
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNoble metal nanowire arrays as an ethanol oxidation electrocatalyst†
Zhenhui
Lam‡
abc,
Cuicui
Liu‡
b,
Dongmeng
Su
 b,
Hua Bing
Tao
c,
Hsin-Yi
Wang
c,
Jiazang
Chen
c,
Weichang
Xu
b,
Liping
Zhang
ac,
Yihan
Zhu
e,
Lingmei
Liu
e,
Yu
Han
e,
Hongyu
Chen
*abd and
Bin
Liu
*ac
b,
Hua Bing
Tao
c,
Hsin-Yi
Wang
c,
Jiazang
Chen
c,
Weichang
Xu
b,
Liping
Zhang
ac,
Yihan
Zhu
e,
Lingmei
Liu
e,
Yu
Han
e,
Hongyu
Chen
*abd and
Bin
Liu
*ac
aEnergy Research Institute of NTU (ERI@N), Interdisciplinary Graduate School, Nanyang Technological University, 639798, Singapore. E-mail: liubin@ntu.edu.sg
bDivision of Chemistry and Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
cSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 637459, Singapore
dInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 Puzhu South Road, Nanjing 211816, China. E-mail: iashychen@njtech.edu.cn
eAdvanced Membranes and Porous Materials Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 11th November 2020
Abstract
Vertically aligned noble metal nanowire arrays were grown on conductive electrodes based on a solution growth method. They show significant improvement of electrocatalytic activity in ethanol oxidation, from a re-deposited sample of the same detached nanowires. The unusual morphology provides open diffusion channels and direct charge transport pathways, in addition to the high electrochemically active surface from the ultrathin nanowires. Our best nanowire arrays exhibited much enhanced electrocatalytic activity, achieving a 38.0 fold increase in specific activity over that of commercial catalysts for ethanol electrooxidation. The structural design provides a new direction to enhance the electrocatalytic activity and reduce the size of electrodes for miniaturization of portable electrochemical devices.
Fuel cells have higher fuel conversion and electrical efficiencies as compared with conventional combustion or power generation technologies.1,2 Liquid fuels such as methanol or ethanol are of particular interest because of their high volumetric energy density and ease of handling as compared with other fuels such as hydrogen, apart from being renewable and less toxic, leading to the development of direct alcohol fuel cells (DAFCs).2 DAFCs rely on reactions between the fuel (methanol or ethanol) at the anode and the oxidant (molecular oxygen) at the cathode.2 Both anodes and cathodes require efficient catalysts to lower the electrochemical overpotential for high energy conversion efficiency.
So far, platinum (Pt) is still the best anode catalyst, but its usage has been greatly limited by its prohibitive cost.3,4 Compared with Pt, palladium (Pd) is more abundant and has good catalytic activity for alcohol oxidation; it works under basic conditions which are more amenable for device fabrication, but its catalytic activity is inferior to that of Pt.5,6 Therefore, how to improve the catalytic activity of Pd for alcohol oxidation to match that of Pt has attracted increasing interest. Various approaches have been attempted to improve the performance of Pd electrodes. For example, facet engineering7,8 and compositional design (e.g., alloying), either with a transition metal9,10 or oxide,11,12 were found to positively influence the catalytic activity of Pd. To increase the electrochemically active surface area (ECSA) while maintaining good electrode conductivity and mass transport properties, Pd electrocatalysts were grown into one-dimensional nanostructures13,14 or loaded onto nanofibers and nanosheets, such as carbon nanotubes (CNTs),15,16 oxide nanotubes,17 or graphene.18,19 Most of the improvements came from the facet engineering and compositional design, whereas the structural design led to only marginal improvements.
Here, we show that standing nanowire arrays have special effects on the electrocatalytic performance of ethanol oxidation, upgrading the Pd catalyst to the level of typical Pt catalysts. The arrays of ultrathin nanowires provide a highly electrochemically active surface, while at the same time improve the electronic charge transport and facilitate the diffusion of reactants as well as products. These improvements collectively lead to superior catalytic activity by more than one order of magnitude as compared to the commercial electrocatalysts. The solution synthesis of nanowire arrays could be readily scaled up, providing a new design to enhance the electrocatalytic activity and to miniaturize the electrode for portable electrochemical devices.
Noble metal (NM) nanowire arrays including Au20 and core–shell Au@Pd were grown on conductive fluorine-doped SnO2 (FTO) substrates (Fig. 1). The entire growth process took less than 2 h at room temperature. As displayed in the scanning electron microscopy (SEM) images (Fig. 1b), the Au nanowires stand densely and vertically on the substrate. The diameter of the Au nanowires is around 3–5 nm (Fig. 2a) while the length can be tuned by changing the growth time (Fig. S1†). After coating Pd on the Au nanowires, the nanowires remain vertically aligned but their diameter increases slightly to 5–8 nm. The crystal structure was examined by transmission electron microscopy (TEM). Both Au and Au@Pd nanowires are quasi-single-crystalline (Fig. 2b and d). Energy-dispersive X-ray spectroscopy line scan and electron energy loss spectroscopy (EELS) elemental mapping were conducted to investigate the Pd distribution in Au@Pd nanowires (Fig. 2e–i). It is clear that Pd has successfully enwrapped the Au nanowires to form a core–shell nanostructure (Fig. 2e and f).
 | ||
| Fig. 1 Digital photograph and SEM images of a vertically aligned noble metal (NM) nanowire forest. (a) A digital photograph showing the bare FTO glass, FTO glass with Au nanowires (NWs), and FTO glass with Au@Pd NWs. (b and c) Large area SEM image of Au and Au@Pd NWs. (d–f) SEM images of Au@Pd NWs grown on conducting carbon cloth. | ||
 | ||
| Fig. 2 Physical characterization of the vertically aligned NM nanowire forest. (a) TEM image of Au NWs. (b) HRTEM image of Au NWs. (c) TEM image of Au@Pd NWs. (d) HRTEM image of Au@Pd NWs. (e and f) HRSTEM image of Au@Pd NWs showing a clear core–shell nanostructure. (g and h) Electron energy loss spectroscopy (EELS) mapping of a single Au@Pd NW as displayed in (f). (i) Energy dispersive X-ray spectroscopy (EDX) line scan along the cross-section of a single Au@Pd NW as shown in (f). | ||
The electrocatalytic properties of the as-prepared nanowire arrays were evaluated and compared to those of the nanoparticle (NP) electrocatalysts. Fig. S2a† displays the cyclic voltammetry (CV) curves, which show that the Au nanowire arrays exhibit significantly higher catalytic activity towards ethanol electrooxidation as compared with the gold nanoparticles (Au NPs), which can be clearly observed from the forward oxidation peak at 1.3 V vs. RHE. The maximum mass specific current of Au nanowire arrays reaches 1345.0 A g−1, which is 11.6 times higher than that of the Au NPs (115.7 A g−1). Additionally, the Au nanowire arrays also exhibit excellent electrocatalytic durability throughout 1 h of continuous electrochemical operation. It is generally believed that Au is not a good electrocatalyst for ethanol oxidation relative to Pd and Pt.21 Even so, our Au nanowire arrays were able to display enhanced catalytic performance, exceeding the performance of the commercially available Pd/C catalyst by 7.8 times.
Furthermore, when the Au nanowires were coated with a thin layer of a more effective ethanol oxidation catalyst – Pd, the electrocatalytic activity was immediately doubled. The forward oxidation peak mass specific current of Au@Pd nanowire arrays at 1.0 V vs. RHE increases to 2237.7 A g−1, which is 12.9 times higher than that of the commercially available Pd/C (172.6 A g−1, Fig. 3a and S4a†), making it the highest among all reported Pd-based electrocatalysts (Table S4†). Control experiments were performed to compare Pd NPs and Au@Pd NPs (Fig. S3†), which showed similar performance and ruled out the possibility of Au–Pd synergistic effects.
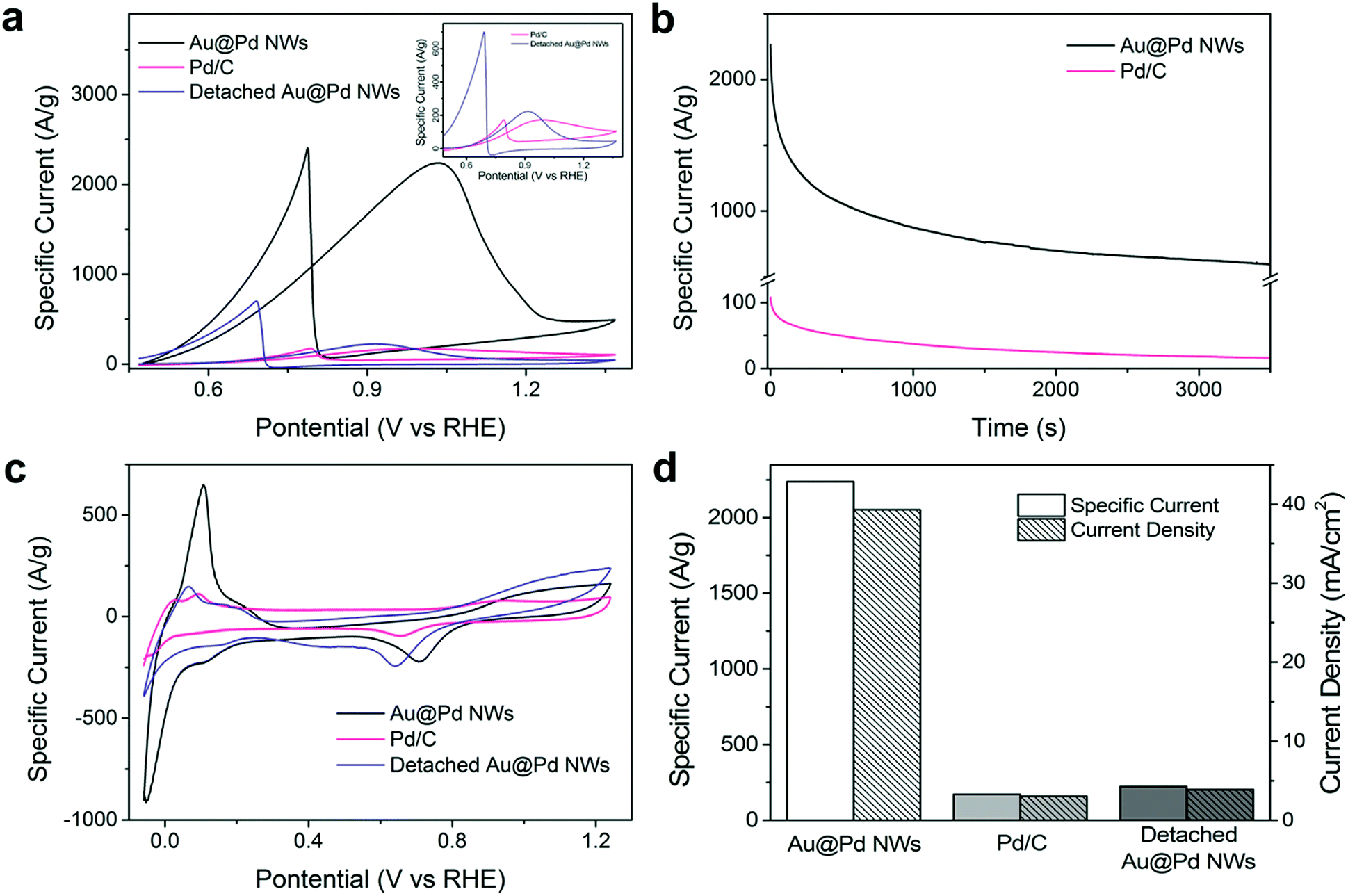 | ||
| Fig. 3 Electrocatalytic properties of Au@Pd NWs. (a) CV curves of Au@Pd NW, Pd/C and detached Au@Pd NW catalysts measured in solutions of 1.0 M NaOH and 1.0 M ethanol at a scan rate of 50 mV s−1. The inset shows the zoom-in CV curves of Pd/C and detached Au@Pd NWs. (b) Chronoamperometry graph measured at 0.87 V vs. RHE. (c) CV curves of Au@Pd NWs, Pd/C and detached Au@Pd NWs measured in solutions of 0.5 M H2SO4 at a scan rate of 50 mV s−1. (d) Specific current and current density comparison of Au@Pd NWs, Pd/C and detached Au@Pd NWs. | ||
To investigate the origin of the enhancement, the ECSA of Au@Pd nanowires and Pd/C was determined from PdO reduction and was 110.1 and 43.3 m2 g−1, respectively (Fig. S5†). Clearly, the Au@Pd nanowires could provide more catalytic sites per unit mass of the catalyst for ethanol electrooxidation as compared to Pd/C. It is important to note that such a high ECSA cannot be achieved simply by stacking Pd/C to the same thickness as the height of nanowire arrays (Table S1†), which offers a huge advantage when the size of the electrode is limited. As the high ECSA can already be achieved on a planar FTO glass electrode, it is conceivable that more active sites could be provided by our nanowire arrays in a highly porous, three-dimensional electrode. The seeded solution growth method of NM nanowire arrays makes it possible for nanowires to be grown on arbitrary conductive supports, such as carbon fiber cloth as shown in Fig. 1d–f and S7,† increasing the possibility to produce electrodes with an ultra-high ECSA.
Apart from the high ECSA, the standing nanowire arrays are also expected to facilitate diffusion of reactants and products, owing to the open channels among the nanowires. The diffusion properties were assessed by measuring the ECSA of nanowire arrays with different nanowire lengths. The ECSA of Au@Pd nanowires remains almost constant up to 600 nm in length (Fig. S8 and Table S1†), indicating that the active surface area of nanowires increases proportionally with their mass. This is a good indication that diffusion should not be a limiting factor in submicron nanowire arrays. On the other hand, Pd/C showed a progressive decrease in the ECSA with increasing electrode thickness (Fig. S8 and Table S1†), suggesting that diffusion is indeed a problem in nanoparticle-based electrocatalysts. This conclusion is further confirmed via comparing the ethanol diffusion coefficient in the Au@Pd nanowires and Pd/C electrodes estimated from the Randles–Sevcik method (Fig. S9 and Table S2†), where the diffusion coefficient in the Au@Pd nanowires (2.8 × 10−14 cm2 s−1) is about two orders of magnitude larger than that in Pd/C (2.9 × 10−16 cm2 s−1).
Besides, the electronic conductivity of the nanowire arrays is also expected to be better than that of their nanoparticle-based counterpart. The aligned nanowires would provide a direct pathway to transport electrons. In contrast, when catalyst-loaded support particles pack into a film, such as Pd/C, electrons face not only scattering, but also multiple junctions among the layers of particles as illustrated in Fig. S10.† These junctions contribute significantly to the high resistance, and thereby severely decrease the electronic conductivity of the nanoparticle film. Typically, the conductivity of nanowires22 is at least one order of magnitude higher than that of their nanoparticle counterpart.23
Most importantly, the orientation of nanowires was found to greatly influence the electrochemical performance. Once the Au or Au@Pd nanowires were detached from the substrate, the electrocatalytic activity dropped drastically to nearly the same level as that of the respective nanoparticles, highlighting the importance of the nanowire alignment (Fig. 3a, S2a and S4a†). We measured the ECSA of both aligned and detached nanowires (110.1 m2 g−1 and 81.8 m2 g−1 respectively, Fig. S6†) and noticed that there was only less than 30% decrease in ECSA after detaching the nanowires. Based on previous studies, metallic junctions were seen to increase the resistivity of metal nanostructures by a few orders of magnitude.24 Thus, the decrease in activity by detaching nanowires from the substrate could be mainly attributed to the greatly increased difficulty in charge transport. In the case of nanowire arrays, the direct contact of individual nanowires with the conductive substrate ensures good electronic conductivity. This high electrode conductivity, together with the facilitated reactant and product diffusion and large ECSA provides the nanowire arrays with unprecedented electrochemical performance. With this insight, it is not surprising that the previous efforts of loading catalysts onto nanowires13,14 and CNTs15,16 could not achieve the same level of performance as our nanowire forest.
The electrocatalytic activity of nanowire arrays can be further optimized by coating the nanowires with catalysts having higher intrinsic catalytic activities. Previously, we demonstrated the Au@Pd core–shell nanowires for ethanol oxidation. If we add Pt into the Pd shell, the electrocatalytic activity can be further enhanced as shown in Fig. 4. Among the tested samples, the Au@Pd2.2Pt1 (Table S3†) core–shell nanowire arrays show the highest activity, achieving a peak mass specific current as high as 6568.1 A g−1, which is 2.9 times higher than that of the Au@Pd nanowire arrays, and 38.0 times higher than that of Pd/C (Fig. 4). We also compared the performance of Au@Pd2.2Pt1 nanowires with commercially available platinum on Vulcan XC-72 (Pt/C), which demonstrated a 20.0 fold enhancement in the mass specific current (Fig. 4).
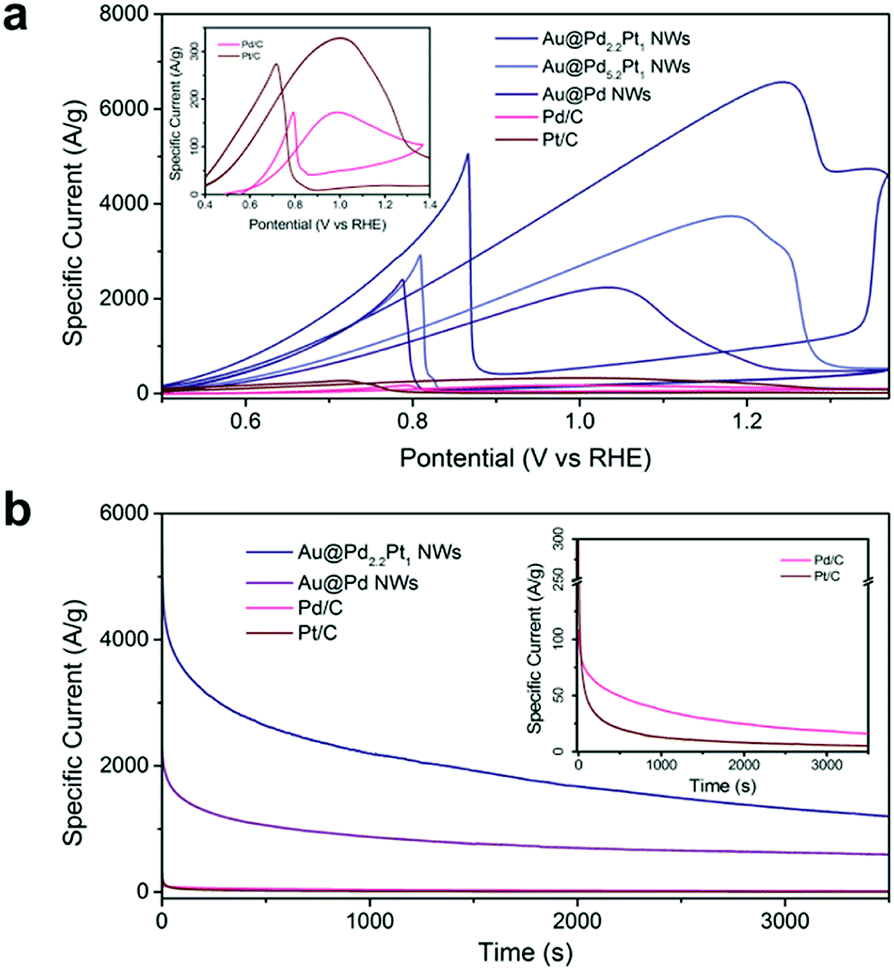 | ||
Fig. 4 Optimization of electrocatalytic properties. (a) CV curves of Au NWs with different Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt ratios, commercial Pd/C, and Pt/C measured in solutions of 1.0 M NaOH and 1.0 M ethanol at a scan rate of 50 mV s−1. The inset shows the zoom-in CV curves of Pd/C and Pt/C. (b) Chronoamperometry graph measured at 0.87 V vs. RHE. The inset shows the zoom in chronoamperometry graph for Pd/C and Pt/C. :Pt ratios, commercial Pd/C, and Pt/C measured in solutions of 1.0 M NaOH and 1.0 M ethanol at a scan rate of 50 mV s−1. The inset shows the zoom-in CV curves of Pd/C and Pt/C. (b) Chronoamperometry graph measured at 0.87 V vs. RHE. The inset shows the zoom in chronoamperometry graph for Pd/C and Pt/C. | ||
Conclusions
In summary, we have demonstrated that by simply changing the morphology of the electrocatalyst from nanoparticles to standing nanowire arrays, we can dramatically improve the catalytic activity by providing a more electrochemically active surface for electrocatalysis, open channels for improved mass transport, and better conductivity. Such a new design principle may provide a simple means to enhance the electrocatalytic activity, to miniaturize the electrode for portable devices, and to improve the effectiveness of existing and emerging electrochemical technologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge funding support from Singapore Ministry of Education Academic Research Fund (AcRF) Tier 1: RG5/16 and RG1/15, Singapore A*Star Science and Engineering Research Council – Public Sector Funding (PSF): 1421200075 and the National Research Foundation (NRF), Prime Minister's Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) program as well as financial support from Nanjing Tech University and SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials, National Natural Science Foundation of China (No. 21673117), Taizhou 500 Talent Program (2016NMS01) and Taizhou Municipal Science and Technology Program (2016NMS02).References
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104(10), 4245–4270 CrossRef CAS.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3(8), 1499–1528 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS.
- M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438 CrossRef CAS.
- C. L. Lu, K. S. Prasad, H. L. Wu, J. A. A. Ho and M. H. Huang, J. Am. Chem. Soc., 2010, 132, 14546 CrossRef CAS.
- N. Tian, Z. Y. Zhou, N. F. Yu, L. Y. Wang and S. G. Sun, J. Am. Chem. Soc., 2010, 132, 7580 CrossRef CAS.
- S. T. Nguyen, H. M. Law, H. T. Nguyen, N. Kristian, S. Wang, S. H. Chan and X. Wang, Appl. Catal., B, 2009, 91, 507 CrossRef CAS.
- S. Y. Shen, T. S. Zhao, J. B. Xu and Y. S. Li, J. Power Sources, 2010, 195, 1001 CrossRef CAS.
- P. K. Shen and C. W. Xu, Electrochem. Commun., 2006, 8, 184 CrossRef CAS.
- C. W. Xu, Z. Q. Tian, P. K. Shen and S. P. Jiang, Electrochim. Acta, 2008, 53, 2610 CrossRef CAS.
- F. Ksar, G. Surendran, L. Ramos, B. Keita, L. Nadjo, E. Prouzet, P. Beaunier, A. Hagège, F. Audonnet and H. Remita, Chem. Mater., 2009, 21, 1612 CrossRef CAS.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703 CrossRef CAS.
- N. Mackiewicz, G. Surendran, H. Remita, B. Keita, G. Zhang, L. Nadjo, A. Hagège, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2008, 130, 8110 CrossRef CAS.
- F. Hu, X. Cui and W. Chen, J. Phys. Chem. C, 2010, 114, 20284 CrossRef CAS.
- F. P. Hu, F. W. Ding, S. Q. Song and P. K. Shen, J. Power Sources, 2006, 163, 415 CrossRef CAS.
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693 CrossRef CAS.
- L. Gao, W. Yue, S. Tao and L. Fan, Langmuir, 2013, 29, 957 CrossRef CAS.
- J. He, Y. Wang, Y. Feng, X. Qi, Z. Zeng, Q. Liu, W. S. Teo, C. L. Gan, H. Zhang and H. Chen, ACS Nano, 2013, 7, 2733 CrossRef CAS.
- A. N. Geraldes, D. F. da Silva, E. S. Pino, J. C. M. da Silva, R. F. B. de Souza, P. Hammer, E. V. Spinacé, A. O. Neto, M. Linardi and M. C. dos Santos, Electrochim. Acta, 2013, 111, 455 CrossRef CAS.
- S. De, T. M. Higgins, P. E. Lyons, E. M. Doherty, P. N. Nirmalraj, W. J. Blau, J. J. Boland and J. N. Coleman, ACS Nano, 2009, 3, 1767 CrossRef CAS.
- Y. Wu, Y. Li and B. S. Ong, J. Am. Chem. Soc., 2006, 128, 4202 CrossRef CAS.
- L. Hu, H. S. Kim, J. Y. Lee, P. Peumans and Y. Cui, ACS Nano, 2010, 4, 2955 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures as described in the main text. See DOI: 10.1039/d0na00804d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |