DOI:
10.1039/D0NA01026J
(Paper)
Nanoscale Adv., 2021,
3, 3100-3106
Theoretical study of the influence of doped oxygen group elements on the properties of organic semiconductors†
Received
6th December 2020
, Accepted 6th April 2021
First published on 27th April 2021
Abstract
Organic semiconductor materials are widely used in the field of organic electronic devices due to their wide variety, low price, and light weight. However, their developments are still restrained by their low stability and carrier mobility. Density functional theory (DFT) was used to study the influence of doped oxygen group elements (O, S, Se, and Te) on the properties of organic semiconductor materials (seven-membered benzothiophene, o-pentacene, thiophene derivatives, and pentacene) in this paper. Based on the calculation of EHOMO, ELUMO, ΔE, and total energy, the performances of organic semiconductor materials without and with doped elements were compared, and it was found that the doping of multi-element Te makes the material have high stability and potential high mobility. For these studied organic semiconductor materials, when the atoms of the doped site change in the order of O, S, Se, and Te, the carrier mobility gradually increases, and the molecules show a tendency of stability. In this paper, promising doping elements and doping methods for these studied molecules are determined through calculations and screening out suitable materials more efficiently and economically without a large amount of repetitive experimental work, which may provide a theoretical basis and guidance for preparing high-performance organic semiconductor materials.
1. Introduction
In the past few decades, organic semiconductors have attracted the attention of many researchers due to their adjustable electronic structure and optical band gap, light weight, and good mechanical flexibility.1–3 Because of their favorable performance, a large number of organic materials have been widely designed and applied in the fields of organic light-emitting diodes,4,5 organic thin film transistors,6 and organic solar cells.7 Compared with inorganic semiconductors, organic semiconductors possess discrete “energy levels” composed of molecular orbitals, and the transfer of charge depends on the ability of carriers to move from one molecule to another.5 Carrier mobility is one of the important indicators for evaluating the performance of organic semiconductors.8 The higher the carrier mobility rate, the better the performance of the prepared device.9,10
Improving carrier mobility is the key to the development of organic semiconductor materials. In the past few decades, researchers have made significant progress in enhancing the carrier mobility of organic semiconductors.11–16 Appropriate molecular design and doping can adjust conductivity and make organic semiconductors potentially useful.17–20 Doping, especially with oxygen group elements,21–26 can lead to the formation of more free charge carriers in organic semiconductors or the passivation of traps, increase carrier mobility, increase the effective injection of electrons, change charge transport properties, and improve the carrier mobility and stability of the material. Di Tian27et al. introduced polarizable and electron-rich sulfur or selenium atoms into electron-deficient nitrogen-containing heteroaromatic compounds, which facilitated the accumulation of adjacent molecules and achieved a π-atom orbital overlap rate of more than 90%.
Common organic semiconductor materials include polycyclic aromatic hydrocarbons (PAHs), metal complexes and small compounds derived from conductive polymers, such as polyolefins, polythiophenes, and pentacene.28–33 The molecular structure and semiconductor properties of these materials have been a hot spot in recent years. Among them, in organic light-emitting devices, the mobility of organic semiconductors based on thiophene and oligoacene is higher than 1.0 cm2 V−1 s−1,34,35 and thiophene derivatives have excellent stability and high carrier mobility on linear oligoacetylene,36 making them common semiconductor materials.
In this paper, we used density functional theory (DFT) to explore the effects of doping oxygen group elements on the conductivity and stability of organic semiconductor materials (seven-membered benzothiophene, o-pentacene, thiophene derivatives, and pentacene), and calculate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of a molecule (corresponding to the valence band and conduction band of the semiconductor37), and the smaller the difference between LUMO and HOMO, the easier the electronic transition between the orbitals.38 By comparing the values of ΔE, total energy and other data, we get the general law of doping oxygen group elements, which provides guidance for screening high-performance organic semiconductor materials.
2. Computational methods
Geometry optimization, electronic properties, and orbital information of organic semiconductor materials were studied by the density functional theory (DFT) method using the DMol3 module in Materials Studio. The generalized gradient approximation (GGA) was treated by the Perdew–Burke–Ernzerhof (PBE) functional. The double numerical atomic orbital plus polarization function (DNP) of basfile v4.4 was chosen to be the basis set. The convergence criteria for the total energy calculation and geometric optimization step were set as below: a self-consistent field (SCF) energy tolerance of 10−5 eV per atom, a maximum force tolerance of 10−1 eV Å−1, and a maximum displacement tolerance of 5 × 10−3 Å. Max self-consistent field cycles were set as 1000 cycles.
3. Results and discussion
3.1 Seven-membered benzothiophene doped with oxygen group elements
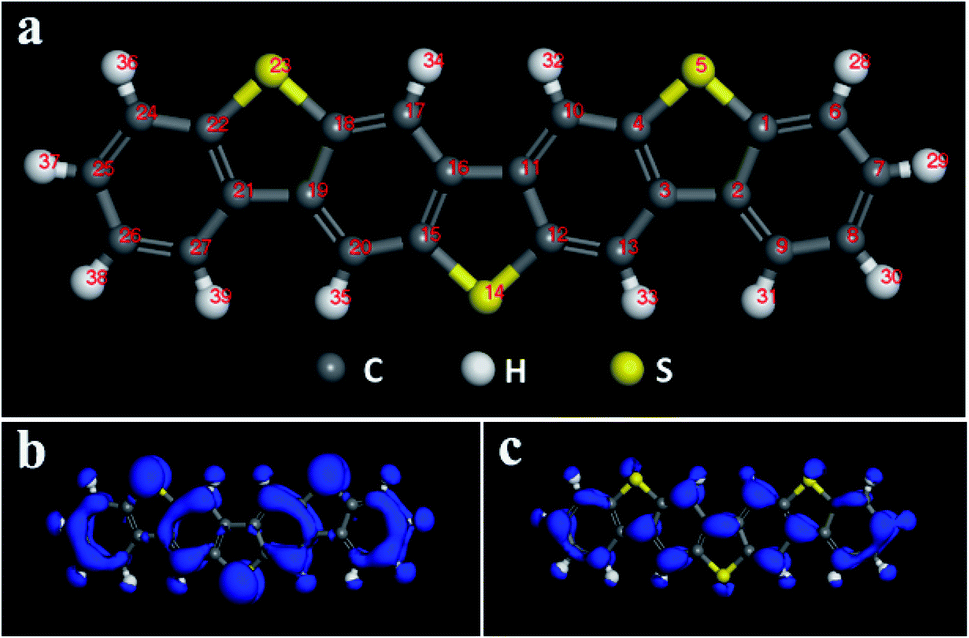
Multi-component benzo-based organic semiconductors tend to form large π-conjugated structures with high carrier mobility, and the introduction of thiophene rings improves the performance of the material to a certain extent,39 so we choose benzothiophene compounds as the research object. Fig. 1 is a schematic diagram of the molecular structure and electron cloud of a seven-membered benzothiophene. It can be seen from Fig. 1b, c and Table S1† that the three sulfur atom electron clouds are densely distributed and the Fukui (−) and Fukui (+) indices have the largest absolute values, indicating that these three sulfur atoms have high activity and are prone to nucleophilic and electrophilic attacks, and they are ideal sites for doping. Therefore, we used sulfur atoms as the starting point27 to study the effect of doping of oxygen group elements on the performance of organic semiconductors. Next, we conducted mono-, binary-, and ternary doping of the seven-membered benzothiophene with oxygen group elements, and the specific structure is shown in Fig. S1.† According to the symmetry of the molecular structure, both single and binary doping adopt symmetry doping methods to make it more stable. The values of EHOMO, ELUMO and ΔE are used to characterize the ability of the organic semiconductor molecule to bind electrons and the electronic transitions between orbitals. The higher the EHOMO, the stronger the electron donating ability of the molecule, which is conducive to accepting holes; the lower the ELUMO, the better the ability to receive electrons, and the smaller the difference between the two, the faster the electron transfer rate between the orbitals.
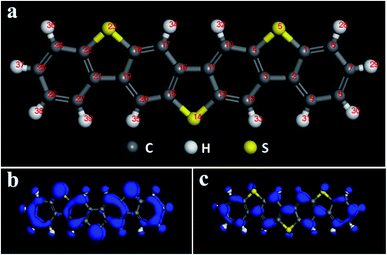 |
| | Fig. 1 (a) Schematic diagram of the seven-membered benzothiophene molecular structure; (b) Fukui (−) electron cloud distribution diagram; (c) Fukui (+) electron cloud distribution diagram. | |
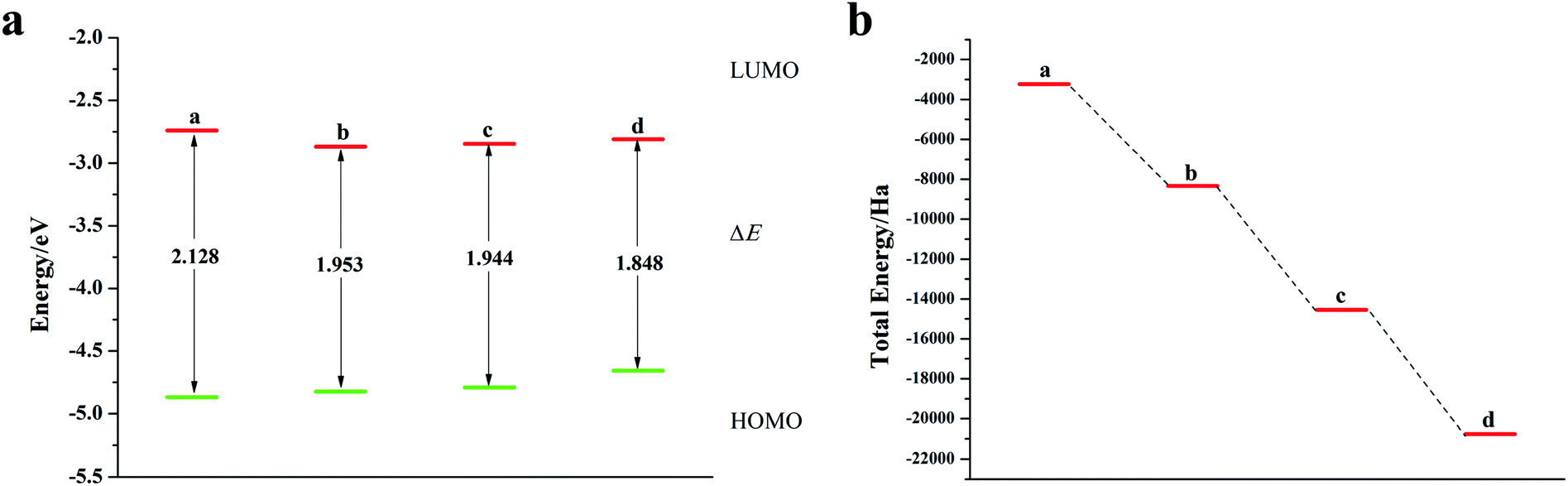
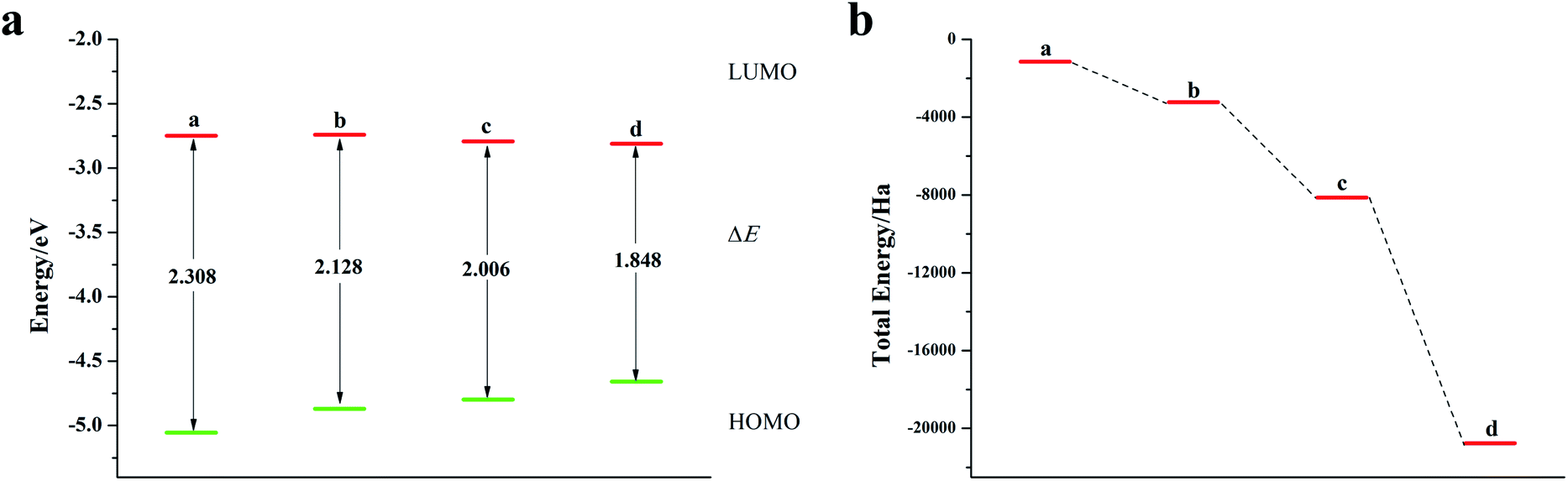
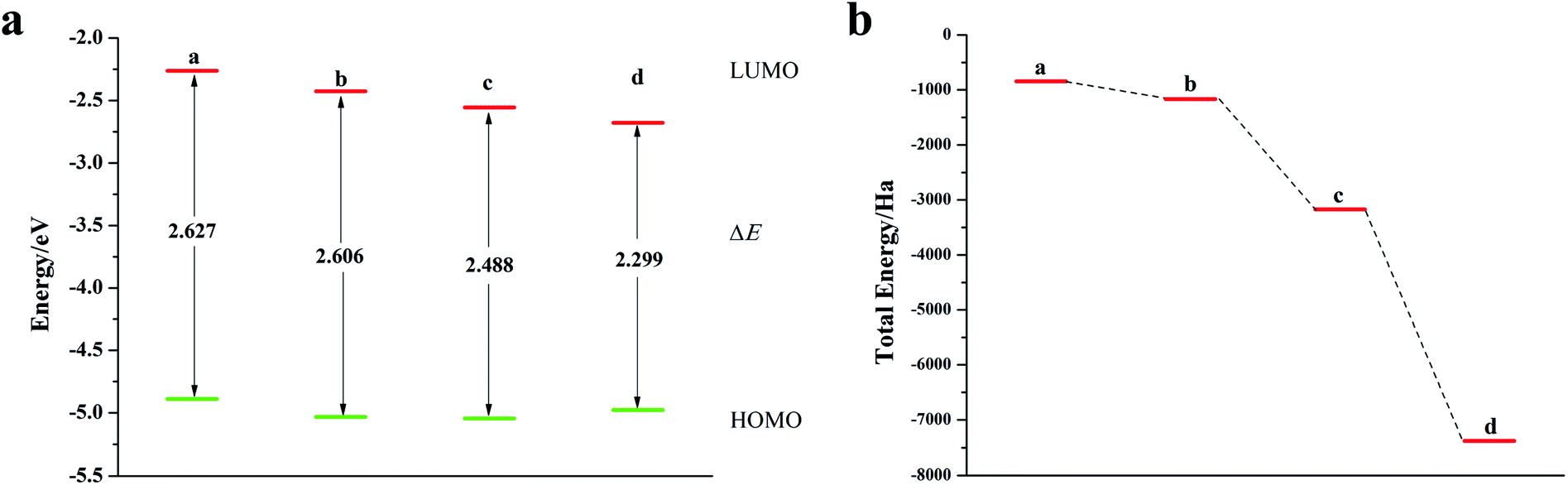
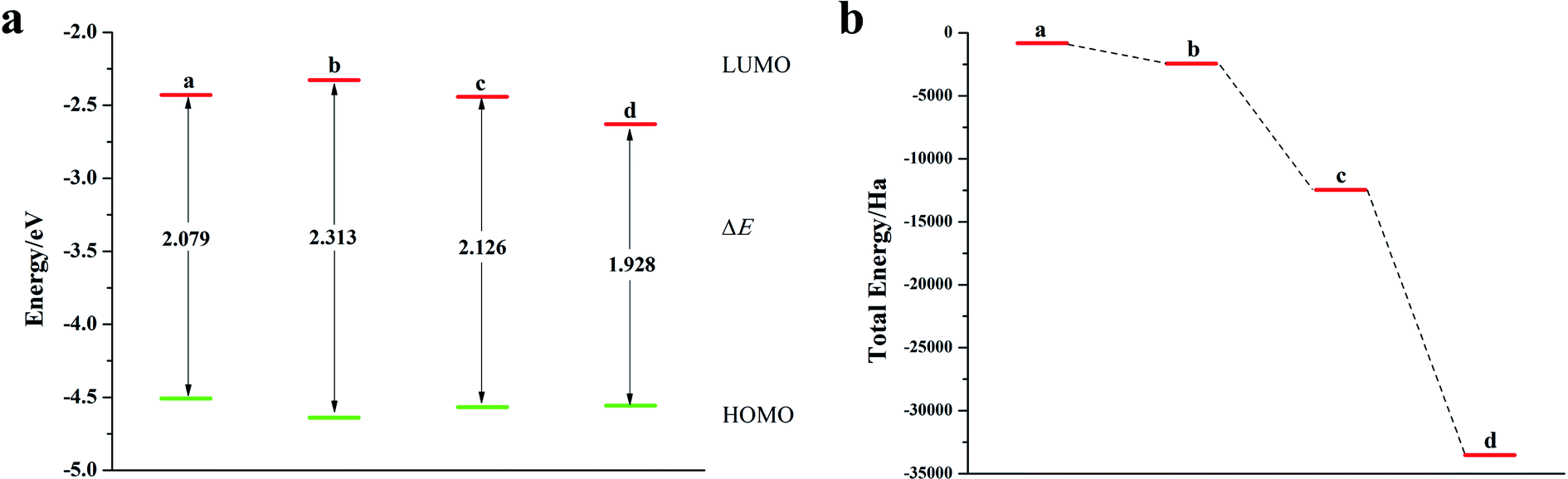
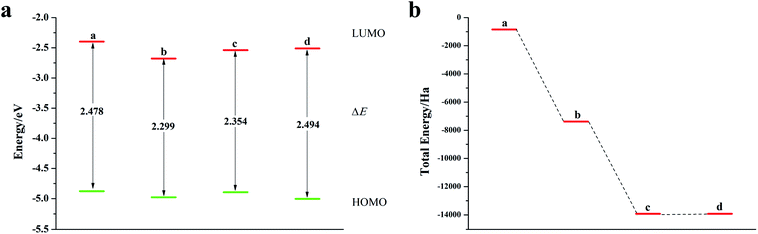
It can be seen from Fig. 2, S2, S3 and Table S2† that the doping of Te has greatly changed the EHOMO and ELUMO values of the seven-membered benzothiophene compared to the undoped benzothiopene and that doped with the other oxygen group elements. The ternary Te doping increases the EHOMO value most significantly (0.211 eV), the ELUMO is also reduced and its electron transport is more favorable, it can be seen that Te is a better oxygen doping element. Fig. 3a shows the ELUMO, EHOMO, and ΔE of the undoped and ternary O, Se, and Te-doped seven-membered benzothiophene molecules. As the relative atomic mass of oxygen group elements increases, ΔE gradually decreases. The interelectron transfer rate becomes faster, and the carrier mobility gradually increases. It can be seen from Fig. 3b that when the atoms of the doping site change in the order of O, Se, and Te, their energy values gradually decrease and the changes are more obvious, and the organic semiconductor molecules show a stable trend.
 |
| | Fig. 2 (a) ELUMO, EHOMO, and ΔE of seven-membered benzothiophene doped with the Te atom; (b) schematic diagram of the total energy of each substance (a: seven-membered benzothiophene; b: mono Te-doped seven-membered benzothiophene; c: binary Te-doped seven-membered benzothiophene; d: ternary Te-doped seven-membered benzothiophene). | |
 |
| | Fig. 3 (a) ELUMO, EHOMO, and ΔE of seven-membered benzothiophene doped with oxygen group elements; (b) schematic diagram of the total energy of each substance (a: ternary O-doped seven-membered benzothiophene; b: seven-membered benzothiophene; c: ternary Se-doped seven-membered benzothiophene; d: ternary Te-doped seven-membered benzothiophene). | |
3.2
o-Pentacene doped with oxygen group elements
O-Pentacene has a special symmetrical structure with five benzene rings. When doped, the six-membered ring breaks to form a five-membered ring and the molecular structure is shown in Fig. S4.†
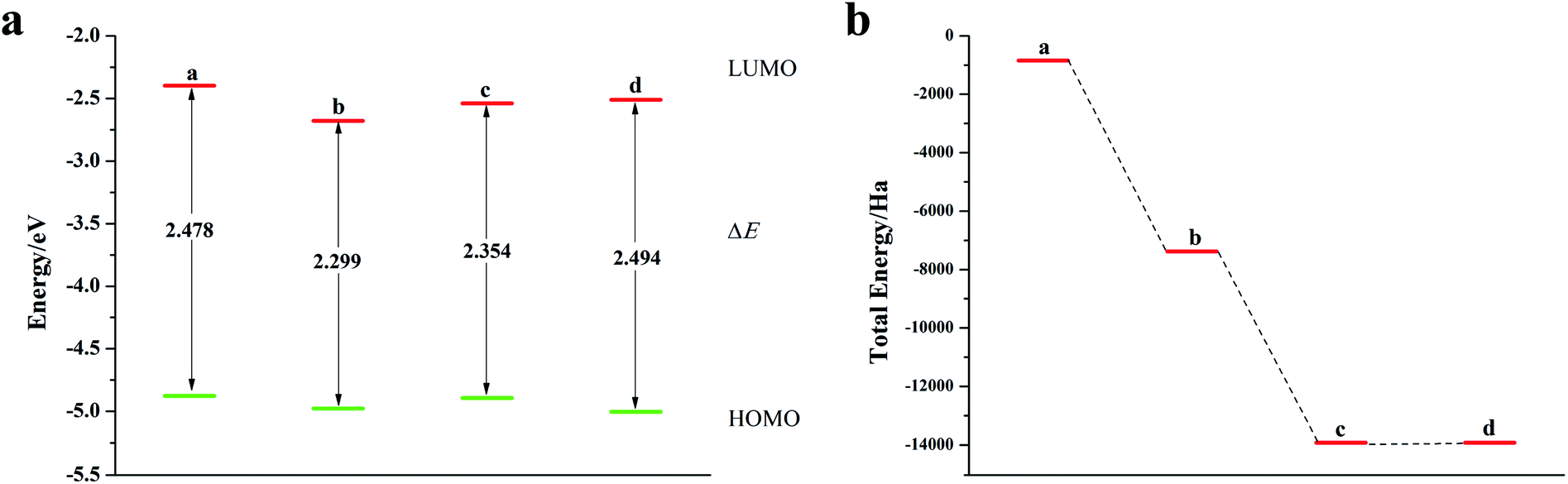
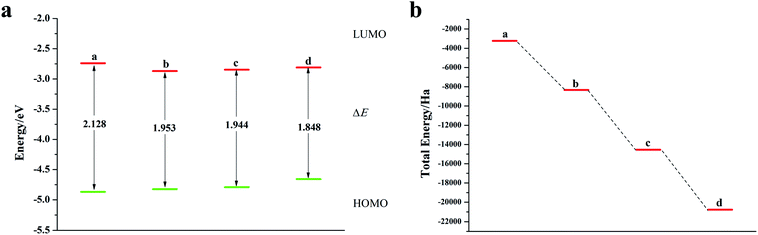
It can be seen from Fig. 4, S5, S7 and Table S3† that the decrease in the ELUMO value of the mono Te-doped o-pentacene is the most obvious (0.279 eV). The lower the ELUMO value, the stronger the ability of the molecule to receive electrons, indicating that the presence of Te atoms improves the ability of the o-pentacene molecule to accept electrons. The binary Te-doped o-pentacene (one-five ring) EHOMO value increased by 0.127 eV, indicating that Te doping enhanced the electron donating ability of the molecule. Based on the analysis of EHOMO, ELUMO, ΔE and other data, although the binary Te-doped o-pentacene molecule (one-five ring) has better stability, its band gap becomes larger, which is not conducive to electron transport. Doping o-pentacene with binary Te (two-four ring) is the best doping method in terms of band gap change and stability. Fig. 5a shows ELUMO, EHOMO, and ΔE of the undoped and mono-O, S, Se, Te doped o-pentacene molecules. As the relative atomic mass of oxygen group elements increases, ΔE gradually decreases, the electron transfer rate between EHOMO and ELUMO orbital becomes faster, and the carrier mobility gradually increases. It can be seen from Fig. 5b that when the atoms at the doping site change in the order of O, S, Se, and Te, their energy values gradually decrease and the changes are more obvious, and the organic semiconductor molecules show a stable trend.
 |
| | Fig. 4 (a) ELUMO, EHOMO, and ΔE of o-pentacene doped with the Te atom; (b) schematic diagram of the total energy of each substance (a: o-pentacene; b: mono Te-doped o-pentacene; c: binary Te-doped o-pentacene (two-four ring); d: binary Te-doped o-pentacene (one-five ring)). | |
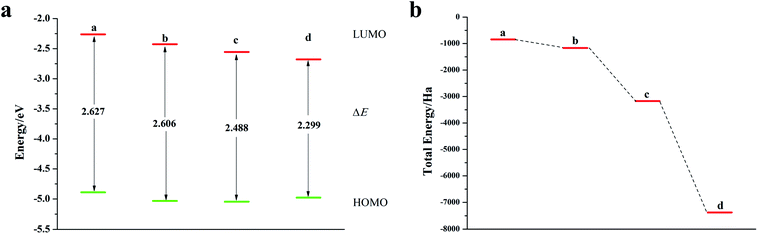 |
| | Fig. 5 (a) ELUMO, EHOMO and ΔE of o-pentacene doped with oxygen group elements; (b) schematic diagram of the total energy of each substance (a: mono O-doped o-pentacene; b: mono S-doped o-pentacene; c: mono Se-doped o-pentacene; d: mono Te-doped o-pentacene). | |
3.3 2,6-Diphenyldithiophene (DP-DTT) doped with oxygen group elements
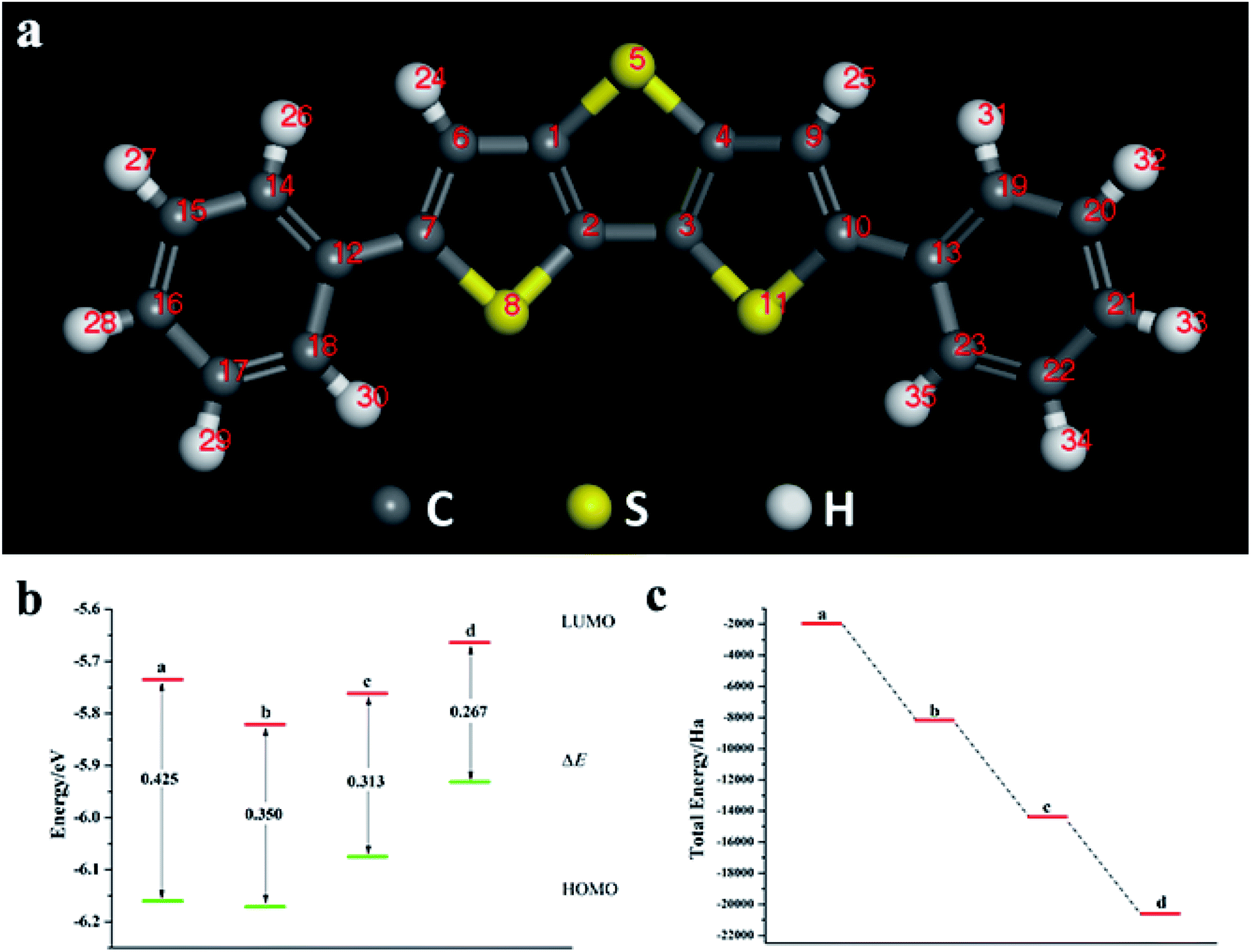
We calculated the Fukui index (Table S4†) of the undoped 2,6-diphenyldithiophene (DP-DTT). The absolute values of the sulfur atoms, Fukui (−) and Fukui (+), are the largest, which makes them prone to nucleophilic and electrophilic attacks, and thus they are ideal sites for doping. According to the symmetry of the DP-DTT molecular structure (Fig. 6a), in order to make it more stable, we adopt a symmetrical doping method, and the structural diagram of the doped structure is shown in Fig. S8.†
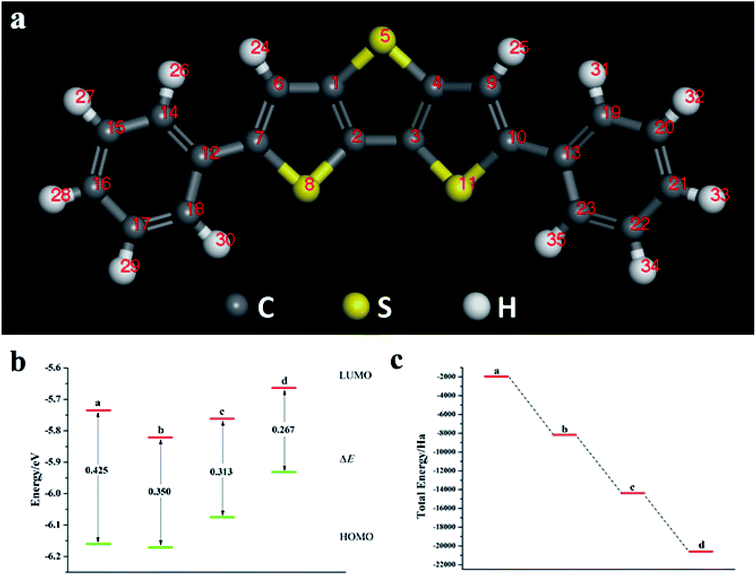 |
| | Fig. 6 (a) Schematic diagram of the DP-DTT molecular structure; (b) ELUMO, EHOMO, and ΔE of DP-DTT doped with the Te atom; (c) schematic diagram of the total energy of each substance (a: DP-DTT; b: mono Te-doped DP-DTT; c: binary Te-doped DP-DTT; d: ternary Te-doped DP-DTT). | |
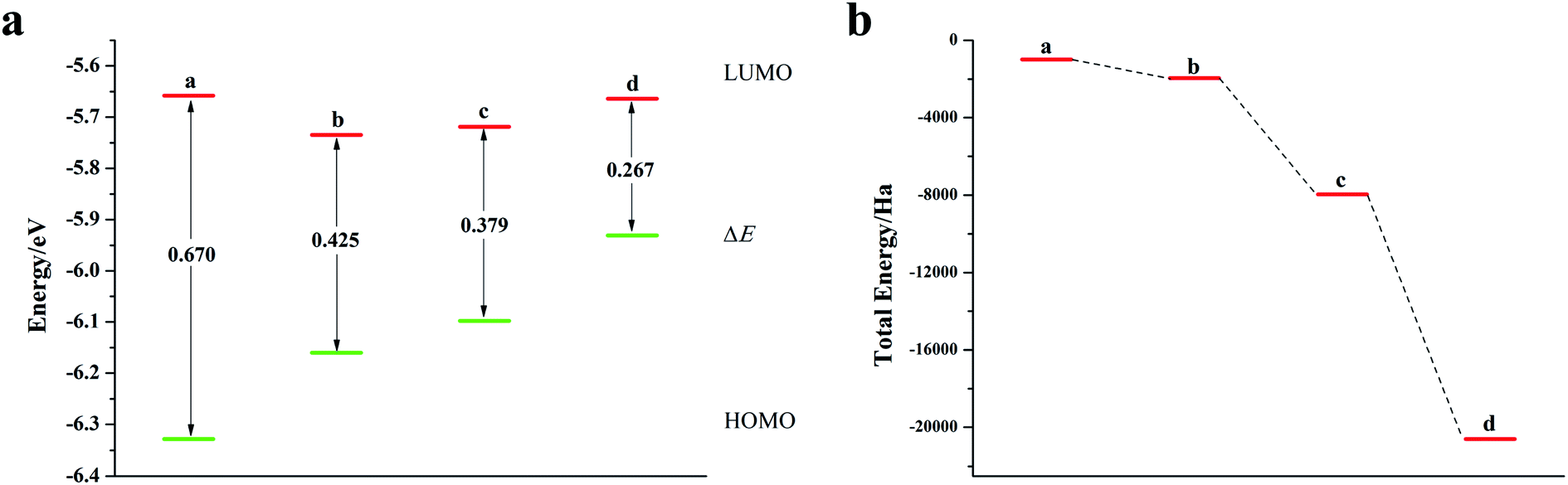
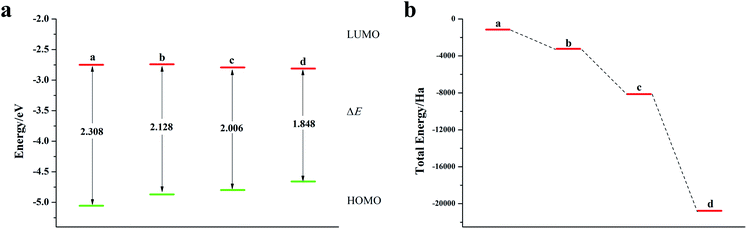
As can be seen from Fig. 6, S9, S10 and Table S5,† the band gap of DP-DTT molecules becomes smaller and the energy becomes lower after doping with Te atoms. Although the band gap of the ternary Te is the smallest, its ELUMO value is significantly increased, and the decline in the ability to accept electrons is not conducive to electron transport. In contrast, binary Te doping is an ideal doping method, which not only improves the electron gaining and losing abilities of DP-DTT molecules, but also reduces its band gap. It can be seen from Fig. 7a that when the doped atoms change in the order of O, S, Se, Te, their ELUMO energy values do not change according to a certain law, but the EHOMO value gradually increases with the increase of the relative atomic mass of the oxygen group elements, indicating that its ability to donate electrons is increasing. The band gap width (ΔE) gradually decreases, indicating that the electrons between orbitals easily undergo transitions. It can be seen from Fig. 7b that when the atoms at the doping site change in the order of O, S, Se, Te, their energy values gradually decrease, and the molecules show a stable trend.
 |
| | Fig. 7 (a) ELUMO, EHOMO, and ΔE of DP-DTT doped with oxygen group elements; (b) schematic diagram of the total energy of each substance (a: ternary O-doped DP-DTT; b: DP-DTT; c: ternary Se-doped DP-DTT; d: ternary Te-doped DP-DTT). | |
3.4 Pentacene doped with oxygen group elements
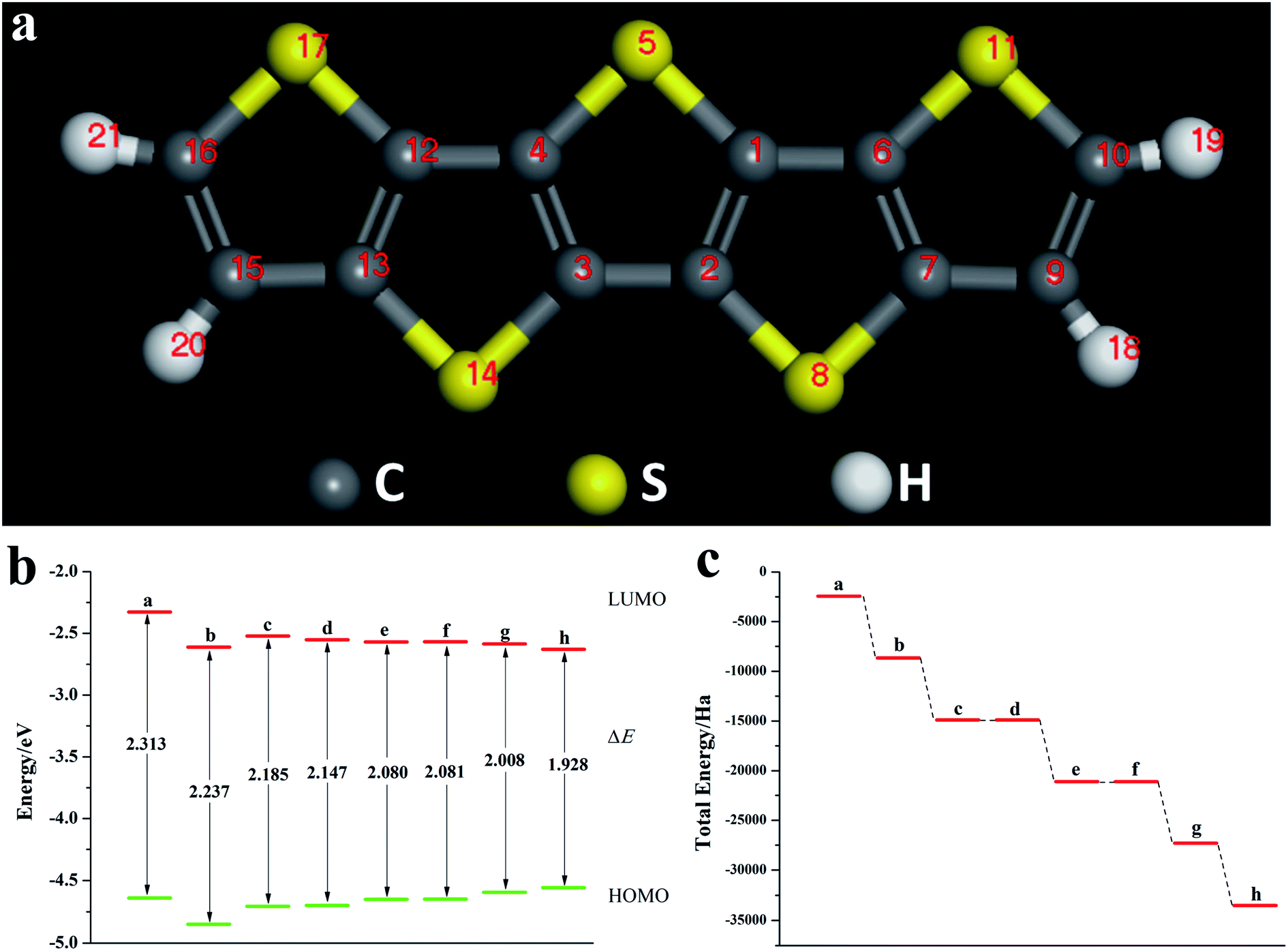
Fig. 8a is a schematic diagram of the structure of a pentacene molecule; the calculation of the Fukui index (Table S6†) shows that the absolute value of the Fukui index of five sulfur atoms is the largest, indicating that the reaction activity is high and they are ideal doping sites. Next, the pentacene molecule is doped with an oxygen group element, and a schematic diagram of the doped molecular structure is shown in Fig. S11.† According to the symmetry of the molecular structure, the binary oxygen group element is doped into a two-four-ring and one-five-ring, and the ternary oxygen group element is doped into a two-three-four ring and one-three-five ring.
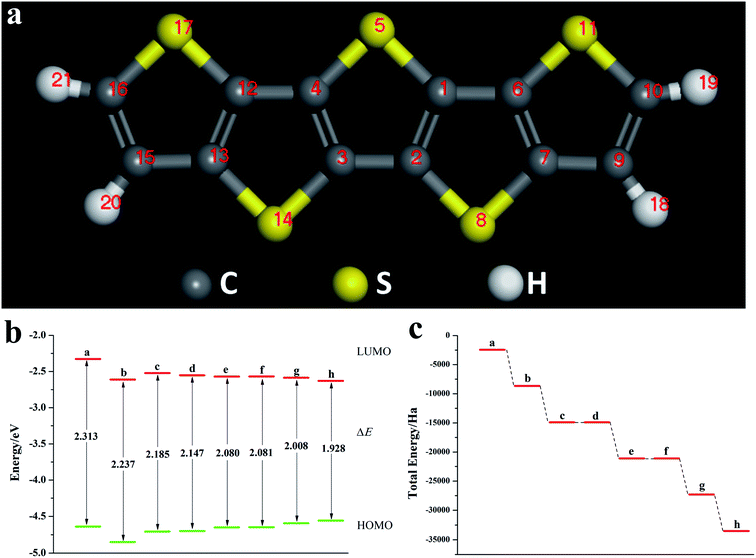 |
| | Fig. 8 (a) Schematic diagram of pentacene; (b) ELUMO, EHOMO, and ΔE of pentacene doped with the Te atom; (c) schematic diagram of the total energy of each substance (a: pentacene; b: mono Te-doped pentacene; c: binary Te-doped pentacene (two-four ring); d: binary Te-doped pentacene (one-five ring); e: ternary Te-doped pentacene (two-three-four ring); f: ternary Te-doped pentacene (one-three-five-ring); g: quaternary Te-doped pentacene; h: five membered Te-doped pentacene). | |
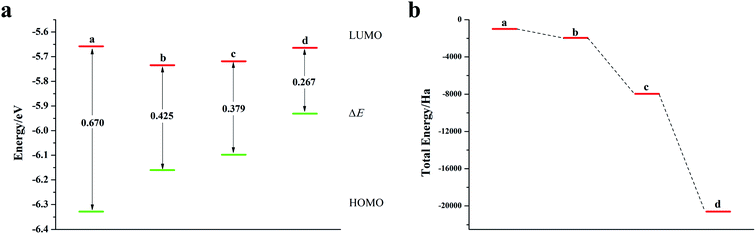
As can be seen from Fig. 8, S12, S13 and Table S7,† the ELUMO value of the mono- and penta-membered Te-doped pentacene decreased the most, being 0.285 eV and 0.302 eV respectively. The quaternary and penta-element Te doping EHOMO values have been increased, and their electron-donating capabilities have been enhanced. With the increase of the number of doping Te atoms, the pentacene molecule shows a stable trend (Fig. 8c), when the number of doping atoms is the same, even if the doping sites are different, they have the same energy. By comparing data such as ELUMO, EHOMO, and ΔE, it was found that the five-membered Te-doped pentacene has a lower ELUMO, higher EHOMO, and smaller band gap, so the best doping method is doping pentacene with five-membered Te. When the doped element changes in the order of S, Se, Te, its ELUMO value gradually decreases, and the EHOMO value changes little, indicating that the doping has little effect on the electron donating ability of the molecule (Fig. 9a). However, the band gap width tends to decrease, which indicates that the electron transfer rate between EHOMO and ELUMO orbitals becomes faster. With the increase of the relative molecular mass of the oxygen group elements (Fig. 9b), the energy value of the organic semiconductor molecule gradually decreases and it is more obvious, showing a stable trend.
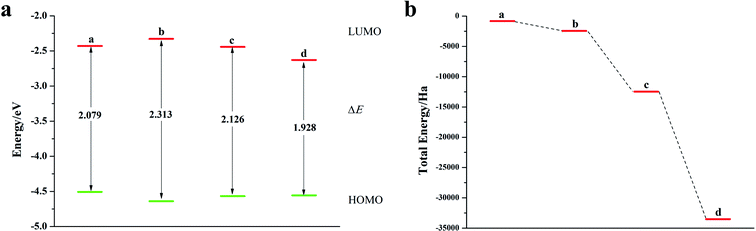 |
| | Fig. 9 (a) ELUMO, EHOMO and ΔE of pentacene doped with oxygen group elements; (b) schematic diagram of the total energy of each substance (a: five-membered O doped with pentacene; b: pentacene; c: five Se-doped with pentacene; d: five-membered Te doped with pentacene). | |
4. Conclusion
In summary, through DFT calculations, the influence of doping of oxygen group elements on the properties of four organic semiconductor materials of seven-membered benzothiophene, o-pentacene, DP-DTT and pentathiophene is explored. By comparing the changes in molecular data before and after doping, we screened the best doping methods for the four materials, all of which are doped with multi-element Te. It can be seen that Te atom doping can significantly improve the carrier mobility and stability of these four organic semiconductor materials, and the molecular band gaps after doping are reduced by 13.16%, 5.00%, 26.35%, and 16.64%, and the energy value is reduced 5.426 times, 15.45 times, 6.36 times and 12.68 times, respectively. Among them, DP-DTT has the largest decrease in the band gap, indicating that the doping of Te has the greatest effect on its carrier mobility, and o-pentacene has the largest decrease in total energy, which proves that the doping of Te atoms has the greatest impact on its stability. By comprehensive comparison, we get the general rule of oxygen group element doping: as the relative atomic mass of oxygen group elements increases, ΔE gradually decreases, and the carrier mobility gradually increases. This research guides experiments from theoretical calculations, provides ideas and directions for specific experimental design, improves the accuracy of research, and accelerates the pace of exploring new organic semiconductor materials.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
Support of the National Natural Science Foundation of China (21902021, 21908017, 51972293, and 51772039), the Joint Research Fund Liaoning-Shenyang National Laboratory for Materials Science (20180510020), the Fundamental Research Funds for the Central Universities (DUT20RC(4)020 and DUT20RC(4)018), and Supercomputing Center of Dalian University of Technology for this work is gratefully acknowledged.
References
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- K. Eswar Srikanth, K. Ramaiah, D. Jagadeeswara Rao, K. Prabhakara Rao, J. Laxman Naik, A. Veeraiah and J. Prashanth, Indian J. Phys., 2019, 94, 1153–1167 CrossRef.
- C. Zhong, C. Duan, F. Huang, H. Wu and Y. Cao, Chem. Mater., 2011, 23, 326–340 CrossRef CAS.
- H. D. Pham, H. Hu, F.-L. Wong, C.-S. Lee, W.-C. Chen, K. Feron, S. Manzhos, H. Wang, N. Motta, Y. M. Lam and P. Sonar, J. Mater. Chem. C, 2018, 6, 9017–9029 RSC.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- V. Dantanarayana, T. Nematiaram, D. Vong, J. E. Anthony, A. Troisi, K. Nguyen Cong, N. Goldman, R. Faller and A. J. Moule, J. Chem. Theory Comput., 2020, 16, 3494–3503 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2013, 26, 647–663 CrossRef.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- B. S. Ong, Y. Wu, Y. Li, P. Liu and H. Pan, Chem.–Eur. J., 2008, 14, 4766–4778 CrossRef CAS PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- A. F. Fernández and K. Zojer, Appl. Phys. Lett., 2017, 111, 173302 CrossRef.
- P. V. Pesavento, R. J. Chesterfield, C. R. Newman and C. D. Frisbie, J. Appl. Phys., 2004, 96, 7312–7324 CrossRef CAS.
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed.
- Y. Zhang, B. de Boer and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 1901–1905 CrossRef CAS.
- B. Lüssem, M. Riede and K. Leo, Phys. Status Solidi A, 2013, 210, 9–43 CrossRef.
- C. Mitsui, T. Okamoto, H. Matsui, M. Yamagishi, T. Matsushita, J. Soeda, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Mater., 2013, 25, 3952–3956 CrossRef CAS.
- X. Liu, Y. Liu and Y. Zheng, Chem. Phys. Lett., 2016, 645, 92–96 CrossRef CAS.
- A. Putta, J. D. Mottishaw, Z. Wang and H. Sun, Cryst. Growth Des., 2014, 14, 350–356 CrossRef CAS.
- J. Hollinger, D. Gao and D. S. Seferos, Isr. J. Chem., 2014, 54, 440–453 CrossRef CAS.
- D. Tian, Z. Ma, L. Gu, C. Zhou, C. Li, Z. Wang and H. Wang, Cryst. Growth Des., 2020, 20, 4479–4490 CrossRef CAS.
- R. Podeszwa, J. Chem. Phys., 2010, 132, 044704 CrossRef PubMed.
- J. E. Norton and K. N. Houk, J. Am. Chem. Soc., 2005, 4162–4163 CrossRef CAS PubMed.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- X. D. Yang, L. J. Wang, C. L. Wang, W. Long and Z. G. Shuai, Chem. Mater., 2008, 20, 3205–3211 CrossRef CAS.
- A. Thomas, R. K. Chitumalla, A. L. Puyad, K. V. Mohan and J. Jang, Comput. Theor. Chem., 2016, 1089, 59–67 CrossRef CAS.
- S. Yoo, B. Domercq and B. Kippelen, Appl. Phys. Lett., 2004, 85, 5427–5429 CrossRef CAS.
- A. L. Appleton, S. M. Brombosz, S. Barlow, J. S. Sears, J.-L. Bredas, S. R. Marder and U. H. F. Bunz, Nat. Commun., 2010, 1, 1–7 Search PubMed.
- A. N. Sokolov, S. Atahan-Evrenk, R. Mondal, H. B. Akkerman, R. S. Sánchez-Carrera, S. Granados-Focil, J. Schrier, S. C. B. Mannsfeld, A. P. Zoombelt, Z. Bao and A. Aspuru-Guzik, Nat. Commun., 2011, 2, 1–8 Search PubMed.
- N. L. Janaki, B. Priyanka, A. Thomas and K. Bhanuprakash, J. Phys. Chem. C, 2012, 116, 22663–22674 CrossRef CAS.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- A. M. Liu, X. F. Ren, Q. Y. Yang, J. Sokolowski, J. Guo, Y. Q. Li, L. G. Gao, M. Z. An and G. Wu, J. Electrochem. Soc., 2018, 165, H725–H732 CrossRef CAS.
- J. G. Laquindanum, H. E. Katz and A. J. Lovinger, J. Am. Chem. Soc., 1998, 120, 664–672 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na01026j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mengfan
Gao
a,
Yan
Ma
*ae,
Xuefeng
Ren
*a,
Mengfan
Gao
a,
Yan
Ma
*ae,
Xuefeng
Ren