
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold plasmonic enhanced luminescence of silica encapsulated semiconductor hetero-nanoplatelets†
Emilio
Garcia
 ab,
Christophe
Arnold
a,
Jean-Pierre
Hermier
a and
Michele
D'Amico
*b
ab,
Christophe
Arnold
a,
Jean-Pierre
Hermier
a and
Michele
D'Amico
*b
aUniversité Paris-Saclay, UVSQ, CNRS, GEMaC, 78000, Versailles, France. E-mail: jean-pierre.hermier@uvsq.fr
bNexdot, 102 Avenue Gaston Roussel, 93230 Romainville, France. E-mail: michele.damico@nexdot.fr
First published on 18th June 2021
Abstract
Optical properties of nanocrystals have the potential to drive the next generation of optoelectronic devices. However, a number of technological limitations remain to be overcome. Nanocrystals' emission is strongly impacted by the chemical environment and high excitation power. In this paper, we present a preparation route to hybrid core–shell nanoplatelets encapsulated in a gold silica shield. The induced plasmon coupling offers a higher brightness at high power excitation. We also detail the highly increased fluorescence stability offered by the silica–gold-shell protection against photobleaching processes.
1. Introduction
Research dedicated to semiconductor nanoparticles has grown tremendously over the last 30 years. They exhibit a broader absorption spectrum and larger absorption cross-section, with higher brightness and resistance to photo-damage under powerful excitation, than conventional organic dyes.1 These properties are proving to be of huge importance for a wide range of applications in nanophotonics, quantum optics2 and more generally in opto-electronics technologies such as lasers,3,4 Q-LEDs,5,6 photodetection,7,8 and photovoltaics.9,10 As an example, spherical quantum dots are already integrated in TV-LED technology as backlight converters, where the sharpness and brightness of their emission allow unparalleled color gamut.11 The key to these unique properties relies on the quantum confinement effect with crystals smaller than the exciton Bohr radius. Thanks to well-developed colloidal synthesis, a fine tuning of the size and shape of nanocrystals allows a precise control of spectroscopic features.12,13 Now nanocrystals (NCs) are made from various semiconductor materials, with emission frequencies peaking from the ultraviolet to the near infrared region of the electromagnetic spectrum.14,15Synthesized for the first time thirteen years ago, two-dimensional nanoplatelets (NPLs) show quantum confinement along one direction only, which offers great control over their spectral properties.16,17 In the last few years, their synthesis became a routine and has been extended to core–shell18 and core–crown19 heterostructures. The latter enhance both the brightness and stability with a surrounding semiconductor of higher bandgap which passivates the surface and protects the crystals from oxidation.18,19
Despite such intrinsic advantages, drawbacks have to be considered. As NCs, NPLs are not chemically inert. Prolonged illumination and reaction with chemical environments are reported to degrade their composition and quench their fluorescence emission.20,21 Particularly, molecules adsorption onto the crystalline surface could give rise to etching22 and create charge carrier traps,23 which behave as fluorescence quenchers and induce strong emission instability. In addition, the diffusion of ions from the environment might induce a chemical modification of the NCs, such as the cation exchange process.24 These characteristics and potential limitations should be overcome by technological advancements in the development of nanoscopic hybrid-heterostructures.
The strategy selected here is to encapsulate hetero-nanoplatelets in the center of an amorphous silica bead, and the latter was surrounded by a chemically deposited gold nano-shell. Hybrid structures coupling fluorescence emission and metallic structures have been studied for decades with an increasing focus.25–28 A barrier of noble metal greatly improves the photochemical robustness29 and gold can act as a plasmon resonator which enhances the local electromagnetic fields. The plasmon coupling strongly modifies the local density of optical states30 and induces modification of the emission: this effect can result in the shortening of the lifetime of emitters (Purcell effect), the modification of the absorption spectrum and the enhancement of the fluorescence emission.31,32 In addition, water solubility of the embedded crystals makes them compatible for a large panel of medical applications.33
In this work, we report the synthesis and optical characterization of CdSe/CdS (core/shell) NPLs encapsulated in gold overcoated silica beads. The synthesis is a four step process first introduced by Brinson et al.29 and more recently transposed to silica–gold-dots by Botao Ji and co-workers.34 We efficiently adapted the protocol to create beads of very monodisperse size distribution, containing a single NPL emitter. The hybrid particles exhibit strong enhancement of both the fluorescence and the resistance to high excitation photobleaching.
2. Experimental section
2.1 Golden-silica NPL synthesis
Core/shell NPLs of CdSe/CdS were supplied by the company Nexdot.35 The cores of CdSe are 12 × 12 × 1.5 nm (1.5 nm thickness) and the final particle size reaches ∼30 × 30 × 20 nm (20 nm thickness) with a giant CdS shell. The formation of a silica shell around the NPLs is achieved by an inverse water-in-oil microemulsion micellar method.34 In particular, 1.0 g of the surfactant Triton X-100 and 0.75 g of 1-hexanol as a co-surfactant are mixed and dissolved in 5 mL of cyclohexane and stirred for 5 minutes with a magnetic bar. Then, a dispersion of almost 0.09 nmol of NPLs in hexane is injected (typical volume of 80 μL) and stirred for other 10 min before injecting 190 μL of water and then 30 μL of ammonia (29% in water). The solution remains clear, which ensures that the nanometric water-in-oil micellar system is correctly formed. To start the reaction of silica formation around the dots, 40 μL of TEOS is added. After five hours, 110 μL more TEOS is injected to have a final diameter of silica of 100 nm after 30 hours of total time of growth (∼30 nm silica thickness). The microemulsion is then broken with acetone addition and after centrifugation, the particles are washed by centrifugation and sonication in different solvents (50% n-butanol–50% hexane, 50% isopropanol–50% hexane, 50% ethanol–50% hexane and three times in pure ethanol, total volume 15 mL). Silica beads are dispersed in 15 mL of ethanol with a final concentration on the order of 4 nM. Then 8 mL of SiO2-NPL ethanol solution was centrifuged and dispersed in methanol (4 mL), washed 3 times with methanol, and finally redispersed in methanol (4 mL) at a concentration of ∼7 nM. The synthesis of PVIS was accordingly done as in ref. 34. The 4 mL SiO2-NPL methanol dispersion is mixed with 5 mL PVIS–methanol solution (1.5 mg, 5 mg m−2 silica). Then the mixture is heated up to 65 °C for 1.5 h. The reacting medium was cooled down to room temperature, washed 3 times with methanol and redispersed in methanol (2 mL).Small gold colloids (“seeds”, 2–3 nm diameter) were prepared, using THPC as the reducing agent. Typically, under rapid stirring, NaOH (450 μL, 0.2 M in water) and THPC (300 μL from a stock solution prepared by diluting 120 μL of a 80 wt% THPC aqueous solution in 10 mL of water) were successively added to water (13.7 mL), and the mixture was stirred for 5 min. HAuCl4 (600 μL, 25 mM in water) was added dropwise and the solution stirred for 15 min. The gold seed solution obtained was then aged at 4 °C for at least 2 weeks before use.
The NPL/SiO2/PVIS particles (150 μL from the previous 2 mL methanol dispersion, ∼14 nM) were dispersed in the aged gold colloids (3 mL) and stirred for 2 h at room temperature. All of the free gold seeds remaining in the solution were removed by washing 3 times in water. Au-seeds-SiO2-NPLs were finally redispersed in water (500 μL) and directly used after preparation to grow the gold nanoshell.
A gold plating solution was prepared by adding HAuCl4 (750 μL, 25 mM in water) to K2CO3 (50 mL, 1.8 mM in water). After 30 min of stirring (the light yellow solution turned colorless), the plating solution was stored at 4 °C for a minimum of 24 h before use. NPL/SiO2/PVIS (30 μL from the 0.5 mL water dispersion, ∼4 nM) was added to the gold plating solution (2 mL), then CH2O (10 μL, 37 wt% in water) was rapidly added and the dispersion was stirred for 45 min. The golden-NPL particles were centrifuged, washed 3 times with water and redispersed in water (1 mL) with a final concentration of ∼0.13 nM.
2.2 Characterization
All optical measurements were carried out at room temperature. Absorption/extinction measurements were performed on a UV-Vis Jasco V-730 spectrometer. Photoluminescence (PL) spectra were collected with a spectrometer Edinburgh Instruments FCS900 mounting both a xenon lamp and a pulsed diode laser – VIS/NIR 375 nm, from the same company. The latter was used to carry out lifetime measurements on a population of nanoparticles in solution. For single molecule measurements, NPLs were diluted in hexane to a concentration of 0.02 nmol L−1 and deposited by drop-casting on a glass substrate. Single NCs were studied on a commercial confocal microscope (Microtime 200, Picoquant) associated with a pulsed laser diode emitting at 405 nm (LDH-D-C-405, Picoquant). The fluorescence was collected through an oil-objective (NA 1.4, 100×, Olympus) and sent into a Hanbury-Brown and Twiss acquisition setup (SPAD PDM; time resolution: 50 ps). The signal was recorded using a HydraHarp 400 module (Picoquant) in a time-tagged, time-resolved mode. Transmission electron microscopy (TEM) images were acquired on a JEOL 2010 field electron gun microscope operated at 200 keV. Scanning electron microscopy (SEM) images were taken using an FEI Magellan.3. Results and discussion
Fig. 1 illustrates the main stages of the sample synthesis. The sketch in the upper part and the TEM images show that we successfully, as far as we know for the first time, encapsulated a single NPL inside a silica spheroid. These pearls were then efficiently covered by a nano-layer of gold.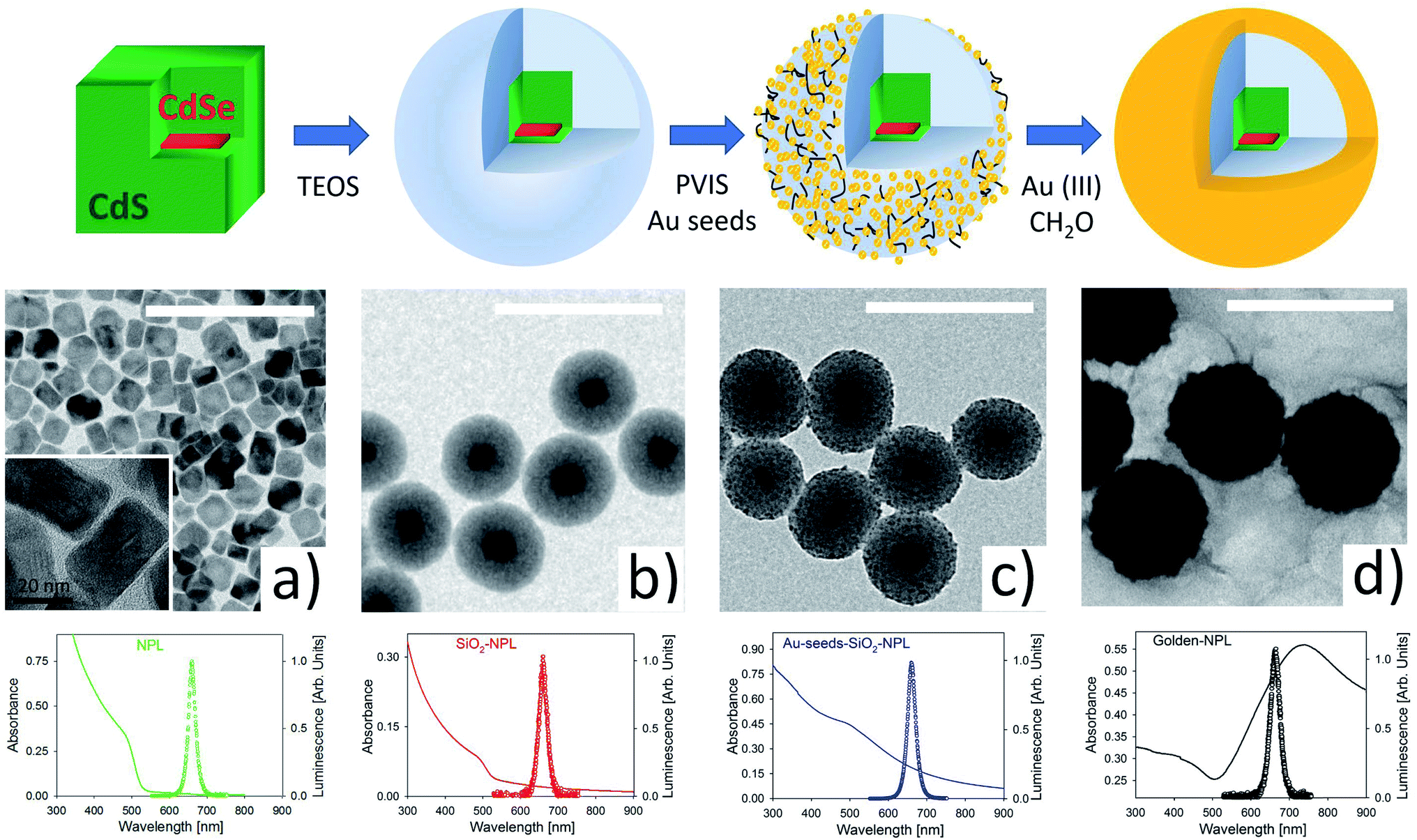 | ||
| Fig. 1 Illustration of the four steps of preparation of golden-NPL emitters: bare-NPLs, SiO2-NPLs, Au-seeds-SiO2-NPLs and golden-NPLs. First row: pictorial view of the different stages. From left to right, core/shell (red/green) NPLs, SiO2-NPLs, polymer attached gold seeds on SiO2-NPLs, a continuous gold shell (golden-NPLs). Second row: from (a) to (d), TEM images of the four stages illustrated respectively in the first row. The white scale bars represent a length of 200 nm. Third row: the absorbance (line) and fluorescence (circles, excitation at λex = 375 nm) spectra for the four steps: NPLs (green), SiO2-NPLs (red), Au-seeds-SiO2-NPLs (blue) and golden-NPLs (black). | ||
TEM images provided in Fig. S1 in the ESI† show high monodispersity of the SiO2 coating (diameter 94 ± 6 nm). The pearls essentially include single NPLs (>90%) and in a minority of case, are empty (∼7%).
Gold shell growth is a two-stage method.29,34 First, the surface of the silica is functionalized with poly(1-vinylimidazole-co-vinyltrimethoxysilane) (PVIS). This polymer is used to improve the adsorption of small gold seeds (2–3 nm) onto the SiO2 surface.34 Then, gold(III) ions are added to the functionalized pearls and reduced by formaldehyde to gold(0). This last step makes the gold seeds grow and merge with each other. This ultimately forms a smooth and continuous shell around SiO2-NPLs. The final thickness of the gold shell is around 20 nm. SEM images of this protocol can be seen in a former communication33 where the same method is applied in quantum dot encapsulation.
Fluorescence spectra (with empty circles) for excitation at 375 nm and absorbance spectra (in a continuous line) are shown in the third row of Fig. 1. The spectra from the four stages provided in Fig. 1 show a fluorescence peak at 658 nm (1.88 eV) with a narrow FWHM of ∼28 nm (∼80 meV). The formation of a continuous gold shell is confirmed by the intense extinction peak centered at 730 nm in the absorbance spectrum (see the black line spectrum in Fig. 1), which comes from the plasmonic resonance of free electrons in the gold layer. The overall spectrum [300–900 nm] is modified with respect to the one measured for a bare NPL. It is very similar to the spectrum modelled in ref. 34. The golden-NPL sample shows a moderate tendency to aggregate due to gold-seeds bridging in the previous synthesis step (see the SEM images in Fig. S2†).
Fig. 2 presents the fluorescence time decays of NPLs in colloidal suspensions associated with the four stages of golden NPL preparation: bare-NPLs (in hexane), SiO2-NPLs (in ethanol), gold-seeds-silica-NPLs and golden-NPLs (both in water). Note that gold-seeds-silica-NPL formation is the initial phase of gold shell growth, as illustrated in Fig. 1c.
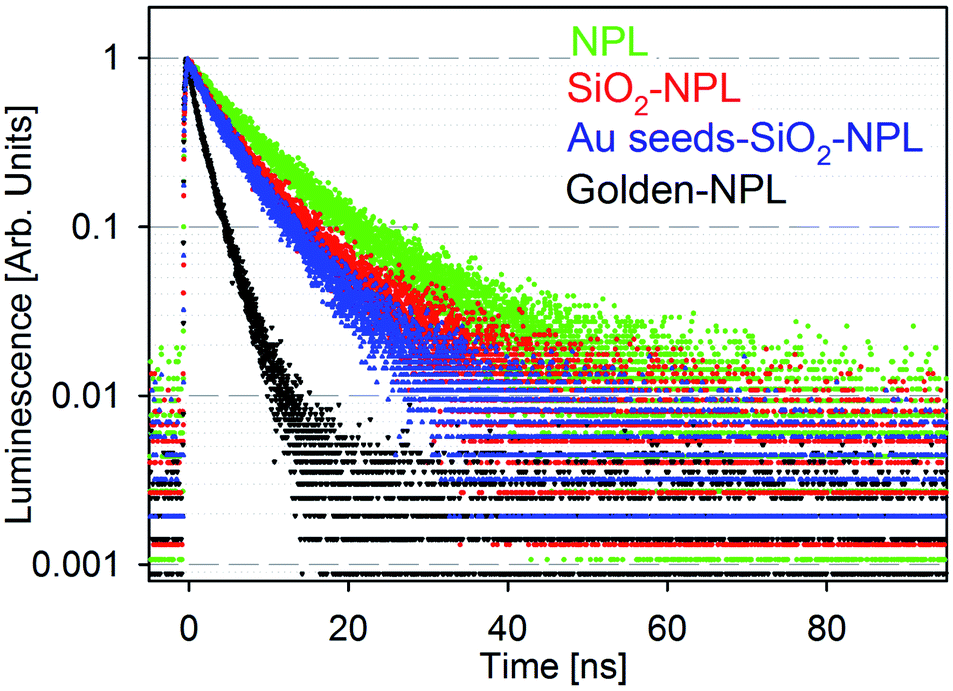 | ||
| Fig. 2 Normalized luminescence decay of solutions of particles excited at 375 nm: bare-NPLs (green), SiO2-NPLs (red), gold-seeds-silica-NPLs (blue) and golden-NPLs (black). | ||
Encapsulation of NCs in silica and other solid matrices is not trivial. The fluorescence of this colloidal semiconductor strongly depends on its surface chemistry. The surface ligand replacement and oxidation due to the water-based environment classically cause a quenching of the fluorescence.23 Indeed, defects on the surface of NCs generally lead to the appearance of electronic traps. The traps introduced by encapsulation typically add non-radiative channels in the de-excitation pathway which also decrease the lifetime. When compared to bare-NPLs (Fig. 2, green points), an increase of the PL decay rate is indeed observed for SiO2-NPLs (Fig. 2, red points). However, the decay features are similar which confirms that ligand replacement with the silica linkage does not quench the emission. This result is probably allowed by the high thickness of CdS shells which efficiently isolate core electronic processes from the surface.
It should be noted that the presence of isolated gold particles (seeds) on silica does not significantly modify the decay either (Fig. 2, blue points). It was also verified that mixing silica-NPLs with gold salts without activating the formation of the gold shell does not affect the decay shape (data not shown). This was carried out by removing the formaldehyde reducing agent. Conversely, it is worth noting that golden-NPLs' decay (Fig. 2, black points) is faster than that of the other samples. Since the NPLs are coated with silica when the continuous gold layer is synthesized, non-radiative recombination channels cannot be generated through the modification of the NPL surface at this step. The increase of PL decay rate (Fig. 2) then comes only from a modification of the electromagnetic environment of the NPLs in the last step of the synthesis. As already observed for the golden-dots by Ji et al.,34 we attribute the decrease of the mean lifetime to the process first reported by E. M. Purcell.40 The gold layer increases the local density of electromagnetic states at the NPL position, which leads to a faster decay. It is important to note that this enhancement is not purely radiative due to optical losses generated by the coupling between the gold nanoshell and the NPLs.
The fluorescence spectra of a single golden-NPL emitter as a function of laser power are presented in Fig. 3a. The excitation power range studied well spans five orders of magnitude from 10−1 to 104 nW and is achieved with a laser diode operating at 405 nm. Note that for a constant laser power, the energy transmitted to an isolated dried particle under a focused beam is much greater than that delivered to each of the emitters dispersed in a spectrophotometer cuvette.
 | ||
| Fig. 3 Panel (a): emission spectra of a single golden-NPL emitter as a function of laser power. The excitation values are provided in the right side of the panel. The breakdown power is indicated in the associated spectrum in red. Panel (b): corresponding emission decays at different laser powers indicated in panel (a). The decay associated with the breakdown power is marked in red. | ||
In low power mode (up to an excitation power of 10 nW), the fluorescence band remains unchanged. For higher laser power, the emission features a clear blue-shifted shoulder whose contribution increases with the increase of power. This higher energy transition tends to shape a second emission band (hereafter named orange band). We provide fits of the spectra with a fixed dual Gaussian distribution in Fig. S3.† The respective band peak values are identified at 1.88 eV (660 nm) and 1.99 eV (620 nm) for all powers used and the evolution of their weight is shown. The global blue-shift of the emission peaks with increasing power results from the variation in the relative weight of the two transitions in the spectrum. The higher the excitation power, the higher the weight of the orange band in the total fluorescence signal, reaching a contribution of almost 40% at the highest power (see Fig. S3†).
High excitation power generates a large number of 405 nm photons, which generate a large number of excitons in the excited state which relax towards the conduction band minimum by phonon coupling. It creates a “bottleneck effect” which statistically favors direct recombinations from higher energy levels (orange band) in addition to non-radiative relaxation toward standard excitons. Equivalent measurements on bare-NPL and SiO2-NPL samples are also performed and shown in Fig. S4 and S5 in the ESI.† The emission spectrum features the same patterns, so this effect must result from an internal NPL optical process. One should note that, since the emission of biexcitons in NPLs is blue-shifted from less than 20 nm with respect to the excitonic emission,36–38 the orange band cannot result from biexcitonic recombinations. A schematic illustration of the fluorescence emission process is provided in Fig. S6 in the ESI.†
This optical response is reversible. After high excitation and the return to 1 nW power, the orange band disappears to leave only the red fluorescence band. However, when the laser power exceeds 1.5 × 104 nW, the time trace starts to oscillate and rapidly falls to very low photon count rates. The associated fluorescence features a strongly blue-shifted band, peaking at 575 nm (2.15 eV). This evolution is illustrated in Fig. 3a (red line). This effect is no longer reversible and the laser power at which it appears is hereafter named breakdown power.
Fig. 3b shows the photoluminescence decay curves for each laser power presented in panel (a). The results match with the previous description: at the low power regime, the decay remains unchanged until reaching the power of 10 nW. For greater excitation energy, the higher the power, the faster the decay. This behavior is probably due to very fast recombinations of the orange band and also possible parallel Auger processes.39 When reaching the irreversible state, the decay features a very fast picosecond decay time, comparable with the laser excitation temporal pulse width. The same behavior is also observable in the bare-NPL and SiO2-NPL samples at their own breakdown levels. The corresponding measurements are provided in Fig. S4 and S5 in the ESI.†
Mean lifetime values as a function of the laser power are provided in Fig. 4a for bare-NPLs (green), SiO2-NPLs (red) and golden-NPLs (black). Fits of the fluorescence decays were performed with a multiple exponential (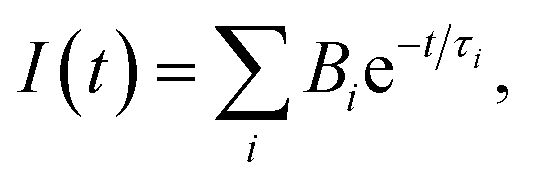 up to i = 4) model. The mean lifetime values shown are the amplitude weighted
up to i = 4) model. The mean lifetime values shown are the amplitude weighted 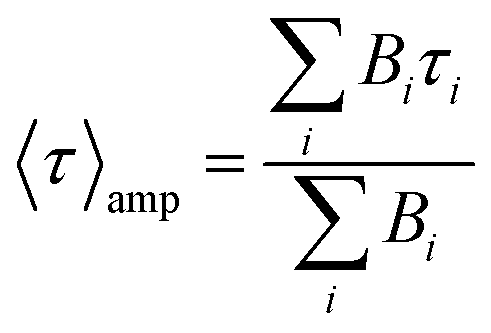 since at high laser power, the presence of really fast τi components on the order of hundreds of picoseconds gives the integral weighted mean lifetime
since at high laser power, the presence of really fast τi components on the order of hundreds of picoseconds gives the integral weighted mean lifetime 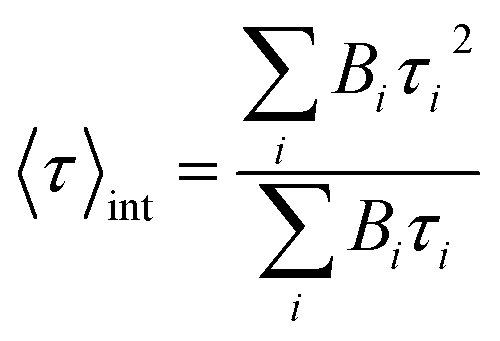 neglecting the faster part. We specify that the data presented in this panel refer only to measurements made at excitation powers lower than the respective breakdown values, where the optical response is reversible (see the discussion above). The fitted decays were measured with both increasing and decreasing excitation power over the reversible domain with no significant difference. The fluorescence intensities of SiO2-NPLs (red) and golden-NPLs (black) are too low to be measured at an excitation power lower than ∼10−1 nW.
neglecting the faster part. We specify that the data presented in this panel refer only to measurements made at excitation powers lower than the respective breakdown values, where the optical response is reversible (see the discussion above). The fitted decays were measured with both increasing and decreasing excitation power over the reversible domain with no significant difference. The fluorescence intensities of SiO2-NPLs (red) and golden-NPLs (black) are too low to be measured at an excitation power lower than ∼10−1 nW.
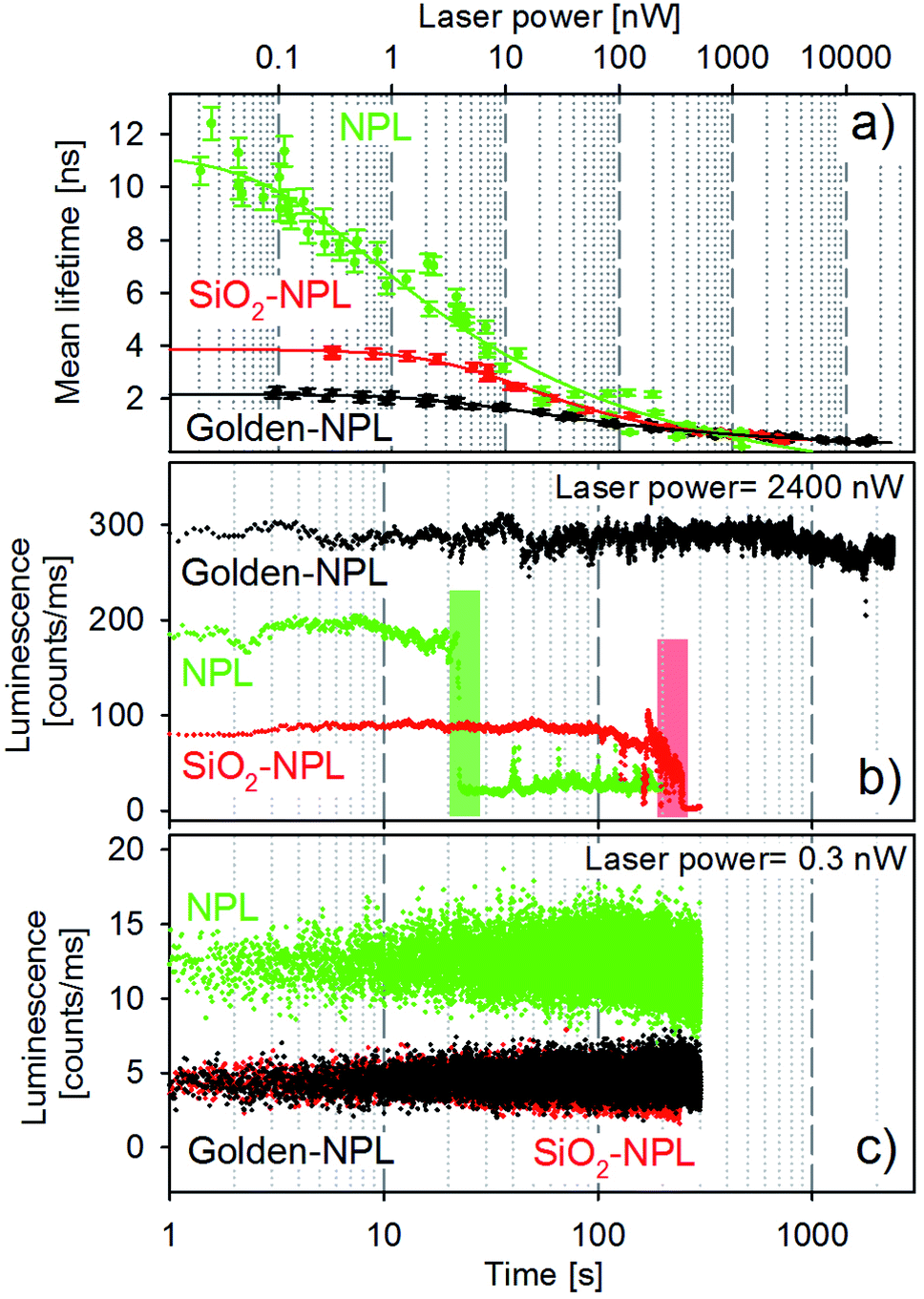 | ||
| Fig. 4 Optical response of single bare-NPLs (green), SiO2-NPLs (red), and golden-NPLs (black) to 405 nm excitation. Panel (a): mean lifetime values as a function of the laser power. The continuous lines are only a guide to the eye. Panel (b): time traces of the photons collected in a typical acquisition at a high laser power (2400 nW). The colored areas highlight the abrupt decrease of the photon count rates. Panel (c): time traces of the photons collected in a typical acquisition at a low laser power (0.3 nW). | ||
The synthesis of the SiO2 beads induces the creation of surface traps and thus competitive non-radiative recombination channels. Non-radiative recombinations also reduce the mean lifetime, which can be observed in Fig. 4a by comparing the values of bare-NPLs (green points) and SiO2-NPLs (red points). Furthermore, the coupling between the plasmon modes of the gold shell and the excitons also lowers the mean lifetime for the golden-NPLs (black dots) compared to that of SiO2-NPLs, according to the well-known Purcell effect.
All three structures feature a strong decrease in their mean lifetimes at excitation powers on the order of above 1 nW. This is attributed to the outcome of both very fast Auger effects and the direct recombination from the orange band. Ultimately, excitation powers higher than the order of 102 nW come with overweighted fast lifetime components (close to the time resolution) in the overall radiative process. Therefore the mean lifetime of all samples dramatically falls (Fig. 4a).
Selected representative time traces of single particle bare-NPLs, SiO2-NPLs and golden-NPLs are reported in Fig. 4 panels (b) and (c) at two representative powers (2400 and 0.3 nW, respectively). We provide in Table 1 the characteristic quantities of these traces: the calculation of the photon count rate per nanowatt of excitation at excitation powers of 0.3 and 2400 nW, the loss factor between these ratios and the temporal stability of the traces at 2400 nW.
| Counts per ms per nW @ | Loss factor | Time stability @ 2400 nW (s) | ||
|---|---|---|---|---|
| 0.3 nW | 2400 nW | |||
| Bare-NPLs | ∼43 | ∼0.08 | ∼1/500 | 20 |
| SiO2-NPLs | ∼13 | ∼0.03 | ∼1/400 | 200 |
| Golden-NPLs | ∼13 | ∼0.12 | ∼1/100 | >2000 |
With the increase of laser power, the photon count rate per nanowatt of excitation decreases for all three samples, which is consistent with the appearance of Auger processes. Moreover, at low excitation power, the emission intensity of the bare-NPLs is higher due to the already described non-radiative channels induced by SiO2 addition. One can also observe that the fluorescence intensity is the same for the SiO2-NPLs and golden NPLs. On the one hand, the presence of the golden shell increases the number of blue photons which are able to excite the inner NPLs (the gold nanoshell enhances the incident intensity for the NPLs as reported by Ji et al.34). On the other hand, the non-radiative contribution of the Purcell effect decreases the quantum efficiency. The two effects compensate each other with a quite perfect balance. Conversely, for higher excitation power, their emission surpasses that of the bare-NPLs. Two effects explain this relative increase. First, the number of excitation–emission cycles per second increases due to PL decay rate enhancement which leads to a higher radiative conversion of incident photon flux (the increase in absorption does not play any role in this case since the operation regime is far above the saturation level). Second, at high power, the Auger recombination32 increases in all three structures, but the acceleration of the radiative emission by the Purcell effect limits its effect for the golden-NPLs. Thus a better radiative efficiency is reached at high excitation power.
In addition, at the excitation power on the order of 103 nW, the traces exhibit significantly different emission stability times before the final photobleaching. Compared to bare-NPLs, a pure silica shell extends the duration by one order of magnitude, while the gold–silica shell does so by at least two orders of magnitude (see Table 1, last column).
We can thus infer that shells offer effective protection against interaction with oxidizing molecules. In particular, the noble metal (Au) acts as a chemically inert barrier. The main drawback of this method is the introduction of non-radiative traps during encapsulation in silica, which makes it less favorable at low power at which the aging speed and the Auger effects are less significant. Future developments in the field of surface treatment will probably allow a more efficient encapsulation by limiting the formation of these traps. It would make this formulation superior in the full range of excitation required for optoelectronic devices.
4. Conclusions
In this work we demonstrated the encapsulation of red emitting core–shell NPLs in silica–gold heterostructures without any significant quenching effect on the emission. The radiative rate of silica–gold-NPLs is enhanced by the coupling between fluorescence and collective oscillations of free electrons in the metallic gold layer. Compared to bare NPLs, this plasmonic coupling offers higher brightness for high excitation energy. Moreover, the shell protects the nano-emitters as it increases their photo-stability over time. An exceptional two orders of magnitude increase can be achieved and is particularly significant at high excitation powers. These performances pave the way for future industrial applications of NPLs as light emitters in optoelectronic technologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Benoit Dubertret for access to the ESPCI-LPEM laboratory, Xianghzhen Xu for performing TEM analysis and Botao Ji for the early training in the synthesis procedure of silica-NPLs.Notes and references
- U. Resch-Genger, M. Grabolle, S. Cavaliere, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- O. Bitton, S. N. Gupta and G. Haran, Nanophotonics, 2019, 8, 559–575 CAS.
- C. She, I. Fedin, D. S. Dolzhnikov, P. D. Dahlberg, G. S. Engel, R. D. Schaller and D. V. Talapin, ACS Nano, 2015, 9, 9475–9485 CrossRef CAS PubMed.
- P. Guo, X. Yang, Q. Ye, J. Zhang, H. Wang, H. Yu, W. Zhao, C. Liu, H. Yang and H. Wang, Adv. Energy Mater., 2019, 9, 1901341 CrossRef.
- V. Colvin, M. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- M. K. Choi, J. Yang, T. Hyeon and D.-H. Kim, npj Flexible Electron., 2018, 2, 10 CrossRef.
- E. Lhuillier, J.-F. Dayen, D. O. Thomas, A. Robin, B. Doudin and B. Dubertret, Nano Lett., 2015, 15, 1736–1742 CrossRef CAS PubMed.
- C. Livache, B. Martinez, N. Goubet, C. Greboval, J. Qu, A. Chu, S. Royer, S. Ithurria, M. Silly, B. Dubertret and E. Lhuillier, Nat. Commun., 2019, 10, 2125 CrossRef PubMed.
- C. Jackson Stolle, T. B. Harvey and B. A. Korgel, Curr. Opin. Chem. Eng., 2013, 2, 160–167 CrossRef.
- M. Que, L. Zhu, Y. Guo, W. X. Que and S. Yun, Toward, J. Mater. Chem. C, 2020, 8, 5321–5334 RSC.
- C. Demers, Wide Color Gamut Coverage of TVs, 2016, www.rtings.com/tv/tests/picture-quality/wide-color-gamut-rec-709-dci-p3-rec-2020, accessed date: 02 02 2021 Search PubMed.
- R. Rossetti, J. L. Ellison, J. M. Gibson and L. E. Brus, J. Chem. Phys., 1984, 80, 4464–4469 CrossRef CAS.
- A. L. Efros and M. Rosen, Annu. Rev. Mater. Sci., 2000, 30, 475–521 CrossRef CAS.
- A. J. Smyder and T. D. Krauss, Mater. Today, 2011, 14, 382–387 CrossRef.
- C. L. J. Paul, J. P. Amalorpavam and C. W. Lee, J. Nanophotonics, 2015, 9, 093069 CrossRef.
- S. Ithurria and B. Dubertret, J. Am. Chem. Soc., 2008, 130, 16504–16505 CrossRef CAS PubMed.
- S. Ithurria, M. D. Tessier, B. Mahler, R. P. S. M. Lobo, B. Dubertret and A. L. Efros, Nat. Mater., 2011, 10, 936–941 CrossRef CAS PubMed.
- B. Mahler, B. Nadal, C. Bouet, G. Patriarche and B. Dubertret, J. Am. Chem. Soc., 2012, 134, 18591–18598 CrossRef CAS PubMed.
- S. Pedetti, S. Ithurria, H. Heuclin, G. Patriarche and B. Dubertret, J. Am. Chem. Soc., 2014, 136, 16430–16438 CrossRef CAS PubMed.
- W. G. J. H. M. Van Sark, P. L. T. M. Frederix, D. J. Van den Heuvel, H. C. Gerritsen, A. A. Bol, J. N. J. Van Lingen, C. de Mello Donega and A. Meijerink, J. Phys. Chem. B, 2001, 105, 8281–8284 CrossRef CAS.
- S. Chen, W. Cao, T. Liu, S.-W. Tsang, Y. Yang, X. Yan and L. Qian, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- S. J. Lim, W. Kim, S. Jung, J. Seo and S. K. Shin, Chem. Mater., 2011, 23, 5029–5036 CrossRef CAS.
- G. Kalyuzhny and R. W. Murray, J. Phys. Chem. B, 2005, 109, 7012–7021 CrossRef CAS PubMed.
- D. H. Son, S. Hughes, Y. Yin and A. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- K. H. Drexhage, H. Kuhn and F. P. Schäfer, Ber. Bunsenges. Phys. Chem., 1968, 72, 329 Search PubMed.
- J. Gersten and A. Nitzan, J. Chem. Phys., 1981, 75(3), 1139–1152 CrossRef CAS.
- P. Törmä and W. L. Barnes, Rep. Prog. Phys., 2015, 78, 013901 CrossRef PubMed.
- H. Yu, Y. Peng, Y. Yang and Z.-Y. Li, npj Comput. Mater., 2019, 5, 1–14 CrossRef.
- B. E. Brinson, J. B. Lassiter, C. S. Levin, R. Bardhan, N. Mirin and N. J. Halas, Langmuir, 2008, 24, 14166–14171 CrossRef CAS PubMed.
- M. Pelton, Nat. Photonics, 2015, 9, 427–435 CrossRef CAS.
- K. T. Shimizu, W. K. Woo, B. R. Fisher, H. J. Eisler and M. G. Bawendi, Phys. Rev. Lett., 2002, 89, 117401 CrossRef CAS PubMed.
- D. Canneson, I. Mallek-Zouari, S. Buil, X. Quélin, C. Javaux, B. Mahler, B. Dubertret and J.-P. Hermier, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245423 CrossRef.
- A. L. Volsi, C. Fiorica, M. D'Amico, C. Scialabba, F. S. Palumbo, G. Giammona and M. Licciardi, Eur. Polym. J., 2018, 105, 38–47 CrossRef.
- B. Ji, E. Giovanelli, B. Habert, P. Spinicelli, M. Nasilowski, X. Xu, N. Lequeux, J.-P. Hugonin, F. Marquier and J.-J. Greffet, Nat. Nanotechnol., 2015, 10, 170–175 CrossRef CAS PubMed.
- Nexdot Company, https://nexdot.fr/, available, 02 02 2021 Search PubMed.
- M. Olutas, B. Guzelturk, Y. Kelestemur, A. Yeltik, S. Delikanli and H. V. Demir, ACS Nano, 2015, 9, 5041–5050 CrossRef CAS PubMed.
- B. Guzelturk, Y. Kelestemur, M. Olutas, S. Delikanli and H. V. Demir, ACS Nano, 2014, 8, 6599–6605 CrossRef CAS PubMed.
- J. Q. Grim, S. Christodoulou, F. Di Stasio, R. Krahne, R. Cingolani, L. Manna and I. Moreels, Nat. Nanotechnol., 2014, 9, 891 CrossRef CAS PubMed.
- J. P. Philbin, A. Brumberg, B. T. Diroll, W. Cho, D. V. Talapin, R. D. Schaller and E. Rabani, J. Chem. Phys., 2020, 153(5), 054104 CrossRef CAS PubMed.
- E. M. Purcell, H. C. Torrey and R. V. Pound, Phys. Rev., 1946, 69, 37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00273b |
| This journal is © The Royal Society of Chemistry 2021 |