
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the use of conducting oligomers and redox molecules in CdS–MoFeP biohybrids†
Alexander W.
Harris
a,
Shambojit
Roy
a,
Saheli
Ganguly
a,
Ashray V.
Parameswar
b,
Francisco W. S.
Lucas
 a,
Adam
Holewinski
ac,
Andrew P.
Goodwin
ab and
Jennifer N.
Cha
*ab
a,
Adam
Holewinski
ac,
Andrew P.
Goodwin
ab and
Jennifer N.
Cha
*ab
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Avenue, Boulder, CO 80303, USA. E-mail: Jennifer.Cha@colorado.edu
bMaterials Science and Engineering Program, University of Colorado Boulder, 3415 Colorado Avenue, Boulder, CO 80303, USA
cRenewable and Sustainable Energy Institute, University of Colorado, Boulder, CO 80303, USA
First published on 28th December 2020
Abstract
In this work we report the effect of incorporating conducting oligophenylenes and a cobaltocene-based redox mediator on photodriven electron transfer between thioglycolic acid (TGA) capped CdS nanorods (NR) and the native nitrogenase MoFe protein (MoFeP) by following the reduction of H+ to H2. First, we demonstrate that the addition of benzidine-a conductive diphenylene- to TGA-CdS and MoFeP increased catalytic activity by up to 3-fold as compared to CdS–MoFeP alone. In addition, in comparing the use of oligophenylenes composed of one (p-phenylenediamine), two (benzidine) or three (4,4′′-diamino-p-terphenyl)phenylene groups, the largest gain in H2 was observed with the addition of benzidine and the lowest with phenylenediamine. As a comparison to the conductive oligophenylenes, a cobaltocene-based redox mediator was also tested with the TGA-CdS NRs and MoFeP. However, adding either cobaltocene diacid or diamine caused negligible gains in H2 production and at higher concentrations, caused a significant decrease. Agarose gel electrophoresis revealed little to no detectable interaction between benzidine and TGA-CdS but strong binding between cobaltocene and TGA-CdS. These results suggest that the tight binding of the cobaltocene mediator to CdS may hinder electron transfer between CdS and MoFe and cause the mediator to undergo continuous reduction/oxidation events at the surface of CdS.
Introduction
In recent years significant effort for generating renewable energy has focused on mimicking or using biological systems through the development of photoactive biohybrid materials. Typically, these biohybrid systems incorporate a synthetic semiconductor photocatalyst that upon photoirradiation from solar light facilitate electron transfer to a biological catalyst (e.g. enzymes and microbes) for electrical power generation or fuel conversion.1–4 In these biohybrid systems, electron transfer between inorganic and biological materials is crucial for improving the feasibility of energy applications like solar-driven fuel production, enzymatic fuel cells, and bioelectrocatalysis.5–8 For this, one method that has been explored extensively is to depend on direct electron transfer from a photoactive nanocrystal to an enzyme to for example reduce protons (H+) to dihydrogen (H2) and dinitrogen (N2) to ammonia (NH3) using hydrogenase and nitrogenase, respectively.6,9 However, because there exists a significant challenge for efficiently transferring high-energy electrons from the semiconductor to the redox active metallocenter of the protein, in order to improve charge transfer between the two, proteins have been physisorbed or covalently conjugated to nanoparticles or electrode surfaces.10–14 For this, a promising strategy used has been to minimize molecular distance by mimicking specific protein–protein interactions or cellular environments.15,16 As an example, we recently showed that the activity of CdS and the MoFe protein (MoFeP) of nitrogenase enzymes can be significantly influenced by understanding how the critical interactions (e.g. electrostatics, site-specific positioning, covalent bonding) between the two can be used to develop generalizable strategies to enable fast and efficient electron transfer between the two materials.17While direct electron transfer between a photocatalyst and enzyme has promise, to improve charge transfer in either photocatalytic or photoelectrocatalytic systems, numerous studies have been run using small molecule diffusional charge transfer agents. To better understand the use of such charge transfer agents in our CdS–MoFeP system, we present here the results of incorporating the use of either short, soluble conductive oligomers or redox-active mediators (Scheme 1). While redox mediators such as methyl viologen and cobaltocene have been tested in electrochemical or photoelectrochemical systems, less success has been demonstrated with photoactive nanoparticles that perform under zero applied bias. Furthermore, studying the use of conductive molecular wires such as oligothiophenes or oligophenylenes to improve electron transfer kinetics between photoactive nanoparticles and enzymes has not been previously reported. Herein, we first show that the incorporation of phenylene-based diamines with CdS nanorods (NR) capped with thioglycolic acid (TGA) and the MoFeP showed significant 3-fold gains in dihydrogen (H2) production as compared to using CdS and MoFeP hybrids alone. Furthermore, in testing a series of phenylene-based oligomers consisting of 1, 2 and 3 phenyl units, we demonstrate that the benzidine oligomers formed of two phenyl groups showed the largest improvement in H2 production as compared to a p-phenylenediamine and 4,4′′-diamino-p-terphenyl(terphenyl) which are composed of one and three phenyl units, respectively. We postulate that these results are due to different electronic HOMO–LUMO states of the individual conducting oligomers that affect the probabilities of electron transport. Furthermore, in testing the redox mediator cobaltocene modified with either diamine or diacid terminal groups, we demonstrate that the addition of soluble mediators lowered the overall activity of the CdS–MoFeP complex which could be attributed to both the strong association of cobaltocene for TGA-CdS and the ease in which such molecules can undergo redox chemistries on the photocatalyst surface, thereby preventing any charge transfer to a neighboring enzyme in the absence of applied bias.
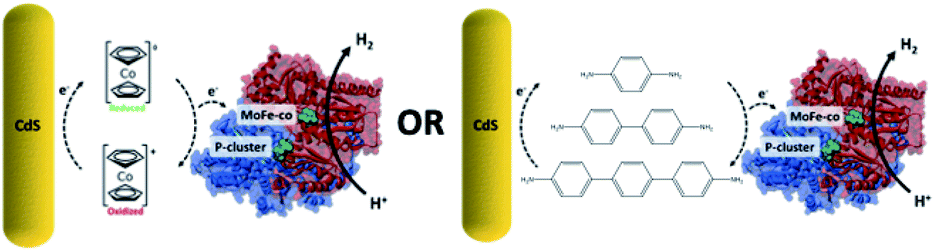 | ||
| Scheme 1 An overall scheme highlighting the focus of this study to investigate diffusional charge transfer agents such as cobaltacene and oligophenylenes for mediating charge transfer between CdS and MoFeP. | ||
Results and discussion
In this work, the effect of incorporating conductive benzene-based oligomers and redox mediators on photodriven electron transfer between CdS NRs and the MoFeP was studied. First, CdS NRs were prepared using a previously reported seeded growth method.6,18 The resulting NRs had dimensions of 3.8 ± 0.1 nm and 26.5 ± 0.3 nm in diameter and length, respectively, with a first absorption peak at 459 nm corresponding to a band gap energy of ∼2.70 eV (Fig. S1†). A ligand exchange procedure previously reported by Dukovic and coworkers was performed to stabilize the CdS NRs in water using TGA ligands.9 Next, the His-tagged wild type MoFeP expressed in A. vinelandii DJ995 were purified under anaerobic conditions using previously established procedures and the purity was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.17We first tested the effect of adding conductive oligomers to the CdS NRs and MoFeP by testing for H+ reduction in the presence of benzidine – a diphenyl diamine – at varying concentrations. In a typical reaction, TGA capped CdS NRs capped with TGA ligands were mixed with ascorbic acid (AA) in PBS (pH 7.4) with benzidine concentrations ranging from 0 to 500 μM. The solutions were next purged with Ar for 30 min followed by injecting in a concentrated aliquot of MoFeP and photoirradiating (9 W, 465 ± 30 nm LED) for 4 h at room temperature. During irradiation, aliquots were removed from the gas phase and analyzed via GC to determine H2 production every hour. Since the conduction band energy of CdS is sufficient for H+ reduction, background H2 produced from CdS and benzidine alone (no MoFeP added) was accounted for by running control reactions in parallel to obtain the net catalytic activity from the MoFeP.
The photocatalytic results of incorporating benzidine to the CdS NR and MoFeP reactions are shown in Fig. 1. First, the baseline for enzymatic activity in the TGA-CdS–MoFeP complex after 4 h without benzidine was 60.8 ± 0.3 μmol H2 (mg protein)−1 and the incorporation of 5 μM benzidine resulted in a negligible difference in enzymatic activity. However, as higher benzidine concentrations were incorporated, a significant increase in enzymatic activity was observed. From benzidine concentrations ranging from 50 to 500 μM, an 82% to 220% increase in H2 was produced, respectively, after 4 h. While the increase in benzidine concentration above 100 μM also contributed to an increase in background H2 from the CdS NRs alone (Fig. S2†), there was still a significant gain in H2 produced from the CdS–MoFeP biohybrids and this was observed for all concentrations of benzidine tested above 5 μM. In addition, with increasing benzidine concentrations, a clear gain in photodriven enzymatic activity was observed indicating that the efficacy of the conductive oligomer plays a significant role in improving charge transfer between the semiconductor and enzyme. Lastly, a plateau or slight decrease in H2 production was observed after 3 h in the presence of 500 μM benzidine, which we attribute to H2 saturation in the vapor phase inhibiting the activity of the photocatalyst, enzyme, or both.
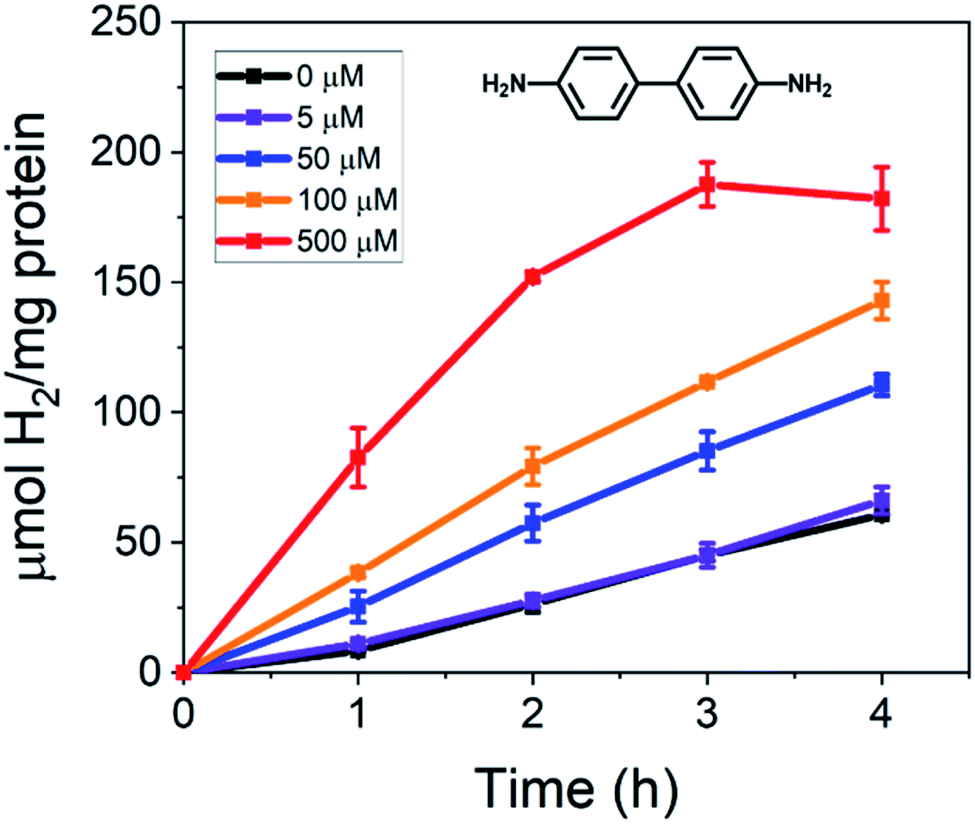 | ||
| Fig. 1 Total amount of H2 produced per mg of protein over time from TGA-CdS–MoFeP biohybrid complexes in the presence of 0 to 500 μM benzidine. CdS NRs act as the electron supplier under irradiation in an Ar purged vial containing 50 mM PB, 100 mM NaCl, 500 mM ascorbic acid (AA), 500 nM CdS, and 500 nM MoFeP at pH 7.4 over 4 h. Error bars indicate one standard deviation (SD) from duplicate measurements. | ||
To probe electron transfer in this biohybrid system further, a series of oligophenylene diamines of varying lengths were added to the CdS NR and MoFeP mixtures all at 100 μM and tested for their effect on electron transfer across the CdS–MoFeP bio-nano interface. In molecularly bridged nanostructures, the measured molecular conductance has been shown to exponentially decay with molecular length and is defined by eqn (1), where Gc is the effective contact conductance, β is the tunneling decay constant, and L is the length of the molecule.19–21
| G = Gce−βL | (1) |
The conductance tunneling decay constants are dependent on the molecular structure of the linker, where the conductance across alkanes (β ≈ 0.6–0.9 Å−1) decreases more rapidly than oligothiophenes (β ≈ 0.1 Å−1) as a function of distance.19 Zhu and coworkers have experimentally illustrated this exponential decay in conductance using a series of oligophenylene diamines (β ∼ 0.5 Å−1) assembled between single-walled carbon nanotubes acting as nanoelectrodes.21 Here, we demonstrate that as shown in Fig. 2, as we change from two phenyl units (benzidine) to one (p-phenylenediamine) or three (terphenyl) that the H2 yields from the CdS–MoFeP hybrids was determined to be highest in the presence of benzidine at 143.0 ± 7.1 μmol H2 (mg protein)−1 followed by terphenyl and p-phenylenediamine at 108.4 ± 9.9 and 102.3 ± 8.8 μmol H2 (mg protein)−1. These results provide support that electron transport is occurring through the conducting oligomers wherein the respective HOMO–LUMO gap and reorganization energy are known to play important roles.19 While an increase in the number of aromatic rings decreases the HOMO–LUMO gap and improves electron transport probabilities with semiconductor-like behavior, a large increase in the number of aromatic rings can weaken coupling strengths since electrons are spread over a larger volume.19 Next, as an additional control, the aliphatic ethylenediamine was tested due to its terminal diamine functionality but overall lower conductance due to its larger HOMO–LUMO gap.19 As shown in Fig. 2, adding ethylenediamine yielded only ≈17% improvement in H2 yields as compared to the CdS–MoFeP photoreaction alone.
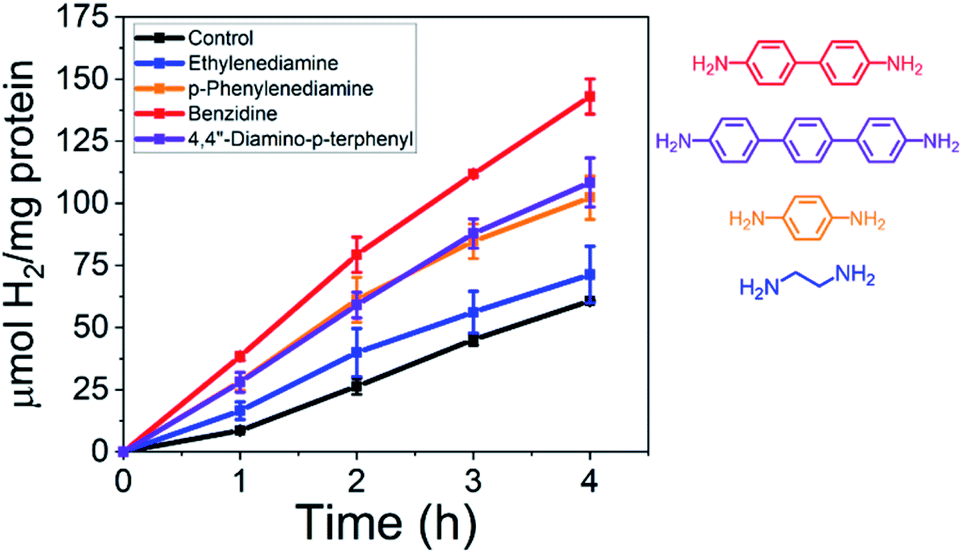 | ||
| Fig. 2 Total amount of H2 produced per mg of protein over time from TGA-CdS–MoFeP biohybrid complexes in the presence of 100 μM oligophenylene diamines and ethylenediamine. CdS NRs act as the electron supplier under irradiation in an Ar purged vial containing 50 mM PB, 100 mM NaCl, 500 mM ascorbic acid (AA), 500 nM CdS, and 500 nM MoFeP at pH 7.4 over 4 h. Error bars indicate one standard deviation (SD) from duplicate measurements. | ||
To better understand the possible mechanisms by which the addition of benzidine aids in improving photocatalysis from the CdS–MoFeP biohybrids, agarose gel electrophoresis was performed using similar techniques to previous studies.17 As shown in Fig. 3 the agarose gel results suggest that only weak interactions exist between benzidine and CdS due to the minimal differences in CdS mobility in the gel. Furthermore, the addition of benzidine to the CdS NRs and MoFeP demonstrated negligible effects on CdS–MoFeP interactions, even at millimolar benzidine concentrations as evidenced by the agarose studies (Fig. 3).
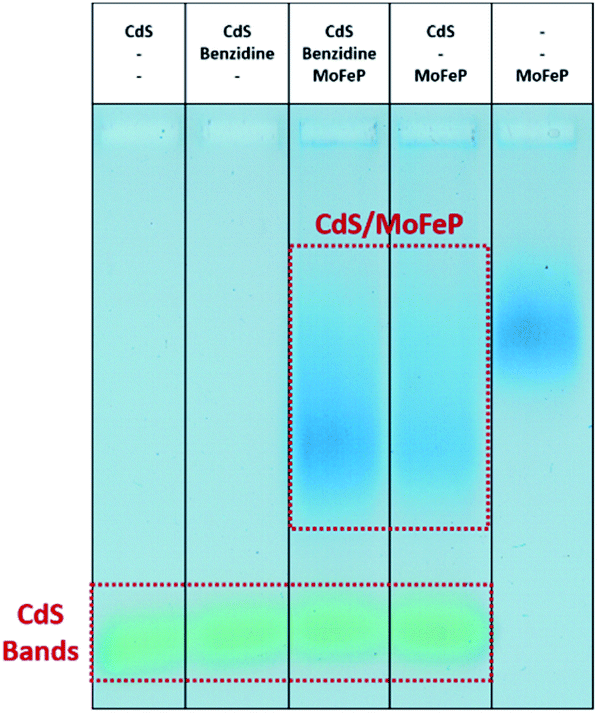 | ||
| Fig. 3 Agarose gel of TGA-CdS NPs and biohybrid complexes with Coomassie stained MoFe protein (MoFeP) with and without the presence of benzidine. From lanes left to right: TGA-CdS only, TGA-CdS and benzidine, TGA-CdS, benzidine and MoFeP, TGA-CdS and MoFeP, MoFeP only. | ||
Next, as a comparison to the use of conductive oligophenylene diamines for aiding in electron transfer between CdS and MoFeP, photocatalytic studies were performed using a diffusional redox active mediator. For this, we chose to study the use of cobaltocene-based redox mediators as these have been recently shown to improve facilitate electron transfer to MoFeP in electrochemical applications.22–24 First, since the redox potential of the MoFeP is −310 mV versus NHE,25 typical electrochemical systems with the MoFeP utilize a cobaltocene mediator due to its redox potential at −958 mV versus NHE.24 However, since the reduction potential of a CdS NR is approximately −800 mV versus NHE,9,26 it was first necessary to modify cobaltocene with electron withdrawing functional groups to make this thermodynamically favorable. For example, a one and two carboxyl-modification of cobaltocene can shift the reduction potential from −958 mV to −840 mV and −690 mV versus NHE, respectively (Fig. S3†).24 Next, to mimic the oligophenylene diamine interactions with TGA-CdS, the 1,1′-dicarboxy-cobaltocenium (di-COOH-cobaltocene) was chemically modified with terminal amines using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-hydroxysuccinimide coupling chemistry with ethylenediamine. The amide reactions were performed in a mixture of DMSO and water for 24 h and the diamine–cobaltocene product was dried using lyophilization then characterized by 1H NMR in DMSO-d6 with 4,4-dimethyl-4-silapentane-1-sulfonic acid as an internal standard (Fig. S4†). The redox potential of the diamine–cobaltacene were measured next and determined to be −740 mV vs. NHE, and thus is reducible by the CdS NRs (Fig. S5†).
The diamine modified cobaltocene mediators were next added to the CdS NRs and MoFeP and tested for H2 production. As shown in Fig. 4, the addition of either 5, 100, or 500 μM diamine–cobaltocene yielded negligible effects on H2 production electron transfer at concentrations under 100 μM and closely matched that of the CdS–MoFeP control without mediator. However, using a 500 μM diamine–cobaltocene concentration caused a significant drop in photodriven activity to yield a 4.5-fold decrease in H2 production. Similar trends in photocatalytic activity were observed when di-COOH-cobaltocene was incorporated into the CdS–MoFeP reactions in place of the diamine modified cobaltocene (Fig. S6†).
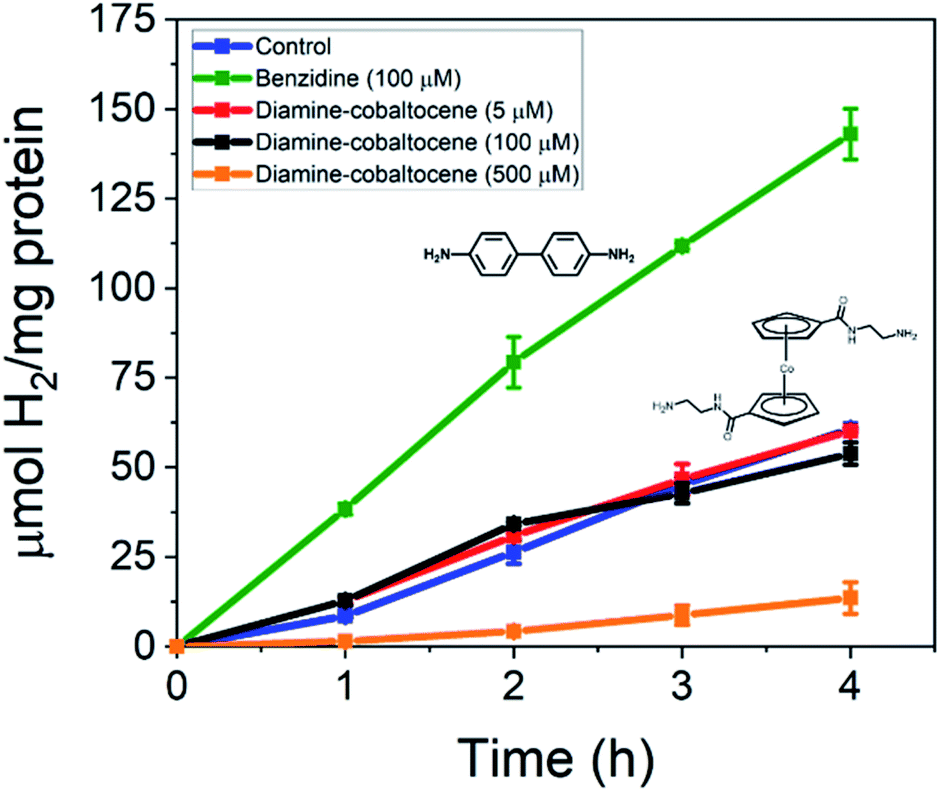 | ||
| Fig. 4 Total amount of H2 produced per mg of protein over time rom TGA-CdS–MoFeP biohybrid complexes in the presence of benzidine and diamine–cobaltocene. CdS NRs act as the electron supplier under irradiation in an Ar purged vial containing 50 mM PB, 100 mM NaCl, 500 mM ascorbic acid (AA), 500 nM CdS, and 500 nM MoFeP at pH 7.4 over 4 h. Error bars indicate one standard deviation (SD) from duplicate measurements. | ||
To determine the net interaction of diamine–cobaltocene with TGA-CdS with and without MoFeP, agarose gel electrophoresis studies were run. As shown in Fig. 5, as opposed to the results with benzidine, there was a clear shift of the TGA-CdS nanorods in the presence of the mediator which was further shifted when the protein was added. These results demonstrate that the mediator showed a strong binding to CdS. Thus, the decrease in activity from the CdS–MoFeP hybrid upon addition of cobaltocene clearly hinders overall electron transfer from the CdS nanoparticles which may be due in part to the mediator undergoing continuous reduction/oxidation events at the surface of CdS. To further support this theory, similar electron mediated results in diffusional systems were observed by Chica et al., where no H2 production from the hydrogenase enzyme was observed when the redox mediator methyl viologen (−440 mV versus NHE) was implemented as an electron shuttling agent from CdSe/CdS dot-in-rod photocatalysts.27 In this system, the authors demonstrated H2 production with a propyl-bridged 2-2′-biphyridinium mediator but it is unclear how this enzymatic activity compares to non-mediated electron transfer from the photocatalyst to hydrogenase. While photoreduction efficiencies are nearly equal for methyl viologen and propyl-bridged 2-2′-biphyridinium, effective electron transfer requires a balance between the driving force for the two forward electron transfer processes, as well as, the structure of the mediator.27,28 The redox potential of di-COOH-cobaltocene (−690 mV versus NHE) is positioned within ≈20 mV of propyl-bridged 2-2′-biphyridinium,28 which suggests that despite the energetic driving force from di-COOH-cobaltocene to the MoFeP, the difference in redox potential between CdS and di-COOH-cobaltocene is not sufficient to drive charge transfer in this CdS–MoFeP complex.
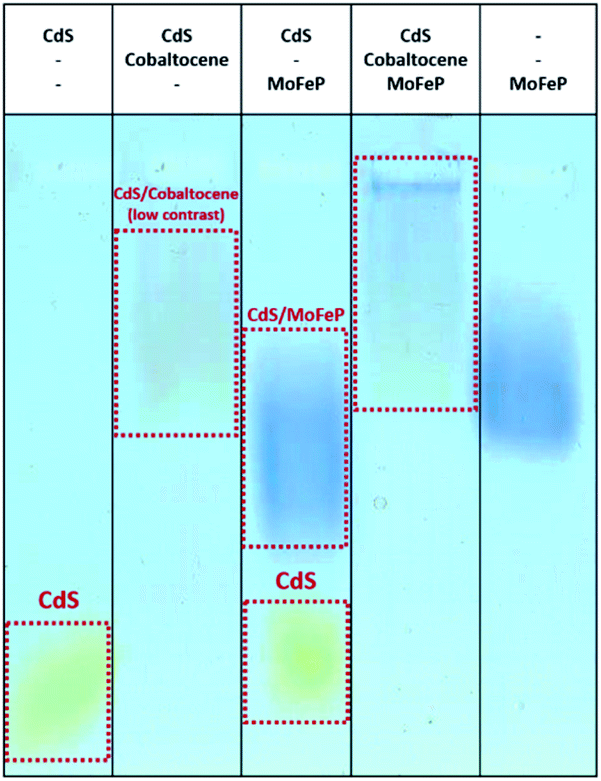 | ||
| Fig. 5 Agarose gel of TGA-CdS NPs and biohybrid complexes with Coomassie stained MoFe protein (MoFeP) with and without the presence of diamine–cobaltocene. From lanes left to right: TGA-CdS only, TGA-CdS and diamine–cobaltocene, TGA-CdS and MoFeP, TGA-CdS and diamine–cobaltocene and MoFeP, MoFeP only. | ||
Conclusions
Here, we have reported the effect of incorporating conductive oligophenylenes and a cobaltocene mediator on electron transfer between CdS photocatalysts and the MoFeP of nitrogenase. First, incorporation of benzidine in this biohybrid complex yielded a significant 3-fold increase in H+ reduction to H2 as compared to the CdS–MoFeP control. Additionally, when phenylene-based oligomers of varying lengths (one, two, and three phenyl units) were tested, the greatest improvement in H2 production were observed from the two-phenyl benzidine oligomer as compared to p-phenylenediamine or terphenyl which can be attributed to increases in the overall HOMO–LUMO energy gap and coupling strength of the oligomers between the photocatalyst and MoFeP. In contrast, although redox mediators such as cobaltocene or methyl viologen have been proven useful for improving electron transfer in many electrochemical and photocatalytic systems, incorporating a diamine- or diacid-cobaltocene redox mediator in our studies with MoFeP and CdS was not effective in enhancing electron transfer and actually lowered the overall activity of the CdS–MoFeP complex as the concentration of the mediator was increased. Agarose gel electrophoresis studies revealed little to no detectable interaction between benzidine and TGA-CdS but strong binding between cobaltocene and TGA-CdS. These results suggest that tight associations of the redox mediator to CdS may hinder electron transfer between CdS and MoFeP due to reversible redox processes that occur at the surface of CdS in the absence of applied bias. The experimental results of these colloidal CdS–MoFeP studies with incorporation of oligophenylenes and cobaltocene mediators provide useful tools and insight into developing methods to improve electron transfer in biohybrid photosystems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Dennis Dean (Virginia Tech) for his generous donation of the MoFeP strains and Prof. John Falconer (CU Boulder) for use of his GC. This work was supported by ACS Petroleum Research Funds (56999-ND10) and the National Science Foundation (DMR 142736) the National Institutes of Health (R21EB027319).References
- M. Frasconi, H. Boer, A. Koivula and F. Mazzei, Electrochim. Acta, 2010, 56, 817–827 CrossRef CAS.
- S. Cosnier, A. J. Gross, A. Le Goff and M. Holzinger, J. Power Sources, 2016, 325, 252–263 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS.
- R. D. Milton, T. Wang, K. L. Knoche and S. D. Minteer, Langmuir, 2016, 32, 2291–2301 CrossRef CAS.
- K. A. Brown, M. B. Wilker, M. Boehm, H. Hamby, G. Dukovic and P. W. King, ACS Catal., 2016, 6, 2201–2204 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS.
- R. D. Milton, R. Cai, S. Abdellaoui, D. Leech, A. L. De Lacey, M. Pita and S. D. Minteer, Angew. Chem., Int. Ed., 2017, 56, 2680–2683 CrossRef CAS.
- V. Fourmond and C. Leger, Angew. Chem., Int. Ed., 2017, 56, 4388–4390 CrossRef CAS.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS.
- J. Klein, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2029–2030 CrossRef CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- C. C. You, S. S. Agasti and V. M. Rotello, Chem.–Eur. J., 2008, 14, 143–150 CrossRef CAS.
- W. Lin and C. J. Murphy, ACS Cent. Sci., 2017, 3, 1096–1102 CrossRef CAS.
- S. Rana, Y. C. Yeh and V. M. Rotello, Curr. Opin. Chem. Biol., 2010, 14, 828–834 CrossRef CAS.
- C. F. Megarity, B. Siritanaratkul, R. S. Heath, L. Wan, G. Morello, S. R. FitzPatrick, R. L. Booth, A. J. Sills, A. W. Robertson, J. H. Warner, N. J. Turner and F. A. Armstrong, Angew. Chem., Int. Ed., 2019, 58, 4948–4952 CrossRef CAS.
- A. Narayan, Nature, 2019, 567, 317–318 CrossRef CAS.
- A. W. Harris, A. Harguindey, R. E. Patalano, S. Roy, O. Yehezkeli, A. P. Goodwin and J. N. Cha, ACS Appl. Bio Mater., 2020, 3, 1026–1035 CrossRef CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS.
- F. Chen and N. J. Tao, Acc. Chem. Res., 2009, 42, 429–438 CrossRef CAS.
- D. J. Wold, R. Haag, M. A. Rampi and C. D. Frisbie, J. Phys. Chem. B, 2002, 106, 2813–2816 CrossRef CAS.
- J. Zhu, J. McMorrow, R. Crespo-Otero, G. Ao, M. Zheng, W. P. Gillin and M. Palma, J. Am. Chem. Soc., 2016, 138, 2905–2908 CrossRef CAS.
- B. Hu, D. F. Harris, D. R. Dean, T. L. Liu, Z. Y. Yang and L. C. Seefeldt, Bioelectrochemistry, 2018, 120, 104–109 CrossRef CAS.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 RSC.
- A. Badalyan, Z. Y. Yang and L. C. Seefeldt, ACS Catal., 2019, 9, 1366–1372 CrossRef CAS.
- W. N. Lanzilotta and L. C. Seefeldt, Biochemistry, 1997, 36, 12976–12983 CrossRef CAS.
- H.-W. Tseng, M. B. Wilker, N. H. Damrauer and G. Dukovic, J. Am. Chem. Soc., 2013, 135, 3383–3386 CrossRef CAS.
- B. Chica, C. H. Wu, Y. Liu, M. W. W. Adams, T. Lian and R. B. Dyer, Energy Environ. Sci., 2017, 10, 2245–2255 RSC.
- M. L. K. Sanchez, C. H. Wu, M. W. W. Adams and R. Brian Dyer, Chem. Commun., 2019, 55, 5579–5582 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00678e |
| This journal is © The Royal Society of Chemistry 2021 |