
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring surface-supported water–melamine complexes by cooperative H-bonding interactions†
Valeria
Lanzilotto
 *abc,
Cesare
Grazioli
c,
Matus
Stredansky
cde,
Teng
Zhang
ae,
Luca
Schio
c,
Andrea
Goldoni
f,
Luca
Floreano
c,
Alessandro
Motta
gb,
Albano
Cossaro
ch and
Carla
Puglia
a
*abc,
Cesare
Grazioli
c,
Matus
Stredansky
cde,
Teng
Zhang
ae,
Luca
Schio
c,
Andrea
Goldoni
f,
Luca
Floreano
c,
Alessandro
Motta
gb,
Albano
Cossaro
ch and
Carla
Puglia
a
aDepartment of Physics and Astronomy, Uppsala University, P.O. Box 516, 751 20 Uppsala, Sweden. E-mail: valeria.lanzilotto@uniroma1.it
bDepartment of Chemistry, Sapienza University of Rome, P.le Aldo Moro 8, 00185 Roma, Italy
cIOM-CNR, Istituto Officina dei Materiali, Basovizza SS-14, Km 163.5, 34149 Trieste, Italy
dDepartment of Physics, University of Trieste, Via A. Valerio 2, 34127 Trieste, Italy
eSchool of Information and Electronics, MIIT Key Laboratory for Low-Dimensional Quantum Structure and Devices, Beijing Institute of Technology (BIT), 100081 Beijing, China
fElettra-Sincrotrone Trieste S.C.p.A., Basovizza SS-14, Km 163.5, 34149 Trieste, Italy
gConsortium INSTM, Via G. Giusti 9, 50121 Firenze, Italy
hDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via Giorgieri 1, 34127 Trieste, Italy
First published on 23rd February 2021
Abstract
The water-splitting photo-catalysis by carbon nitride heterocycles has been the subject of recent theoretical investigations, revealing a proton-coupled electron transfer (PCET) reaction from the H-bonded water molecule to the CN-heterocycle. In this context, a detailed characterization of the water–catalyst binding configuration becomes mandatory in order to validate and possibly improve the theoretical modeling. To this aim, we built a well-defined surface-supported water/catalyst interface by adsorbing water under ultra-high vacuum (UHV) conditions on a monolayer of melamine grown on the Cu(111) surface. By combining X-ray photoemission (XPS) and absorption (NEXAFS) spectroscopy we observed that melamine adsorbed onto copper is strongly tilted off the surface, with one amino group dangling to the vacuum side. The binding energy (BE) of the corresponding N 1s component is significantly higher compared to other N 1s contributions and displays a clear shift to lower BE as water is adsorbed. This finding along with density functional theory (DFT) results reveals that two adjacent melamine molecules concurrently work for stabilizing the H-bonded water–catalyst complex: one melamine acting as a H-donor via the amino-N (NH⋯OHH) and another one as a H-acceptor via the triazine-N (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N⋯HOH).
N⋯HOH).
Introduction
Melamine (triamino-s-triazine) and its condensed derivative, melem (triamino-s-heptazine), are the building blocks of polymeric carbon nitride materials, p-CN(H), a new class of photocatalysts with the ability to catalyze the water-splitting reaction under visible light irradiation.1,2 Specifically, melamine-based carbon nitrides, such as poly(triazine) imide (PTI), are characterized by a graphitic-layered structure where the single triazine units are connected through secondary amino groups, –NH– (see Fig. 1c).3,4 On the other hand, melem-based polymers are usually identified with melon, a linear polymer where the –NH– bridging heptazine units also preserve a primary amino group –NH2 (see Fig. 1d).5 Melon chains can arrange in a tightly hydrogen-bonded 2D array further stabilized by van der Waals interlayer interactions.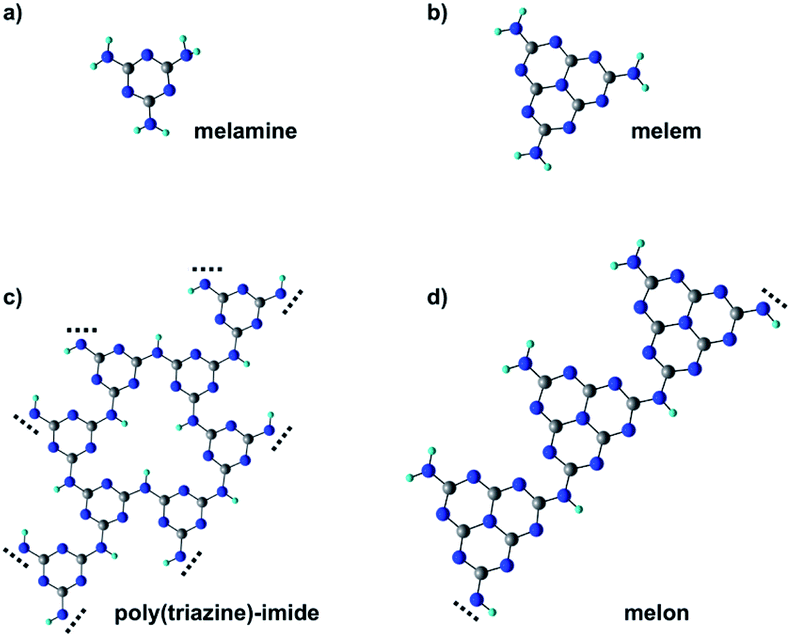 | ||
| Fig. 1 Molecular structure of melamine (a), melem (b) and the corresponding polymeric derivatives poly(triazine) imide (c) and melon (d). Carbon, nitrogen and hydrogen atoms correspond, respectively, to gray, blue and white spheres. | ||
p-CN(H) are usually regarded as semiconductors with a band gap between 2.5 and 2.8 eV, as estimated by UV/vis absorption spectra.1 Consequently, the water-splitting mechanism is often treated in the framework of heterogeneous photo-catalysis6,7 that involves several steps: (i) exciton generation by light absorption, (ii) exciton dissociation, (iii) migration of charge carriers (hole and electrons) toward the active sites of the catalyst, and (iv) electron and hole extraction from the catalyst for driving the surface reduction of protons and the oxidation of water, respectively. In this context, a layered structure, like that featured by PTI or 2D arrays of crystalline melon, would be beneficial for increasing photoactivity as the π–π inter-planar interaction between the heterocyclic units would improve exciton separation and charge transport along the stacking direction.7 Indeed, both inter-chain and in-plane charge transport are quite unlikely given the absence of conjugated linkers between the triazine/heptazine units. However, in contrast to this expectation, nanosheets of PTI have been found to outperform by 17 times the photoactivity of bulk (not exfoliated) crystalline PTI,3 which in turn has a photoactivity comparable to that of amorphous melon.4 At the same time, negligible activity is observed for crystalline melon.8 The lack of correlation between the photoactivity and mesoscopic structure is often explained by the fact that the active sites, crucial for H-binding the water molecules, are not sufficiently exposed in the crystalline phases of p-CN(H).8,9 This is consistent with the higher photo-activity reported for heptazine oligomers with respect to melon that has been explained by attributing the main reducing sites to primary and/or secondary amines. The latter are indeed not only more abundant in the oligomers than in the polymeric case but also better exposed due to the stacking disorder and conformational flexibility of the less condensed oligomer aggregates.8 Moreover, as the H2 evolution reaction is often conducted with small amounts of co-catalysts, usually platinum, these amines would also allow better coordination of the platinum centers for efficient electron transfer.8 A similar role has also been attributed to other functionalities, such as cyanamides10 and urea groups,11 which have been used to modify the periphery of the CN backbone.
At variance with the semiconductor-like mechanism, recent theoretical investigations have shown that water splitting with p-CN(H) may be better described as a photochemical reaction essentially confined to a single molecular unit.12–14 The mechanism, also known as proton-coupled electron transfer (PCET), relies on the H-bond formation between the water molecule and the lone pair of triazine-N. Upon photo-excitation, an electron–proton transfer occurs from the H-bonded water molecule to the heterocycle with the formation of two neutral radicals: an OH radical and a hypervalent heterocycle radical. Successively, two heterocyclic radicals can recombine via an exothermic dark reaction to form molecular hydrogen.
The detection of the reaction intermediates, i.e. OH radicals or hypervalent heterocyclic radicals, would be crucial to shed light on the p-CN(H) water-splitting mechanism. In this context, the molecular-level description of surface sensitive techniques like X-ray photoemission spectroscopy (XPS) could be exploited for probing the evolution of the reactant chemical state. Chemical state variations can, indeed, largely affect the XPS binding energy (BE) of the involved heteroatoms. However, the accurate detection of binding energy (BE) shifts often requires well-defined interfaces as those prepared in ultra-high vacuum conditions by means of surface science methods.
From the perspective of performing photochemistry experiments and understanding the role of all p-CN(H) functionalities, we built up a well-defined surface-supported water/catalyst interface by exploiting a monolayer of standing up melamine molecules on the Cu(111) surface.15 In this configuration, some of the N-functional groups would be exposed toward the vacuum side, in a favorable position to interact with the possible reactants. By combining DFT simulations with XPS and NEXAFS measurements, we precisely assess both the melamine adsorption geometry on Cu(111) and the water–melamine binding configuration. In agreement with the STM images of ref. 15, we find that the adsorbed melamine molecules are strongly tilted through two partially dehydrogenated amino groups (NH–Cu), while the third one points away from the surface. Interestingly, the water–catalyst complex involves two melamine molecules, one acting as a H-donor via the free amino group (NH⋯OHH) and another one as a H-acceptor via the triazine-N (CN⋯HOH). Although the triazine-N is considered the active site of the PCET reaction, our results evidence how the co-presence of different N-functional groups, as those featured by p-CN(H) materials, may be beneficial for achieving a more steady H-bonded water–catalyst interface and, in turn, a more efficient water-splitting process.
Results and discussion
1 ML melamine/Cu(111)
According to Lin et al.,15 melamine would adsorb onto Cu(111) in a standing-up orientation with two partially de-hydrogenated amino groups binding to the surface. Fig. 2 shows synchrotron radiation-based N 1s XP spectra for 1 ML of melamine adsorbed on Cu(111) taken at different photon energies. Similarly to the Al Kα XP spectrum reported in ref. 15, we observe a main component (peak a) at 398.2 eV and a smaller one (peak b) at 400.1 eV. The latter corresponds to the amino-N pointing away from the surface, while the former is contributed by the triazine-N and by the singly de-hydrogenated amino nitrogens bound to the Cu substrate. The partial de-hydrogenation is indeed responsible for a significant shift to lower BEs of the NH–Cu component, as unambiguously shown for a sub-monolayer of hexaaminotriphenylene/Cu(111).16 The standing-up orientation is further confirmed by the a/b intensity ratio variation observed as a function of the photon energy (for further details, see the ESI†). The relative intensities a and b deviate from a rigorous 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio because of (i) the different attenuation experienced by the photoelectrons coming from different depths and (ii) photoelectron diffraction (PED) effects (especially on locally ordered systems).17–19 Actually, the experimental trend of the a/b ratio compares well with that estimated by escape depth arguments (see Table S1†), in particular, the deviations due to PED effects are minimum at the photon energy of 650 eV and maximum at 750 eV.
:1 stoichiometric ratio because of (i) the different attenuation experienced by the photoelectrons coming from different depths and (ii) photoelectron diffraction (PED) effects (especially on locally ordered systems).17–19 Actually, the experimental trend of the a/b ratio compares well with that estimated by escape depth arguments (see Table S1†), in particular, the deviations due to PED effects are minimum at the photon energy of 650 eV and maximum at 750 eV.
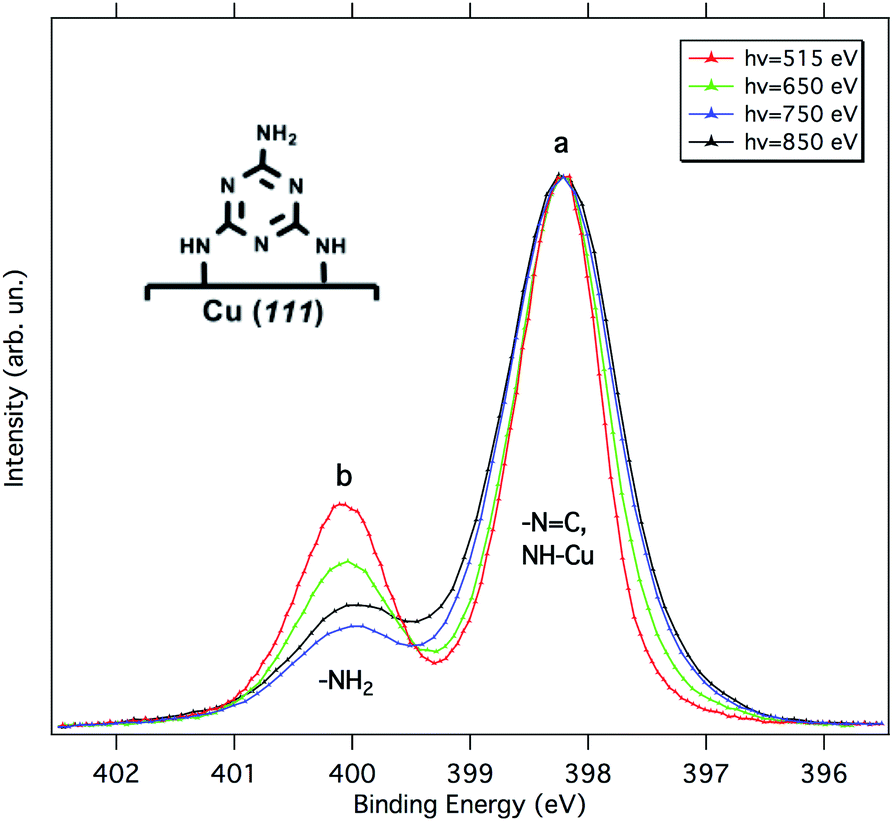 | ||
| Fig. 2 N 1s XP spectra of 1 ML of melamine on Cu(111) acquired at different photon energies. All spectra were acquired at normal emission and grazing incidence (4°). The spectra are normalized to the intensity of peak a. A schematic of the melamine adsorption configuration is shown in the inset. | ||
A direct evaluation of the molecular tilt off the surface can be obtained from the study of the NEXAFS resonances of the molecular orbitals at the C K-edge, as a function of the surface orientation with respect to the (linear) polarization of the X-ray beam. Due to the selection rules, the NEXAFS resonances of π*-symmetry orbitals have a maximum intensity when the electric field is perpendicular to the π*-symmetry orbital plane and zero when the electric field lies in the nodal plane. Providing that the rehybridization with the substrate does not alter the molecular orbital symmetry (at least not the nodal planes), the spatially averaged molecular tilt is obtained from the intensity ratio of the π*-symmetry resonances (the so-called NEXAFS dichroism), as measured with the surface oriented either parallel or orthogonal to the electric field of the incoming X-ray beam (s- and p-polarization, respectively), according to the formula reported in ref. 20. The observed degree of NEXAFS dichroism at the C K-edge (see Fig. 3) indicates a standing-up molecular orientation with an average molecular tilt off the surface of about 65°. Contrarily, the X-ray absorption spectra at the N K-edge of 1 ML on Cu(111) (shown in Fig. S2†) displays a significant change with respect to the gas phase one, making a clear-cut identification of the main resonances in both polarizations difficult and thus preventing a quantitative analysis of the NEXAFS dichroism.
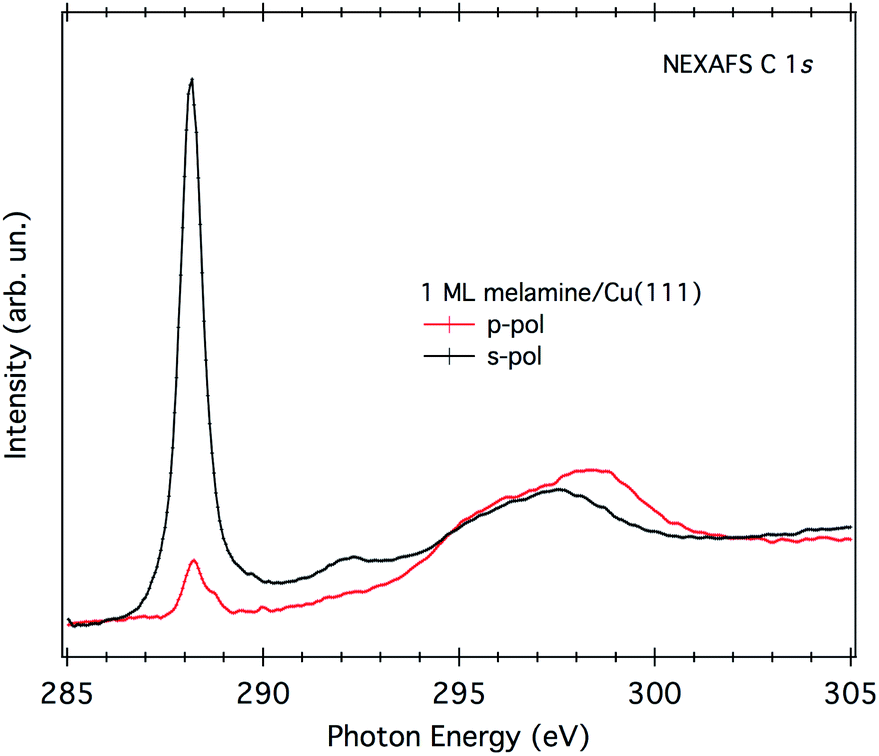 | ||
| Fig. 3 NEXAFS C K-edge of 1 ML of melamine/Cu(111). Both p- and s-polarization spectra have been normalized to the spectra acquired on the clean substrate. | ||
Fig. 4 shows a top and a side view of the DFT model for 1 ML of partially de-hydrogenated melamine molecules on Cu(111), as computed on the basis of the STM images of ref. 15. Accordingly, the molecules arrange in a herringbone distribution forming rows running along the 〈11−2〉 direction. Finally, DFT calculations predict the molecules to have an average tilt angle of 75° off the surface, in good agreement with the average value determined by NEXAFS dichroism.
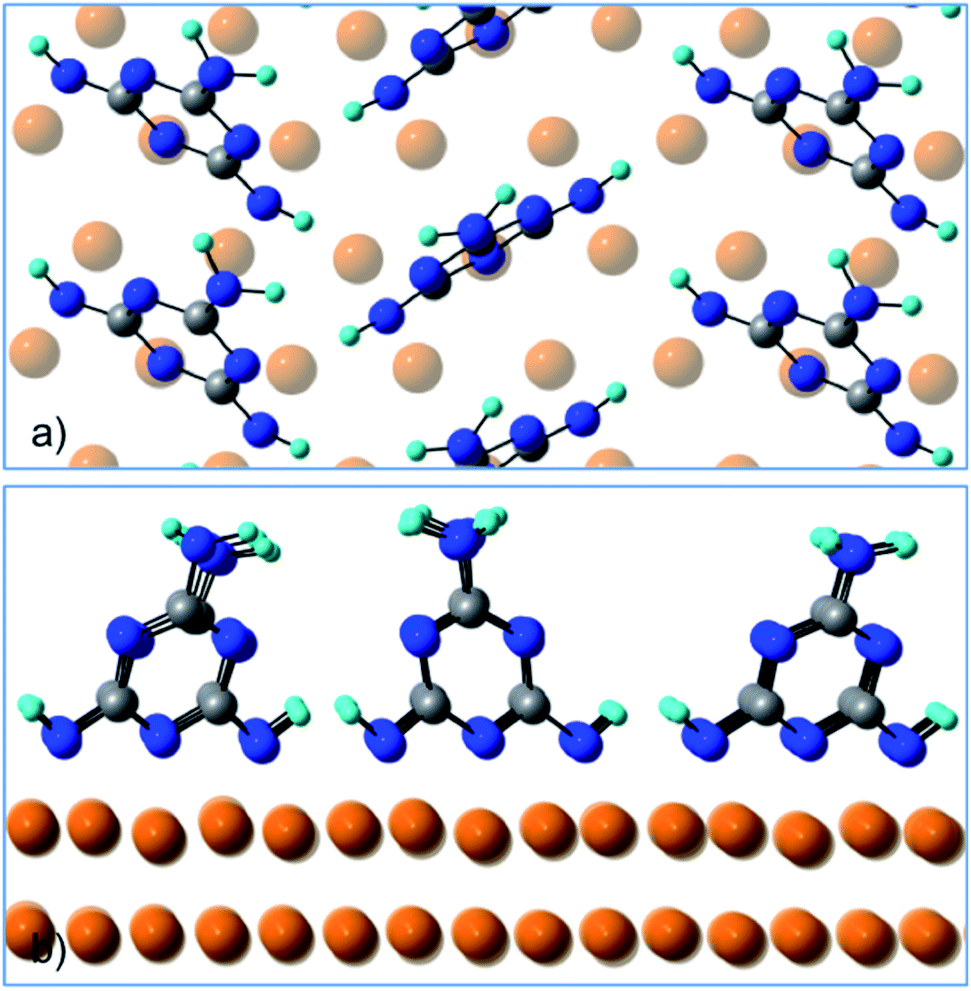 | ||
| Fig. 4 (a) Top view and (b) side view of the DFT model for 1 ML of partially de-hydrogenated melamine molecules on Cu(111). | ||
D2O/melamine/Cu(111)
As evidenced by the above analysis, the 1 ML melamine/Cu(111) sample represents a perfect candidate for probing the interaction between the water molecule and the N-functional groups typical of p-CN(H) heterocycles. At variance with the melamine/Au(111) interface,21 where all functionalities are locked-up by in-plane intermolecular H-bonds, the Cu(111) surface allows the N-functional groups (that are pointing toward the vacuum side) to form H-bonding with the water molecules.As mentioned in the Experimental section, water uptake was performed by keeping the sample at a low temperature (173 K) while dosing heavy water (D2O) at a partial pressure of 10−8 mbar for a few minutes (see Experimental details). Both N 1s and O 1s XP spectra were measured after restoring the UHV chamber base pressure (3 × 10−10 mbar). After a first dosing of 2.4 Langmuir (L), the b component of the N 1s spectrum shifts to lower BEs by 0.2–0.3 eV, whereas the peak a remains practically unaffected (see Fig. 5). No further shifts are observed upon additional dosing of 2.4 L, suggesting that all water anchoring sites have been saturated with the first dosing. The annealing of the sample at 203 K already causes water desorption (see Fig. S3†).
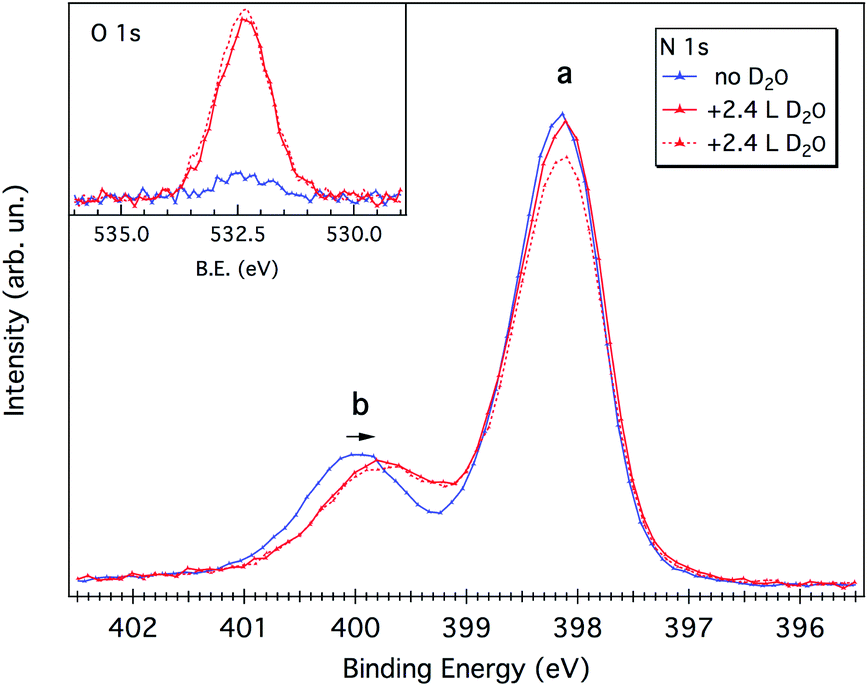 | ||
| Fig. 5 N 1s spectra of 1 ML of melamine/Cu(111) before and after two consecutive D2O dosing (2.4 Langmuir, 2.4 L). The corresponding O 1s XP spectra are reported in the inset. During dosing, the sample temperature was kept at 173 K. The spectra were acquired with a photon energy of 650 eV and performed at the grazing incidence (∼10°) and at an emission angle of 45° from the surface normal. | ||
Water adsorption thus shifts the b component corresponding to the protruding –NH2 amino group. The sign of the energy shift is consistent with the amino-N acting as a H-donor with respect to the water molecule, NH⋯OHH.21,22
In order to discriminate among the possible water binding configurations, DFT simulations were performed by positioning the water molecule between two melamine molecules of the same molecular row (model 1, Fig. 6) or between two melamine molecules of two adjacent rows (model 2, Fig. 6). In the former case, the water molecule is found to be coordinated by two amino groups, one acting as a H-donor (H-bond #1) and another one as a H-acceptor (H-bond #2). In the second model, one melamine acts as a H-donor through the amino group (H-bond #1), while the other melamine acts as a H-acceptor through the triazine-N (H-bond #2). The latter configuration (model 2) was found to be 7 kcal mol−1 (300 meV) more stable than model 1. The difference in stability is mainly related to geometrical constraints (see Table 1). In model 2, the H-bond lengths (#1, #2) are shorter compared to those found in model 1 and the H-bond angles are on average closer to 180°. Moreover, model 2 can count on a third, although weak, H-bonding interaction (H-bond #3) provided by the close proximity of the water molecule to the amino group of the H-acceptor melamine.
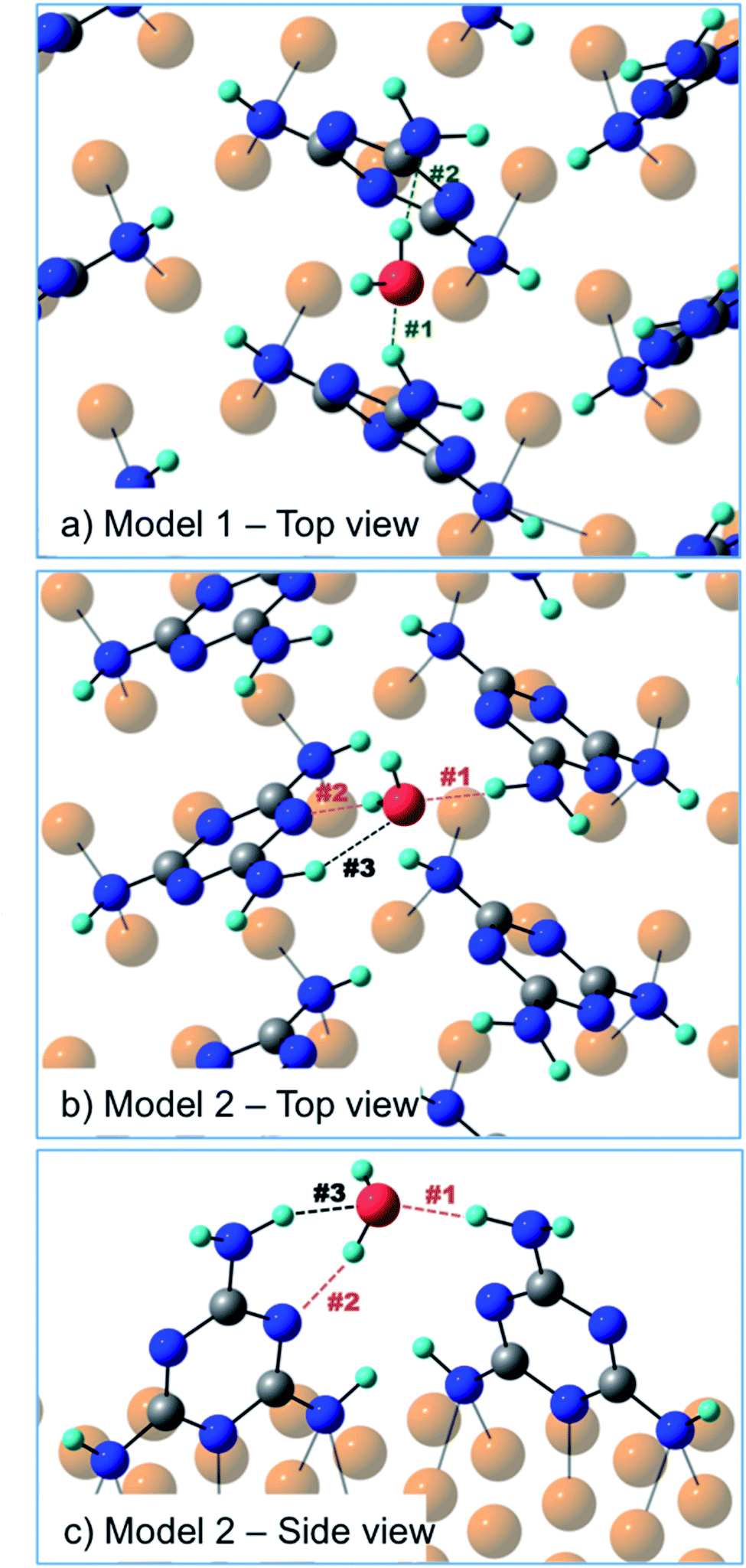 | ||
| Fig. 6 (a) Top view of model 1 – one water molecule coordinated by two melamine molecules of the same molecular row; (b) top view and (c) side view of model 2 – one water molecule bridging two melamine molecules of two adjacent rows. | ||
| H-Bond length (Å) | H-Bond angle (°) | |||||
|---|---|---|---|---|---|---|
| #1 H-donor | #2 H-acceptor | #3 H-donor | #1 | #2 | #3 | |
| Model 1 | 2.0 | 1.9 | — | 146 | 167 | — |
| Model 2 | 1.8 | 1.8 | 2.1 | 172 | 158 | 140 |
Actually, the three-point H-bonding assembly favors a dynamic water adsorption since, at the temperature used for the experiment, the thermal energy is likely sufficient for the water molecule to constantly flip, thus inverting the donor/acceptor roles of the two melamine molecules. A similar dynamic absorption was also proposed for water molecules adsorbed onto the anatase TiO2(101) surface at 190 K.23
On the more stable model 2, we performed simulations of the N 1s BE shifts at two different levels of theory (see ESI, Fig. S6 and Table S3†). Although both methods underestimate the BE shift of the free amino-N component with respect to the triazine/NH–Cu ones, the changes observed upon water coordination are in qualitative agreement with the XPS measurements of Fig. 5. Both theoretical methods predict a significant shift of 0.5–0.6 eV to lower BEs for the strongly H-bonded amino-N component of the H-donor molecule (H-bond #1), whereas only a minor shift of 0.2 eV to higher BEs is expected for the triazine-N component of the H-acceptor melamine (H-bond #2). All the other N components of both molecules remain practically unchanged, including that associated with the weakly interacting amino-N of the H-acceptor melamine (H-bond #3). Although quantitatively limited, these calculations are consistent with the measured N 1s spectra. We remark that, within the proposed dynamic model, the observed BE shift of the XPS b component upon water uptake results from the combination of two contributions, albeit unresolved, one from the strongly interacting amino-N (shifted) of the H-donor molecule and the other from the weakly interacting amino-N of the H-acceptor molecule, which is overall almost unperturbed. In fact, the BE of the H-acceptor is known to be less influenced by H-bonding interactions compared to the H-donor.21,22 As an example, we recall the case of the double H-bond occurring in a dimer of melamine molecules, i.e. NH⋯NC. In this case, the chemical shift of the amino-N is about −0.6/−0.7 eV, while that of triazine-N is only +0.1 eV.21
Further support to this water binding configuration is provided by the amino-N 1s to O 1s XPS intensity ratio (normalized to the corresponding cross section), which is 1.8 at hν = 650 eV. This value can be regarded as a representative of the stoichiometry of the melamine/water complex, in good agreement with that expected by the proposed H-bonding model (2 amino groups for 1 oxygen atom). In fact, both amino-N and O atoms correspond to the most protruding species and, according to the DFT model, the water molecule is in-between the two amino groups and not above. This geometry excludes attenuation effects due to escape depth, as well as significant PED modulations for both O and amino-N species.
Finally, we investigated the effect of increased amount of water uptake by monitoring in real time the N 1s XP spectrum, while dosing water in the 10−6 mbar range (see Fig. S4†). Under these conditions, we observed a significant increase of the oxygen 1s peak together with a shift of the main N 1s peak (a) by +0.1 eV without any shift of peak b. At the same time, the C 1s peak also slightly shifts to higher BEs. Although the origin of these shifts is not clear yet, they do not look associated with the formation of H-bonds as the new interface resulted to be thermodynamically unstable. In fact, the water content (i.e. the intensity of the O 1s peak) started to decrease just after stopping the water dosing. The system recovered its initial configuration and the corresponding BE of N and C 1s peaks after 1 h. We exclude that water molecules adsorb onto the bare Cu(111) surface as no sticking is observed upon similar dosing on the clean Cu(111) at the same temperature (173 K). Excess water is rather physisorbed atop the D2O-saturated melamine film, as witnessed by the slight attenuation of peak a upon dosing in the 10−6 mbar range (see Fig. S4†).
Conclusions
In this work, we addressed the adsorption geometry of the melamine molecule on the Cu(111) surface and the binding configuration of the surface-supported water/melamine complex. XPS and NEXAFS data showed that melamine adsorbs onto Cu(111) in a standing-up orientation (∼65° tilted off the surface), binding to the substrate through two singly dehydrogenated amino groups. The NH–Cu contribution to the N 1s XPS closely overlaps with that of the triazine nitrogen atoms. Instead, the remaining amino-N, which points outward, yields a large core level shift by about 1.8 eV to higher BEs. Upon water adsorption, the latter XPS component shows a BE decrease by 0.2–0.3 eV, which indicates that the amino group acts as a H-donor toward the water molecule. DFT modeling of the monolayer film predicts the alignment of molecules in homogeneous stripes, which are arranged in a herringbone packing, in agreement with the present experimental findings and former microscopic investigations.15 Simulations at T = 0 K of water adsorption shows evidence that the water molecule linked to the dangling amino group also establishes a second H-bond with the triazine-N of a nearby melamine molecule (thus acting as a H-acceptor) of the adjacent molecular stripe. Actually, at the temperature used for this experiment, the water configuration is not frozen but dynamically changes, continuously inverting the roles of the two melamine molecules. With reference to the PCET reaction,12–14 which is usually modeled by only considering the H-bond between water and triazine-N, our study highlights how the amino groups (or other moieties) may play an important role in stabilizing the water-catalyst H-bonded complex and, in turn, improving the photoactivity efficiency. This hypothesis could explain the fact that the H2 evolution rates with p-CN(H) materials significantly depend on the nature of the dangling functional groups, i.e. –NH2, anionic cyanamides, and urea containing functional groups.8,10,11Experimental
Experimental details
All the experiments were performed at the CNR-IOM beamline ALOISA of the Elettra Synchrotron (Trieste, Italy). Melamine (C3H6N6) was purchased from Sigma-Aldrich (99%) and sublimed from a boron nitride crucible held at the temperature of ∼393 K. As playground for water adsorption, we used one monolayer (ML) of melamine adsorbed onto Cu(111). The 1 ML sample was prepared by annealing a multilayer up to the molecule sublimation temperature. Optimization of the sample preparation and its characterization were performed in the ALOISA main chamber by means of X-ray photoemission spectroscopy (XPS) and near-edge X-ray absorption fine structure spectroscopy (NEXAFS) measurements. The N 1s photoemission spectra were acquired at normal emission and grazing incidence (∼4°) with photon energies of 515, 650, 750 and 850 eV. The energy scale was calibrated by setting the Cu 3p3/2 peak to the reference binding energy of 75.1 eV. NEXAFS measurements were performed at the C and N K-edges in the partial electron yield (PEY) mode. The orientation of the surface with respect to the linear polarization of the photon beam was changed from Transverse Magnetic (TM, closely p-polarization) to Transverse Electric (TE, s-polarization) by sample rotation around the photon beam axis while keeping a constant grazing angle of 6°. Details about calibration and normalization of the NEXAFS spectra can be found in ref. 24 and 25.The study of water uptake was carried out at the ANCHOR-SUNDYN end-station26 of the ALOISA branchline, where real-time XPS can be performed while exposing the sample to gas dosing. Heavy water, D2O (Sigma-Aldrich, 99.9 atom% D), was used as an extra precaution against synchrotron light induced dissociation.27 D2O dosing was performed through a leak valve by keeping the total pressure at 2.0 × 10−8 mbar for a couple of minutes (base pressure = 3 × 10−10 mbar). O 1s and N 1s photoemission spectra were measured after closing the leak valve and restoring the chamber base pressure. We also performed photoemission measurements while dosing water at 1.0 × 10−6 mbar (see ESI, Fig. S4†). Dosing and XPS measurements were performed with the sample held at ∼173 K (minimum temperature accessible at the ANCHOR-SUNDYN end-station). XPS spectra were acquired with a photon energy of 650 eV and performed at grazing incidence (∼10°) and with a take-off angle of 45°.
Computational details
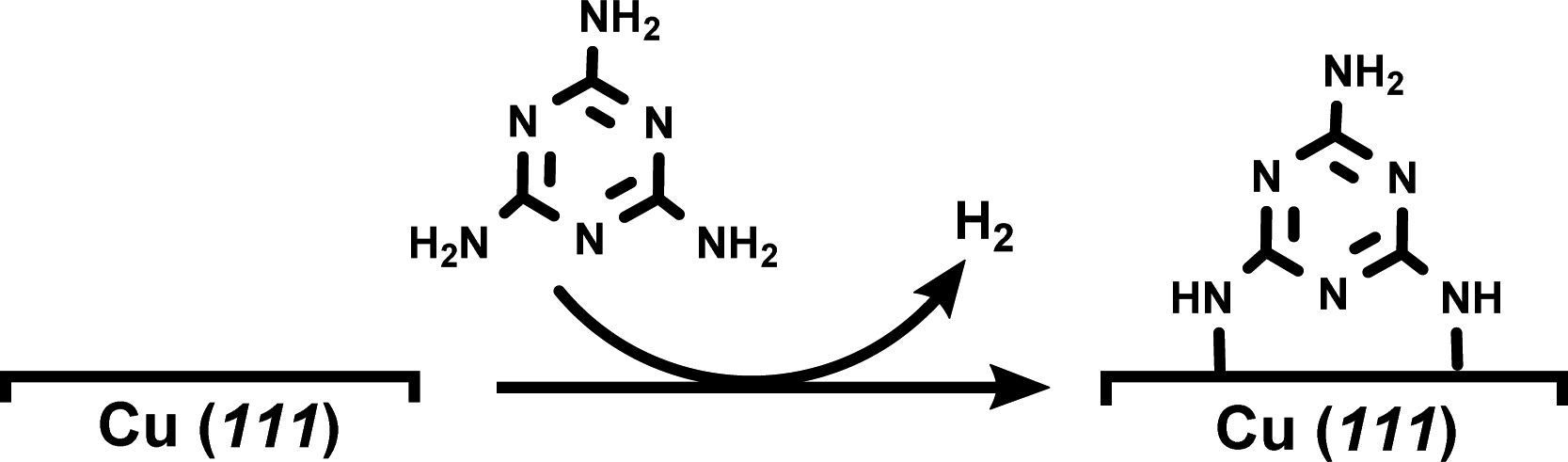
DFT-based simulations were performed with the CP2K/Quickstep package, using a hybrid Gaussian and plane-wave method.28 A double-quality DZVP Gaussian basis set29 was employed for all the atoms. The Goedecker–Teter–Hutter pseudopotentials30 together with a 400 Ry plane-wave cut-off were used to expand the densities obtained with the Perdew–Burke–Ernzerhof (PBE)31 exchange–correlation density functional, and vdW forces were taken into account with the Grimme D3 method.32 Only the gamma point was considered in a supercell approach. Periodic boundary conditions are applied in all directions of space.The copper (111) surface was constructed using a slab model having four Cu layers with a 22.011 Å × 25.418 Å surface unit cell (400 Cu atoms). A 20 Å vacuum region between the slabs was adopted in order to minimize unrealistic slab interactions. A monolayer (ML) of melamine molecules was then modeled on the copper surface by means of the dehydrogenation process shown in eqn (1) (for further details, see Fig. S5†):
 | (1) |
The water adsorption process was modeled by moving forward one water molecule on the melamine ML at different surface positions (model 1 and model 2) in order to find the most stable and representative configuration. Simulation of N 1s BEs was performed at two different levels of theory (HF and B3LYP) by considering a representative portion of the melamine decorated surface (see Fig. S6 and Table S3† for further details).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Carl Trygger Foundation for financial contribution. V. L. is grateful for the financial support provided by the Equal Opportunity Program of the Department of Physics and Astronomy at Uppsala University. Computational resources were provided by CINECA High Performance Computing Cluster award no. HP10CC5WSY 2020 under the ISCRA initiative.References
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Carbon nitrides: synthesis and characterization of a new class of functional materials, Phys. Chem. Chem. Phys., 2017, 19(24), 15613–15638 RSC.
- X. Wang, et al., A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8(1), 76–80 CrossRef CAS PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, Crystalline carbon nitride nanosheets for improved visible-light hydrogen evolution, J. Am. Chem. Soc., 2014, 136(5), 1730–1733 CrossRef CAS PubMed.
- K. Schwinghammer, et al., Triazine-based carbon nitrides for visible-light-driven hydrogen evolution, Angew. Chem., Int. Ed., 2013, 52(9), 2435–2439 CrossRef CAS PubMed.
- B. V Lotsch, et al., “Unmasking Melon by a Complementary Approach Employing Electron Diffraction , Solid-State NMR Spectroscopy , and Theoretical Calculations — Structural Characterization of a Carbon Nitride Polymer, Chem.–Eur. J., 2007, 13, 4969–4980 CrossRef PubMed.
- J. Wen, J. Xie, X. Chen and X. Li, A review on g-C3N4-based photocatalysts, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- C. Merschjann, et al., Complementing Graphenes: 1D Interplanar Charge Transport in Polymeric Graphitic Carbon Nitrides, Adv. Mater., 2015, 27, 7993–7999 CrossRef CAS PubMed.
- V. W. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V Lotsch, Low-Molecular-Weight Carbon Nitrides for Solar Hydrogen Evolution, J. Am. Chem. Soc., 2015, 137, 1064–1072 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Polymeric Graphitic Carbon Nitride as a Heterogeneous Organocatalyst: From Photochemistry to Multipurpose Catalysis to Sustainable Chemistry, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- V. W. Lau, et al., Rational design of carbon nitride photocatalysts by identification of cyanamide defects as catalytically relevant sites, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- V. W. Lau, et al., Urea-Modified Carbon Nitrides: Enhancing Photocatalytic Hydrogen Evolution by Rational Defect Engineering, Adv. Energy Mater., 2017, 1602251 CrossRef.
- J. Ehrmaier, T. N. V. Karsili, A. L. Sobolewski and W. Domcke, Mechanism of Photocatalytic Water Splitting with Graphitic Carbon Nitride: Photochemistry of the Heptazine-Water Complex, J. Phys. Chem. A, 2017, 121(25), 4754–4764 CrossRef CAS PubMed.
- J. Ehrmaier, M. J. Janicki, A. L. Sobolewski and W. Domcke, Mechanism of photocatalytic water splitting with triazine-based carbon nitrides: Insights from: Ab initio calculations for the triazine-water complex, Phys. Chem. Chem. Phys., 2018, 20(21), 14420–14430 RSC.
- W. Domcke, J. Ehrmaier and A. L. Sobolewski, Solar Energy Harvesting with Carbon Nitrides and N-Heterocyclic Frameworks: Do We Understand the Mechanism?, ChemPhotoChem, 2019, 3(1), 10–23 CrossRef CAS.
- Y. P. Lin, et al., Self-assembled melamine monolayer on Cu(111), J. Phys. Chem. C, 2013, 117(19), 9895–9902 CrossRef CAS.
- M. Lischka, et al., Competitive Metal Coordination of Hexaaminotriphenylene on Cu(111) by Intrinsic Copper Versus Extrinsic Nickel Adatoms, Chem.–Eur. J., 2019, 25(8), 1975–1983 CrossRef CAS PubMed.
- D. Woodruff, Adsorbate structure determination using photoelectron diffraction: Methods and applications, Surf. Sci. Rep., 2007, 62(1), 1–38 CrossRef CAS.
- F. Allegretti, M. Polcik and D. P. Woodruff, Quantitative determination of the local structure of thymine on Cu (1 1 0) using scanned-energy mode photoelectron diffraction, Surf. Sci., 2007, 601(17), 3611–3622 CrossRef CAS.
- D. C. Jackson, et al., Structure of cytosine on Cu(110): A scanned-energy mode photoelectron diffraction study, J. Phys. Chem. C, 2010, 114(36), 15454–15463 CrossRef CAS.
- A. Cossaro, M. Puppin, D. Cvetko, G. Kladnik, A. Verdini, M. Coreno, M. de Simone, L. Floreano and A. Morgante, Tailoring SAM-on-SAM Formation, J. Phys. Chem. Lett., 2011, 2, 3124–3129 CrossRef CAS.
- V. Lanzilotto, et al., Spectroscopic Fingerprints of Intermolecular H-Bonding Interactions in Carbon Nitride Model Compounds, Chem.–Eur. J., 2018, 24(53), 14198–14206 CrossRef CAS PubMed.
- S. Garcia-Gil, A. Arnau and A. Garcia-Lekue, Exploring large O 1s and N 1s core level shifts due to intermolecular hydrogen bond formation in organic molecules, Surf. Sci., 2013, 613, 102–107 CrossRef CAS.
- A. Schaefer, V. Lanzilotto, U. Cappel, P. Uvdal, A. Borg and A. Sandell, First layer water phases on anatase TiO2(101), Surf. Sci., 2018, 674, 25–31 CrossRef CAS.
- L. Floreano, et al., Periodic Arrays of Cu-Phthalocyanine Chains on Au(110), J. Phys. Chem. C, 2008, 112(29), 10794–10802 CrossRef CAS.
- G. Bavdek, et al., Pentacene nanorails on Au(110), Langmuir, 2008, 24(3), 767–772 CrossRef CAS PubMed.
- R. Costantini, et al., “ANCHOR-SUNDYN: a novel endstation for time resolved spectroscopy at the ALOISA beamline, J. Electron Spectrosc. Relat. Phenom., 2018, 229(July), 7–12 CrossRef CAS.
- K. Andersson, A. Nikitin, L. G. M. Pettersson, A. Nilsson and H. Ogasawara, Water dissociation on Ru(001): An activated process, Phys. Rev. Lett., 2004, 93(19), 1–4 CrossRef PubMed.
- T. D. Kühne, et al., CP2K: An electronic structure and molecular dynamics software package -Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152(19), 194103 CrossRef PubMed.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127(11), 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(3), 1703–1710 CrossRef CAS PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Tables S1–S3. See DOI: 10.1039/d0na01034k |
| This journal is © The Royal Society of Chemistry 2021 |