
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast high-capacity capture and release of uranium by a light-switchable nanotextured surface†
Ella
Borberg
a,
Reut
Meir
ab,
Larisa
Burstein
c,
Vadim
Krivitsky
a and
Fernando
Patolsky
 *ad
*ad
aSchool of Chemistry, Faculty of Exact Sciences, Tel Aviv University, Tel Aviv, 69978, Israel. E-mail: fernando@post.tau.ac.il
bDepartment of Analytical Chemistry, Nuclear Research Centre Negev, Beer-Sheva, 84190, Israel
cThe Wolfson Applied Materials Research Centre, Tel-Aviv University, Tel-Aviv 69978, Israel
dDepartment of Materials Science and Engineering, The Iby and Aladar Fleischman Faculty of Engineering, Tel Aviv University, Tel Aviv 69978, Israel
First published on 17th May 2021
Abstract
Nuclear power is growing in demand as a promising sustainable energy source, its most prevalent source being uranium salts. The resulting processing and transportation of uranium raise concerns regarding the environmental impact and risks for human health. Close proximity to uranium mines puts populations at higher risk for exposure due to elevated uranium concentrations. As the main form of uranium in aqueous solutions, uranyl (UO22+) has been the focus of many methods of uranium sieving; most fall short by being time-consuming or lacking a retrieval mechanism for the captured uranium. Here, we demonstrate the ultrafast and selective uranyl-capturing properties of aptamer-modified branched silicon nanopillar (BSiNP) arrays. Our nanostructured surfaces demonstrate an ultrahigh surface area modified with a uranyl-specific DNA aptamer, allowing for high uranyl-capturing capacity, reaching up to 550 mg g−1. Uranyl capture is followed by the activation of a covalently bonded photoacid, causing a light-triggerable, ultrafast release. This capture-and-release cycle results in the collection of over 70% of the uranium found in the original samples within less than one hour.
Alongside the rise of nuclear power as a sustainable energy source, uranium handling has increased, bringing forth greater environmental and human health risks linked to the higher uranium concentrations in soils.1,2
Various methods aimed at lowering uranium concentrations in aqueous solutions include chelating resins,3–6 porous materials,7–11 reductive precipitation,12–14 and adsorbent materials.15–19 Chelating methods, while energy efficient, highly selective, and achieving high separation factors, result in third-phase formation, secondary waste generation, and solvent loss. Adsorbent materials and reductive precipitation methods are easily operated and widespread, both displaying high capacities. However, these surface-based processes are sensitive to contamination and oxidative inhibition.20 Porous materials can remove uranium ions with high efficiency, but limitations such as process complexity, low selectivity, clogging, and low capacity cause these materials to be less predominant in the field. Mainly, most techniques are time consuming, with days-long procedures. Additionally, though the capturing of uranium species has been greatly researched, their post-capture release is less established and can be difficult, expensive, or damaging to the capturing platform.3,21
DNA aptamers are well known as a special class of polynucleic acids that could be easily manufactured and bind specifically, with high affinity, to a given target molecule. DNA aptamers have been implemented in various bioanalytical applications, such as the specific detection of proteins, metal ions, and small molecules, as well as target-specific delivery. For instance, a uranyl-binding aptamer was developed, referred to as HS-DNA1,22 with high uranyl affinity and good extraction capabilities. HS-DNA1 was derived from a DNAzyme shown to be extremely specific against other metals.23–27 Recently, a carboxyl-terminated HS-DNA1 derivative was used by our group for highly specific and extremely sensitive sensing of uranyl in urine, a highly complex biosample.28
Here, we implemented the use of this carboxyl-terminated derivative of HS-DNA1 as an on-surface capturing agent for uranyl on nanostructured silicon nanopillar (SiNP) arrays. Cavities created by the confined interpillar space, in which uranyl ions experience limited diffusion, delay their release while repeatedly adsorbing and desorbing to and from the surface.29,30 This occurrence inside the interpillar aqueous space leads to extremely prolonged effective dissociation rates; the complete desorption process could take weeks.29
SiNPs, as controlled nanostructures, are fabricated in a bottom-up31,32 or top-down33–35 approach, both resulting in an array of NPs with extremely high surface areas. Presenting many distinctive qualities such as tunable porosity and pore size, well-known surface chemistry, high loading capacity, and biocompatibility, SiNPs have been implemented on many fronts, mostly in the fields of optics36–38 and theranostics.39–44 SiNPs could easily be integrated with other on-chip technologies and have shown great promise.
Previous works from our group29,45 presented the potential of SiNP arrays as a platform for on-chip, light-controlled, reusable, selective and quantitative separation, desalting, and preconcentration in the direct analysis of complex biosamples. On-chip selective separation of analytes from raw biosamples was first performed using antibody-photoacid-modified SiNPs with ultralarge binding surface area and enormously high binding affinity, followed by light-controlled rapid release of the tightly bound target molecules in a controlled liquid medium.
Triggerable fast release of the target is achieved by activating a covalently bonded photoacid-molecular monolayer, causing a drastic pH change near the SiNP surface. Photoacids are aromatic organic molecules that in their first excited electronic state exhibit acidity greater by many orders of magnitude than their weak acid properties in their ground electronic state. These materials have been widely studied in several applications,46–50 including the light-triggered pH drop caused by photoactivation of photoacid molecules applied in silicon nanowire-based field-effect transistor (SiNW-FET) devices for the on-surface modulation of protein affinity to an antibody.51 The resulting controlled on-surface pH drop immediately transforms the highly receptive binding surface into a highly reflective antibinding surface, due to the dissociation of antibody–antigen pairs. This light-triggered step transforms the SiNPs' capturing surface into a fast-releasing unit, thus exploiting the best advantages from these two worlds.
Importantly, photoactivation of the photoacid creates a pH change that is triggerable, reversible, and does not involve buffer-handling limitations. In addition, the modulation of the photoacid molecules' surface density, along with the applied light intensity, achieves a controlled surface pH and the subsequent rapid seconds-long release of the tightly captured species from the inter-SiNP cavity into the bulk solution.
In this work, we demonstrate the fast and selective uranyl-capturing properties of branched-SiNP (BSiNP) arrays modified with a uranyl-specific aptamer, implementing an ultrahigh surface area alongside triggerable ultrafast release into a selective medium.
Results
BSiNP array fabrication and chemical modification
A monolayer of polystyrene beads52 was used as an etching mask for a metal-assisted wet-etching step with HF/H2O2 mixture as etchant and oxidant, respectively,53 and a film of metallic silver as a catalyst, forming vertical SiNP arrays of 3–20 μm SiNP height (see Materials and methods in the ESI† for details).As previously shown in atomic force microscopy and transmission electron microscopy measurements,45 SiNPs of surface areas up to ∼400 m2 g−1 and pore sizes up to 15 nm could be fabricated with mild tuning of this fabrication process. This correlates to an increase of geometrical surface area from a planar area of 1 cm2 to 1200 cm2 after the etching of a SiNP array comprising SiNPs of 5 μm height, 250 nm diameter, 250 nm inter-NP distance, and a roughness factor of 16. This represents a dramatic increase of more than a 1200-fold in active surface area compared to a planar device of identical geometrical area.
To further increase the surface area, silicon nanobranches (SiNBs) were fabricated on the surface of SiNPs to form branched SiNP (BSiNP) arrays, as shown schematically in Fig. 1a. Electroless deposition of gold was achieved by soaking SiNP arrays in AuCl4/HF solution, followed by chemical vapor deposition (CVD) using gold to catalyze the growth of SiNWs via the vapor–liquid–solid (VLS) mechanism.54 The SiNB growth process used SiH4 and B2H6 (100 ppm in argon balance gas) as reactants, in which boron served as a p-type dopant with a boron/silicon ratio of 1/4000. The rate of SiNW growth was ∼1 μm min−1. Assuming a SiNP of 5 μm height, 250 nm diameter, SiNWs with 20 nm diameter, and surface occupancy of 1/3, the surface area is increased by a factor of 41.
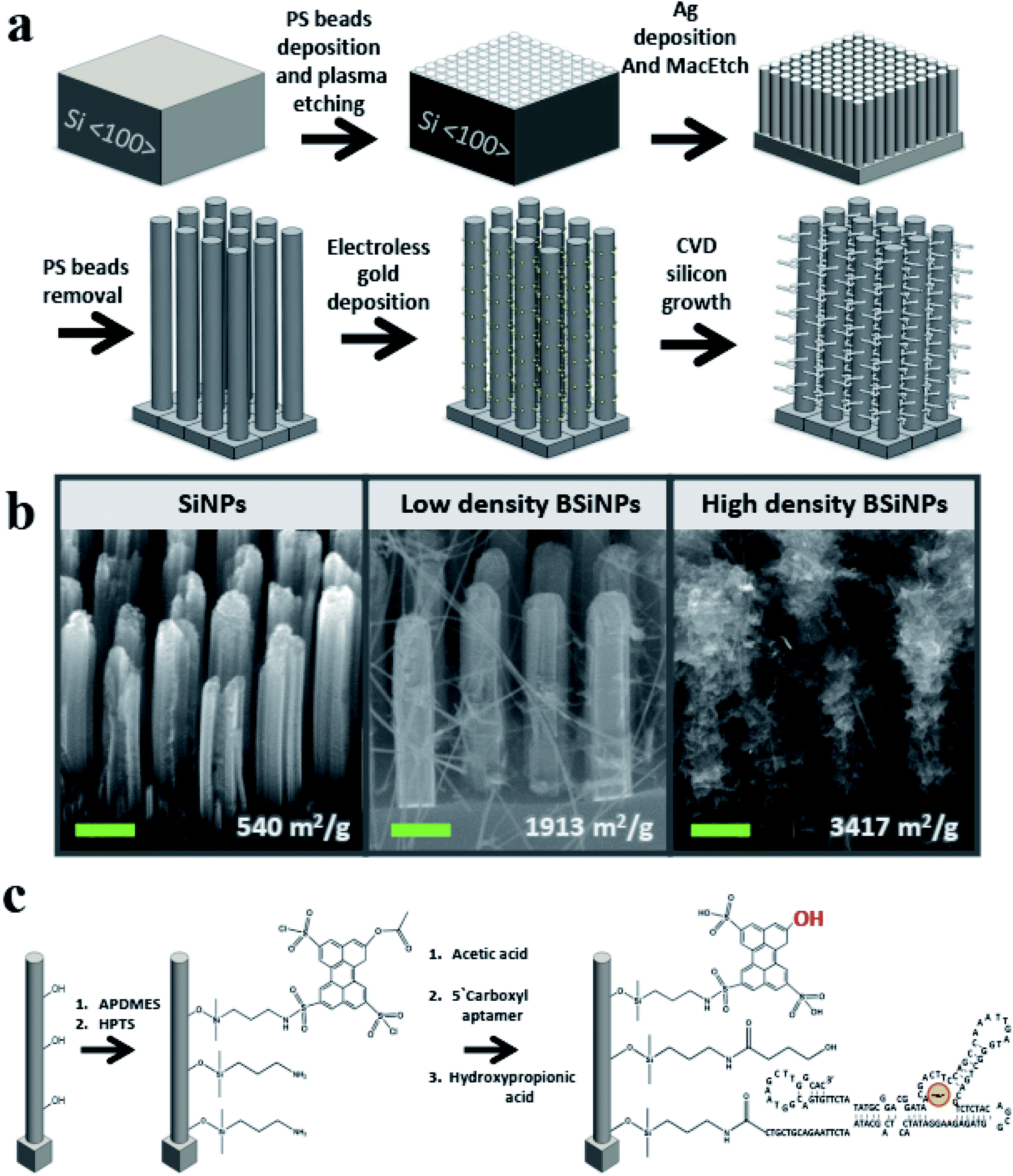 | ||
| Fig. 1 BSiNP array filter fabrication and modification analysis using scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). (a) Schematic representation of the BSiNP fabrication process. (b) SEM images of the SiNP array with increasing branch density, left to right: SiNPs, BSiNPs with low branch density and BSiNPs with high branch density; 30-degree view, scale bars: 0.5 μm. The values below each image are the corresponding BET SSA values. (c) Schematic representation of the HPTS and aptamer modification on the BSiNP surface. | ||
The active capturing area could be further amplified by the fabrication of taller SiNP arrays displaying increased roughness and a more densely packed growth of SiNBs, achieved by slight adaptation of the gold deposition and/or the CVD process. This trend of surface area amplification has been confirmed by Brunauer–Emmett–Teller (BET) measurements, and the results are summarized in Table 1. SiNP surface area reached a value of 540 m2 g−1, which is similar to the reported surface areas of porous silicon,55,56 while the BSiNP arrays reached a value of ∼3400 m2 g−1. Scanning electron microscopy (SEM) images of the different arrays in Fig. 1b show the difference in SiNB density that could be reached with adjustment of the electroless deposition time and CVD process, with surface area values shown in the bottom of Fig. 1b.
| Sample | Sample mass (mg) | R 2 | Slope (cm3 g−1 STP) | Y-Intercept (cm3 g−1 STP) | Q m (cm3 g−1 STP) | BET surface area (m2 g−1) |
|---|---|---|---|---|---|---|
| SiNPs | 1.0123 | 0.99803 | 0.2797 ± 0.0072 | 0.0402 ± 0.0013 | 3.1268 | 540 |
| Low-density BSiNPs | 1.0204 | 0.99986 | 0.0871 ± 0.0005 | 0.0031 ± 0.0001 | 11.0852 | 1913 |
| High-density BSiNPs | 1.0933 | 0.99996 | 0.0476 ± 0.0001 | 0.0029 ± 0.0002 | 19.7981 | 3417 |
Surfaces of BSiNP arrays were then chemically modified as outlined in Fig. 1c, with 3-aminopropyldimethylethoxysilane (APDMES), followed by a derivative of 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS), 8-acetoxy-pyrene-1,3,6-trisulfonyl chloride.29,49 Frequently applied as a light-triggered source of protons in various studies,29,48,49,51,57–59 HPTS has a pKa of ∼7.3 at the ground state and is exceptionally more acidic when photoexcited, with pKa as low as ∼0.4. Previous fluorescence experiments verified that the photoactivated pH decrease is confined to the surface, depending on the light intensity of the source. Upon activation, surface pH was ∼3.3–3.5, while bulk pH stayed at ∼7.5.51
Following the photoacid modification, surfaces were modified to immobilize the DNA aptamer units. This short DNA sequence of 117 bases, Tm = 70.9 °C and Mw = 36.2 kDa, was characterized with specificity and high affinity against uranyl with Kd = 84.6 pM,22 and used as a binding agent for uranyl ion (see Materials and methods in the ESI† for details).
Finally, 3-hydroxypropionic acid was used in order to block free APDMES amino groups. The modification was confirmed by X-ray photoelectron spectroscopy (XPS), as shown in Tables 2 and 3 (see the Experimental methods section in the ESI† for detailed information). XPS is a well-known chemical analysis technique for surface chemical composition and properties, as it measures ∼10 nm into the surface of a sample and is able to determine quantitative elemental composition alongside chemical state information.60
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Unmodified Si/SiO2 surface-referencea | Surface modified with amino-silanea | Surface modified with amino-silane and photoacida | Surface modified with amino-silane, photoacid, and DNA aptamera | |
| a The calculations error values are specified in ESI section Table S1. | ||||
| C | 1.69 | 7.22 | 19.41 | 21.25 |
| O | 64.17 | 58.06 | 50.93 | 49.81 |
| Si | 31.96 | 30.49 | 25.53 | 24.00 |
| N | 0.14 | 1.34 | 1.75 | 2.05 |
| S | — | — | 1.39 | 1.85 |
| C | N | ||||||
|---|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/O–Si–C C/O–Si–C |
C–S/C–N | C–O | CO |
C–N | –NH2\NH3+ | –N–SO2 | |
| (2) Surface modified with amino-silane | 76.03 | 23.97 | — | — | 85.41 | 14.59 | — |
| (3) Surface modified with amino-silane and photoacid | 69.2 | 18.23 | 10.33 | 2.23 | 71.19 | — | 28.81 |
| (4) Surface modified with amino-silane, photoacid, and DNA aptamer | 60.33 | 19.45 | 17.56 | 2.66 | 76.73 | — | 23.27 |
Surface coverage was determined by quartz crystal microbalance (QCM) showing ∼1.6 × 1014 molecules per cm2 of APDMES and 0.1–3 × 1013 molecules per cm2 of HPTS, depending on the time given for the chemical linking to the amino groups.48 This indicates that about 85–97% of the APDMES amino groups are available for subsequent chemical coupling with DNA aptamers. Immobilization of DNA onto the BSiNP array surface was achieved by first activating the 5′ carboxylic residue of the DNA using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS). Then, the amino-modified BSiNP array was soaked in the DNA/EDC/sulfo-NHS solution (see the Experimental methods section in the ESI† for detailed information). XPS analysis of the modified planar Si wafer determined surface coverage to be ∼3.0 × 1013 molecules per cm2 of APDMES, ∼8.6 × 1012 molecules per cm2 of HPTS, and ∼1.6 × 1013 molecules per cm2 of DNA aptamer. Theoretically, more than one of the three sulfonyl chloride groups of HPTS could bind to the amines on the surface, meaning HPTS surface coverage could be between 2.9–8.6 × 1012 molecules per cm2.
Assuming BSiNPs of low to high branch density, a maximum aptamer density can be theoretically expected at 2.4 × 1017 molecules per cm2 (1 cm2 geometric flat area converts into ∼15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2). Higher aptamer densities (per cm2 geometrical area) could be reached by using taller and rougher SiNP arrays or more densely grown SiNBs. These 3D-BSiNP arrays will ultimately boost the capturing capabilities of the resulting separation device.
000 cm2). Higher aptamer densities (per cm2 geometrical area) could be reached by using taller and rougher SiNP arrays or more densely grown SiNBs. These 3D-BSiNP arrays will ultimately boost the capturing capabilities of the resulting separation device.
Uranium collection efficiency
To evaluate the capturing efficiency of the BSiNPs, uranyl separation from urine and concentrated salt solutions spiked with different uranyl concentrations was performed as described below.Uranyl acetate dihydrate (UO2(CH3COO)2·2H2O) was dissolved in deionized water (DIW) to obtain 1.6 mM and 19 mM uranyl stock solutions. Uranyl concentrations of 1.2–1900 ppm (5 μM–8 mM) were quantified using its emission at 515 nm;61 all measurements were performed at room temperature (25 °C). Adsorption of uranyl, or other species, was calculated using eqn (1) and (2):
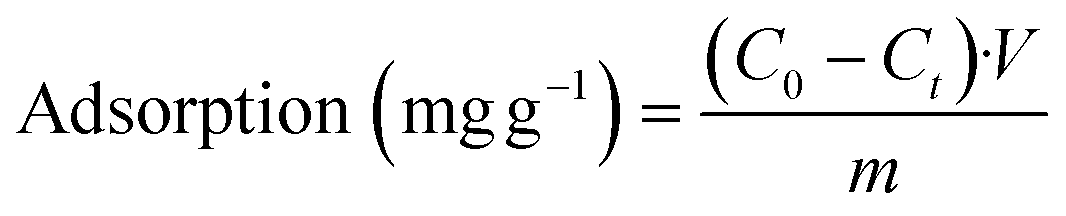 | (1) |
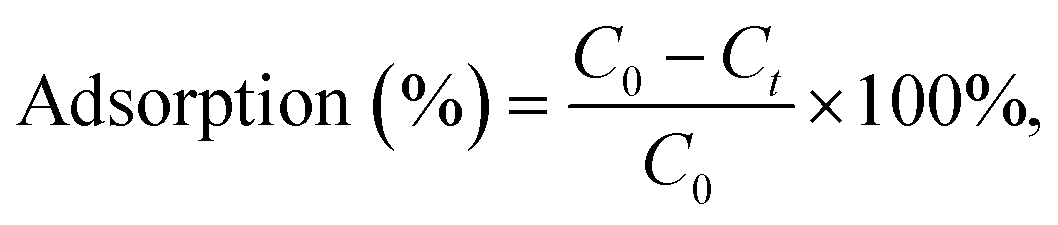 | (2) |
Fig. 2a shows the increase of uranyl-capturing capacity as a result of increased surface area, reaching up to 1.1 mg cm−2 within 30 minutes, which correlates to an adsorption capacity of 456 mg g−1 (uranyl per 10 μm × 1 cm2 Si wafer base for BSiNP). This result for adsorption capacity is moderately high, as reported adsorption capacities range from 0.1–1550 mg g−1.62–64 However, when accounting for the extremely short time of adsorption exhibited, 30 minutes compared to adsorption reactions that take 12 hours to several weeks,62–64 the capturing abilities of the BSiNP array are remarkable.
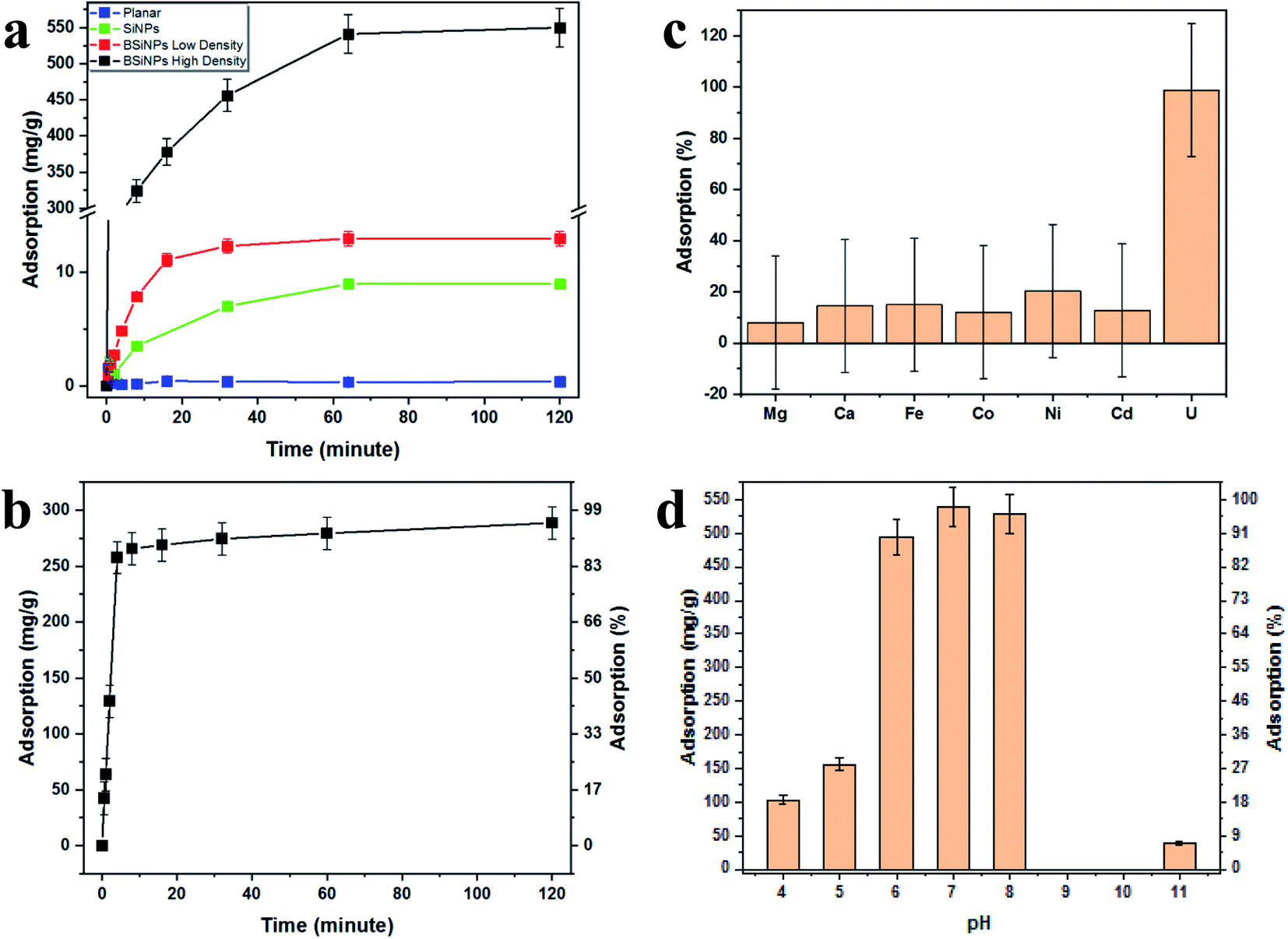 | ||
| Fig. 2 Capturing capabilities of the BSiNP light-controlled array. (a) Adsorption of uranyl from phosphate buffer (PB, 10 mM, pH 8.5) spiked with 11 mM (∼3000 ppm) uranyl onto planar Si wafer (blue curve), SiNP arrays (green curve), BSiNWs of low density (red curve) and high density (black curve) modified with uranyl-specific aptamer. (b) Adsorption of uranyl from concentrated salt solution spiked with 5.4 mM (∼1500 ppm) uranyl. All measurements were performed at room temperature. (c) ICP-MS results of adsorption from DIW spiked with 5 ppm U(VI), Mg(II), Ca(II), Fe(II), Co(II), Ni(II), and Cd(II), after 2 hours. (d) Maximal adsorption of uranyl from solutions spiked with 5.4 mM (∼1500 ppm) at different pH, after 2 hours. All measurements were performed at room temperature. | ||
As mentioned previously, a larger surface area, and therefore higher capturing capacity, could be easily achieved with mild alterations to the fabrication process. This is shown in Fig. 2a, as the planar wafer reaches a maximal adsorption of 0.09 mg g−1 after 2 h, four orders of magnitude lower than that of high-density BSiNP arrays.
Fig. 2b shows the fast depletion of uranyl from concentrated salt solution spiked with 5.4 mM uranyl (1500 ppm), thus displaying competitiveness in ultrahigh concentrations of other ionic species (Ca, Na, K, etc.). This is further exhibited alongside adsorption specificity in Fig. 2c, where concentrations of uranyl and competing ions were determined using ICP-MS, showing that nonspecific adsorption of competing ions is lower than 20% and does not affect uranyl capturing efficiency, reaching over 95% in both cases.
Measurement of the uranyl capturing capacity in different pH solutions has shown that the best results are achieved at pH values of 6–8, as shown in Fig. 2d. Importantly, minimal precipitation was observed in phosphate buffer (10 mM, pH 8.5) over 2 hours, (see ESI Fig. S2†).
Adsorption isotherms are plotted in Fig. 3. Equilibrium data points were fitted to three adsorption models to evaluate the BSiNP's ability to absorb uranyl.
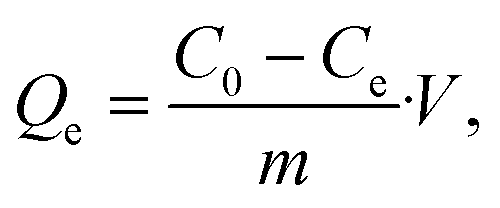 | (3) |
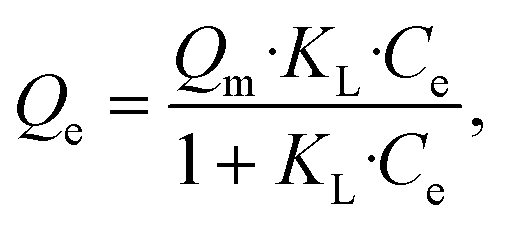 | (4) |
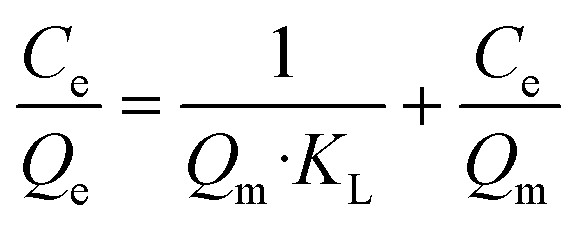 | (5) |
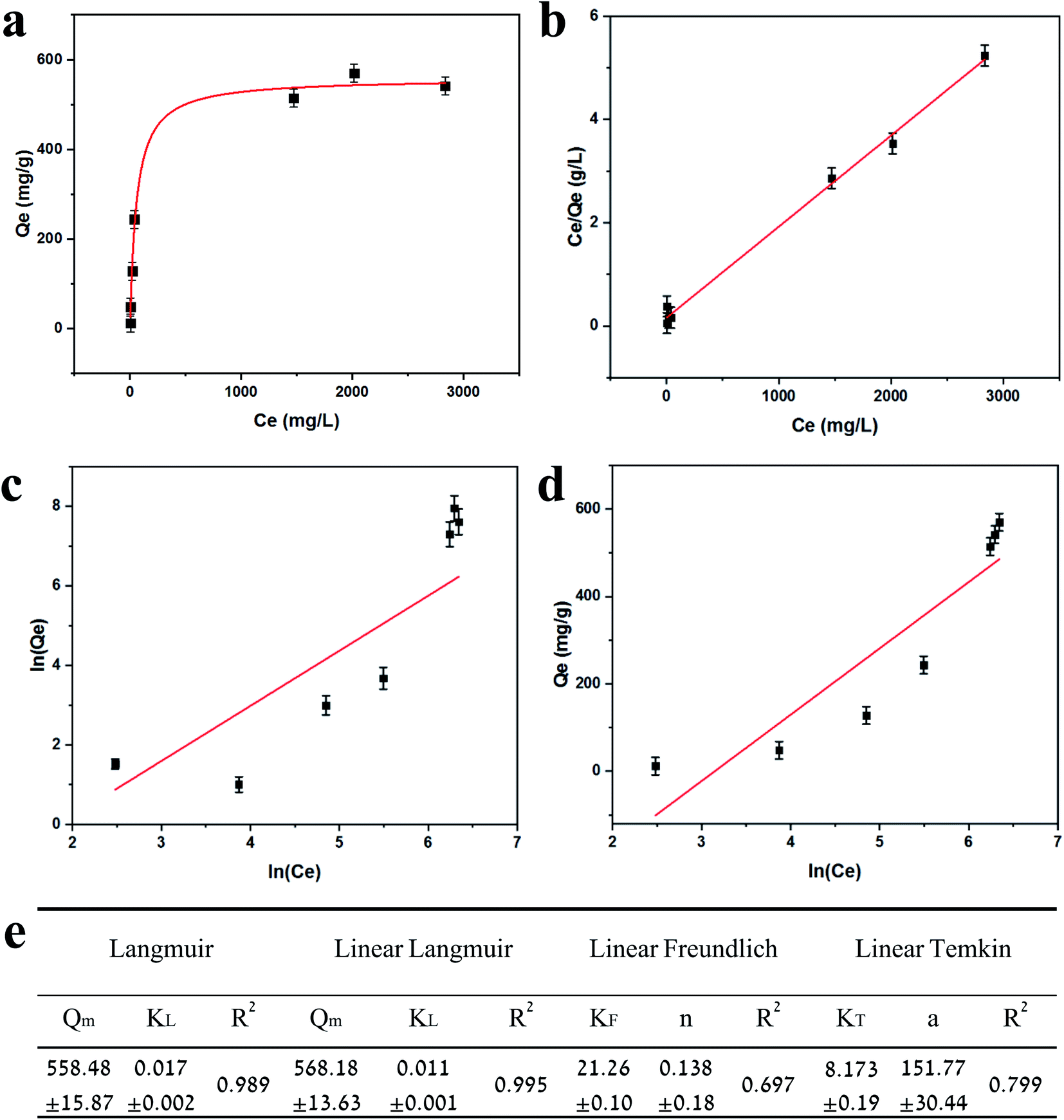 | ||
| Fig. 3 Adsorption isotherms for uranyl. (a) Equilibrium data for uranyl adsorption from PB (10 mM, pH 8.5) spiked with various uranyl concentrations; the red curve represents the fitting to the Langmuir isotherm plot. (b) Linear Langmuir isotherm plot. (c) Freundlich isotherm plot. (d) Temkin isotherm plot. (e) Fitting parameters table. All measurements were performed at room temperature. | ||
The linear Freundlich isotherm model equation is:
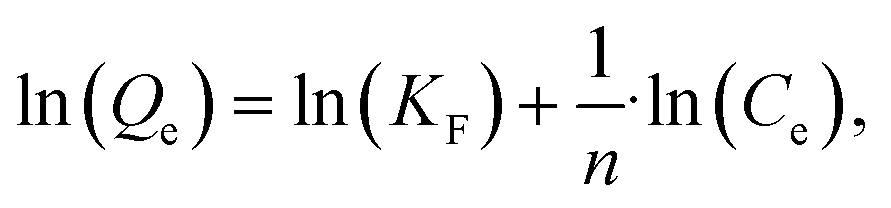 | (6) |
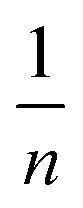 is sorption intensity, also indicating the relative distribution of the energy and the heterogeneity of the adsorbate sites. It was reported that the Freundlich isotherm constant can be used to explore the favorability of the adsorption process. The linear Temkin isotherm model equation is:
is sorption intensity, also indicating the relative distribution of the energy and the heterogeneity of the adsorbate sites. It was reported that the Freundlich isotherm constant can be used to explore the favorability of the adsorption process. The linear Temkin isotherm model equation is:| Qe = a·ln(KT) + a·ln(Ce), | (7) |
Fig. 3e summarizes the calculated fitting parameters, indicating that the best fitting is found with the Langmuir model (R2 = 0.995 for the linear model), suggesting monolayer adsorption behavior, which correlates well to the expected monolayer of capturing moieties achieved in the chemical modification shown in Fig. 1c. The fitting parameters indicate the theoretical maximal adsorption of 558.48 ± 15.87 mg g−1 and 568.18 ± 13.63 mg g−1 for nonlinear and linear Langmuir model fittings, respectively.
The essential characteristics of the Langmuir isotherm can be expressed by a dimensionless constant called the separation factor RL:65
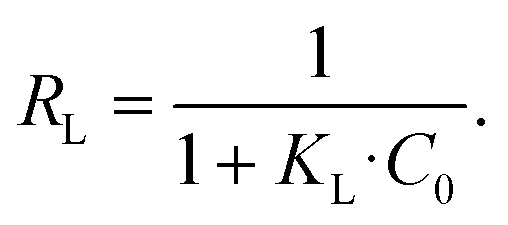 | (8) |
R L values indicate the adsorption to be linear when RL = 1, and favorable when 0 < RL< 1. RL values calculated from fitting parameters for uranyl concentrations from 1 ppt to 5 ppm are 0.921 < RL < 0.999; therefore, the uranyl adsorption is favorable to linear in environmental uranyl concentrations.
Fig. 4 demonstrates the effectiveness of our uranium collection device for rapid and quantitative capturing and preconcentration. Uranyl capturing showed to be extremely efficient, Fig. 4a, with full depletion (96%, up to ca. 1.1 μmol cm−2) of uranyl from spiked urine samples occurring very fast, achieving a plateau after about 10–60 minutes of incubation.
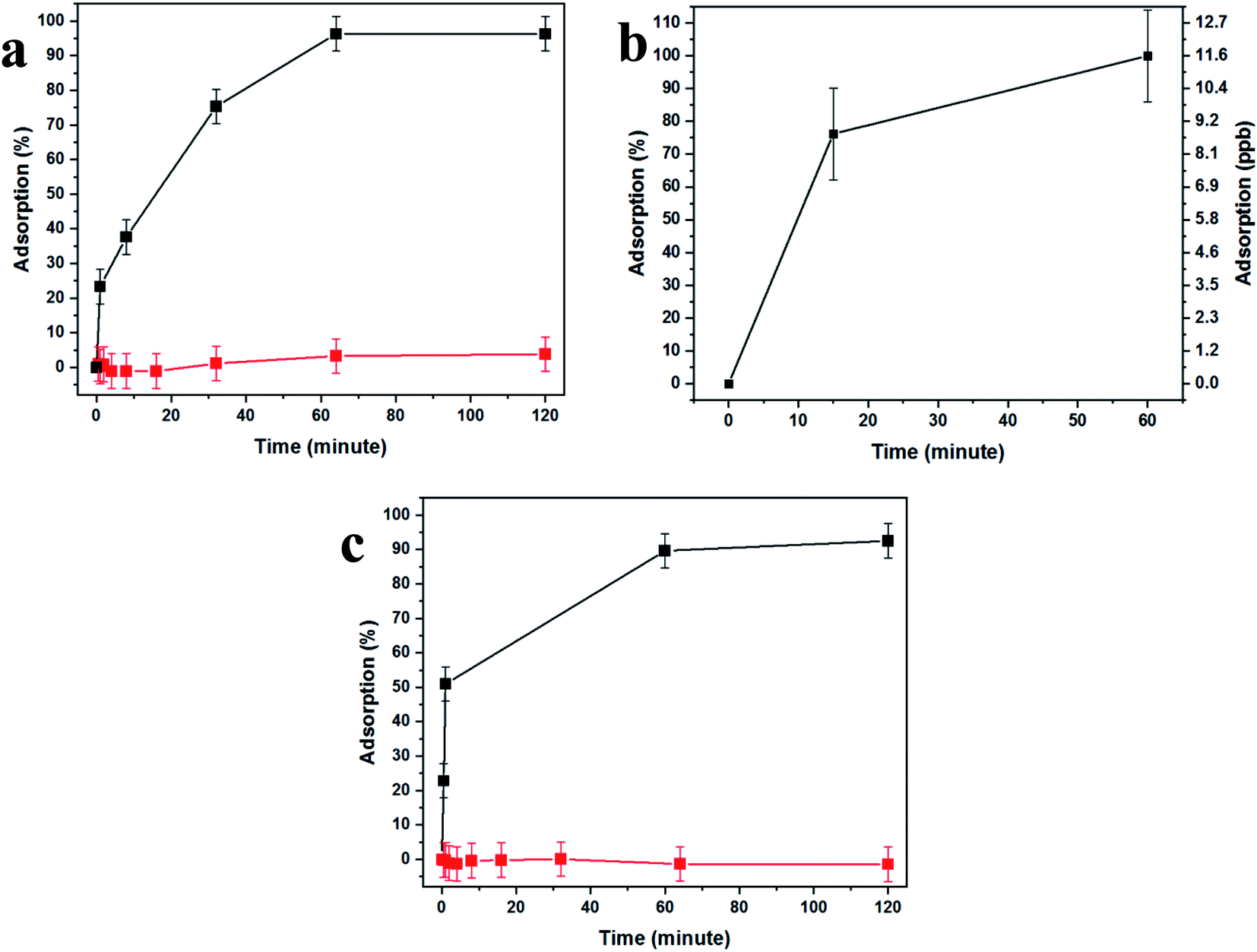 | ||
| Fig. 4 BSiNP specificity and versatility. (a) Specific adsorption of uranyl from untreated urine spiked with 0.11 mM (∼30 ppm) uranyl to a BSiNP array modified with a uranyl-specific aptamer (black curve) versus nonspecific adsorption to an array modified with arsenic-specific aptamer (red curve). (b) ICP-MS results of uranyl-specific adsorption from DIW spiked with 40 nM (∼12 ppb) uranyl to a BSiNP array modified with a uranyl-specific aptamer. (c) Specific adsorption of arsenic from DIW spiked with 0.13 mM (∼10 ppm) arsenic to a BSiNP array modified with arsenic-specific aptamer (black curve) versus nonspecific adsorption of arsenic to an array modified with uranyl-specific aptamer (red curve). All measurements were performed at room temperature. | ||
These results are expected due to the extreme magnification of the surface area, which dramatically increases the abundance of aptamer-capturing units and contributes to the rapid depletion of uranyl from the sample, as the surface coverage was shown to be as high as ∼1.3 × 1017 aptamer molecules per cm2, 102-fold higher than the number of uranyl molecules in the sample. Additionally, the lack of nonspecific adsorption was demonstrated by the use of a uranyl-spiked sample with a DNA aptamer specific to arsenic and nonspecific to uranyl (Fig. 4a, red curve).
In order to test uranyl adsorption at concentrations comparable to uranyl sea concentrations (ca. 3–5 ppb), we performed ICP-MS uranium quantification of samples. Adsorption results in Fig. 4b show that approximately 80% of uranyl is adsorbed after only 15 minutes, with full depletion reached within 1 hour.
This method could be applied successfully to other species, as shown in Fig. 4c, where the BSiNP array was modified with an arsenic-specific DNA aptamer and showed the same depletion efficiency (black curve) and high specificity (red curve).
Light-triggered rapid and quantitative release
Irradiation at a wavelength of 400 nm brings HPTS molecules to their excited state, where they display a dramatic change in their pKa, from 7.4 to ∼0.4, becoming super-acidic molecules that rapidly expel their phenolic protons, thus lowering the surface pH. This light-activated mechanism of uranyl release is schematically shown in Fig. 5a. Light excitation causes HPTS to release protons, lowering the surface pH. The light-triggered pH drop caused by photoactivation of the covalently bonded photoacid molecules may be applied for the on-surface activation of pH-dependent chemical and biological processes, such as the dissociation of antigen–antibody pairs.51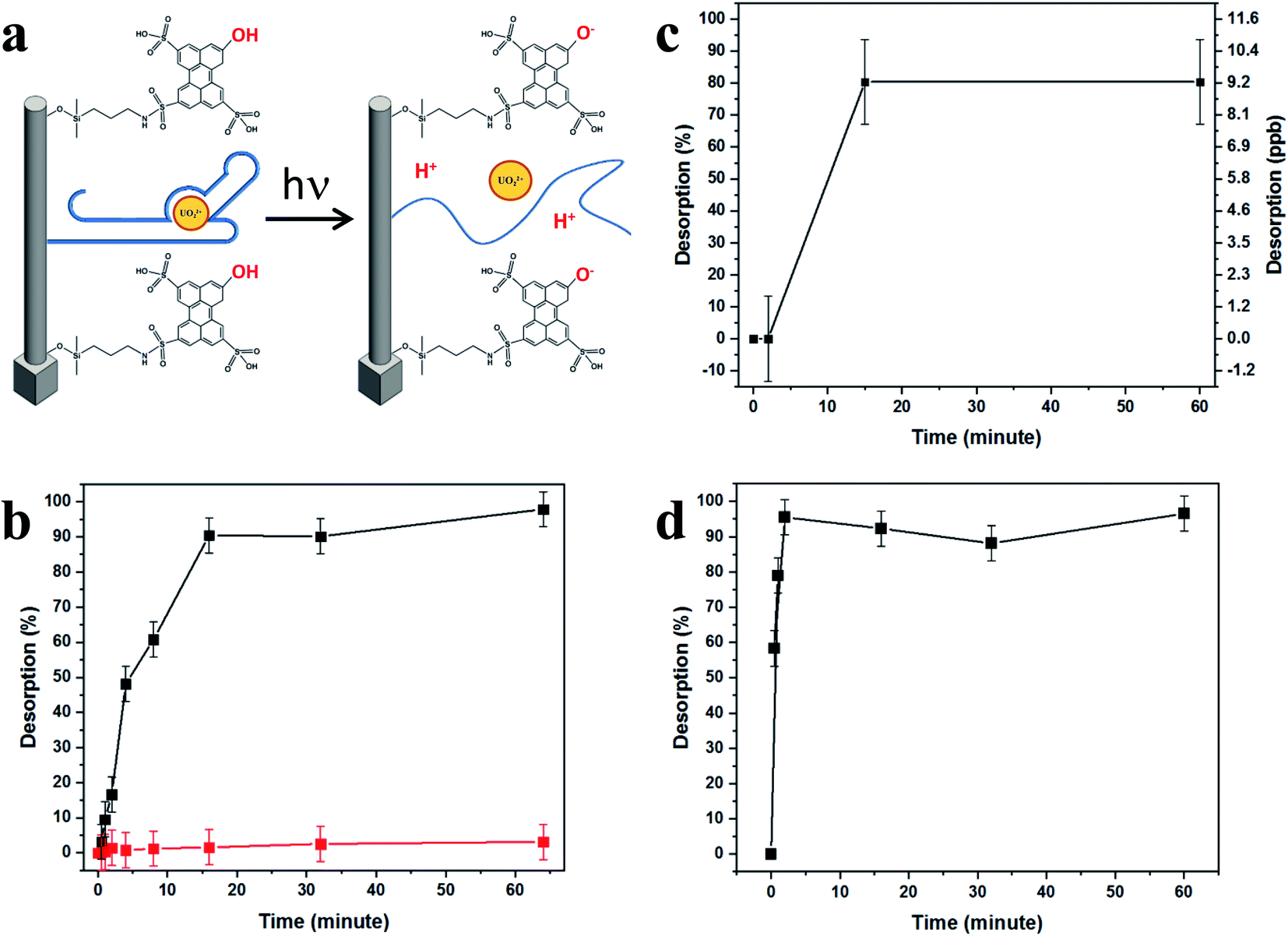 | ||
| Fig. 5 Light-controlled array release to a controlled medium. (a) Schematic of the light-activated mechanism of uranyl desorption. (b) Desorption of uranyl under illumination at a wavelength of 400 nm light (black curve) and under dark conditions (red curve). (c) ICP-MS results of light-activated uranyl desorption from BSiNP array. (d) Desorption of arsenic under light illumination. All measurements were performed in PB 100 nM pH 8.5, at room temperature. | ||
We accordingly propose that the lower surface pH influences the DNA aptamer structure and lowers its uranyl-binding capabilities. Reported to be most efficient at pH 8.01,22 the aptamer binding efficiency was expected to decrease immensely after the pH drop induced by light irradiation of the HPTS-modified SiNP arrays, as was shown to be the case with surface-attached antibody–antigen pairs, by decreasing the pH around and within close vicinity of the SiNP surfaces.29,49,51 This effect is demonstrated in Fig. 5b–d, showing the release characterization of uranyl and arsenic.
As expected, the fast and quantitative uranyl capture is followed by prolonged release; only up to ca. 4% of the captured molecules desorb spontaneously from the aptamer-modified BSiNP arrays after 2 hours (Fig. 5b, red curve), meaning that the dissociation rate is koff ≈ 2.6 × 10−4% min−1; therefore, full release would be achieved in ca. 507000 minutes (about 351 days). As previously discussed,29 we believe that the molecules bound at the top of the BSiNP array are fast-releasing, making up the ca. 4% of spontaneous release. The slow release cannot be explained by potential nonspecific adsorption of species to the surface of the BSiNP array, as negligible nonspecific adsorption was observed (Fig. 4a, red curve).
Activation of HPTS by light irradiation (400 nm, 50 mW cm−2) allows for immediate retrieval of the captured uranyl species, with koff = ∼0.15% min−1, five orders of magnitude greater than the koff measured under dark conditions. Hence, the complete quantitative release of captured uranyl is observed in less than 20 minutes. Over 90% of captured uranyl is desorbed within 16 minutes. Since ca. 80% is adsorbed within 30 minutes, this correlates to the retrieval of approximately 70% of uranyl found in the sample in less than 1 hour. Remarkably, no decrease in the effectiveness of the BSiNP arrays was observed after performing several cycles of capturing and light-triggered release (ESI Fig. S1†). Previous experiments have brought us to the conclusion that the pH drop is experienced in close proximity to the SiNP surface, controlled by light intensity and photoacid surface concentration.49
The integration of photoactivated pH drop and the consequent fast release of the target molecule achieves controlled capturing and retrieval of over 85% of the uranyl found in the sample in less than 20 minutes, without resulting in secondary waste and with an easily operable and reusable capturing apparatus. Notably, this is the first time pH triggering of DNA aptamer deactivation has been demonstrated. Moreover, this work shows the first use of photoacid activation for the on-surface modulation of aptamer activity and target affinity. To date, there has been no report of DNA switching by pH change, activated by a light source.
Discussion
We hypothesized that molecules experience limited diffusion inside nanocavities of the nanostructured SiNP arrays, causing them to delay while being adsorbed to, and desorbed from, capturing agents on the surface into the confined interpillar space repeatedly, until ultimately being released to the above medium. This ultimately leads to extremely prolonged effective dissociation rates, allowing our highly receptive SiNP arrays to effectively and very efficiently capture molecules from bulk solutions, further preventing their release back to the bulk by this novel nanoconfinement-related capturing mechanism.Theoretical examination of this effect29 yields a calculated N, the mean number of collisions of a particle with the sidewalls while it is traveling along a channel closed on one end and open on the other, with length L and square cross-section b × b. The diffusion coefficients along and perpendicular to the channel axis are given by D‖ and D⊥. N was shown to be calculated by:
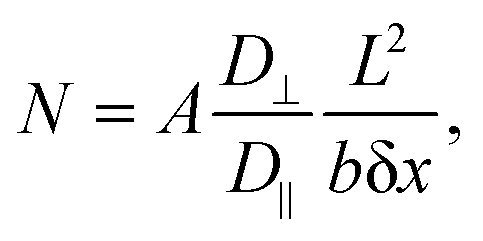 | (9) |
For b = 250 nm, L = 5 μm, 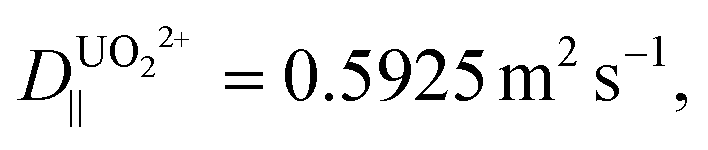 δx = 1 nm, and A = 0.50, a particle is expected to collide/rebind to the cavity's walls ca. 5 × 104 times before finally escaping the cavity into the bulk solution. Considering the aptamer's reported Kd of ca. 84.6 pM,22 even if only a small fraction of these collisions lead to rebinding of the uranyl species to the walls of the NP array, this correlates to theoretically delaying the captured uranyl from escaping from the nanocavity to the bulk by a 104-fold factor compared to its bare dissociation rate. When accounting for the additional surface area created by the growth of SiNWs over the SiNPs to comprise the BSiNP array, N is expected to increase, depending on the density of the branches.
δx = 1 nm, and A = 0.50, a particle is expected to collide/rebind to the cavity's walls ca. 5 × 104 times before finally escaping the cavity into the bulk solution. Considering the aptamer's reported Kd of ca. 84.6 pM,22 even if only a small fraction of these collisions lead to rebinding of the uranyl species to the walls of the NP array, this correlates to theoretically delaying the captured uranyl from escaping from the nanocavity to the bulk by a 104-fold factor compared to its bare dissociation rate. When accounting for the additional surface area created by the growth of SiNWs over the SiNPs to comprise the BSiNP array, N is expected to increase, depending on the density of the branches.
In addition, the geometry of the SiNP array facilitates uranyl preconcentration, as the interpillar void volume is only 0.1 μL for 1 cm2 SiNP array devices with an NP height of 5 μm. The incubation of a 10 μL sample will lead to preconcentration in the interpillar void volume of 0.1 μL, a factor of 100. The use of a smaller capturing void volume could be expected for BSiNP arrays, and larger sample volumes will further increase the achievable preconcentration factors.
Generally, this delayed-release mechanism from SiNP arrays greatly contributes to the collection capabilities of these devices; however, it strongly impedes the release of the captured species. Thus, a mechanism for the effective switching from a highly sticky, nonreleasing state into a highly reflective, fast-releasing state is highly desirable. This is achieved by activating a pH drop near the surface of the BSiNPs using light activation of the photoacid molecules. We deduce that lower pH causes the uranyl-aptamer units to be mostly nonbinding, resulting in apparent koff greater by five orders of magnitude than the apparent koff without light activation.
In conclusion, here we demonstrated the ultrafast high-capacity collection, up to ca. 550 mg g−1, of uranium from samples, including complex samples such as urine and concentrated salt solution. The adsorption capability of BSiNPs is a result of the extremely high surface area and high surface coverage of DNA-aptamer capturing units, trapping uranyl inside the NP cavities. This approach showed high specificity and selectivity, with the ability to be applied to other target ions with similar adsorption capabilities. Uranyl capturing is coupled with the light-triggered rapid and quantitative uranyl release, harvesting more than 70% of the uranyl found in a given sample within one cycle (1 hour) of capturing and light-activated release. Overall, our device is able to capture uranyl efficiently in less than 1 hour and allows for easy retrieval of the captured uranyl in a controlled medium with the switch of a light source.
Author contributions
Ella Borberg: conceptualization, methodology, investigation, visualization, data curation, formal analysis, and writing – original draft. Reut Meir: resources, methodology, and writing – review and editing. Larisa Burstein: investigation, formal analysis. Vadim Krivitsky: validation, supervision. Fernando Patolsky: conceptualization, supervision, validation, funding acquisition, project administration, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Legacy Foundation (Israel Science Foundation) for the funding of this project.References
- E. S. Craft, A. W. Abu-Qare, M. M. Flaherty, M. C. Garofolo, H. L. Rincavage and M. B. Abou-Donia, Depleted and Natural Uranium: Chemistry and Toxicological Effects, J. Toxicol. Environ. Health, Part B, 2004, 7(4), 297–317, DOI:10.1080/10937400490452714.
- S. Keith, O. Faroon, N. Roney, F. Scinicariello, S. Wilbur, L. Ingerman, F. Llados, D. Plewak, D. Wohlers and G. Diamond, Toxicological Profile for Uranium; Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles, Agency for Toxic Substances and Disease Registry (US), Atlanta (GA), 2013 Search PubMed.
- B. F. Parker, Z. Zhang, L. Rao and J. Arnold, An Overview and Recent Progress in the Chemistry of Uranium Extraction from Seawater, Dalton Trans., 2018, 47(3), 639–644, 10.1039/C7DT04058J.
- A. C. Sather, O. B. Berryman and J. Rebek, Selective Recognition and Extraction of the Uranyl Ion from Aqueous Solutions with a Recyclable Chelating Resin, Chem. Sci., 2013, 4(9), 3601, 10.1039/c3sc51507a.
- A. C. Sather, O. B. Berryman and J. Rebek, Selective Recognition and Extraction of the Uranyl Ion, J. Am. Chem. Soc., 2010, 132(39), 13572–13574, DOI:10.1021/ja1035607.
- C. Liu, P.-C. Hsu, J. Xie, J. Zhao, T. Wu, H. Wang, W. Liu, J. Zhang, S. Chu and Y. Cui, A Half-Wave Rectified Alternating Current Electrochemical Method for Uranium Extraction from Seawater, Nat. Energy, 2017, 2(4), 1–8, DOI:10.1038/nenergy.2017.7.
- M.-L. Feng, D. Sarma, X.-H. Qi, K.-Z. Du, X.-Y. Huang and M. G. Kanatzidis, Efficient Removal and Recovery of Uranium by a Layered Organic–Inorganic Hybrid Thiostannate, J. Am. Chem. Soc., 2016, 138(38), 12578–12585, DOI:10.1021/jacs.6b07351.
- L. Xiong, K. Lv, M. Gu, C. Yang, F. Wu, J. Han and S. Hu, Efficient Capture of Actinides from Strong Acidic Solution by Hafnium Phosphonate Frameworks with Excellent Acid Resistance and Radiolytic Stability, Chem. Eng. J., 2019, 355, 159–169, DOI:10.1016/j.cej.2018.08.118.
- M. Montaña, A. Camacho, I. Serrano, R. Devesa, L. Matia and I. Vallés, Removal of Radionuclides in Drinking Water by Membrane Treatment Using Ultrafiltration, Reverse Osmosis and Electrodialysis Reversal, J. Environ. Radioact., 2013, 125, 86–92, DOI:10.1016/j.jenvrad.2013.01.010.
- J. H. Kim, H. I. Lee, J.-W. Yeon, Y. Jung and J. M. Kim, Removal of Uranium(VI) from Aqueous Solutions by Nanoporous Carbon and Its Chelating Polymer Composite, J. Radioanal. Nucl. Chem., 2010, 286(1), 129–133, DOI:10.1007/s10967-010-0624-3.
- R. Villalobos-Rodríguez, M. E. Montero-Cabrera, H. E. Esparza-Ponce, E. F. Herrera-Peraza and M. L. Ballinas-Casarrubias, Uranium Removal from Water Using Cellulose Triacetate Membranes Added with Activated Carbon, Appl. Radiat. Isot., 2012, 70(5), 872–881, DOI:10.1016/j.apradiso.2012.01.017.
- K. J. Cantrell, D. I. Kaplan and T. W. Wietsma, Zero-Valent Iron for the in Situ Remediation of Selected Metals in Groundwater, J. Hazard. Mater., 1995, 42(2), 201–212, DOI:10.1016/0304-3894(95)00016-N.
- J. N. Fiedor, W. D. Bostick, R. J. Jarabek and J. Farrell, Understanding the Mechanism of Uranium Removal from Groundwater by Zero-Valent Iron Using X-Ray Photoelectron Spectroscopy, Environ. Sci. Technol., 1998, 32(10), 1466–1473, DOI:10.1021/es970385u.
- B. Gu, W.-M. Wu, M. A. Ginder-Vogel, H. Yan, M. W. Fields, J. Zhou, S. Fendorf, C. S. Criddle and P. M. Jardine, Bioreduction of Uranium in a Contaminated Soil Column, Environ. Sci. Technol., 2005, 39(13), 4841–4847, DOI:10.1021/es050011y.
- Q. Sun, B. Aguila, J. Perman, A. S. Ivanov, V. S. Bryantsev, L. D. Earl, C. W. Abney, L. Wojtas and S. Ma, Bio-Inspired Nano-Traps for Uranium Extraction from Seawater and Recovery from Nuclear Waste, Nat. Commun., 2018, 9(1), 1644, DOI:10.1038/s41467-018-04032-y.
- J.-H. Jang, B. A. Dempsey and W. D. Burgos, A Model-Based Evaluation of Sorptive Reactivities of Hydrous Ferric Oxide and Hematite for U(VI), Environ. Sci. Technol., 2007, 41(12), 4305–4310, DOI:10.1021/es070068f.
- A. Mellah, S. Chegrouche and M. Barkat, The Removal of Uranium(VI) from Aqueous Solutions onto Activated Carbon: Kinetic and Thermodynamic Investigations, J. Colloid Interface Sci., 2006, 296(2), 434–441, DOI:10.1016/j.jcis.2005.09.045.
- S. M. Webb, C. C. Fuller, B. M. Tebo and J. R. Bargar, Determination of Uranyl Incorporation into Biogenic Manganese Oxides Using X-Ray Absorption Spectroscopy and Scattering, Environ. Sci. Technol., 2006, 40(3), 771–777, DOI:10.1021/es051679f.
- S. Brown, Y. Yue, L.-J. Kuo, N. Mehio, M. Li, G. Gill, C. Tsouris, R. T. Mayes, T. Saito and S. Dai, Uranium Adsorbent Fibers Prepared by Atom-Transfer Radical Polymerization (ATRP) from Poly(Vinyl Chloride)-Co-Chlorinated Poly(Vinyl Chloride) (PVC-Co-CPVC) Fiber, Ind. Eng. Chem. Res., 2016, 55(15), 4139–4148, DOI:10.1021/acs.iecr.5b03355.
- T. B. Scott, O. Riba Tort and G. C. Allen, Aqueous Uptake of Uranium onto Pyrite Surfaces; Reactivity of Fresh versus Weathered Material, Geochim. Cosmochim. Acta, 2007, 71(21), 5044–5053, DOI:10.1016/j.gca.2007.08.017.
- L.-J. Kuo, H.-B. Pan, C. M. Wai, M. F. Byers, E. Schneider, J. E. Strivens, C. J. Janke, S. Das, R. T. Mayes, J. R. Wood, N. Schlafer and G. A. Gill, Investigations into the Reusability of Amidoxime-Based Polymeric Adsorbents for Seawater Uranium Extraction, Ind. Eng. Chem. Res., 2017, 56(40), 11603–11611, DOI:10.1021/acs.iecr.7b02893.
- J. Kim, M. Y. Kim, H. S. Kim and S. S. Hah, Binding of Uranyl Ion by a DNA Aptamer Attached to a Solid Support, Bioorg. Med. Chem. Lett., 2011, 21(13), 4020–4022, DOI:10.1016/j.bmcl.2011.04.139.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, A Catalytic Beacon Sensor for Uranium with Parts-per-Trillion Sensitivity and Millionfold Selectivity, Proc. Natl. Acad. Sci., 2007, 104(7), 2056–2061, DOI:10.1073/pnas.0607875104.
- A. K. Brown, J. Liu, Y. He and Y. Lu, Biochemical Characterization of a Uranyl Ion-Specific DNAzyme, ChemBioChem, 2009, 10(3), 486–492, DOI:10.1002/cbic.200800632.
- J. H. Lee, Z. Wang, J. Liu and Y. Lu, Highly Sensitive and Selective Colorimetric Sensors for Uranyl (UO22+): Development and Comparison of Labeled and Label-Free DNAzyme-Gold Nanoparticle Systems, J. Am. Chem. Soc., 2008, 130(43), 14217–14226, DOI:10.1021/ja803607z.
- H. Zhang, X. Cheng, L. Chen, F. Mo, L. Xu and F. Fu, Magnetic Beads-Based DNA Hybridization Chain Reaction Amplification and DNAzyme Recognition for Colorimetric Detection of Uranyl Ion in Seafood, Anal. Chim. Acta, 2017, 956, 63–69, DOI:10.1016/j.aca.2016.12.021.
- B. Zhou, L.-F. Shi, Y.-S. Wang, H.-X. Yang, J.-H. Xue, L. Liu, Y.-S. Wang, J.-C. Yin and J.-C. Wang, Resonance Light Scattering Determination of Uranyl Based on Labeled DNAzyme–Gold Nanoparticle System, Spectrochim. Acta, Part A, 2013, 110, 419–424, DOI:10.1016/j.saa.2013.03.036.
- R. Meir, M. Zverzhinetsky, N. Harpak, E. Borberg, L. Burstein, O. Zeiri, V. Krivitsky and F. Patolsky, Direct Detection of Uranyl in Urine by Dissociation from Aptamer-Modified Nanosensors Arrays, Anal. Chem., 2020, 92, 12528–12537, DOI:10.1021/acs.analchem.0c02387.
- E. Borberg, M. Zverzhinetsky, A. Krivitsky, A. Kosloff, O. Heifler, G. Degabli, H. P. Soroka, R. S. Fainaro, L. Burstein, S. Reuveni, H. Diamant, V. Krivitsky and F. Patolsky, Light-Controlled Selective Collection-and-Release of Biomolecules by an On-Chip Nanostructured Device, Nano Lett., 2019, 19(9), 5868–5878, DOI:10.1021/acs.nanolett.9b01323.
- N. H. Finkel, B. G. Prevo, O. D. Velev and L. He, Ordered Silicon Nanocavity Arrays in Surface-Assisted Desorption/Ionization Mass Spectrometry, Anal. Chem., 2005, 77(4), 1088–1095, DOI:10.1021/ac048645v.
- A. I. Hochbaum, R. Fan, R. He and P. Yang, Controlled Growth of Si Nanowire Arrays for Device Integration, Nano Lett., 2005, 5(3), 457–460, DOI:10.1021/nl047990x.
- H. J. Joyce, Q. Gao, H. Hoe Tan, C. Jagadish, Y. Kim, J. Zou, L. M. Smith, H. E. Jackson, J. M. Yarrison-Rice, P. Parkinson and M. B. Johnston, III–V Semiconductor Nanowires for Optoelectronic Device Applications, Prog. Quantum Electron., 2011, 35(2), 23–75, DOI:10.1016/j.pquantelec.2011.03.002.
- H. Alhmoud, D. Brodoceanu, R. Elnathan, T. Kraus and N. H. Voelcker, A MACEing Silicon: Towards Single-Step Etching of Defined Porous Nanostructures for Biomedicine, Prog. Mater. Sci., 2019, 100636, DOI:10.1016/j.pmatsci.2019.100636.
- L. Sun, Y. Fan, X. Wang, R. A. Susantyoko and Q. Zhang, Large Scale Low Cost Fabrication of Diameter Controllable Silicon Nanowire Arrays, Nanotechnology, 2014, 25(25), 255302, DOI:10.1088/0957-4484/25/25/255302.
- S. Chang, V. P. Chuang, S. T. Boles and C. V. Thompson, Metal-Catalyzed Etching of Vertically Aligned Polysilicon and Amorphous Silicon Nanowire Arrays by Etching Direction Confinement, Adv. Funct. Mater., 2010, 20(24), 4364–4370, DOI:10.1002/adfm.201000437.
- S. Mouli Thalluri, J. Borme, D. Xiong, J. Xu, W. Li, I. Amorim, P. Alpuim, J. Gaspar, H. Fonseca, L. Qiao and L. Liu, Highly-Ordered Silicon Nanowire Arrays for Photoelectrochemical Hydrogen Evolution: An Investigation on the Effect of Wire Diameter, Length and Inter-Wire Spacing, Sustainable Energy Fuels, 2018, 2(5), 978–982, 10.1039/C7SE00591A.
- H. Bao and X. Ruan, Optical Absorption Enhancement in Disordered Vertical Silicon Nanowire Arrays for Photovoltaic Applications, Opt. Lett., 2010, 35(20), 3378–3380, DOI:10.1364/OL.35.003378.
- C. Lin and M. L. Povinelli, Optical Absorption Enhancement in Silicon Nanowire Arrays with a Large Lattice Constant for Photovoltaic Applications, Opt. Express, 2009, 17(22), 19371–19381, DOI:10.1364/OE.17.019371.
- R. Elnathan, B. Delalat, D. Brodoceanu, H. Alhmoud, F. J. Harding, K. Buehler, A. Nelson, L. Isa, T. Kraus and N. H. Voelcker, Maximizing Transfection Efficiency of Vertically Aligned Silicon Nanowire Arrays, Adv. Funct. Mater., 2015, 25(46), 7215–7225, DOI:10.1002/adfm.201503465.
- S. Qi, C. Yi, S. Ji, C.-C. Fong and M. Yang, Cell Adhesion and Spreading Behavior on Vertically Aligned Silicon Nanowire Arrays, ACS Appl. Mater. Interfaces, 2009, 1(1), 30–34, DOI:10.1021/am800027d.
- C. Yi, C.-W. Li, H. Fu, M. Zhang, S. Qi, N.-B. Wong, S.-T. Lee and M. Yang, Patterned Growth of Vertically Aligned Silicon Nanowire Arrays for Label-Free DNA Detection Using Surface-Enhanced Raman Spectroscopy, Anal. Bioanal. Chem., 2010, 397(7), 3143–3150, DOI:10.1007/s00216-010-3889-z.
- Y. Guerfi and G. Larrieu, Vertical Silicon Nanowire Field Effect Transistors with Nanoscale Gate-All-Around, Nanoscale Res. Lett., 2016, 11(1), 210, DOI:10.1186/s11671-016-1396-7.
- T. Tieu, M. Alba, R. Elnathan, A. Cifuentes-Rius and N. H. Voelcker, Advances in Porous Silicon–Based Nanomaterials for Diagnostic and Therapeutic Applications, Adv. Ther., 2019, 2(1), 1800095, DOI:10.1002/adtp.201800095.
- H. Alhmoud, B. Delalat, X. Ceto, R. Elnathan, A. Cavallaro, K. Vasilev and N. H. Voelcker, Antibacterial Properties of Silver Dendrite Decorated Silicon Nanowires, RSC Adv., 2016, 6(70), 65976–65987, 10.1039/C6RA13734B.
- V. Krivitsky, L.-C. Hsiung, A. Lichtenstein, B. Brudnik, R. Kantaev, R. Elnathan, A. Pevzner, A. Khatchtourints and F. Patolsky, Si Nanowires Forest-Based On-Chip Biomolecular Filtering, Separation and Preconcentration Devices: Nanowires Do It All, Nano Lett., 2012, 12(9), 4748–4756, DOI:10.1021/nl3021889.
- Y. Luo, C. Wang, P. Peng, M. Hossain, T. Jiang, W. Fu, Y. Liao and M. Su, Visible Light Mediated Killing of Multidrug-Resistant Bacteria Using Photoacids, J. Mater. Chem. B, 2013, 1(7), 997–1001, 10.1039/C2TB00317A.
- X. Yue, C. O. Yanez, S. Yao and K. D. Belfield, Selective Cell Death by Photochemically Induced PH Imbalance in Cancer Cells, J. Am. Chem. Soc., 2013, 135(6), 2112–2115, DOI:10.1021/ja3122312.
- H. P. Soroka, R. Simkovitch, A. Kosloff, S. Shomer, A. Pevzner, O. Tzang, R. Tirosh, F. Patolsky and D. Huppert, Excited-State Proton Transfer and Proton Diffusion near Hydrophilic Surfaces, J. Phys. Chem. C, 2013, 117(48), 25786–25797, DOI:10.1021/jp4087514.
- H. Peretz-Soroka, A. Pevzner, G. Davidi, V. Naddaka, R. Tirosh, E. Flaxer and F. Patolsky, Optically-Gated Self-Calibrating Nanosensors: Monitoring PH and Metabolic Activity of Living Cells, Nano Lett., 2013, 13(7), 3157–3168, DOI:10.1021/nl401169k.
- M. T. Guardado-Alvarez, M. M. Russell and J. I. Zink, Nanovalve Activation by Surface-Attached Photoacids, Chem. Commun., 2014, 50(61), 8388–8390, 10.1039/C4CC03293D.
- H. Peretz-Soroka, A. Pevzner, G. Davidi, V. Naddaka, M. Kwiat, D. Huppert and F. Patolsky, Manipulating and Monitoring On-Surface Biological Reactions by Light-Triggered Local PH Alterations, Nano Lett., 2015, 15(7), 4758–4768, DOI:10.1021/acs.nanolett.5b01578.
- D. Wang and H. Möhwald, Rapid Fabrication of Binary Colloidal Crystals by Stepwise Spin-Coating, Adv. Mater., 2004, 16(3), 244–247, DOI:10.1002/adma.200305565.
- Z. Huang, N. Geyer, P. Werner, J. de Boor and U. Gösele, Metal-Assisted Chemical Etching of Silicon: A Review, Adv. Mater., 2011, 23(2), 285–308, DOI:10.1002/adma.201001784.
- F. Patolsky, G. Zheng and C. M. Lieber, Fabrication of Silicon Nanowire Devices for Ultrasensitive, Label-Free, Real-Time Detection of Biological and Chemical Species, Nat. Protoc., 2006, 1(4), 1711–1724, DOI:10.1038/nprot.2006.227.
- Properties of Porous Silicon, ed. C. Leigh, and L. T. Canham, Inst of Engineering & Technology, 1997, vol. 18 Search PubMed.
- A. Loni, D. Barwick, L. Batchelor, J. Tunbridge, Y. Han, Z. Y. Li and L. T. Canham, Extremely High Surface Area Metallurgical-Grade Porous Silicon Powder Prepared by Metal-Assisted Etching, Electrochem. Solid-State Lett., 2011, 14(5), K25, DOI:10.1149/1.3548513.
- R. L. A. Timmer, M. J. Cox and H. J. Bakker, Direct Observation of Proton Transfer in Ice Ih Using Femtosecond Spectroscopy, J. Phys. Chem. A, 2010, 114(5), 2091–2101, DOI:10.1021/jp908561h.
- M. M. A. Kelson, R. S. Bhosale, K. Ohkubo, L. A. Jones, S. V. Bhosale, A. Gupta, S. Fukuzumi and S. V. Bhosale, A Simple Zinc-Porphyrin-NDI Dyad System Generates a Light Energy to Proton Potential across a Lipid Membrane, Dyes Pigm., 2015, 120, 340–346, DOI:10.1016/j.dyepig.2015.04.040.
- R. Simkovitch, D. Pines, N. Agmon, E. Pines and D. Huppert, Reversible Excited-State Proton Geminate Recombination: Revisited, J. Phys. Chem. B, 2016, 120(49), 12615–12632, DOI:10.1021/acs.jpcb.6b09035.
- B. Strohmeier, R. White, T. Nunney, P. Mack and A. Wright, Chemical Characterization of Material Surfaces Using X-Ray Photoelectron Spectroscopy (XPS): The Perfect Complement to Electron Microscopy Techniques, Microsc. Microanal., 2014, 20, 2062–2063, DOI:10.1017/S1431927614012045.
- M. W. Tuijtel, A. A. Mulder, C. C. Posthuma, B. van der Hoeven, A. J. Koster, M. Bárcena, F. G. A. Faas and T. H. Sharp, Inducing Fluorescence of Uranyl Acetate as a Dual-Purpose Contrast Agent for Correlative Light-Electron Microscopy with Nanometre Precision, Sci. Rep., 2017, 7(1), 10442, DOI:10.1038/s41598-017-10905-x.
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Recovery of Uranium from Seawater: A Review of Current Status and Future Research Needs, Sep. Sci. Technol., 2013, 48(3), 367–387 CrossRef CAS.
- J. Lehtonen, J. Hassinen, A. A. Kumar, L.-S. Johansson, R. Mäenpää, N. Pahimanolis, T. Pradeep, O. Ikkala and O. J. Rojas, Phosphorylated Cellulose Nanofibers Exhibit Exceptional Capacity for Uranium Capture, Cellulose, 2020, 27, 10719–10732, DOI:10.1007/s10570-020-02971-8.
- H.-B. Pan, C. M. Wai, L.-J. Kuo, G. A. Gill, J. S. Wang, R. Joshi and C. J. Janke, A Highly Efficient Uranium Grabber Derived from Acrylic Fiber for Extracting Uranium from Seawater, Dalton Trans., 2020, 49(9), 2803–2810, 10.1039/C9DT04562G.
- Modelling and Interpretation of Adsorption Isotherms, accessed Jan 20, 2021, https://www.hindawi.com/journals/jchem/2017/3039817/.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00277e |
| This journal is © The Royal Society of Chemistry 2021 |