
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-term effects of impurities on the particle size and optical emission of carbon dots†
Nasir
Javed
a and
Deirdre M.
O'Carroll
 *ab
*ab
aDepartment of Materials Science and Engineering, Rutgers, The State University of New Jersey, 607 Taylor Road, Piscataway, NJ 08854, USA. E-mail: ocarroll@rutgers.edu
bDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 123 Bevier Road, Piscataway, NJ 08854, USA
First published on 24th September 2020
Abstract
Carbon dots (CDs) are fluorescent nanoparticles that exhibit strong photoluminescence (PL) emission throughout the visible range of the electromagnetic spectrum. Recent studies highlight the presence of fluorescent impurities in CD dispersions. Here, the long-term impact of these impurities on the stability of the physical and optical properties of CDs synthesized by the solvothermal method is studied. A significant increase in particle size is observed as a function of time after synthesis from transmission electron microscopy analysis of CDs. Furthermore, the quantum yield of blue PL emission, which is mostly caused by impurities that contain carboxyl groups, gradually decays from 30% to ∼3% over 13 weeks. The reduction in quantum yield is attributed to decomposition of impurities that, consequently, deposit on the particles and increase particle size. Finally, it is observed that the blue emission decreases considerably when CDs are properly purified and a solvent-dependent yellow emission arises. The yellow emission is almost negligible when CDs are dispersed in water; however, the intensity of yellow emission increases significantly when the concentration of ethanol is increased.
Introduction
Carbon dots (CDs) are nanoparticles that exhibit strong photoluminescence (PL) emission throughout the visible spectrum.1–4 In addition to strong PL emission, they possess several other promising properties, e.g., high water solubility, easy functionalization, excellent biocompatibility, noticeable electron donating and accepting capabilities, and low toxicity.4,5 Due to these attractive properties, with easy and low-cost synthesis, and abundance of the raw materials, they have been extensively studied as an alternative to conventional semiconductor quantum dots.1–4 Different top-down and bottom-up synthesis methods have been utilized to synthesize carbon dots. CDs were discovered during the purification of single-walled carbon nanotubes, derived from arc-discharge by Xu et al.6 Therefore, early research on the synthesis of carbon dots was focused on top-down methods, for instance: oxidation of graphite;7 laser irradiation;8–10 and electrochemical synthesis.11,12 However, in the last decade wet-chemistry-based bottom-up methods to synthesize CDs became more popular due to possibilities for one-step synthesis and functionalization. Hydrothermal synthesis,13–15 microwave assisted pyrolysis,16,17 carbonization of organic compounds18–21 and ultrasonication techniques22,23 are a few notable examples.During synthesis of CDs, especially by chemical methods, various reaction pathways from precursors to products are possible. Therefore, it is natural to assume that several molecular byproducts can also form during synthesis of CDs. It has been reported that byproducts formed during the synthesis of CDs could be molecular fluorophores.24,25 Hinterberger et al. reported the synthesis of blue- and yellow-emitting fluorophores during the microwave assisted synthesis of blue-emitting CDs form citric acid and o-phenylenediamine.26 They further stated that these fluorophores were not only attached on the CDs but were also found to be freely floating in the solution. In another study, they reported different degrees of functionalization of CDs by the fluorophores.27 They were able to separate CDs with fluorophores attached to them from CDs without fluorophores on them. Further, they determined that the PL quantum yield of the free-floating fluorophores is the highest, whereas the CDs without fluorophores attached on them have the lowest quantum yield. In another similar study, Wang et al. synthesized CDs by microwave-assisted heating of citric acid and urea, and compared the fluorescence properties of CDs after dialysis with those of the dialysate. They found that the dialysate showed a similar fluorescence response to that of dialyzed CDs.25 Furthermore, a few researchers have investigated the structure of molecular fluorophores formed along with CDs. Mostly, fluorophores are reported to have pyridine-based structures.24,27–29
Interestingly, in most of the reports about formation of fluorophores with CDs, synthesis was carried out in water and citric acid was one of the precursor.30,31 This may give an impression that molecular fluorophores only form in water. There are a few reports available in literature about the formation of fluorophores while the reaction was carried out in ethanol. For instant, Khan et al. reported that CDs synthesized in ethanol by solvothermal synthesis from urea and p-phenylenediamine possess photophysical properties of a mixture of nanocrystals and dye molecules.32 Similarly, Zhu et al. synthesized CDs by solvothermal reaction of naphthalenediol and K2S2O8 in ethanol, and showed that PL originates from organic fluorophores.33 Therefore, formation of molecular fluorophores can be anticipated during bottom-up synthesis of CDs in organic solvents as well.
In order to remove impurities or reaction byproducts from CDs, various purification techniques have been employed and reported in the literature. Dialysis, centrifugation, solvent extraction and chromatography are among the most widely used purification methods. Recently, Essner et al. reviewed the purification techniques of more than 550 publications on fluorescent CDs and brought to attention that the literature of CDs is continuously being filled by “uncritical, unsubstantiated, and detrimental claims and misconceptions” which mostly arose from “inadequate, unsubstantiated” sample purification methods.34 Surprisingly, they found that more than 10% of surveyed literature did not use any purification method. Poor purification, together with the possibility of formation of molecular fluorophores having fluorescence similar to that of CDs implies that emission from impurities could be misunderstood as being from CDs and may have been mistakenly studied by many researchers in the context of CDs.34,35
In short, it is undeniable that impurities or reaction byproducts can be present in CDs. This raises the question of how these impurities effect the properties of CD over time. There are hardly any reports available on the study of the effects of impurities/byproducts on the properties of CDs over the long-term. Herein, we report the post-synthesis increase in the particle size of CDs caused by decomposition and accumulation of impurities on particles. The stability of the optical properties of CDs synthesized using the solvothermal method in the presence of impurities or reaction byproducts is also studied.
In addition, the PL mechanism of CDs is still strongly debated. Sun et al.10 proposed that the excitation-dependent PL emission is due to quantum confinement as well as emission from surface energy traps. Jiang et al.36 attributed the PL emission to particle size effects and nitrogen content; while Hu et al.37 observed that the PL emission color change was not supported by particle size effects and depended on the composition of the CDs. In most reports, PL emission from CDs is attributed to surface electronic states caused by surface functional groups,1–4,38 yet some researchers argue that it is due to inter-band electronic transitions of crystalline carbon.39,40 Therefore, in addition to the role of impurities on the optical and physical properties of CDs, we also briefly investigate the origin of PL emission from purified CDs, and find that it originates from solvent-induced surface states.
Experimental
Synthesis of carbon dots
CDs were synthesized by modifying a method published by Yuan et al. for synthesis of carbon quantum dots.40 Briefly, 10 mg of citric acid (Sigma-Aldrich; ≥99.5%) and 20 mg of 1,5-diaminonaphthalene (Tokyo Chemical Industry Co., Ltd; >98.0%) were dissolved in 10 mL ethanol (Koptec; 190 proof) by sonication for 15 minutes. Then the solution was heated at 200 °C for 5 hours in a 25 mL Teflon-lined stainless-steel autoclave. After heating, the resulting black solution was diluted with deionized water (milli-Q, 18.2 MΩ cm) and centrifuged at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 hour to remove larger particles. The sample was stored at room temperature in the dark/amber vial for further characterization. The synthesis process for preparation of CDs is shown in Scheme 1a; whereas the structure of CDs is depicted in Scheme 1b. For dialyzed CDs, the as-synthesized product was diluted with deionized water and dialyzed for 48 hours in deionized water using Regenerated Cellulose (RC) dialysis membrane having molecular weight cutoff (MWCO) of 1 kDa (Spectrum Laboratories Inc. manufacturer part number: 132640). Dialysis water was changed every three hours from 9:00 am to 9:00 pm (five time in one day); however, no water was changed between 9:00 pm to 9:00 am.
000g for 1 hour to remove larger particles. The sample was stored at room temperature in the dark/amber vial for further characterization. The synthesis process for preparation of CDs is shown in Scheme 1a; whereas the structure of CDs is depicted in Scheme 1b. For dialyzed CDs, the as-synthesized product was diluted with deionized water and dialyzed for 48 hours in deionized water using Regenerated Cellulose (RC) dialysis membrane having molecular weight cutoff (MWCO) of 1 kDa (Spectrum Laboratories Inc. manufacturer part number: 132640). Dialysis water was changed every three hours from 9:00 am to 9:00 pm (five time in one day); however, no water was changed between 9:00 pm to 9:00 am.
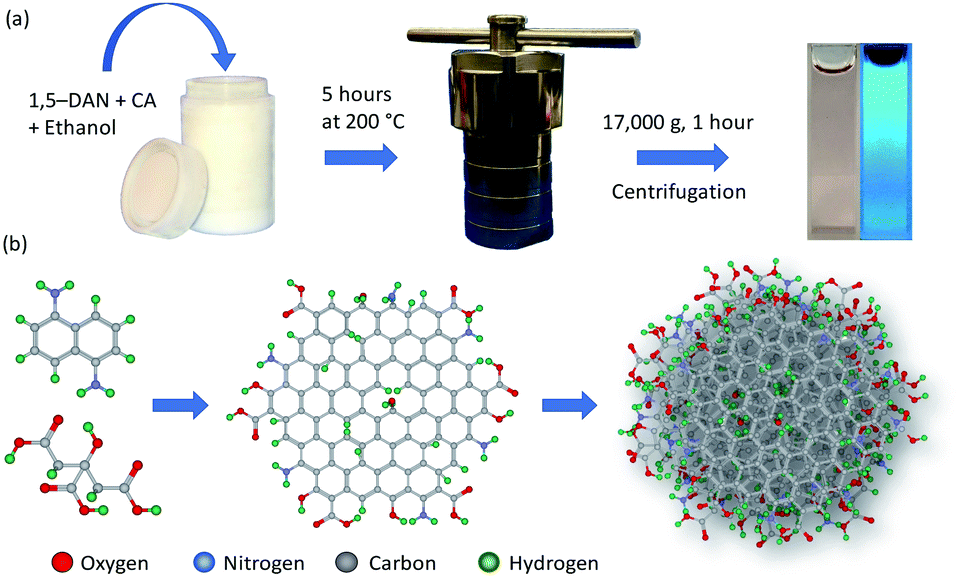 | ||
| Scheme 1 Schematic representation of (a) solvothermal synthesis of CDs and (b) the envisioned reaction pathway and particle structure. | ||
Characterization
Absorption spectra were recorded on a UV-visible spectrometer (SI Photonics CCD Array UV-vis spectrometer) equipped with deuterium and tungsten lamps for UV and visible light analysis, respectively. Quartz cuvettes of 10 mm path length were used to hold the liquid samples. Photoluminescence (PL) emission was measured on a Horiba's Fluorolog-3 spectrofluorometer. The spectrofluorometer is equipped with of a 450 watt xenon lamp source, a double excitation monochromator, an iHR 320 emission monochromator and Hamamatsu R928P photomultiplier tube (PMT) detector. The synthesis product (with/without purification) was held in a standard 10 mm square quartz cuvette. PL emission was measured using the front-face collection mode, i.e., the PL signal was detected at 22.5° to the incident light path. As the PMT doctor does not respond linearly when counts per second (cps) increase beyond few millions, therefore, to keep the cps in linear range excitation and emission slit widths were adjusted depending on the photoluminescence intensity of samples, e.g., narrow slit widths were used for sample emitting strongly. The excitation and emission slit widths used for each sample are mentioned in the figure captions. For studying the excitation dependence of PL emission, spectra were recorded by using difference excitation wavelengths. Absolute photoluminescence quantum yield (PLQY) was measured with a calibrated integrated sphere (F-3018) on the same spectrofluorometer. PLQY was measured at two different excitation wavelengths (λexc): 340 nm and 440 nm. Scattered spectra were integrated from λexc − 5 nm to λexc + 5 nm. Integration ranges of emission spectra varies with samples and are mention with the results. Raman Spectroscopy was carried out on a Renishaw Raman Spectrometer using a 514 nm laser and a 50× objective lens. For the measurement, the sample was drop cast on a zinc selenide (ZnSe) substrate and dried at room temperature. For Fourier Transform Infrared (FTIR) spectroscopy, synthesis product was drop cast on a ZnSe substrate, and measurements were carried out on a Bruker Tensor 37 spectrometer in transmission mode. A clean zinc selenide substrate was used to record the background signal. Transmission Electron Microscope (TEM) images were taken using a Philips CM12 electron microscope with AMT-XR11 digital camera. An acceleration voltage of 80 kV was used for the imaging. Samples were dip coated on a copper grid coated with an ultrathin plane carbon film (Electron Microscopy Science, CF400-CU-UL). Dynamic Light Scattering (DLS) particle size measurements were carried out using a Malvern Panalytical's Zetasizer Ultra. Colloidal samples were held in a standard 10 mm square glass cuvette (Malvern Panalytical Inc. PCS1115) compatible with the instrument.Results and discussion
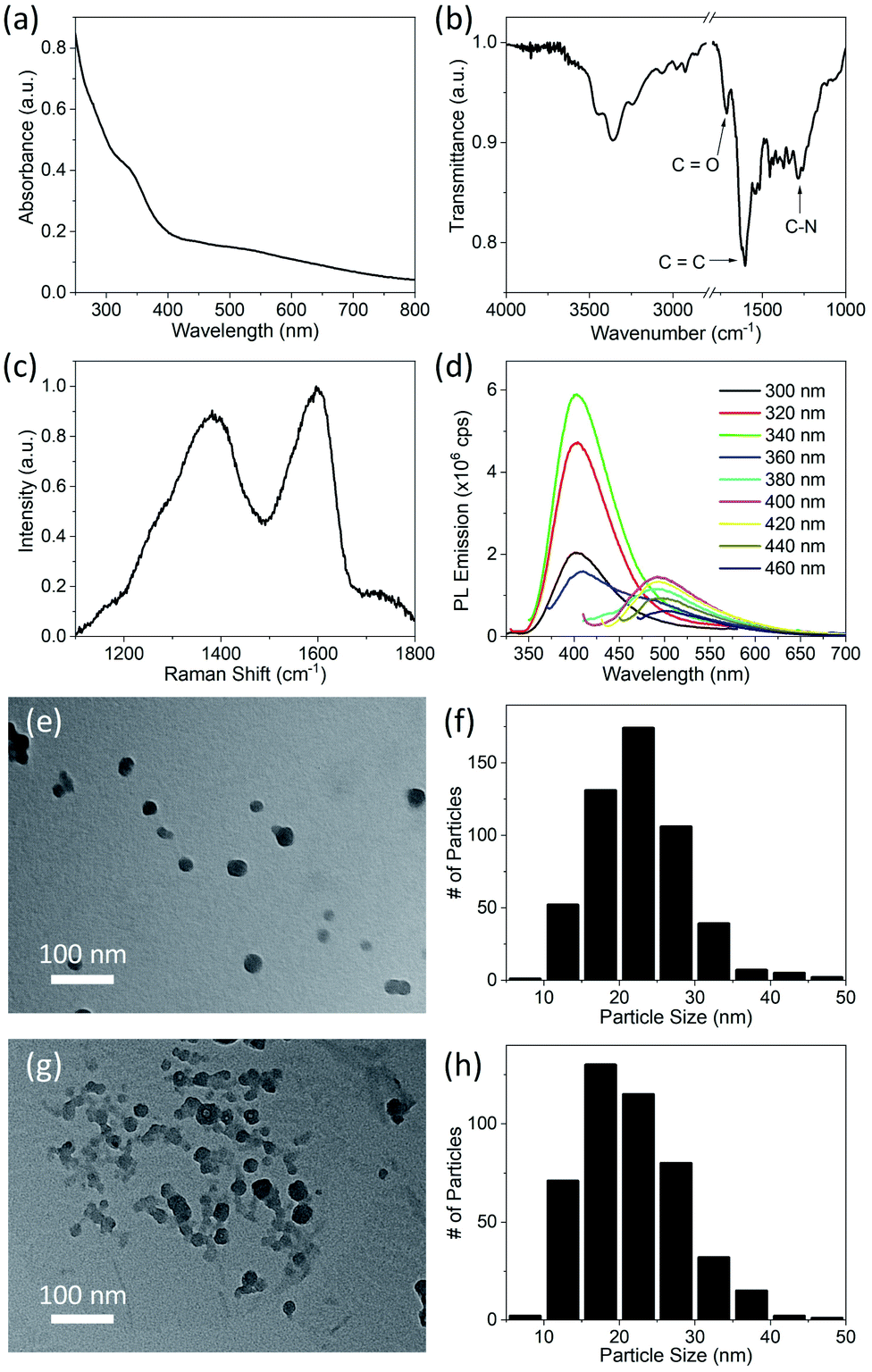
CDs were synthesized by the solvothermal method (see Experimental section), which allows control of particle size because pressure and the solvent's properties, e.g., density, can be adjusted by temperature which ultimately can affect the nucleation of particles.41 Precursors, 1,5-diaminonaphthalene (1,5-DAN) and citric acid (CA) were chosen so that as-synthesized CDs would not require any further surface modification. Under solvothermal conditions the 1,5-DAN molecules pyrolyze and after giving away the amine functional group provide a graphitic core for the growth of CDs. However, the amine and carboxyl functional groups, provided by pyrolysis of citric acid, soon attach on the carbon core, which not only act as capping agents but also help stabilize particles in a variety of solvents. The envisaged structure of CDs is depicted in Scheme 1b. It is also expected that the cyclic aromatic byproducts containing amine, carboxyl and amine function groups also form during the synthesis.25,27 Further we anticipate that these byproducts are soluble and cannot be removed by centrifugation; therefore, they remain in the dispersion.CDs form clear dispersions in water as well as organic solvents. The dispersion emits strong blue light when irradiated with UV light. Fig. 1a inset shows the optical photographs of CDs dispersed in a water/ethanol mixture (3:1) under room light (left) and under UV-light of 365 nm wavelength (right). The UV-visible absorption spectrum of the CDs dispersion (Fig. 1a) shows a strong absorption band at 315 nm. This absorption is attributed to the n → π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.37,42,43 The photoluminescence (PL) emission of the CDs dispersion was measured using different excitation wavelengths from 300 nm to 460 nm (Fig. 1b). A typical PL spectrum shows a strong emission peak at ∼400 nm when the excitation wavelength is less than 360 nm. The PL emission peak moves toward longer wavelengths with increase in excitation wavelength from 360 nm to 400 nm. For excitation wavelengths longer than 400 nm, the PL emission spectra show multiple peaks at 461 nm, 482 nm and 514 nm (Fig. 1b, inset). The PL emission at short wavelengths (blue emission) is attributed to electronic transition from a π* state of CO bond to non-bonding orbitals of oxygen. However, the multiple emission peaks at longer wavelengths may be attributed to vibronic transitions in which electronic energy states of a molecule undergo transitions simultaneously with vibrational energy states.44 As this is typically a molecular phenomenon, the long wavelength emission may be attributable to fluorescent byproducts.
O.37,42,43 The photoluminescence (PL) emission of the CDs dispersion was measured using different excitation wavelengths from 300 nm to 460 nm (Fig. 1b). A typical PL spectrum shows a strong emission peak at ∼400 nm when the excitation wavelength is less than 360 nm. The PL emission peak moves toward longer wavelengths with increase in excitation wavelength from 360 nm to 400 nm. For excitation wavelengths longer than 400 nm, the PL emission spectra show multiple peaks at 461 nm, 482 nm and 514 nm (Fig. 1b, inset). The PL emission at short wavelengths (blue emission) is attributed to electronic transition from a π* state of CO bond to non-bonding orbitals of oxygen. However, the multiple emission peaks at longer wavelengths may be attributed to vibronic transitions in which electronic energy states of a molecule undergo transitions simultaneously with vibrational energy states.44 As this is typically a molecular phenomenon, the long wavelength emission may be attributable to fluorescent byproducts.
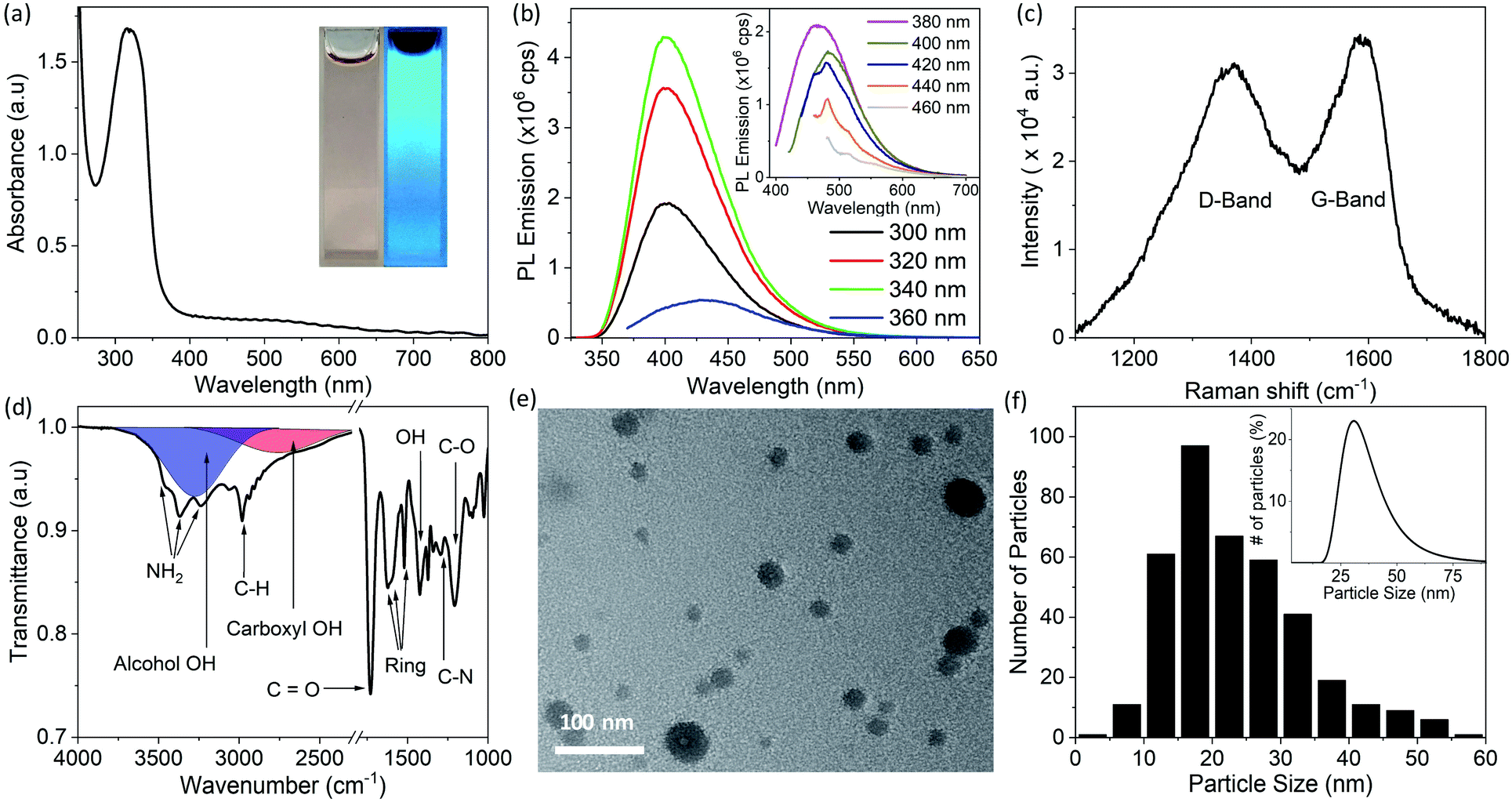 | ||
| Fig. 1 Optical and physical properties of CDs synthesized in ethanol at 200 °C measured in the first week after synthesis. (a) UV-visible absorption spectrum of CDs dispersed in a water/ethanol mixture, inset shows the optical photograph of CDs dispersion under room light (left) and under UV-light of 365 nm wavelength (right). (b) Photoluminescence emission of the CDs measured at different excitation wavelengths from 300 nm to 460 nm. Excitation and emission slit width was set to 1 nm for spectra shown in main panel. Spectra shown in the inset were recorded with excitation and emission slit width of 2 nm. (c) Raman spectrum recorded by drop casting CDs on a zinc selenide (ZnSe) disc. (d) FTIR transmittance spectrum of drop-casted CDs on ZnSe disc; area in red represents the IR absorption of OH group in carboxyl group (COOH) and area in blue shows the IR absorption caused by hydrogen bonded OH group. (e) TEM image and (f) histogram of particle size of CDs made by measurements of the size of 383 individual particles in TEM images. The inset in (f) shows the DLS particle size measurements. All of these measurements were carried out within one week after synthesis except for the FTIR data which were acquired at a later stage. | ||
Raman spectroscopy was used to investigate the structure of the CDs. The Raman spectrum (Fig. 1c) of CDs drop cast on a zinc selenide (ZnSe) substrate shows a D-band (due to disordered carbon) at 1370 cm−1 and a G-band (due to graphitic carbon) at 1585 cm−1. The relative intensity of these bands (IG/ID) is 0.8, which indicates that the CDs are mostly composed of disordered carbon.45,46 It is pertinent to mention here that for Raman spectra, a straight line was used as a baseline. The relative intensity of IG/ID is used to quantify the crystallinity (or degree of sp2 and sp3 hybridization) in carbon materials. The same ratio has also been used to determine the degree of graphitic structure in carbon (quantum) dots.40 Fluorescent CDs show a strong PL background in Raman spectra which makes calculation of peak intensities dependent on baseline selection. For instance, IG/ID increases significantly if an asymmetric least square smoothing method is used for baseline creation instead of a straight baseline. IG/ID calculated from the same spectrum after different baseline selection is shown in Fig. S1 in ESI.† Due to this variation it is difficult to directly compare the crystallinity of CDs synthesized in this work with reported values in the literature.
The FTIR spectrum of the CDs drop cast on a ZnSe substrate is shown in Fig. 1d. The absorption bands at 3450 cm−1 and 3370 cm−1 are attributed to antisymmetric and symmetric stretching of NH2, and the band at 3240 cm−1 is due to the interaction of NH2 deformation with the N–H stretching band.38,40,47 These three bands are superimposed on the broad absorption band of hydrogen-bonded OH groups (shown by the blue band in Fig. 1d). The bands at 2984 cm−1 and 1295 cm−1 are attributed to vibrations of C–H and C–N bonds, respectively. The absorption bands at 1623 cm−1, 1602 cm−1, 1520 cm−1, are due to stretching of the aromatic ring.47 The pronounced band at 1730 cm−1 indicates the presence of CO. The absorption band at 1423 cm−1 and a broad hump at 3000 cm−1 (shown by the red band in Fig. 1d) are due to vibration of the OH bond of carboxyl groups (COOH). Strong absorption at 1207 cm−1 is attributed to the C–O bond of the carboxyl group. Collectively, it is concluded from FTIR studies that hydroxyl, carboxyl and amine functional groups are present on the CDs or possibly on byproducts present in the dispersion prior to sample deposition.40,47
A transmission electron microscope (TEM) image of the synthesized CDs dip coated on a copper grid coated with an ultrathin planar carbon film is shown in Fig. 1e. A histogram of the particle size distribution for CDs (made by measurements of the size of 383 individual particles in TEM images) shows that the CDs mostly have sizes between 15 to 20 nm (Fig. 1f); however, the average CD size is 23.5 nm due to a small fraction of larger particles. The inset of Fig. 1f shows a dynamic light scattering (DLS) particle size measurement of the CD dispersion. The peak of the DLS particle size measurement is at ∼30 nm. The discrepancy in TEM and DLS particle size results is attributed to agglomeration, larger hydrodynamic size of CDs or the relatively smaller number of particles sampled in TEM measurements.
After initial characterization, CDs dispersed in water/ethanol (3:1) were stored in dark/amber vials at room temperature, and TEM images of the CDs were recorded as a function time after synthesis. Fig. 2 shows TEM images of the CDs taken during the 1st, 7th, 47th and 58th week after synthesis. It is apparent that the average particle size increases from 23 nm to 63 nm in a duration of ∼13 months. It is also observed that the increase in particle size is quicker in the beginning and slows down with the passage of time as shown in Fig. S2 in ESI.† As mentioned earlier, the CDs were collected by centrifugation after solvothermal synthesis, and it is possible that byproducts of the reaction were still present in the dispersion. We believe that these reaction byproducts slowly attach to the surface of particles and cause particle growth. Another possible mechanism of the particle size increase is coalescence of particles.48 However, after carefully analyzing TEM images of more than a thousand particles, partly coalesced particles were not observed, which eliminates the mechanism of particle growth by coalescence.
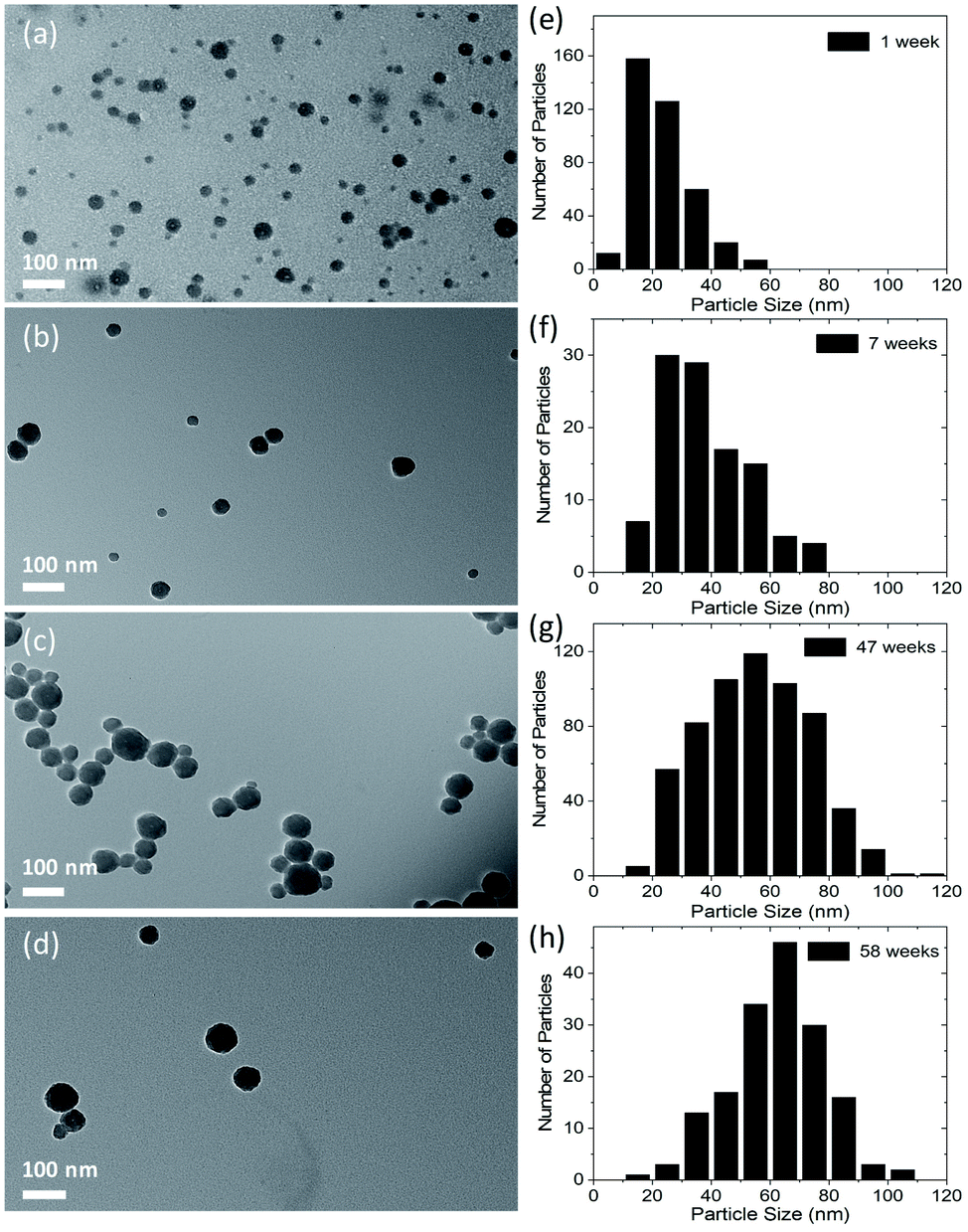 | ||
| Fig. 2 TEM images of CDs along with histograms showing the number of particles vs. size of particles. Images were taken: (a) within 1 week of synthesis of particles; (b) in the 7th week after synthesis; (c) in the 47th week; and (d) in the 58th week. Corresponding histograms of images (a), (b), (c) and (d) are shown in (e), (f), (g) and (h), respectively. The total number of particles that were measured to plot the histograms were 383, 107, 610, and 166 for (e), (f), (g) and (h), respectively. | ||
In order to investigate the origin of this increase in particle size, optical characterization of CDs was carried out at different times after synthesis. Absorption, PL emission, FTIR and Raman spectra measured at different times after synthesis are shown in Fig. 3. It is clear from Fig. 3a that the absorption intensity of CDs in the UV region gradually decreases with the passage of time. An intense absorption peak gradually turns into a weak shoulder in the scattering background. In addition to a decrease in absorption, a slight redshift of the absorption peak is also observed. The absorption peak is at 315 nm for freshly synthesized CDs and moves to 324 nm in 8 weeks and to 327 nm in 13 weeks. As we attribute this absorption to an n → π* transition of the CO bond, from the decrease in absorption with the passage of time it is deduced that the CO bond is being decomposed with time. The red shift in absorption wavelength with time indicates the change in environment of the absorbing species. In addition to this, an increase in the absorbance in the longer wavelength region is observed. This increase in absorbance is attributed to Mie scattering of light, which increases with the increase in particle size. PL emission spectra measured as a function of time are shown in Fig. 3b. Although there is no change in the shape of PL emission spectra taken over time, it is observed that the PL emission intensity of CDs in the shorter wavelength range decreases with time. The absolute photoluminescence quantum yield (PLQY) of CDs was measured using an excitation wavelength of 340 nm, and PL emission spectra were integrated from 350 nm to 550 nm. The PLQY of the freshly made CDs was 30%. The value of the PLQY decreased to 14% after 8 weeks and to 3% after 13 weeks. Therefore, we assume that the byproducts which decompose and cause the particle sized increase are blue emitting fluorophores. The PLQY of the longer wavelength emission caused by longer wavelength excitation was also measured; however, its value was less than the detection limit of the equipment even for freshly synthesized CDs. Despite its weakness, the longer wavelength emission stays in CDs for a much longer duration compared to the short wavelength emission, and it appears that this emission does not vary with particles size and time of synthesis. Therefore, this long wavelength emission may be due to vibronic progression in a different molecule which neither decomposes with time nor causes the particle size increase.
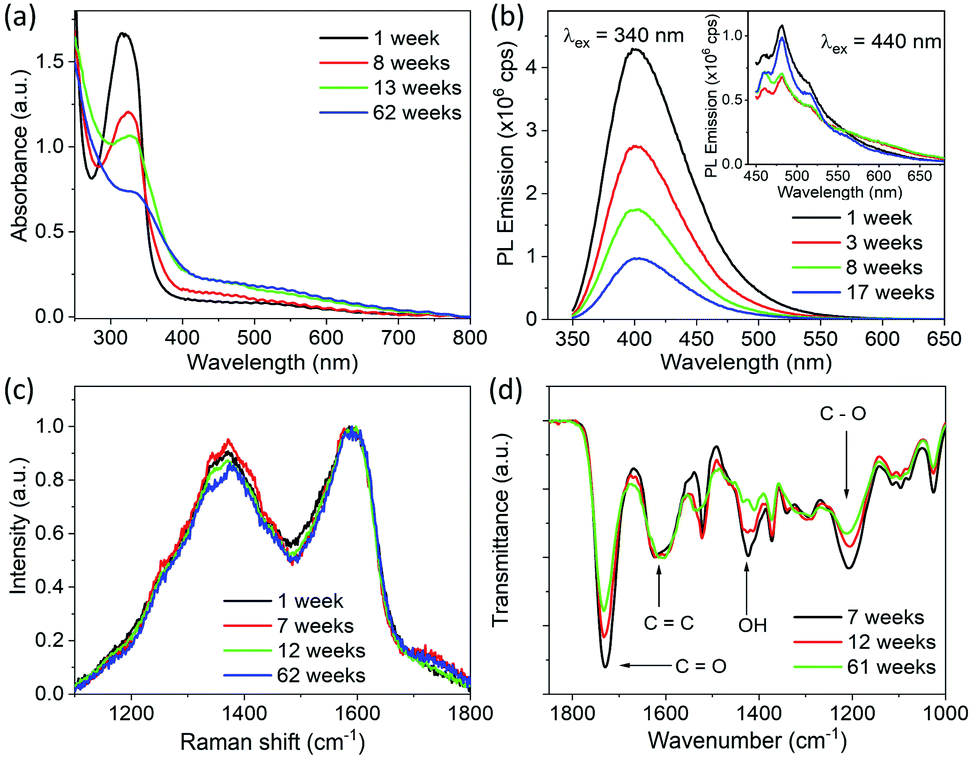 | ||
| Fig. 3 Optical and physical properties of CDs measured at different time after solvothermal synthesis in ethanol at 200 °C. (a) UV-visible absorption spectra of CDs dispersed in a water/ethanol mixture. (b) PL emission spectra of dispersion of CDs measured using an excitation wavelength of 340 nm and excitation and emission slit width of 1 nm; inset shows the PL spectra measured with an excitation wavelength of 440 nm with excitation and emission slit width of 2 nm. (c) Raman spectra of CDs drop casted on ZnSe substrate. (d) FTIR spectra recorded by drop casting CDs on a ZnSe disc. Complete FTIR spectra from 1000 cm−1 to 3800 cm−1 are shown in Fig. S3.† | ||
Raman and FTIR spectra of CDs drop cast on a ZnSe disc measured as a function of time after synthesis are shown in Fig. 3c and d, respectively. No considerable change in the relative intensities of G- and D-bands, i.e., IG/ID, is observed over time. However, the relative intensity of the FTIR absorption band at 1730 cm−1 decreases gradually with time compared to that of the absorption band at 1625 cm−1. In addition to this, the intensities of bands at 1423 cm−1 and 1207 cm−1 also decrease with the passage of time. As mention above, the absorption bands at 1730 cm−1, 1423 cm−1 and 1207 cm−1 were attributed to CO, O–H and C–O bonds in carboxyl groups and the band at 1625 cm−1 is attributed to CC bond vibrations. This further confirms that the molecular byproducts having carboxyl groups on them decompose with the passage of time.
To further confirm that particle size increased by deposition of byproducts (the blue emitting fluorophores) which were not initially attached to the CDs, we dialyzed the as-synthesized product after the synthesis. After synthesis, the product was immediately diluted with water and dialyzed for 48 hours in deionized water using a dialysis membrane having molecular weight cutoff (MWCO) of 1 kDa. To make sure the purification was complete, UV-visible absorption spectra of the dialyzed product (shown in Fig. S4†) was measured after regular intervals of time. It was noticed that the intensity of the peak attributed to CO states in impurities decreased considerably in first 12 hours, and after that there was no appreciable change in its intensity, which showed that purification was complete in 12 hours. However, the dialysis was run for 48 hours to ensure the maximum purification. Dialyzed CDs show a weak shoulder at ∼330 nm instead of a strong absorption band in the absorption spectrum, as shown in Fig. 4a. This confirms that the absorption at 315 nm in CDs (Fig. 1a) was due to CO bonds of carboxyl groups that were mostly present on byproducts freely floating in the solvent, in addition to a relatively small quantity on the particles as well. The FTIR spectrum of dialyzed CDs (shown in Fig. 4b) also shows a considerable reduction in the intensity of the absorption band at 1730 cm−1, which was previously attributed to CO bonds. Besides this, absorption bands due to C–N (at 1286 cm−1) and NH2 are still present, which indicates that amine, hydroxyl and carboxyl groups are present on dialyzed CDs. The Raman spectrum of dialyzed CDs was also measured (Fig. 4c). The Raman spectrum and the relative intensity of the G- and D-band (IG/ID) of dialyzed CDs is the same as that of CDs without dialysis. A representative TEM image of dialyzed CDs and a histogram of the CD size distribution are shown in Fig. 4e and f, respectively. The average size of dialyzed CDs is 22.5 nm with a standard deviation of 6.1 nm, which is similar to that of freshly synthesized CDs without dialysis. In order to confirm that there is no particle size increase in dialyzed CDs, TEM images of the dialyzed CDs were taken during the 8th week after synthesis (Fig. 4g) and the size of the CDswas analyzed again. It was found that the average particle size of dialyzed CDs does not increase with time. The average size of 448 individual dialyzed CDs measured from TEM images taken during the 8th week after synthesis is 21.7 nm with a standard deviation of 6.7 nm, which is similar to that of the dialyzed CDs measured in the first week. This further supports the attribution of the size increase in CDs without dialysis to adsorption of reaction byproducts onto the CD surface over time.
 | ||
| Fig. 4 Optical and physical properties of dialyzed CDs synthesized in ethanol at 200 °C measured after synthesis. (a) UV-visible absorption spectrum of dialyzed CDs dispersed in a water. (b) FTIR transmittance spectrum of dialyzed CDs drop cast on a ZnSe substrate. (c) Raman spectrum of dialyzed CDs drop cast on a ZnSe substrate. (d) Photoluminescence emission of dialyzed CDs dispersed in water measured with different excitation wavelengths, with excitation and emission slit width of 1 nm. (e) TEM image of dialyzed CDs. (f) Histogram of the particle size of dialyzed CDs made by measurements of the size of 518 individual particles in TEM images. The data in (a) to (f) were acquired in the first week after synthesis. (g) TEM image of dialyzed CDs taken in the 8th week after synthesis. (h) Histogram of particle size of dialyzed CDs in the 8th week after synthesis made by measurements of the size of 448 individual particles in TEM images. | ||
PL emission spectra of dialyzed CDs dispersed in water/ethanol (3:1) mixture were also measured using different excitation wavelengths (Fig. 4d). Multiple peaks (461 nm, 482 nm and 514 nm), which were observed in the PL emission of CDs without dialysis (Fig. 1b) are no longer present in the emission of dialyzed CDs. However, PL emission of dialysate (spectra shown in Fig. S5;† QY = 39.2%) is like that of the impure CD dispersion, which indicates that the PL of CDs without dialysis mostly originated from molecular fluorophores.25–27 The PL emission spectra of the dialyzed CDs is similar to that of many CDs reported in literature.1–4 Although carbon dots have been extensively studied due to their promising PL emission, the PL emission mechanism of CDs is still not clearly understood, and different researchers have explained it differently. Some researchers argue that it is due to inter-band electronic transitions of crystalline carbon39,40 or due to quantum confinement.10 Other report that the PL emission in CDs depends on the particular composition37 of the dots such as the nitrogen content.36 In most reports, PL emission in carbon dots is attributed to surface electronic states caused by surface functional groups.1–4 Recently, Wang et al. observed that PL emission in carbon dots depends on the solvent environment and was attributed to solvent-induced surface electronic states.38
To further investigate the origin of PL emission, PL emission spectra of dialyzed CDs dispersed in deionized water were obtained; see Fig. 5a. They show a weak PL emission at 405 nm with no emission in the visible region, which is also clear from the optical photograph (Fig. 5a, inset). The PLQY of dialyzed CDs dispersed in water is less than the measurement limit of the instrument. On the other hand, dialyzed CDs dispersed in ethanol show significant PL emission in the yellow wavelength range, in addition to blue emission; see Fig. 5b. The blue emission, which does not appear to be affected by solvent, is attributed to the fluorophores attached on the surface of the CDs without carbonization.26–28,49 The PLQY of dialyzed CDs was measured in ethanol at two different excitation wavelengths: 340 nm and 440 nm. When excited at a wavelength of 340 nm, and PL emission integrated from 370 nm to 650 nm, the PLQY was 2.6%. The PLQY was 2.5% when excited at a wavelength of 440 nm and with PL emission integrated from 490 nm to 690 nm. The dependence of PL emission of dialyzed CDs on the solvent suggests that the yellow PL emission of the CDs is from surface states which are activated by solvents.38 It is important to mention that the solvent dependent yellow emission, which arise from the surface states of CDs, is very weak compared to the emission from the fluorophores. Therefore, in the presence of fluorophores, it is difficult to detect emission form the CDs (solvent dependent emission) separately from the fluorophores. Fig. S6† shows the PL emission spectra of CDs without purification dispersed in ethanol and in water for comparison. The PL emission intensity around 550 nm (where solvent dependent emission is observed in purified CDs) was found to be slightly higher in ethanol compared to that in water. This shows that solvent dependent emission exists in impure CDs as well, but it is difficult to detect. Furthermore, the strong PL emission from dialyzed CDs of relatively large particle size reported here, as compared with many smaller CDs reported in literature, rules out the possibility of PL emission caused by quantum confinement. However, further work is required to elucidate the exact structure of solvent-induced emissive states.
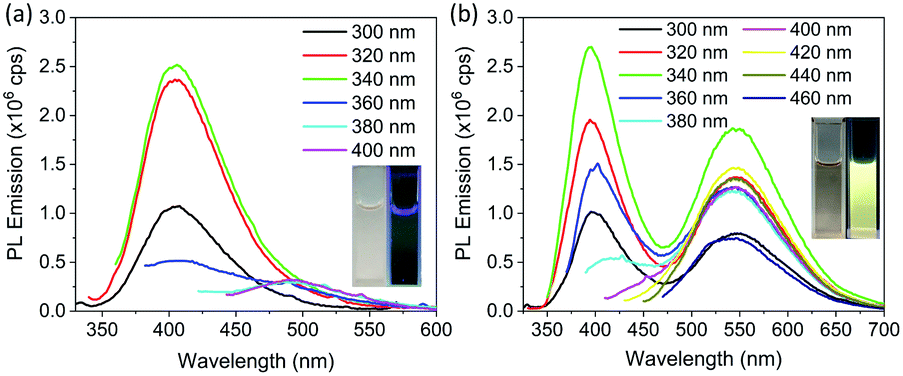 | ||
| Fig. 5 PL emission spectra of dialyzed CDs dispersed in different liquids and measured at different excitation wavelengths. (a) Dialyzed CDs dispersed in deionized water with excitation and emission slit with of 3 nm. (b) Dialyzed CDs dispersed in ethanol with excitation and emission slit width of 2 nm. Insets of both (a) and (b) show the corresponding optical photographs of dialyzed CDs in room light (left) and under UV light (right). | ||
Conclusions
CDs were synthesized using citric acid and 1,5-diaminonaphthlene by a solvothermal synthesis method, and their physical and optical properties were studied. Based on Raman analysis it was determined that CDs have an amorphous internal structure. From FTIR studies, it was revealed that hydroxyl, carboxyl and amine functional groups are present on the CDs, with molecular fluorophores containing the same functional groups/bonds floating feely in the dispersion. The CDs without dialysis showed strong blue emission with a quantum yield of 30% which was attributed to CO states mostly associated with molecular fluorophores. In addition to blue emission, CDs also showed a weak green emission which became distinctive only at excitation wavelengths longer than 400 nm. With storage time, the blue emission was substantially reduced (quantum yield 3.1% after 13 weeks) and CO states decreased. This reduction in CO states was accompanied by an increase in the average size of CDs from ∼20 nm to ∼60 nm after 58 weeks. From these observations it was concluded that the blue emission was mostly from the molecular fluorophores which slowly decomposed and attached to the CDs and caused an increase in particle size. Considerable reduction of blue PL emission with CO states after dialysis was accompanied by stable particle size, further strengthening our conclusions about the origin of the blue emission and particle size increase. An altogether different yellow emission arose in dialyzed CDs that showed a strong dependence on the solvent environment. In water, the yellow emission was negligible; in ethanol it was dramatically increased. This emission was attributed to solvent-activated surface states on CDs. Further work is required to elucidate the exact origin of these solvent-activated states. In summary, a decay in PL emission of carbon dots along with an increase in particle size over time was reported. Furthermore, emission from relatively larger particles and the dependence of longer wavelength emission on the local environment of the particle ruled out the possibility that the PL emission originated from quantum size effects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kun Zhu for assistance with purification of CDs and XRD characterization. We are also thankful to Prof. Richard Haber for access to particle characterization facilities and the Rheology Laboratory for dynamic light scattering measurements. Nasir Javed acknowledges funding from the Institute of International Education, Fulbright grant program. This work was supported in part by funding from the U. S. National Science Foundation, grant DMR-1554954.References
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- A. Sciortino, A. Cannizzo and F. Messina, C, 2018, 4, 67 CAS.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- S.-T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J.-H. Liu, Y. Liu, M. Chen, Y. Huang and Y.-P. Sun, J. Phys. Chem. C, 2009, 113, 18110–18114 CrossRef CAS.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS.
- Q.-L. Zhao, Z.-L. Zhang, B.-H. Huang, J. Peng, M. Zhang and D.-W. Pang, Chem. Commun., 2008, 5116–5118 RSC.
- S.-L. Hu, K.-Y. Niu, J. Sun, J. Yang, N.-Q. Zhao and X.-W. Du, J. Mater. Chem., 2009, 19, 484–488 RSC.
- S. Hu, J. Liu, J. Yang, Y. Wang and S. Cao, J. Nanopart. Res., 2011, 13, 7247–7252 CrossRef CAS.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS.
- L. Zheng, Y. Chi, Y. Dong, J. Lin and B. Wang, J. Am. Chem. Soc., 2009, 131, 4564–4565 CrossRef CAS.
- D. B. Shinde and V. K. Pillai, Chemistry, 2012, 18, 12522–12528 CrossRef CAS.
- B. Hu, K. Wang, L. Wu, S.-H. Yu, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 813–828 CrossRef CAS.
- L. Liu, Y. Li, L. Zhan, Y. Liu and C. Huang, Sci. China: Chem., 2011, 54, 1342–1347 CrossRef CAS.
- Z. Zhang, J. Hao, J. Zhang, B. Zhang and J. Tang, RSC Adv., 2012, 2, 8599–8601 RSC.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- S. Mitra, S. Chandra, T. Kundu, R. Banerjee, P. Pramanik and A. Goswami, RSC Adv., 2012, 2, 12129–12131 RSC.
- J. Zhang, W. Shen, D. Pan, Z. Zhang, Y. Fang and M. Wu, New J. Chem., 2010, 34, 591–593 RSC.
- S. F. Chin, S. N. A. M. Yazid, S. C. Pang and S. M. Ng, Mater. Lett., 2012, 85, 50–52 CrossRef CAS.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- H. Li, X. He, Y. Liu, H. Huang, S. Lian, S.-T. Lee and Z. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS.
- H. Li, X. He, Y. Liu, H. Yu, Z. Kang and S.-T. Lee, Mater. Res. Bull., 2011, 46, 147–151 CrossRef CAS.
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V. Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC.
- T. Wang, A. Wang, R. Wang, Z. Liu, Y. Sun, G. Shan, Y. Chen and Y. Liu, Sci. Rep., 2019, 9, 10723 CrossRef.
- V. Hinterberger, W. Wang, C. Damm, S. Wawra, M. Thoma and W. Peukert, Opt. Mater., 2018, 80, 110–119 CrossRef.
- V. Hinterberger, C. Damm, P. Haines, D. M. Guldi and W. Peukert, Nanoscale, 2019, 11, 8464–8474 RSC.
- W. Wang, B. Wang, H. Embrechts, C. Damm, A. Cadranel, V. Strauss, M. Distaso, V. Hinterberger, D. M. Guldi and W. Peukert, RSC Adv., 2017, 7, 24771–24780 RSC.
- A. Das, V. Gude, D. Roy, T. Chatterjee, C. K. De and P. K. Mandal, J. Phys. Chem. C, 2017, 121, 9634–9641 CrossRef CAS.
- S. Zhu, X. Zhao, Y. Song, S. Lu and B. Yang, Nano Today, 2016, 11, 128–132 CrossRef CAS.
- D. Qu and Z. Sun, Mater. Chem. Front., 2020, 4, 400–420 RSC.
- S. Khan, W. Li, N. Karedla, J. Thiart, I. Gregor, A. M. Chizhik, J. Enderlein, C. K. Nandi and A. I. Chizhik, J. Phys. Chem. Lett., 2017, 8, 5751–5757 CrossRef CAS.
- W. Zhu, X. Meng, H. Li, F. He, L. Wang, H. Xu, Y. Huang, W. Zhang, X. Fang and T. Ding, Opt. Mater., 2019, 88, 412–416 CrossRef CAS.
- J. B. Essner, J. A. Kist, L. Polo-Parada and G. A. Baker, Chem. Mater., 2018, 30, 1878–1887 CrossRef CAS.
- K. Mishra, S. Koley and S. Ghosh, J. Phys. Chem. Lett., 2019, 10, 335–345 CrossRef.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS.
- S. Hu, A. Trinchi, P. Atkin and I. Cole, Angew. Chem., Int. Ed., 2015, 54, 2970–2974 CrossRef CAS.
- H. Wang, C. Sun, X. Chen, Y. Zhang, V. L. Colvin, Q. Rice, J. Seo, S. Feng, S. Wang and W. W. Yu, Nanoscale, 2017, 9, 1909–1915 RSC.
- X. Wang, L. Cao, S.-T. Yang, F. Lu, M. J. Meziani, L. Tian, K. W. Sun, M. A. Bloodgood and Y.-P. Sun, Angew. Chem., Int. Ed., 2010, 49, 5310–5314 CrossRef CAS.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. a. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef.
- H. Hayashi and Y. Hakuta, Materials, 2010, 3, 3794–3817 CrossRef CAS.
- M. Liu, Y. Xu, F. Niu, J. J. Gooding and J. Liu, Analyst, 2016, 141, 2657–2664 RSC.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS.
- W. Barford and M. Marcus, J. Chem. Phys., 2017, 146, 130902 CrossRef.
- A. Ferrari, J. Robertson, S. Reich and C. Thomsen, Philos. Trans. R. Soc. A, 2004, 362, 2271–2288 CrossRef.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley and Sons, Chichester, West Sussex, England, 3rd edn, 2004 Search PubMed.
- M. Grouchko, I. Popov, V. Uvarov, S. Magdassi and A. Kamyshny, Langmuir, 2009, 25, 2501–2503 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00479k |
| This journal is © The Royal Society of Chemistry 2021 |