material Chemistry and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300350, P. R. China
First published on 6th November 2020
Abstract
Progress toward the development of wearable electromechanical sensors with durable and reliable sensing performance is critical for emerging wearable integrated electronic applications. However, it remains a long-standing challenge to realize mechanically stretchable sensing materials with extremely durable and high-performing sensing ability due to the fundamental dilemma lying in the sensing mechanism. In this work, we proposed an in situ and rapid self-healing strategy through nano-confining a dynamic host–guest supramolecular polymer network in a graphene-based multilevel nanocomposite matrix to fabricate a mechanically stretchable and structurally healable sensing nanocomposite which is provided with intriguing sensing durability and sensitivity simultaneously. When repeatedly stretching and releasing the nanocomposite sensing film, the fast association kinetics of cyclodextrin and adamantane host–guest inclusion complexes and good polymer chain dynamics in the supramolecular polymer network endowed by the nanoconfinement effect enable autonomous and rapid repair of the micro-cracks in situ generated in the sensing material. As a result, our strain sensing devices can achieve an extremely high durability and retain stable sensing performance even after over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 stretching-releasing cycles at large strain of 50%. Moreover, the brittle nature originated from the inorganically dominated structure in conjunction with the thermodynamically stable host–guest interactions and dynamic hydrogen bonds inside the multilevel nanocomposite allow the sensing material to exhibit an ultrahigh gauge factor over 1500 with a large working strain of 58%. This work presents a reliable approach for the construction of ultradurable and high-performing wearable electronics.
000 stretching-releasing cycles at large strain of 50%. Moreover, the brittle nature originated from the inorganically dominated structure in conjunction with the thermodynamically stable host–guest interactions and dynamic hydrogen bonds inside the multilevel nanocomposite allow the sensing material to exhibit an ultrahigh gauge factor over 1500 with a large working strain of 58%. This work presents a reliable approach for the construction of ultradurable and high-performing wearable electronics.
New conceptsStretchable electromechanical sensors should be ultradurable and ultrasensitive to meet the requirements of emerging wearable electronic applications. However, it remains a challenge to couple high sensitivity with long-term cycling stability. The dilemma lies in the fundamental sensing mechanism that high sensitivity demands drastic structural or morphological deformation, which inevitably hastens mechanical fracture of the sensing materials. To address this conundrum, we propose an in situ self-healing strategy based on nanoconfinement of a host–guest supramolecular polymer network and high-boiling point solvent in a multilevel nanocomposite matrix to improve the durability of strain-sensing materials, without compromising the sensitivity and stretchability. The brittle nature originated from the inorganically dominated structure can induce considerable crack formation when stretching, guaranteeing high sensitivity. The rapid association kinetics of host–guest inclusion complexes and appropriate polymer chain dynamics in the supramolecular polymer network enable autonomous, rapid, and in situ repair of the cracks, suppressing crack propagation. The rupture of thermodynamically stable host–guest interactions and dynamic hydrogen bonds inside the multilevel nanostructure can dissipate loading energy, ensuring large stretchability. Our strain sensor achieves extremely high durability of over one hundred thousand stretching-releasing cycles at 50% strain, and exhibits working strain of 58% with a gauge factor over 1500. |
Introduction
In recent years, stretchable and skin-mountable strain sensors, which transduce mechanical deformation into readable electrical signals, have been regarded as the central part of emerging intelligent medical applications, spanning personal health monitoring systems, implantable biosensors, soft robotics, and electronic skins.1,2 In principle, the working mechanism for resistive-type strain sensors is derived from the structural deformation or morphological change in conductive sensing materials, such as crack generation and propagation in the sensing film, or disconnection of overlapped or connected sensing networks.3–6 This sensing mechanism inevitably brings about two fundamental dilemmas when developing high performance strain sensors for practical applications. First, the strain-sensing materials are faced with the dilemma of how to achieve both high sensitivity (gauge factor >100) and large stretchability (working strain range >50%).6–10 Generally, high sensitivity demands considerable structural deformation or disconnection in the sensing materials, whereas large stretchability requires structural integrity for sensing materials under specific strains.3 Thus, there is a trade-off between sensitivity and stretchability in conventional strain-sensing materials.3,4,11,12 Cracks generating and propagating in brittle or rigid conductive materials during stretching and thus greatly limiting the electrical conduction through the sensing materials is the main mechanism exploited to design strain sensors with high sensitivity.4,7,13–18 Although a variety of strain sensors based on brittle elastomer-free or inorganic material-dominated sensing materials with good response linearity and gauge factor (GF) higher than 100 were reported following the crack-propagation mechanism, the intrinsic lack of flexibility in those sensing materials greatly restricted their working strain range, normally narrower than 20%.17,19,20Second, the durability of a strain sensor is severely limited by the dilemma of mechanical fractures generated in the sensing materials over repeated structural deformation processes.6,21–25 Large structural deformation in conductive sensing materials is typically unrecoverable to a certain degree, which fundamentally leads to structure instability or permanent damage of the sensing materials, ultimately causing malfunction to the sensing devices (Fig. 1a). One feasible approach to address this cycle-life issue is to repair the fractures and damages by incorporating healable capability into the strain-sensing materials.26–29 Several healable material systems, including hydrogels,30,31 elastomers,5,8,32 and ionic liquids,33 have been successfully developed to construct stretchable strain sensors with extended working lifetime and cycling stability. However, these elastomer or organic material-matrixed sensing materials tend to have exceptionally low sensitivity due to the incorporation of soft or low-viscosity materials into the sensing structures.3,34 We previously reported a strategy to circumvent the above issues by developing a nacre-inspired and inorganic material-matrixed hierarchical nanocomposite system with both rigid and diffusible components containing dynamic hydrogen and coordination bonding, which gives rise to stretchable strain sensors with high sensitivity and effective healing capability to expand the lifespan and cycling durability.15 However, the triggers of the healing behavior for this sensing material required both applications of water and heating at 80 °C, which severely hinders their applications in real wearable electronics. Moreover, since most reported healable structures are not dynamic enough, the reformation of the ruptured bonds in those healable sensing materials is usually at a time scale much longer than that of the device stretching and releasing rates, which makes it difficult to rapidly and in situ repair the cracks and fractures generated during the stretch-release cycling process.26,35–38 The issues of durable and stable operation remain, and the cycle-life for most reported healable strain sensors is limited to thousands of stretch-release cycles,7,24,39 far insufficient to meet the daily activity needs for wearable electronics. For instance, humans will typically bend their knee joints about 100000 times per week. Therefore, integrating the highly desirable attributes of ultra-durability, ultra-sensitivity, and large stretchability into strain sensors remains challenging.
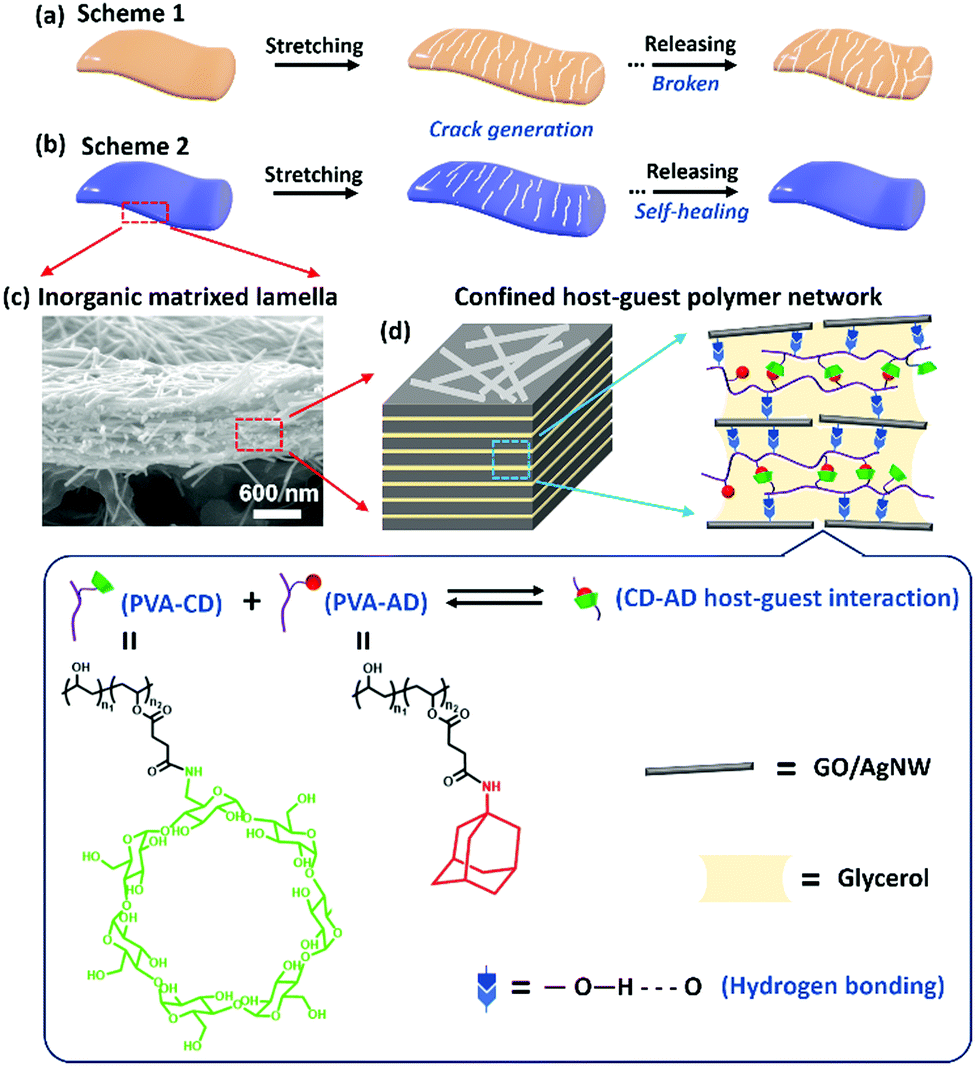 | ||
| Fig. 1 (a) Schematic illustration of the cyclic sensing behavior of a crack-based strain sensor that shows rupture of the sensing film due to gradual propagation of cracks in the sensing materials, which results in the failure of the sensing device. (b) Schematic illustration of the cyclic sensing behavior of our self-healable strain sensor that maintains integrity in the sensing material. (c) Cross-sectional SEM image of the GO:AgNW/PVA-AD:PVA-CD nanocomposite film. (d) Schematic illustration showing that the PVA network crosslinked by reversible CD–AD host–guest interaction was dissolved in glycerol and confined in the nano-spacing between the GO/AgNW nanofillers. | ||
In principle, for most self-healing materials based on dynamic reversible chemical bonds, their self-healing properties are determined by the molecular mobility and bond dynamics of the reversible sites,40i.e., higher molecular mobility and dynamics of the reversible bonds often result in faster self-healing behavior.36,41 However, high molecular mobility, together with weak dynamic interaction, usually yields materials with soft and viscoelastic features, which are in conflict with the structural requirements of high strain-sensing sensitivity.26,30 Such contradicting requirements make the design of strain-sensing materials with combined fast self-repair, ultrahigh sensitivity, and large stretchability fundamentally challenging. To this end, herein, we propose a spontaneous, rapid, and in situ self-healing strategy to boost both the durability and sensitivity of stretchable strain-sensing materials by physically confining a supramolecular poly(vinyl alcohol) (PVA)-based network bearing dynamic cyclodextrin (CD)–adamantane (AD) host–guest interaction and high boiling point glycerol in an inorganic graphene oxide (GO)/silver nanowire (AgNW)-matrixed nanocomposite with multilevel lamellar architecture.
Fig. 1a shows the crack propagation sensing mechanism of a typical resistive-type strain sensor without self-healing capability, in which the microcracks generated in the conductive sensing film when applying strain would gradually propagate to permanent fractures during the stretch-release cycles, resulting in rupture of the sensing film. In our current design, the physical nanoconfinement of the glycerol and supramolecular polymer network induced by the GO/AgNW matrix endows the healable polymer network with suitable polymer chain dynamics in the nanocomposite. This, together with the highly dynamic nature of CD–AD host–guest interaction, enables the rapid, autonomous, and in situ repair of the cracks and damages induced by structure deformation during stretch-release cycles (Fig. 1b). As a result, the in situ self-healable strain sensors attain an extremely long cycle-life of more than 200000 and 110000 stretch-release cycles at 35% and 50% strains, respectively. Moreover, the mechanically brittle nature of the inorganically-dominated material system (>98 wt% of inorganic components) facilitates considerable crack generation when stretching the sensing materials, while the thermodynamically stable host–guest interactions in conjunction with the kinetically labile hydrogen bonding formed between poly(vinyl alcohol) (PVA) and GO can effectively dissipate the loading energy to suppress crack propagation during stretching. These synergistic effects enable the ultradurable sensor to be stretched up to 58% with a high gauge factor up to 1591. This nanocomposite design concept can provide a feasible and strong impetus to the development of durable and wearable electronics for practical use.
Results and discussion
Fig. S1 (ESI†) and Fig. 1 show the overall material design for our strain sensitive and self-healable nanocomposite and its construction into a stretchable strain sensor.13,15 Briefly, a typical construction of the strain sensor begins with the synthesis of PVA bearing β-cyclodextrin groups (PVA-CD) and poly(vinyl alcohol) bearing adamantine groups (PVA-AD) following the reported method.37 Subsequently, a viscous and shear-thinning nanocomposite gel-like ink consisting of GO nanosheets, AgNWs, PVA-AD, PVA-CD, glycerol, and water was formulated.14 The stretchable strain sensor was then constructed by directly screen printing the nanocomposite gel onto an O2 plasma-treated polyurethane substrate, followed by ambient drying to evaporate the water but maintain the high boiling point glycerol confined in the nanocomposite.GO nanosheets were employed as the inorganic matrix in the nanocomposite due to their ability to form lamella structure in the sensing film (Fig. 1c) and abundant oxygen-containing active groups on their surface.42–45 AgNWs were chosen to assemble highly conductive networks on the GO nanosheets, allowing the nanocomposite to have resistive-type strain-sensing ability. The combination of 2D GO and 1D AgNW is able to form a conductive and brittle nanocomposite with a multilevel nanostructure, which is the foundation of constructing a highly-sensitive mechanical crack-based sensor.42–45 PVA-CD and PVA-AD were chosen as “bridging materials” to be incorporated into the nanocomposite due to the following two considerations: (1) adamantane and its derivatives can form stable inclusion complexes with β-CD molecules dissolved in glycerol with a high association constant in the range of 104 M−1,46–48 which leads to the formation of supramolecular polymer networks cross-linked by the dynamic host–guest inclusion complexes in the nanocomposite; (2) the reversible nature of the CD–AD host–guest interaction along with the fast binding dynamic (picosecond timescales) of adamantane guest to β-CD host allows rapid and spontaneous healing damages in the nanocomposite.49–51 However, the polymer chain dynamics of the neat supramolecular polymer network could be severely limited due to its physical confinement in the inorganic component-matrixed nanocomposite, which is unfavorable to self-healing.40 Glycerol was thus selected as a high boiling point solvent to dissolve the host–guest polymer networks, greatly reducing the viscosity but avoiding solvent evaporation in the nanocomposite. This low viscosity of polymer/glycerol can be balanced by the nanoconfinement effects caused by the high loadings of GO nanosheets and AgNWs in the nanocomposite, which result in suitable polymer chain dynamics in the host–guest polymer network favorable for self-healing.40
Based on these material selections, a series of strain sensors with different weight ratios of these individual constituents were fabricated, with the optimal weight ratio of GO/AgNW/PVA-AD/PVA-CD/glycerol found to be 5:40:0.05:0.1:0.5. Increasing the contents of PVA-AD and PVA-CD would increase the stretchability at the cost of sensitivity of the nanocomposite (Fig. S2, ESI†). The final self-healable nanocomposite-based strain-sensing device was denoted as GO:AgNW/PVA-AD:PVA-CD sensor. For comparison purposes, four additional controlled strain-sensing nanocomposites with different compositions were also fabricated on the basis of the optimized component ratio and denoted as GO-AgNW:PVA sensor (GO/AgNW/PVA/glycerol = 5:40:0.15:0.5), GO:AgNW sensor (GO/AgNW/glycerol = 5:40:0.5), GO:AgNW/PVA-AD sensor (GO/AgNW/PVA-AD/glycerol = 5:40:0.15:0.5), and GO:AgNW/PVA-CD sensor (GO/AgNW/PVA-CD/glycerol = 5:40:0.15:0.5), respectively.
The nanoconfinement of the host–guest polymer network in the GO:AgNW/PVA-AD:PVA-CD nanocomposite was first characterized through various methodologies. The X-ray diffraction (XRD) patterns of the pure GO film and GO:AgNW/PVA-AD:PVA-CD nanocomposite film exhibit quintessential diffraction peaks for d-spacing of GO nanosheets at ∼13.0° and 11.9°, respectively (Fig. 2a). The increase in d-spacing between GO nanosheets in the GO:AgNW/PVA-AD:PVA-CD sample was based on the intercalation of the PVA-AD:PVA-CD complex into GO nanosheets via hydrogen bonding.52 From the view of supramolecular structure, PVA-AD can form strong host–guest complexes with PVA-CD via host–guest interaction. As revealed in Fig. 2b, the Fourier-transformed infrared spectrum (FT-IR) of the PVA-CD, PVA-AD and PVA-AD:PVA-CD mixture shows an absorption peak at 1725.1 cm−1, which is attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the side chains of PVA-AD and PVA-CD.53,54 Moreover, the absorption peaks at 1030.0 cm−1 corresponding to the stretching vibration of C–OH in the PVA-CD red-shifted to 1014.6 cm−1 after PVA-AD inclusion, which is caused by the host–guest interaction between CD and AD.55–57 Moreover, the 1H NMR spectrum comparison of PVA, PVA-acid, PVA-CD, PVA-AD, and the PVA-AD:PVA-CD mixture in Fig. 2c also provides strong evidence of the existence of CD–AD host-guest inclusion complexes in the PVA-AD:PVA-CD mixture. In region 1, a newly emerged peak at a chemical shift of 2.60 ppm in the PVA-acid spectra was ascribed to the protons of methylene near the CO group suggesting the successful grafting of succinic anhydride onto PVA.37 Essentially, in region 2, the –OH proton peak of β-CD in PVA-CD red-shifted from 3.77 ppm to 3.71 ppm as well as broadening compared with PVA-AD:PVA-CD spectra.56,58 In addition, the characteristic proton peak of AD moieties located at 1.80 ppm in the PVA-AD spectra becomes less obvious after PVA-CD addition in region 3.59,60 This above evidence suggests the existence of a host–guest interaction in the GO:AgNW/PVA-AD:PVA-CD nanocomposite.
O in the side chains of PVA-AD and PVA-CD.53,54 Moreover, the absorption peaks at 1030.0 cm−1 corresponding to the stretching vibration of C–OH in the PVA-CD red-shifted to 1014.6 cm−1 after PVA-AD inclusion, which is caused by the host–guest interaction between CD and AD.55–57 Moreover, the 1H NMR spectrum comparison of PVA, PVA-acid, PVA-CD, PVA-AD, and the PVA-AD:PVA-CD mixture in Fig. 2c also provides strong evidence of the existence of CD–AD host-guest inclusion complexes in the PVA-AD:PVA-CD mixture. In region 1, a newly emerged peak at a chemical shift of 2.60 ppm in the PVA-acid spectra was ascribed to the protons of methylene near the CO group suggesting the successful grafting of succinic anhydride onto PVA.37 Essentially, in region 2, the –OH proton peak of β-CD in PVA-CD red-shifted from 3.77 ppm to 3.71 ppm as well as broadening compared with PVA-AD:PVA-CD spectra.56,58 In addition, the characteristic proton peak of AD moieties located at 1.80 ppm in the PVA-AD spectra becomes less obvious after PVA-CD addition in region 3.59,60 This above evidence suggests the existence of a host–guest interaction in the GO:AgNW/PVA-AD:PVA-CD nanocomposite.
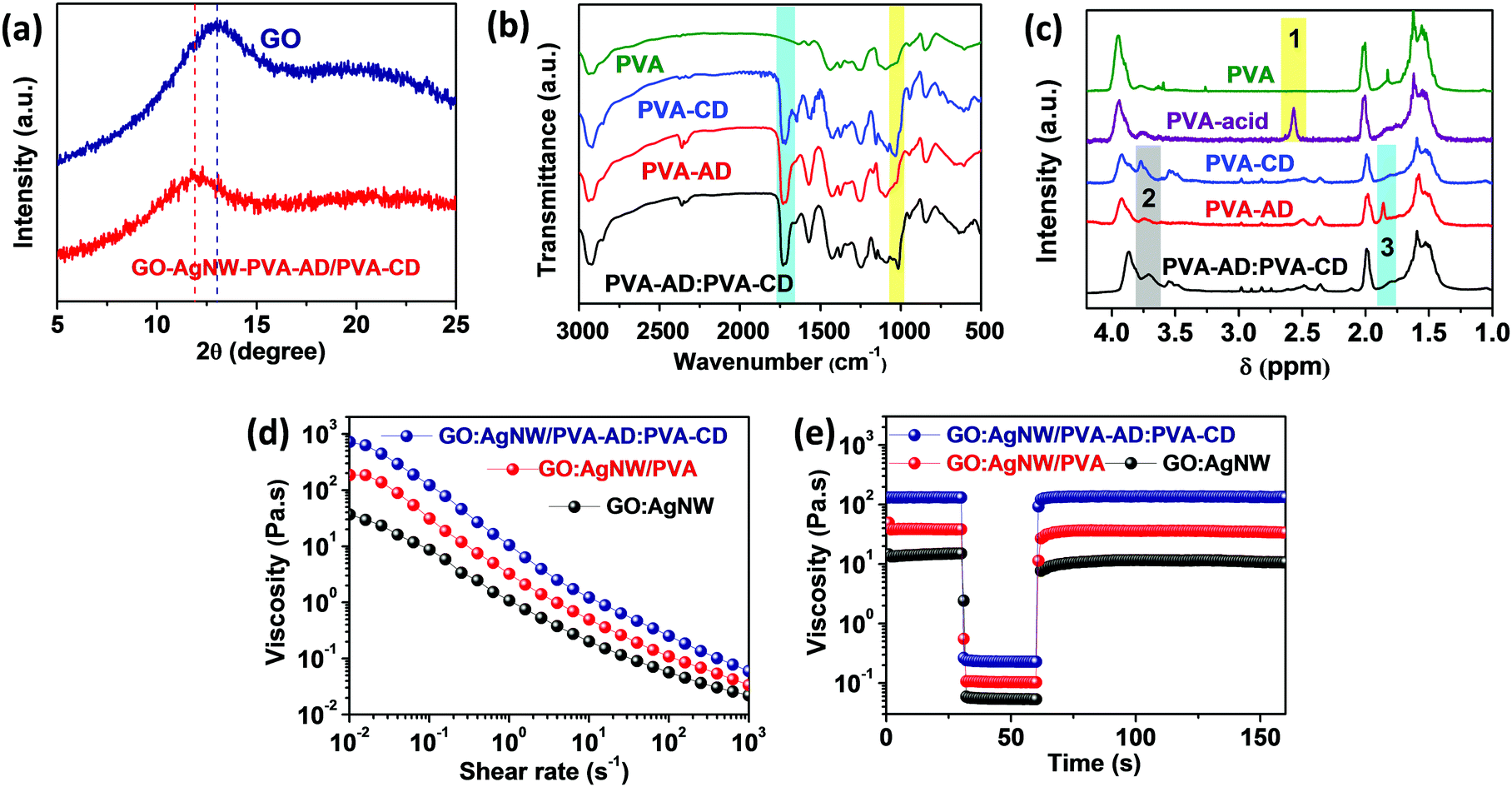 | ||
| Fig. 2 (a) XRD patterns for the pure GO film and GO/PVA-AD:PVA-CD nanocomposite film. (b) FT-IR spectra of PVA, PVA-AD, PVA-CD, and PVA-AD:PVA-CD mixtures. (c) 1H NMR spectra of PVA, PVA-acid, PVA-AD, PVA-CD, and PVA-AD:PVA-CD in D2O at 25 °C and the assignments of their characteristic peaks. (d) Viscosity as a function of shear rate for GO:AgNW, GO:AgNW/PVA, and GO:AgNW/PVA-AD:PVA-CD nanocomposite inks. (e) Rheological behavior of GO:AgNW, GO:AgNW/PVA, and GO:AgNW/PVA-AD:PVA-CD nanocomposite inks during simulation of screen printing. A peak hold step test was performed with constant shear rates for three intervals (0.1 s−1 shear rate for 30 s, 200 s−1 for 30 s, and 0.1 s−1 for 100 s). | ||
Furthermore, the host–guest interaction also greatly influences the viscous properties of the nanocomposite inks. As observed in Fig. 2d, all the GO:AgNW, GO:AgNW/PVA, and GO:AgNW/PVA-AD:PVA-CD nanocomposite inks exhibit shear-thinning behavior based on the physically cross-linked network formed between 1D AgNWs and 2D GO nanosheets.61 It is interesting to note that the incorporation of a tiny amount (about 0.33 wt%) of PVA-AD:PVA-CD into the nanocomposite ink can greatly increase the viscosity from 39 Pa s at 0.01 s−1 for GO:AgNW to 753 Pa s at 0.01 s−1 for the GO:AgNW/PVA-AD:PVA-CD nanocomposite ink. Such a significant viscosity increase is attributed to the formation of thermodynamically stable host–guest interactions between CD and AD in the polymer network.37 In addition to rheological behavior, all the nanocomposite inks show rapid viscous recovery in the peak hold step (PHS) experiment with different shear rates (Fig. 2e). When the shear rate was increased from 0.1 to 200 s−1 for 30 s, the viscosity of the nanocomposite inks immediately decreased but recovered rapidly when the shear rate was returned to 0.1 s−1. This high elasticity, together with the high viscosity, of the nanocomposite inks enables the construction of strain sensors with diverse geometries using screen-printing technology.
To evaluate the sensing performance of the strain-sensing nanocomposites, the typical strain sensors were constructed at a length of 20 mm, width of 3 mm, and thickness of ∼600 nm through screen printing as revealed in Fig. S3 (ESI†). All nanocomposite-based strain sensors were mounted on a linear motorized stage to control the testing uniaxial strain range. A Keithley 2000 digital multimeter was connected to record the resistance changes of the devices during the stretch-release processes, and the device sensitivity (gauge factor, marked as GF) was calculated by normalized resistance change ((R − R0)/R0, marked as ΔR/R0). Fig. 3a illustrates the typical relative resistance change as a function of applied strain curves and sensing linear behavior of different nanocomposite-based strain sensors. The GO:AgNW strain sensor without the confinement of any polymeric “bridging” material exhibits a high gauge factor up to 1557 due to its brittle structure that can induce considerable crack generation when stretching.15 However, as expected, this pure inorganic nanocomposite suffers from the dichotomy between sensitivity and stretchability and exhibits a limited working strain range of only 36%, far lower than the 50% value desired for wearable electronic applications.13 The separate confinement of PVA, PVA-CD, and PVA-AD as bridging materials into the layered structure of the nanocomposites can all greatly extend the working strain range to higher than 50% (Fig. 3a). This stretchability enhancement is mainly ascribed to the rich interfacial hydrogen bonding formed between GO nanosheets and the hydrophilic polymer chains, which, together with the polymer chain stretching, can accommodate partially applied stress during stretching.14 However, this stretchability improvement was achieved at the cost of sensitivity, and the maximum gauge factor for the GO:AgNW/PVA, GO:AgNW/PVA-CD, and GO:AgNW/PVA-AD sensors was significantly decreased to 138.1, 183.7, and 139.9, respectively.
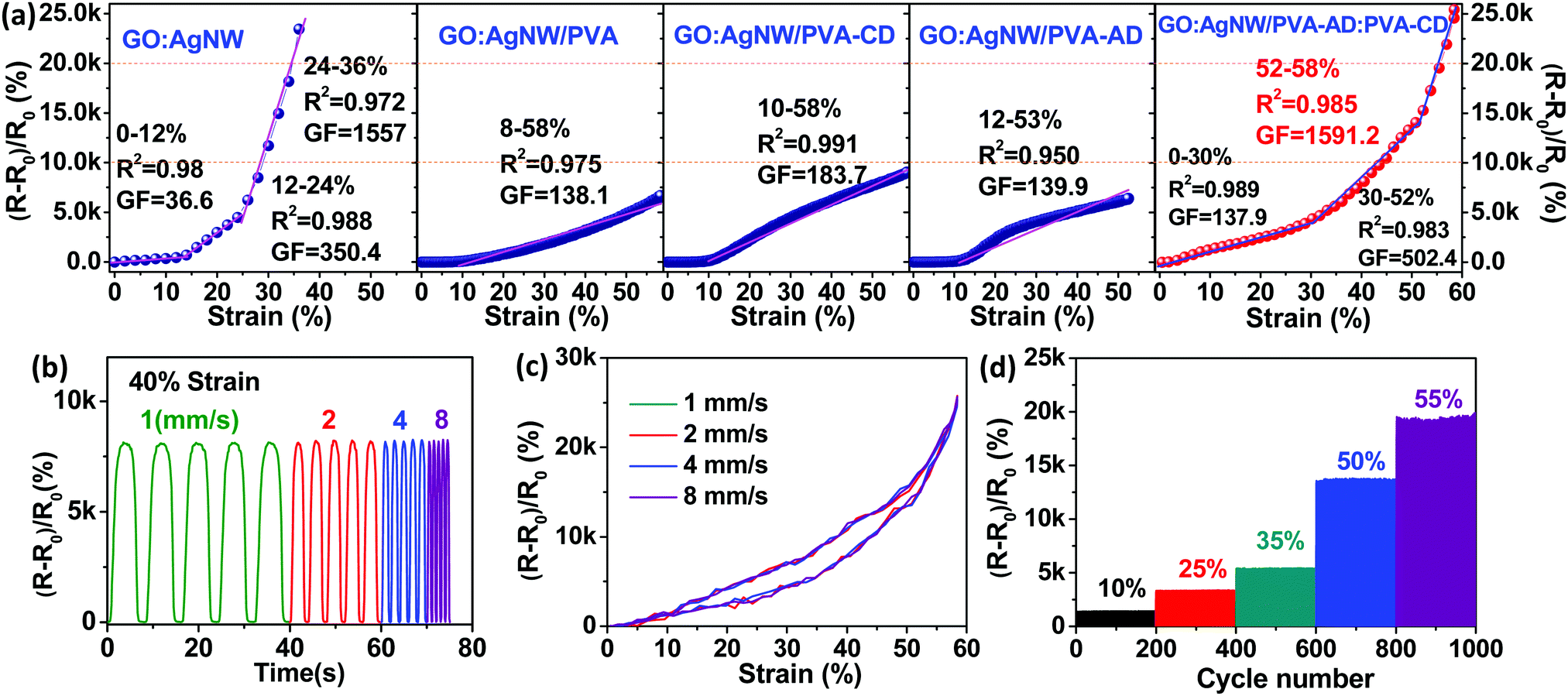 | ||
| Fig. 3 (a) Typical relative resistance change as a function of applied strain curves showing the GF and linear behavior of GO:AgNW, GO:AgNW/PVA, GO:AgNW/PVA-CD, GO:AgNW/PVA-AD, and GO:AgNW/PVA-AD:PVA-CD sensors. (b) Relative resistance change of the GO:AgNW/PVA-AD:PVA-CD sensor under incremental strain rates cycling between 0 and 40% strain. (c) Typical relative resistance changes versus applied strain curves under one stretch and release cycle for the GO:AgNW/PVA-AD:PVA-CD sensor at strain rates of 1, 2, 4, and 8 mm s−1. (d) Relative resistance variations under various cyclical maximum strains (10, 25, 35, 50, and 55) for the GO:AgNW/PVA-AD:PVA-CD sensor. | ||
When looking at the sensing curve for the GO:AgNW/PVA-AD:PVA-CD sensor, three subdivided linear regions are observed: 0–30, 30–52, and 52–58% (Fig. 3a). The corresponding linearities and gauge factors for these regions are 0.989, 0.983, and 0.985, and 137.9, 502.4, and 1591.2, respectively. This indicates that the strain-sensing nanocomposite with the addition of a host–guest polymer network can exhibit a gauge factor over 100 over a large working strain range of up to 58%, successfully eliminating the dilemma between large stretchability and high sensitivity. The maximum gauge factor of 1591.2 in the strain range of 52–58% is higher than all other four control nanocomposite-based sensors and is comparable to the state-of-the-art of sensitivity of reported stretchable strain sensors (Table S1, ESI†). The simultaneous improvement of both sensitivity and stretchability in the GO:AgNW/PVA-AD:PVA-CD sensor is mainly attributed to the formation of the CD-AD host–guest interaction in the nanocomposite. Compared with the control samples, the incorporation of CD-AD host–guest interaction into the nanocomposite can enrich the interfacial interaction among the multilevel structure of the nanocomposite, resulting in improved tensile strength for the nanocomposite. The thermodynamically stable but kinetically labile host–guest interaction can be broken upon applied strain, dissipating considerable amounts of loading energy to retard the crack propagation.14 Moreover, typical tensile stress–strain curves of freestanding GO:AgNW and GO:AgNW/PVA-AD:PVA-CD nanocomposites films are further evaluated to manifest their mechanical difference in Fig. S4 (ESI†). The addition of PVA-AD:PVA-CD greatly increases the elongation at break and tensile strength by almost 226% and 73% compared with those of the GO:AgNW film, respectively. This further confirms that the existence of PVA-AD:PVA-CD could improve the stretchability of the sensing film on the premise of high sensitivity.
As a practical wearable signal collection device, the accuracy and reliability of the strain sensor must be ensured. To show the reliability of the GO:AgNW/PVA-AD:PVA-CD strain sensor during the stretching process, its sensing curves under various strain rates were measured and are demonstrated in Fig. 3b. Under a maximum strain of 40%, the peak values of ΔR/R0 are steady and remain unchanged under strain rates increasing from 1 to 8 mm s−1. ΔR/R0-applied strain curves at various strain rates (1, 2, 4, 8 mm s−1) between 0 and 58% applied strain are also illustrated in Fig. 3c. Despite the different stretching velocities, the ΔR/R0-applied strain curves coincident with each other validated the accuracy and reliability of the ultradurable self-healing strain sensor. As depicted in Fig. 3d, the ΔR/R0 values under 10%, 25%, 35%, 50%, and 55% cyclic strains at an identical strain rate of 2 mm s−1 were 12.61, 33.22, 54.57, 137.53, and 190.39, matching the corresponding ΔR/R0 values of 12.95, 32.41, 57.02, 137.34, and 190.14 calculated from Fig. 3a. These results indicate that our GO:AgNW/PVA-AD:PVA-CD strain sensor exhibited good functioning reliability.
Next, we proceeded to test the durability and cycle-life of our nanocomposite-based strain sensors under various cyclical maximum strains, and direct comparison between the control samples and GO:AgNW/PVA-AD:PVA-CD nanocomposite reveals the critical role of the host–guest polymer network for durability improvements. All cycling tests were performed under ambient conditions without any external stimulus at a strain rate of 2 mm s−1. As shown in Fig. 4a–c, and Fig. S5 (ESI†), the GO:AgNW/PVA-AD:PVA-CD sensor exhibited intriguing cycling stability: both the peak and baseline resistance changes remain relatively stable over more than 200000, 200000, and 110000 stretch-release cycles under maximum strains of 15%, 35%, and 50%, respectively. Specifically, the peak values in the relative resistance changes for the GO:AgNW/PVA-AD:PVA-CD sensor are about 5500, 6700, 7300, 8440, 9080, and 9300 at the 1st, 1000th, 50000th, 100000th, 150000th, and 200000th cycles, respectively, under applied strain between 0 and 35%, showing only a mild increase upon continual cyclical stretch and release. Compared to the peak value in the relative resistance changes in the 1st cycle, the peak value at the 200000th cycle merely increases about 70%. This is in strong contrast to the GO:AgNW sensor and control nanocomposite sensors with the addition of PVA, PVA-AD, or PVA-CD, which demonstrate rather poor stability (Fig. 4d and Fig. S6–S8, ESI†). The peak values of the inorganic component-based GO:AgNW sensor at the 3rd, 5th, and 10th cycles reach 2.6×, 5.6×, and 11.7× the value in the 1st cycle, respectively, and turn into a malfunctioning state after following a few stretch-release cycles under 35% strain (Fig. 4d). The control sample of the GO:AgNW/PVA sensor with the incorporation of unhealable PVA shows 1.6×, 2.7×, and 6.5× increase in the peak value of resistance changes at the 1000th, 2000th, and 4000th cycles, respectively, and becomes completely damaged after about 7000 stretch-release cycles under 35% strain (Fig. 4e). Thus, we introduce the dynamic and in situ self-healing ability into an inorganic material-matrixed nanocomposite system without compromising the high sensitivity (GF > 100) and large stretchability (working strain range >50%) of the sensing material.
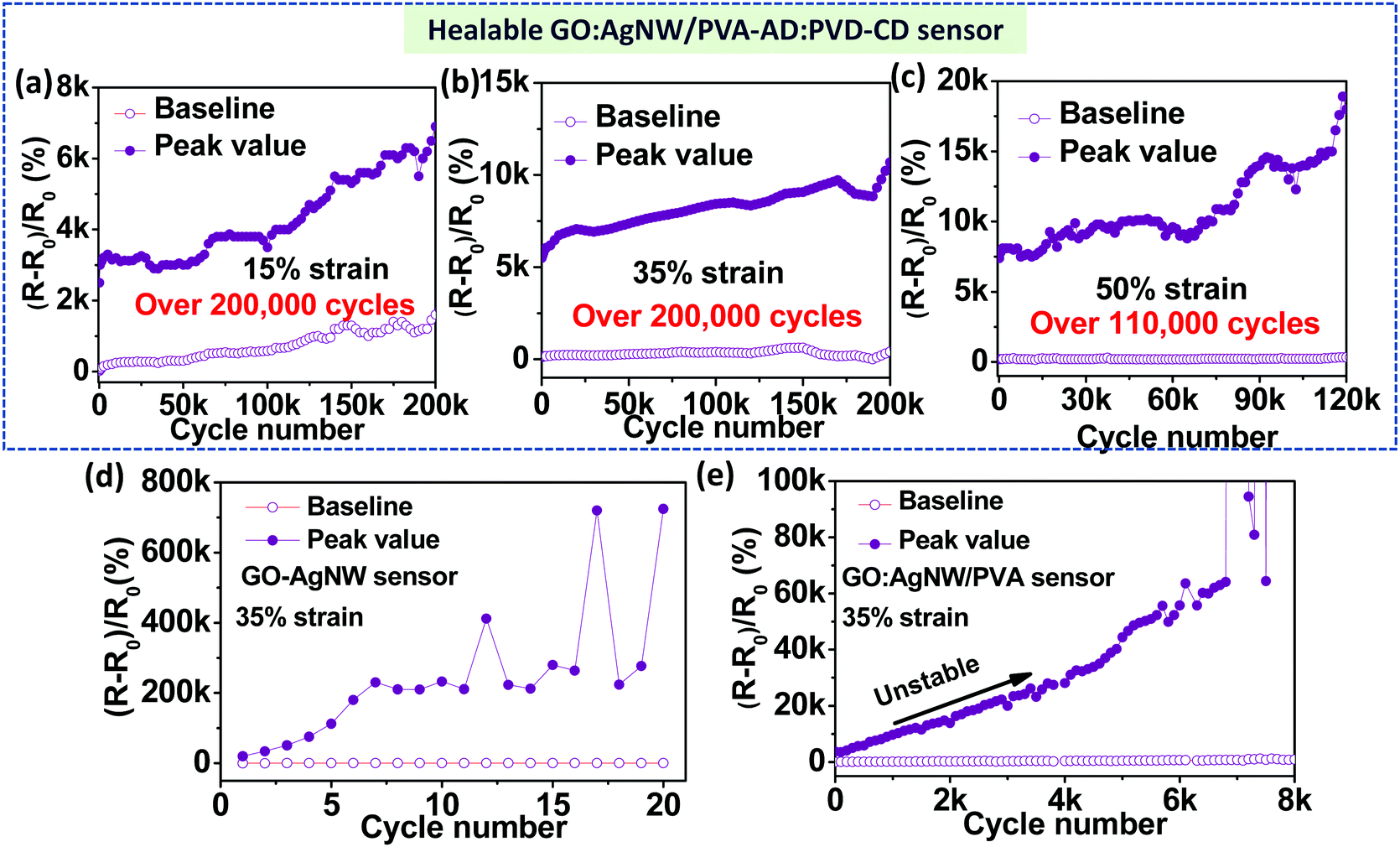 | ||
| Fig. 4 Cyclic durability of GO:AgNW/PVA-AD:PVA-CD sensors under various applied strain ratios between (a) 0 and 15%, (b) 0 and 35%, and (c) 0 and 50%. (d) Cyclic durability of GO:AgNW sensors under applied strain between 0 and –35%. (e) Cyclic durability of GO:AgNW/PVA sensors under applied strain between 0–35%. The strain rate for all measurements was 2 mm s−1. | ||
Such significant durability enhancement is suggested to be attributed to the nanoconfined self-healable supramolecular polymer networks that have suitable polymer chain mobility and fast dynamic host–guest interaction. Fig. 5a illustrates how the cracked GO:AgNW/PVA-AD:PVA-CD sensing film was spontaneously, rapidly, and in situ repaired by the host–guest polymer networks during cyclical stretching and releasing processes. At their original state, the polymer networks, cross-linked by the CD–AD host–guest interaction, are dissolved in glycerol confined between GO/AgNW nanofillers. After being stretched, cracks are generated in the sensing film due to the brittle nature of the inorganic-matrixed nanocomposite; in the meantime, the glycerol ruptures, and the AD-CD host–guest interactions in the PVA based polymer networks, as well as the hydrogen bonding between PVA and GO, are broken to accommodate the partially applied stress. It should be noted that the nanoconfinement effects can confine the polymer/glycerol into the nanocomposite without flowing into the cracks. When released to the unstretched state, the separated glycerol immediately merges once two edges of the cracks are brought into close contact. As such, the dissolved PVA-AD and PVA-CD molecular chains can rapidly diffuse across the damaged interfaces, allowing the spontaneous and in situ reformation of CD–AD host–guest interaction in the polymer network. Importantly, the fast dynamics of the association of AD to CD and the movable polymer chains in glycerol enable the rapid reformation CD–AD host–guest interaction within a few hundreds of microseconds.49–51 In turn, this can lead to the in situ self-repairing of the microcracks and damages in the sensing films during the stretch and release cycles, thus greatly increasing the cycle-life and durability of the sensing device.
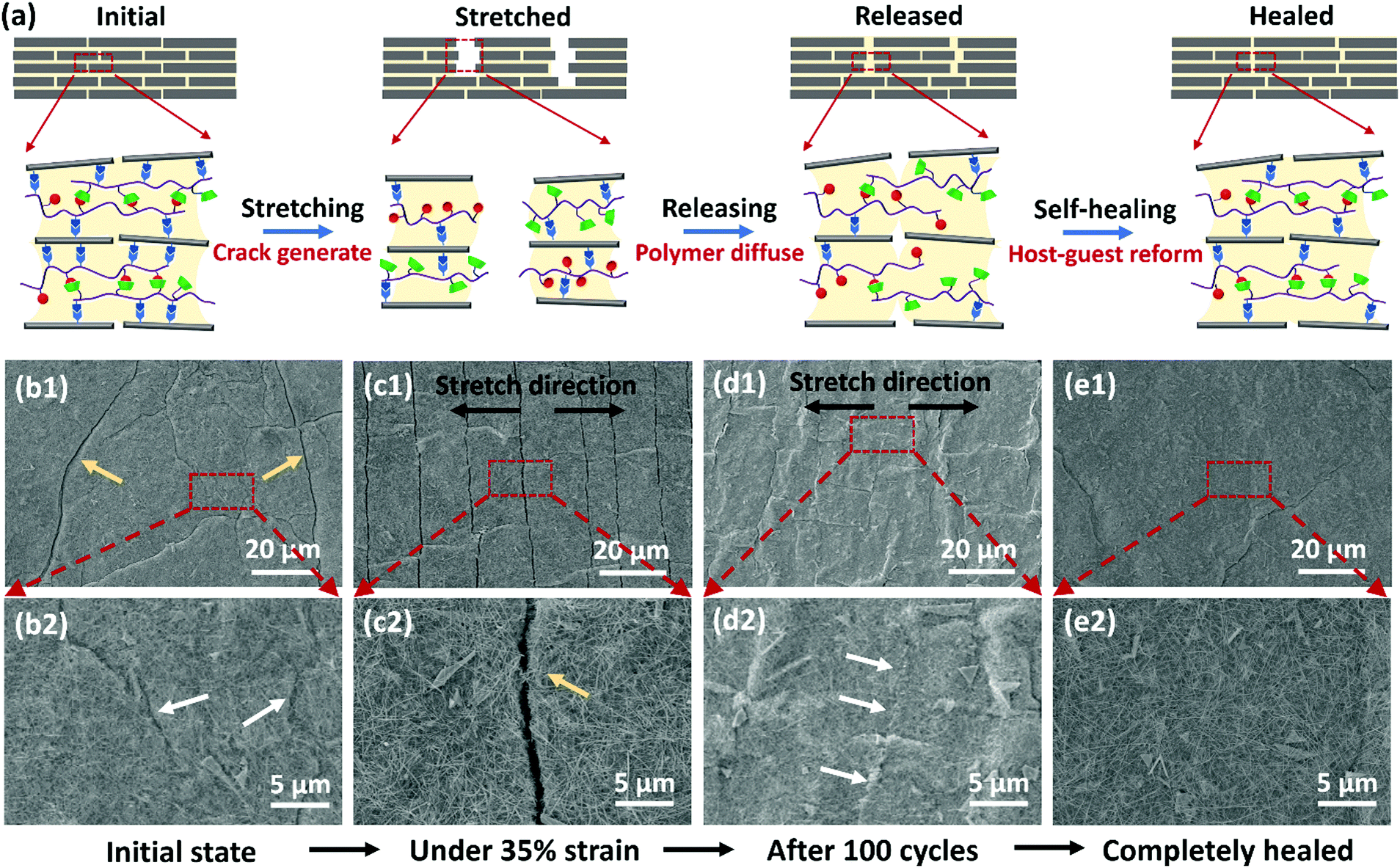 | ||
| Fig. 5 (a) Schematic illustration of the damage-healing process for the GO:AgNW/PVA-AD:PVA-CD sensor under stretch-release cycles. (b1) SEM image and (b2) magnified SEM image showing the surface morphology of the GO:AgNW/PVA-AD:PVA-CD sensor at an initial state (after pre-strained) before cycling; cracks and scar-like structures indicated by yellow and white arrows, respectively. (c1) SEM image and (c2) magnified SEM image showing the surface morphology of the GO:AgNW/PVA-AD:PVA-CD sensor under a 35% strain state; crack indicated by a yellow arrow. (d1) SEM image and (d2) magnified SEM image showing the surface morphology of the GO:AgNW/PVA-AD:PVA-CD sensor under a released state after 100 stretch-release cycles with a 35% strain; scar-like structures (indicated by white arrows) can be found on the nanocomposite, which appear to be cracks that subsequently healed. (e1) SEM image and (e2) magnified SEM image showing the surface morphology of the GO:AgNW/PVA-AD:PVA-CD sensor after 100 stretch-release cycles and kept at a released state under ambient conditions for 3 h; most of the cracks were completely healed. | ||
To provide further evidence, the surface morphology of the GO:AgNW/PVA-AD:PVA-CD sensing film under repeated stretch and release was monitored using scanning electron microscopy (SEM). Before cycling, pre-strain was first applied to the sensing film, and some microcracks and scar-like structures appeared throughout the nanocomposite film (Fig. 5b1 and b2). When stretching to 35% strain, the cracks propagated along the orientation perpendicular to the stretching direction (Fig. 5c1), and opening gap of the cracks was about 1 μm (Fig. 5c2). After 100 stretch-release cycles and releasing to the unstretched state, some long scar-like structures can be found to vertically extend in the stretching direction, which appeared to be the microcracks formed under the stretched state that subsequently self-healed by the host–guest polymer networks under the released state (Fig. 5d1 and d2). Moreover, when the cycled nanocomposite film was left at its released state for 3 h, it was observed that most of the initial microcracks along with long scar-like structures were completely self-healed and disappeared, and the nanocomposite film displayed a relatively flat and smooth surface morphology (Fig. 5e1 and e2), strongly indicating the presence of self-healing effects. In contrast, it can be clearly observed from the SEM images (Fig. S9, ESI†) that microcracks generated during the stretching process still existed, and no scar-like structures appeared on the control nanocomposite films with the incorporation of unhealable polymer PVA, PVA-AD, or PVA-CD, even when the samples were left in their released state for 10 days.
The contribution of the self-healing effects from the host–guest supramolecular polymer networks to the cycling durability of the nanocomposite sensing film was further confirmed by a control sample prepared through adding a certain amount of AD small molecules as a guest inhibitor into the GO:AgNW/PVA-AD:PVA-CD nanocomposite system. These AD small molecules could replace PVA-AD to form the CD–AD host–guest complex with PVA-CD to prevent the polymer network cross-linking. As demonstrated in Fig. S10 (ESI†), the sensing film fabricated from the control sample showed a rapid decay behavior in the cycling stability, which is similar to those of the nanocomposites without self-healing ability (Fig. 4e and Fig. S7, ESI†), and is much poorer than that of the GO:AgNW/PVA-AD:PVA-CD sensor (Fig. 4a–c). Moreover, the GO:AgNW/PVA-AD:PVA-CD nanocomposite without the addition and confinement of high boiling point glycerol solvent did not exhibit self-healing behavior, and its resistance changes over repeated stretch-release cycles also displayed rather poor cycling stability (Fig. S11, ESI†). Thus, good polymer chain mobility and dynamic host–guest interaction formed in the supramolecular polymer networks are two key enablers for intrinsic rapid self-healing in the nanocomposite-based strain-sensing system. Irreparable cracks would appear when the GO:AgNW/PVA-AD:PVA-CD sensor sensing materials were stretched larger than ∼80% strain.
Conclusions
In summary, we demonstrated that the nanoconfinement of a dynamic host–guest supramolecular polymer network together with high boiling point glycerol solvent in the inorganically-matrixed nanocomposite system with multilevel structure enabled the fabrication of a stretchable and ultrasensitive strain sensor with self-healing ability and significantly improved cycling durability. More specifically, the dynamic CD-AD host–guest complex in the PVA system has a large affinity constant and fast association dynamic; the nanoconfinement effect induced by the inorganic nanofillers endowed the host–guest polymer network with suitable molecular mobility in glycerol; these effects work together to enable the spontaneous, rapid, and in situ repair of the damages generated in the predominantly inorganic structural nanocomposite system during the continual stretch-release cycling process. In addition, the simplicity of the nanocomposite and subsequent sensing device fabrication implies that our strategy has great potential for application of wearable and durable strain sensors in full-range human-health diagnostics and human-motion monitoring. Finally, this investigation points to a challenging strategy of designing ultradurable, ultrasensitive, and stretchable strain sensors for practical wearable electronic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work reported here was supported by the National Key R&D Program of China (2016YFA0200200) and NSFC (51872146, 51673099, 51633002, and 21421001).References
- W. Wu and H. Haick, Adv. Mater., 2018, 30, 1705024 CrossRef.
- W. Liu, M.-S. Song, B. Kong and Y. Cui, Adv. Mater., 2017, 29, 1603436 CrossRef.
- Z. Liu, D. Qi, P. Guo, Y. Liu, B. Zhu, H. Yang, Y. Liu, B. Li, C. Zhang, J. Yu, B. Liedberg and X. Chen, Adv. Mater., 2015, 27, 6230–6237 CrossRef CAS.
- Q. Liu, J. Chen, Y. Li and G. Shi, ACS Nano, 2016, 10, 7901–7906 CrossRef CAS.
- M. Hempel, D. Nezich, J. Kong and M. Hofmann, Nano Lett., 2012, 12, 5714–5718 CrossRef CAS.
- Y. Cai, J. Shen, G. Ge, Y. Zhang, W. Jin, W. Huang, J. Shao, J. Yang and X. Dong, ACS Nano, 2018, 12, 56–62 CrossRef CAS.
- K. K. Kim, S. Hong, H. M. Cho, J. Lee, Y. D. Suh, J. Ham and S. H. Ko, Nano Lett., 2015, 15, 5240–5247 CrossRef CAS.
- M. Amjadi, A. Pichitpajongkit, S. Lee, S. Ryu and I. Park, ACS Nano, 2014, 8, 5154–5163 CrossRef CAS.
- Z. Liu, D. Qi, G. Hu, H. Wang, Y. Jiang, G. Chen, Y. Luo, X. J. Loh, B. Liedberg and X. Chen, Adv. Mater., 2018, 30, 1704229 CrossRef.
- J. Zhou, X. Xu, Y. Xin and G. Lubineau, Adv. Funct. Mater., 2018, 28, 1870101 CrossRef.
- M. Amjadi, K.-U. Kyung, I. Park and M. Sitti, Adv. Funct. Mater., 2016, 26, 1678–1698 CrossRef CAS.
- X. Xiao, L. Yuan, J. Zhong, T. Ding, Y. Liu, Z. Cai, Y. Rong, H. Han, J. Zhou and Z. L. Wang, Adv. Mater., 2011, 23, 5440–5444 CrossRef.
- X. Shi, S. Liu, Y. Sun, J. Liang and Y. Chen, Adv. Funct. Mater., 2018, 28, 1800850 CrossRef.
- X. Shi, H. Wang, X. Xie, Q. Xue, J. Zhang, S. Kang, C. Wang, J. Liang and Y. Chen, ACS Nano, 2019, 13, 649–659 CrossRef CAS.
- Y. Liu, X. Shi, S. Liu, H. Li, H. Zhang, C. Wang, J. Liang and Y. Chen, Nano Energy, 2019, 63, 103898 CrossRef CAS.
- G. Shi, Z. Zhao, J.-H. Pai, I. Lee, L. Zhang, C. Stevenson, K. Ishara, R. Zhang, H. Zhu and J. Ma, Adv. Funct. Mater., 2016, 26, 7614–7625 CrossRef CAS.
- D. Kang, P. V. Pikhitsa, Y. W. Choi, C. Lee, S. S. Shin, L. Piao, B. Park, K.-Y. Suh, T.-i. Kim and M. Choi, Nature, 2014, 516, 222–226 CrossRef CAS.
- B. Park, J. Kim, D. Kang, C. Jeong, K. S. Kim, J. U. Kim, P. J. Yoo and T.-i. Kim, Adv. Mater., 2016, 28, 8130–8137 CrossRef CAS.
- X. Li, R. Zhang, W. Yu, K. Wang, J. Wei, D. Wu, A. Cao, Z. Li, Y. Cheng, Q. Zheng, R. S. Ruoff and H. Zhu, Sci. Rep., 2012, 2, 870 CrossRef.
- Y. Heo, Y. Hwang, H. S. Jung, S.-H. Choa and H. C. Ko, Small, 2017, 13, 1700070 CrossRef.
- Y. Wang, J. Hao, Z. Huang, G. Zheng, K. Dai, C. Liu and C. Shen, Carbon, 2018, 126, 360–371 CrossRef CAS.
- L. Li, H. Xiang, Y. Xiong, H. Zhao, Y. Bai, S. Wang, F. Sun, M. Hao, L. Liu, T. Li, Z. Peng, J. Xu and T. Zhang, Adv. Sci., 2018, 5, 1800558 CrossRef.
- F. Pan, S.-M. Chen, Y. Li, Z. Tao, J. Ye, K. Ni, H. Yu, B. Xiang, Y. Ren, F. Qin, S.-H. Yu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1803221 CrossRef.
- T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296–301 CrossRef CAS.
- B. Park, S. Lee, H. Choi, J. U. Kim, H. Hong, C. Jeong, D. Kang and T.-i. Kim, Nanoscale, 2018, 10, 4354–4360 RSC.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef.
- J. Wu, L.-H. Cai and D. A. Weitz, Adv. Mater., 2017, 29 Search PubMed.
- D. Son, J. Kang, O. Vardoulis, Y. Kim, N. Matsuhisa, J. Y. Oh, J. W. F. To, J. Mun, T. Katsumata, Y. Liu, A. F. McGuire, M. Krason, F. Molina-Lopez, J. Ham, U. Kraft, Y. Lee, Y. Yun, J. B. H. Tok and Z. Bao, Nat. Nanotechnol., 2018, 13, 1057–1065 CrossRef CAS.
- M. Khatib, O. Zohar, W. Saliba, S. Srebnik and H. Haick, Adv. Funct. Mater., 2020, 30, 1910196 CrossRef CAS.
- G. Cai, J. Wang, K. Qian, J. Chen, S. Li and P. S. Lee, Adv. Sci., 2017, 4, 1600190 CrossRef.
- M. A. Darabi, A. Khosrozadeh, R. Mbeleck, Y. Liu, Q. Chang, J. Jiang, J. Cai, Q. Wang, G. Luo and M. Xing, Adv. Mater., 2018, 30, 1705922 CrossRef.
- Y. Tang, Z. Zhao, H. Hu, Y. Liu, X. Wang, S. Zhou and J. Qiu, ACS Appl. Mater. Interfaces, 2015, 7, 27432–27439 CrossRef CAS.
- W. Miao, D. Wang, Z. Liu, J. Tang, Z. Zhu, C. Wang, H. Liu, L. Wen, S. Zheng, Y. Tian and L. Jiang, ACS Nano, 2019, 13, 3225–3231 CrossRef CAS.
- D. J. Lipomi, M. Vosgueritchian, B. C. K. Tee, S. L. Hellstrom, J. A. Lee, C. H. Fox and Z. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef.
- Y.-G. Jia, J. Jin, S. Liu, L. Ren, J. Luo and X. X. Zhu, Biomacromolecules, 2018, 19, 626–632 CrossRef CAS.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J.-C. Lai, L. Liu, C.-H. Li, X. Yan, C. Liu, J. B.-H. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, 30, 1801435 CrossRef.
- C. Wang, X. Li, E. Gao, M. Jian, K. Xia, Q. Wang, Z. Xu, T. Ren and Y. Zhang, Adv. Mater., 2016, 28, 6640–6648 CrossRef CAS.
- G. Du, A. Mao, J. Yu, J. Hou, N. Zhao, J. Han, Q. Zhao, W. Gao, T. Xie and H. Bai, Nat. Commun., 2019, 10, 800 CrossRef.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381–4395 RSC.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS.
- S. Wan, F. Xu, L. Jiang and Q. Cheng, Adv. Funct. Mater., 2017, 27, 1605636 CrossRef.
- S. Wan, Y. Li, J. Peng, H. Hu, Q. Cheng and L. Jiang, ACS Nano, 2015, 9, 708–714 CrossRef CAS.
- H. Cheng, Y. Huang, Q. Cheng, G. Shi, L. Jiang and L. Qu, Adv. Funct. Mater., 2017, 27, 1703096 CrossRef.
- M. Paolino, F. Ennen, S. Lamponi, M. Cernescu, B. Voit, A. Cappelli, D. Appelhans and H. Komber, Macromolecules, 2013, 46, 3215–3227 CrossRef CAS.
- M. R. Eftink, M. L. Andy, K. Bystrom, H. D. Perlmutter and D. S. Kristol, J. Am. Chem. Soc., 1989, 111, 6765–6772 CrossRef CAS.
- F. Schibilla, J. Voskuhl, N. A. Fokina, J. E. P. Dahl, P. R. Schreiner and B. J. Ravoo, Chem. – Eur. J., 2017, 23, 16059–16065 CrossRef CAS.
- S. Nishikawa and T. Ugawa, J. Phys. Chem. A, 2000, 104, 2914–2918 CrossRef CAS.
- A. Douhal, Cyclodextrin Materials Photochemistry, Photophysics and Photobiology, 2006 DOI:10.1016/B978-0-444-52780-6.X5000-1.
- D. Granadero, J. Bordello, M. J. Pérez-Alvite, M. Novo and W. Al-Soufi, Int. J. Mol. Sci., 2010, 11, 173–188 CrossRef CAS.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- Y. Zhang, W. Wang, Q. Li, Q. Yang, Y. Li and J. Du, Talanta, 2015, 141, 33–40 CrossRef CAS.
- W.-F. Pu, Y. Yang, B. Wei and C.-D. Yuan, Ind. Eng. Chem. Res., 2016, 55, 8679–8689 CrossRef CAS.
- M. Zhang, M. Jiang, L. Meng, K. Liu, Y. Mao and T. Yi, Soft Matter, 2013, 9, 9449–9454 RSC.
- C. Yuan, Z. Jin and X. Xu, Carbohydr. Polym., 2012, 89, 492–496 CrossRef CAS.
- X. Ma, N. Zhou, T. Zhang, W. Hu and N. Gu, Mat. Sci. Eng. C-Mater., 2017, 73, 357–365 CrossRef CAS.
- M. Miyauchi and A. Harada, J. Am. Chem. Soc., 2004, 126, 11418–11419 CrossRef CAS.
- B. Knudsen, B. E. Kergl, H. Paulsen, V. Durnev and H. Ritter, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2472–2482 CrossRef CAS.
- S. Liu, Y. Wang, J. Cai, L. Ren, L. Wang and Y. Wang, Polym. Int., 2014, 63, 1930–1935 CrossRef CAS.
- S. Liu, J. Li, X. Shi, E. Gao, Z. Xu, H. Tang, K. Tong, Q. Pei, J. Liang and Y. Chen, Adv. Electron. Mater., 2017, 3, 1700098 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh01539c |
| This journal is © The Royal Society of Chemistry 2021 |