
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent and non-covalent coupling of a Au102 nanocluster with a fluorophore: energy transfer, quenching and intracellular pH sensing†
Eero
Hulkko
a,
Tanja
Lahtinen
 a,
Varpu
Marjomäki
b,
Emmi
Pohjolainen
c,
Ville
Saarnio
a,
Karolina
Sokolowska
a,
Ardra
Ajitha
a,
Mikael
Kuisma
a,
Lauri
Lehtovaara
a,
Gerrit
Groenhof
a,
Hannu
Häkkinen
ac and
Mika
Pettersson
*a
a,
Varpu
Marjomäki
b,
Emmi
Pohjolainen
c,
Ville
Saarnio
a,
Karolina
Sokolowska
a,
Ardra
Ajitha
a,
Mikael
Kuisma
a,
Lauri
Lehtovaara
a,
Gerrit
Groenhof
a,
Hannu
Häkkinen
ac and
Mika
Pettersson
*a
aDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014, Finland. E-mail: mika.j.pettersson@jyu.fi
bDepartment of Biology and Environmental Science, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014, Finland
cDepartment of Physics, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014, Finland
First published on 24th September 2021
Abstract
Interactions between an atomically precise gold nanocluster Au102(p-MBA)44 (p-MBA = para mercaptobenzoic acid) and a fluorescent organic dye molecule (KU, azadioxatriangulenium) are studied. In solution, the constituents form spontaneously a weakly bound complex leading to quenching of fluorescence of the KU dye via energy transfer. The KU can be separated from the complex by lowering pH, leading to recovery of fluorescence, which forms a basis for an optical reversible pH sensor. However, the sensor is not a stable entity, which could be delivered inside cells. For this purpose, a covalently bound hybrid is synthesized by linking the KU dye to the ligand layer of the cluster via an ester bond. Covalent linking facilitates entry of the cluster–dye hybrids into cells via endocytosis. Inside cells, the hybrids accumulate in endosomes where Au102 releases its cargo via hydrolysis of the ester bond. Changes of the local pH inside endosomes regulate reversible fluorescence due to variations in the interactions between the Au102 cluster and the dye. This work presents a concept for delivering reporter molecules into cells by using atomically precise gold nanoclusters as carriers and paves the way for future developments of cluster-reporter sensors for in vivo measurements of e.g. absolute pH values or ion concentrations.
Introduction
Monolayer-protected gold nanoclusters are fundamentally interesting because their size (typically 1–2 nm for the metal core) covers transition from molecular to metallic behavior.1,2 The structures of many nanoclusters have been determined to atomic precision enabling studies of exact structure–property relationships.2,3 Nanoclusters are synthesized with wet-chemistry methods and they can be made water soluble, which expands the scope of applications to biology. Compared to large plasmonic nanoparticles, gold nanoclusters are much smaller, but they can still be easily seen in electron microscopy, providing information on the structures of bionanoparticles or biomolecules inside cells. Recent studies have shown how gold nanoclusters can be used as markers for electron microscopy enabling structural studies of enteroviruses and virus-like particles4–6 and intracellular tracking of proteins.7Using gold nanoclusters as fluorescence imaging labels is currently of high interest. Smallest nanoclusters with few or few tens of metal atoms are fluorescent themselves and can be used for bioimaging.8–10 Metal cores of nanoclusters are usually stabilized with some protecting layer, such as dendrimers.11,12 While dendrimer-encapsulated clusters exhibit high quantum yields of up to ∼40%, the values are much lower for monolayer-protected nanoclusters. Typical values for the quantum yield are 10−5 to 10−3 for nanoclusters with few tens of atoms.13 The mechanism of luminescence in nanoparticles is currently not well understood.13,14
Functionalization of nanoclusters with dye molecules is an alternative approach to tune their fluorescent properties. Varnholt et al. attached porphyrin molecules covalently to a Au38L24 cluster (L = phenylethenethiolate) by ligand exchange.15 They observed a shoulder in the electronic absorption spectrum of porphyrin, which they attributed to excitonic coupling. The fluorescence intensity of porphyrin decreased by a factor of 10 upon attachment to the gold cluster and this was explained by energy transfer to the gold cluster. Pyo et al. conjugated fluorescein to Au22 nanoclusters which led to a large enhancement of the pH-dependent emission intensity of the dye-molecule.16 The conjugate was tested for imaging in cells and high photostability was observed compared to the bare dye, showing that dye-functionalized nanoclusters exhibit beneficial properties for bioimaging. The electronic properties of Au25–pyrene hybrid was studied theoretically by TD-DFT methods and it was found that the coupling between the dye and the cluster is very weak.17
Cheng et al. studied dynamic and static quenching of fluorophores by small monolayer-protected gold nanoclusters with diameters of 1–4 nm.18 They observed that the fluorophores were dynamically quenched by energy transfer in all cases and that the quenching efficiency increased with the nanocluster core size. They also observed static quenching of an electrostatically bound [Ru(bpy)3]2+ dye due to energy transfer from the dye molecule to the nanocluster. Energy transfer between a non-plasmonic Au20 cluster and a fluorophore was studied by TD-DFT and compared to a hypothetical plasmonic particle. It was found that the energy transfer rate was similar in both cases underlining that plasmonic effects are not essential for efficient energy transfer between metal particles and molecules.19 Quenching of fluorophores by energy transfer is beneficial for development of optical sensors when interactions between the nanocluster and the fluorophore depend on a quantity to be measured.
In this paper, we report a study of interactions between the atomically precise para-mercaptobenzoic acid protected gold nanocluster Au102(p-MBA)44 and an azaoxotriangulenium dye molecule. Au102 cluster was chosen for the study because it is one of the few water-soluble gold clusters for which structure and properties are known, providing excellent background knowledge for the study.3 In water solution, the nanocluster–dye pair forms a weakly bound complex, for which the binding constant depends on pH. In the complex, fluorescence of KU is totally quenched while the free unbound molecule exhibits strong fluorescence. This scheme forms the basis for a spectroscopic pH sensor. However, the sensor cannot be loaded directly into living cells due to dissociation of the complex at physiological salt concentration. To avoid dissociation, the complex was covalently coupled which enabled loading of the sensor into living cells. We demonstrate that the hybrid functions as an intracellular pH sensor by performing live-cell imaging of endosome acidification dynamics. The results are important for developing new probes for bioimaging. In addition to fluorescence imaging, the hybrid could be used for high-resolution electron microscopy of proteins and other biomolecules. The concept can potentially be extended to probe also other intracellular properties, such as ion concentrations by tuning the chemistry of the ligand layer as well as the overall size of the nanocluster.
Experimental
All materials were commercial unless otherwise mentioned and used without further purification. Au102(pMBA)44 (2) was prepared according to literature procedures.20 The KU-dye used in the complex studies was obtained as a courtesy of Thomas Just Sørensen and Bo W. Larsen from the University of Copenhagen and was used without further modifications.Synthesis of covalently bound Au102–KU (3)
A solution of azadioxatriangulenium dye (1) (1.0 mg, 2.32 μmol) in dry DCM (1 mL) was added to a pre-sonicated solution of Au102(pMBA)44 (2) (2.5 mg, 0.0931 μmol) in dry DMSO (5 mL) in a 25 mL round-bottom flask. The reaction mixture was vigorously stirred for 20 minutes at RT. After this, a solution of N,N′-dicyclohexylcarbodiimide (1 mg, 4.84 μmol) in dry DCM (1 mL) was added dropwise to the cooled reaction mixture on an ice-bath while stirring. The reaction mixture was then allowed to warm to RT and continued to stir overnight. After this, the reaction mixture was centrifuged at 3500 rpm for 5 min. Supernatant was collected and transferred to a new 50 mL conical and the functionalized nanoparticles were precipitated from the solution by adding NH4OAc (73 mg, 0.82 mmol) and MeOH (20 mL). The conical was shaken to mix all the contents and then centrifuged at 3500 rpm for 15 min. The precipitates were collected, dried and dissolved in ultrapure water.The amount of linker molecules per cluster could not be accurately controlled but it was kept sufficiently low so that the functionalized clusters remained water-soluble. Substitution of all p-MBA ligands with KU would lead to decrease in water solubility.
PAGE
PAGE was run on a 15% polyacrylamide gel (29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acrylamide:bisacrylamide) using a 1× TBE run buffer in Bio-Rad Mini-Protean Tetra System gel electrophoresis apparatus at 130 V. Page gels were imaged using a Olympus E-620 camera. UV-illumination PAGE gels were done with 254 nm UV-lamp.
:1 acrylamide:bisacrylamide) using a 1× TBE run buffer in Bio-Rad Mini-Protean Tetra System gel electrophoresis apparatus at 130 V. Page gels were imaged using a Olympus E-620 camera. UV-illumination PAGE gels were done with 254 nm UV-lamp.
Optical spectroscopy methods
Sample absorbance and photoemissions were collected using a commercial PerkinElmer Lambda 850 UV/vis absorption, and LS 55 luminescence spectrophotometers. For Au102 complex samples, fluorescence intensities were collected with Horiba AquaLog spectrophotometer. Sample solutions were measured at ambient condition using quartz absorption or fluorescence cuvettes (Hellma). We used nanopure water as a background in all of our solution spectroscopy experiments. Fluorescence emission spectra were collected using λexc = 500 nm excitation. Excitation spectra were collected by detecting fluorescence intensity at λdet = 600 nm, while scanning the λexc.. Lifetime of KU fluorescence was measured with a PicoQuant HydraHarp400 time-correlated single-photon counting module, combined to a Hamamatsu microchannel plate photomultiplier tube. Pulsed 485 nm laser diode (pulse duration ∼ 100 ps) was used as an excitation source. Additional details of the pH-dependent spectroscopic measurements and sample preparation are given in ESI1.†Transmission electron microscopy (TEM) sample was prepared by drop-casting ∼10 μL of dilute (basic) cluster sample solution on a glow discharged 400 mesh holey carbon copper grid (Ted-Pella ultrathin c). The hybrid sample solution was allowed to deposit for ∼15 min, after which excess sample was removed and the grid was allowed to dry in glass desiccator under vacuum overnight. Samples were imaged using a JEOL JEM-1400HC TEM operated at 120 kV, equipped with an 11 Megapixel CCD camera (Olympus SIS Quemesa).
Molecular dynamics simulations
Simulations were performed with Gromacs 5.1.4 simulation package21 using AMBER compatible force field parameters previously derived for the Au102(pMBA)44 nanocluster.22 The AMBER-compatible parameters for the KU-dye were derived using the Antechamber software of the Ambertools package.23,24 Simulations were performed for four main systems including fully protonated or fully deprotonated gold nanocluster with covalently (production simulations 250 ns) or non-covalently bound (production simulations 100 ns) KU-dye. For technical details of both Gromacs simulations see the ESI.†Live imaging and confocal microscopy
HeLa MZ cells (a kind gift from Marino Zerial, Max Planck Institute, Dresden) were grown on Ibidi coverglass-bottom 8-well imaging plates to subconfluency using DMEM containing 10% fetal bovine serum and antibiotics (penicillin/streptomycin). For live imaging, internalization medium consisting of the CO2-independent medium (Gibco), 0.2% bovine serum albumin and Glutamax (Invitrogen) was changed on cells. Ten to twenty minute pulse of 1 μM Au102–KU (1 volume of 2 μM probe mixed with 1 volume of internalization medium) was added on cells and incubated at 37 °C. Thereafter, the cells were briefly washed with the internalization medium for 3 times and continued incubating in the same medium at the Olympus microscope IX81 with a FluoView-1000 confocal setup at 37 °C (with plate and objective heater and ambient temperature set to 37 °C). The cells were imaged using 488 nm excitation (argon ion laser) and emission recorded at 572 nm. Bafilomycin A1 (Calbiochem) was added at 50 nM concentration while incubating the cells in Olympus microscope to revert pH to neutrality in endosomes as observed before.25 Regarding the possible cytotoxicity, during experiments when Au102 was introduced to cells and followed in overnight incubations, we did not observe any cell detachment, which is a typical readout in cytotoxicity assays. Furthermore, we did not observe any change in the cell morphology (e.g. rounding up before possible detachment). Thus, there is no evidence of cytotoxicity in these experiments.Results and discussion
Non-covalently bound Au102–KU complex
We initially studied properties of a non-covalently bound complex system consisting of the N-propyl alcohol-azadioxatriangulenium tetrafluoroborate salt (1), hereafter called KU, and the para-mercaptobenzoic acid protected gold nanocluster Au102(p-MBA)44 (2), hereafter called Au102. Molecular structures of the KU and the p-MBA acid ligands of the cluster are presented in Fig. 1a. The KU is a cationic dye with a fluorescence lifetime τF = 23 ns and a fluorescence quantum yield ΦF = 0.69.26–28 Au102 was synthesized and purified according to the procedure described in ref. 20. X-ray crystallographic analysis has revealed that the gold nanocluster has a core diameter of ∼1.3 nm.3 Comparison of relative sizes of the complex constituents are shown as atomic structures for non-covalently interacting Au102 and KU in Fig. 1b.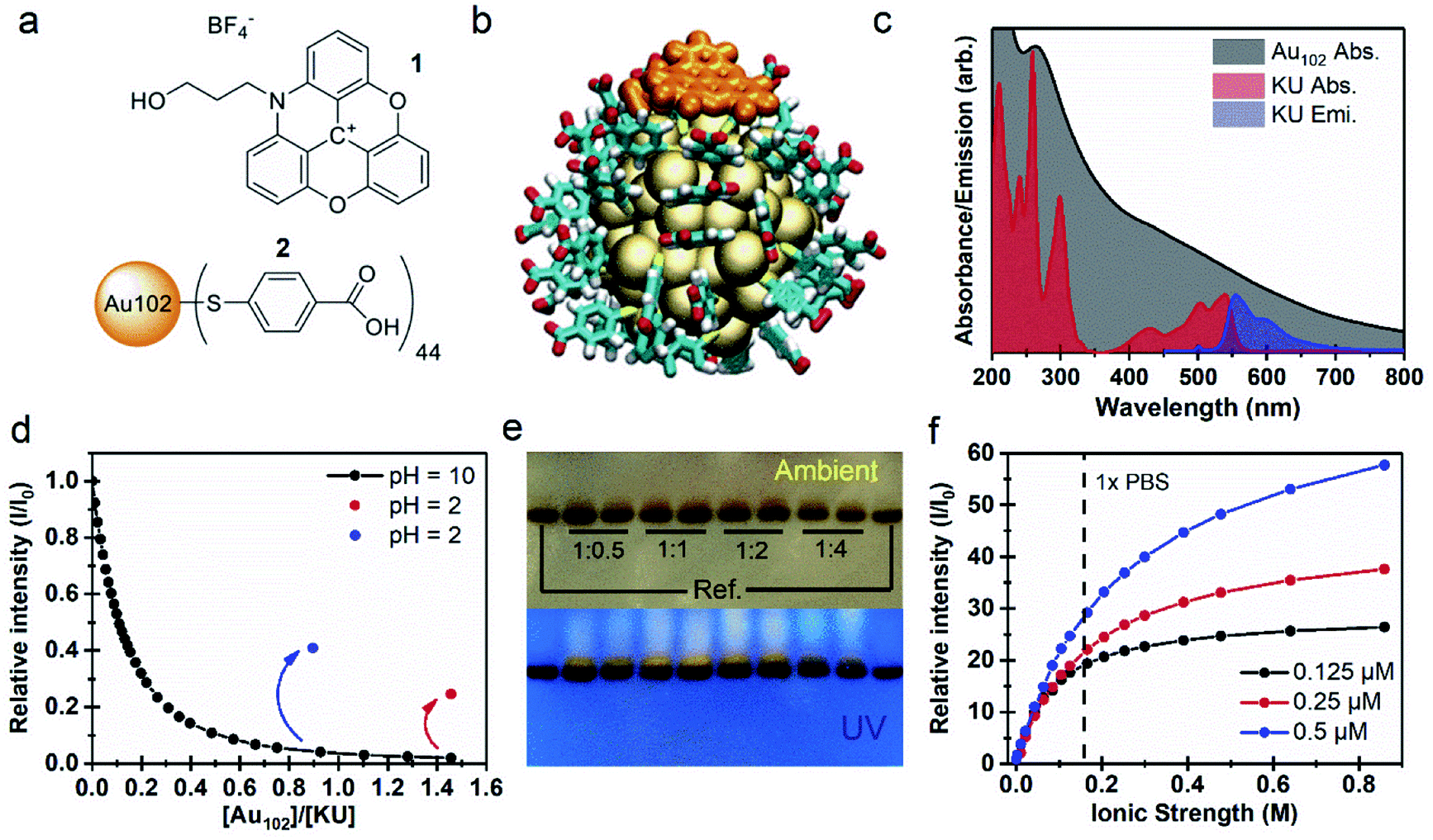 | ||
| Fig. 1 (a) Molecular structures of (1) KU and (2) p-MBA ligands. (b) Atomic structure of Au102–KU complex, with deprotonated p-MBA ligands. Colour coding (orange) KU atoms (yellow) Au (cyan) p-MBA C (white) H (red) O. (c) Absorption spectra of Au102 and KU in water solution in comparison with the emission spectrum of KU with λex = 500 nm. (d) Relative intensity of KU emission as function of [Au102]/[KU] concentration ratio in basic conditions (pH = 10, black dots) and after acidification of the solution (pH = 2, red and blue dots). (e) Image of dried PAGE gel in ambient room lighting and under UV light (λ = 254) for different Au102:Ku ratios in comparison with two Au102 references. (f) Relative intensity of KU emission with respect to the initial fluorescence of the Au102–KU mixture with different concentrations plotted as a function of increasing ionic strength. | ||
UV-vis absorption spectra of Au102 and KU in water solutions are presented in Fig. 1c. The Au102 cluster has a pseudo-continuous absorption spectrum spanning the visible and the UV-region, without significant resolvable electronic transitions, apart from the peak around 265 nm belonging to hybridized transitions localized mainly on the p-MBA ligands.29 The KU-dye with C2v symmetry exhibits several resolvable singlet-to-singlet S0 → Sn absorption bands in the vis/UV region, where the corresponding transition dipoles are located in the plane of the molecule.27 While Au102 is not luminescent due to rapid non-radiative relaxation,30 KU emits fluorescence in the wavelength range of 550–700 nm, as shown in Fig. 1c for 500 nm excitation (blue spectrum). Note that the electronic absorption of the cluster is strongly overlapping with the dye absorption and emission bands, forming the basis of the fluorescence control by energy transfer.
When Au102 is added to an aqueous solution of KU in basic conditions (pH = 10), fluorescence intensity of KU drops rapidly (Fig. 1d). The surface of Au102 gold nanocluster is covered by a p-MBA monolayer with 44 carboxylic acid groups. In basic conditions (pH above 8) the acid groups are in the deprotonated (–COO−) state, and the cluster has a nominal surface charge of −44e.31 The KU-dye is a positively charged (+1) cation. Therefore, formation of an electrostatically bound complex between the cluster and the dye is expected in basic environment. The fluorescence intensity behavior in Fig. 1d supports this argument, where strong quenching of fluorescence is observed when Au102 cluster is added to the basic dye solution. The complexation involves several KU molecules interacting with one Au102 cluster. From the initial slope of the Au102 titration curve (Fig. 1d) we determined approximately that at the low Au102 concentrations ([Au102]/[KU] < 0.1), five KU molecules bind on average to one cluster (see Fig. SI2†). When the [Au102]/[KU] ratio reaches approximately 1, fluorescence is almost totally quenched, indicating that most KU molecules are complexed with Au102. In principle, the average number of KU molecules bound to Au102 can be controlled by varying the concentrations of the complex constituents, which shifts the equilibrium.
If the pH of the water solution is acidic (pH below 4) the cluster ligands are in a protonated (–COOH) state and the electrostatic binding in Au102–KU complex is expected to be reduced due to disappearance of coulombic interaction. We verified the complex dissociation in acidic environment by lowering the pH to 2 with HCl at the end of two separate Au102-titration experiments. Acidification resulted in increase of fluorescence intensity (see Fig. 1d, red and blue dots), indicating that KU molecules were released from complexes due to the change in the protonation state of the clusters. The magnitude of the fluorescence increase depends on the total amount of Au102 added. With a final Au102 concentration of 0.29 μM we observed 13-fold increase and 25% recovery of the initial intensity (red dot). In a separate experiment with a lower concentration of Au102 (0.16 μM), the recovered fluorescence intensity is notably higher (41%, blue dot). Note that only the intensity increase is presented for the 0.16 μM solution in Fig. 1d. The full titration data is shown in Fig. SI2.† The incomplete recovery of the fluorescence intensity can be due to two effects: (i) also at low pH (protonated ligands) there is non-covalent binding between the dye and the Au102, but with a lower affinity than at higher pH (deprotonated ligands); (ii) part of this effect can be due to dynamic quenching, i.e. randomly moving acceptor and donor coming to sufficiently close range for long-distance energy transfer to take place. The pH dependency of the fluorescence, observed here, demonstrates that the Au102–KU complex can be utilized as a reversible pH-sensor.
Formation of the Au102–KU complex was verified using polyacrylamide gel electrophoresis (PAGE) where Tris–borate–EDTA (TBE), used as a running buffer (pH = 8.3), controls the protonation state of the p-MBA ligands and the Au102–KU complex formation equilibria (see Fig. 1e). On the left- and rightmost lanes, only bare cluster was loaded, while other lanes contained a mixture of Au102 and KU at different ratios (indicated in the figure). The upper PAGE image taken in ambient room lighting shows two results: (1) the synthesized Au102 is monodisperse, since only one well defined band is observed; (2) in the lanes containing KU, a diffuse band is observed just above the cluster band, indicating a species with smaller charge/mass ratio than for the bare Au102. Because the charge of the KU is positive while the cluster is negative, the isolated Au102 and KU are pulled in opposite directions under the influence of the static electric field (see ESI3†). The lower image in Fig. 1e, shows the same gel under UV-light. The KU is fluorescent making it selectively visible. The dye is localized in a distinct band above the gold cluster band confirming that the KU is complexed with the gold nanocluster and the complex diffuses slower than the bare cluster. Note that the wet gel does not show fluorescence from the complex due to quenching (see ESI3†). We hypothesize that drying in methanol leads to a change in the protonation of the cluster ligands as well as rearrangement of the complex in a dissociated form that activates the KU fluorescence. There is also weak fluorescence all the way up to the loading point of the samples indicating that some KU molecules escape from the complex during the PAGE run and are mobile in the opposite direction from the cluster. This perfectly fits the picture of a reversibly bound complex with an equilibrium strongly on the side of the complex in the basic pH environment of the gel.
An important consideration for bio-applications is the salt concentration of the environment. The fluorescence titration of 1:1 Au102–KU mixtures with 25 × PBS solution (phosphate-buffered saline) shows a strong correlation between the integrated emission intensity and the ionic strength of the resulting solution (Fig. 1f). While the Au102–KU mixtures at the starting point (pH = 9) are only weakly fluorescent, increasing the concentration of PBS leads to a prominent increase of the emission intensity (I) relative to the initial fluorescence intensity (I0), suggesting the complex dissociates at increasing ion concentrations. We observed no significant effect on the shape of the KU fluorescence spectra by the increasing ionic strength (Fig. SI4†). In the physiological conditions (1 × PBS), the lowest concentration of the Au102–KU complex (0.125 μM) has almost completely dissociated, shown as the slowly saturating emission intensity curve (black dots). It can be estimated that the complete dissociation of the dye from the cluster surface requires a 104 molar excess of ions with respect to the Au102–KU complex. At the higher concentration of the complexes (0.25 μM and 0.5 μM), the emission intensity continues to rise with increasing ionic strength, but does not reach saturation with the tested ionic strengths. In titrations with sodium chloride, the behavior of the fluorescence intensity is similar as in the titrations with PBS (see ESI4†). The emission enhancement with NaCl was slightly smaller compared to PBS and also the differences between tested concentrations are less prominent. This result could be due to effects from the differently charged ions in PBS or the slight effect that the buffer can have on the pH of the solution. Nevertheless, the increase of fluorescence intensity with increasing ionic strength is clearly observed in both cases.
We also investigated the non-covalent binding between the Au102 cluster and KU, by performing molecular dynamics (MD) simulations in explicit solvent. The results of these simulations show that for both fully deprotonated and protonated cluster, binding of the KU is thermodynamically favorable. In both situations, KU interacts with the p-MBA layer, with virtually identical distances to the center of the gold core (1.39 ± 0.04 nm for the fully protonated cluster and 1.29 ± 0.03 nm for the deprotonated cluster). A representative average structure from MD simulations of fully deprotonated Au102 interacting with non-covalently bound KU-dye was shown in Fig. 1b. For further details of simulations and representative structures for both protonation states, see ESI5.†
Fluorescence quenching in triangulenium dyes is known to occur through a photoinduced electron transfer (PET) mechanism, in which the chromophore acts as an electron acceptor. The PET induced quenching is observed in the presence of electron donor molecules, or by intramolecular electron transfer from electron rich functional groups, such as phenols or morpholines.32,33 In the present case the electron transfer mechanism is most likely not the dominant quenching mechanism. Instead, the fluorescence quenching is more likely dominated by the energy transfer due to the strong spectral overlap between the KU fluorescence spectrum and the cluster absorption (Fig. 1c). We estimated an energy transfer rate for the complex based on Förster theory34 and available spectroscopic data for the Au102 and the KU.26–28,35 The procedure is described in detail in ESI6.† The estimated energy transfer rates (and fluorescence quantum yields) for 1.39 nm and 1.29 nm donor–acceptor distances are 2.1 × 1012 s−1 (1.4 × 10−5) and 1.3 × 1012 s−1 (2.2 × 10−5), respectively, confirming that energy transfer is highly efficient. Thus, the very low fluorescence quantum yield seems due the close proximity of the dye and the metal cluster (for simulations of donor–acceptor distances, see ESI6†). In the Au102–KU complex the KU fluorescence is expectedly totally quenched and the system is in a “fluorescent off-state”.
Covalently bound Au102–KU hybrid
Next, we developed a covalently bound cluster–dye hybrid system, where KU was covalently bound to the p-MBA ligand layer of Au102 clusters via Steglich esterification reaction.3,4,20 The covalently bound dye–cluster system is shown in Fig. 2a and it is hereafter called Au102–KU hybrid (3). Synthesis was done in dry DMSO/DCM in the presence of dicyclohexylcarbodiimide (DCC). For details of synthesis, see Experimental section. The used strategy is similar as in the covalent linking of gold nanoclusters to viruses that we have shown previously.4,5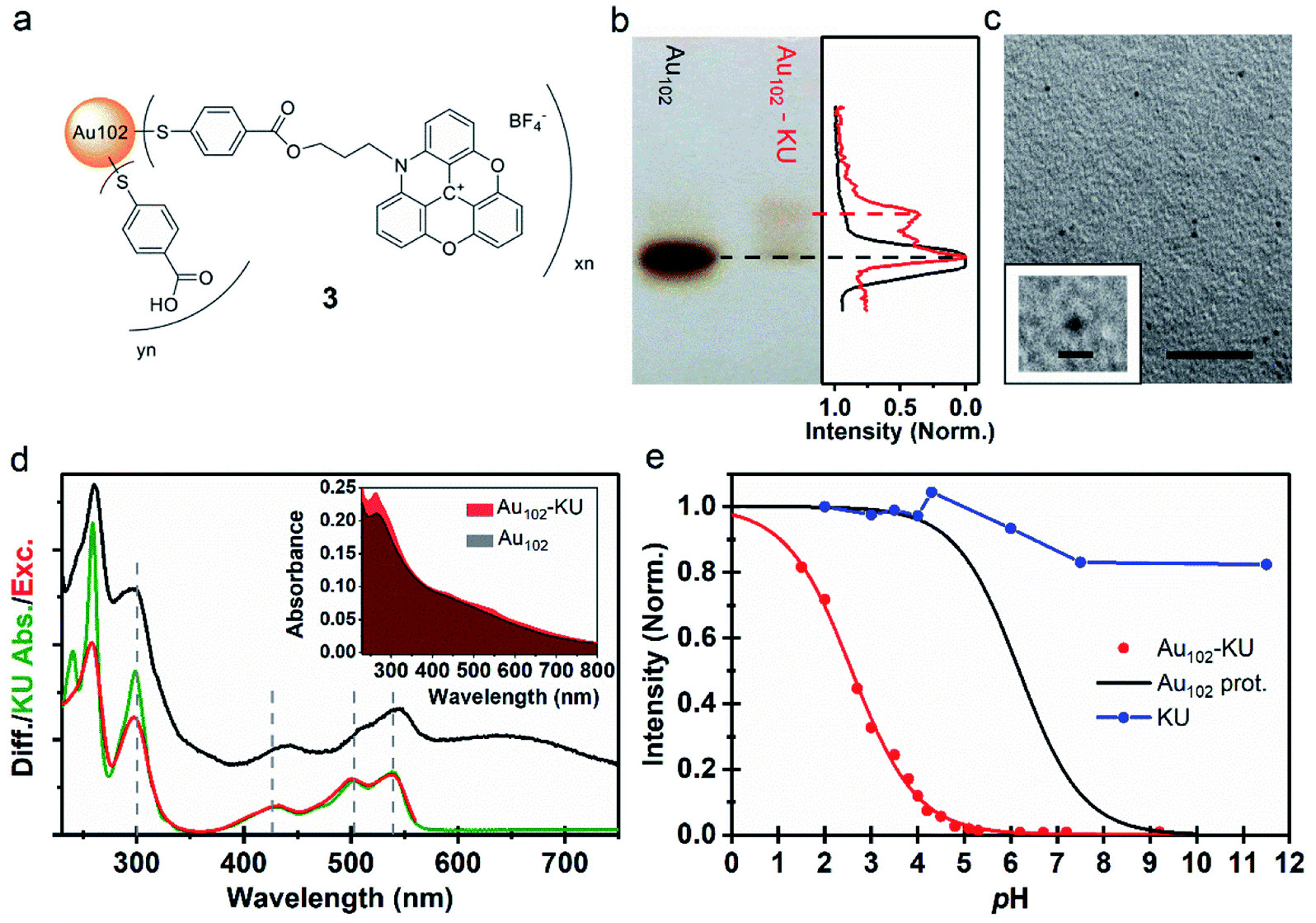 | ||
| Fig. 2 (a) Proposed molecular structure of Au102–KU hybrid (3). (b) (Left) Image of PAGE run of Au102–KU hybrid synthesis product with Au102 reference. (Right) Normalized grayscale intensity cross-section of the Au102 reference (black) and Au102–KU hybrid (red) PAGE lanes. (c) TEM image of Au102–KU hybrid sample, scale = 20 nm. (Inset) Close-up of cluster, scale = 2 nm. (d) (Inset) Full absorption spectrum of Au102–KU-hybrid in comparison to Au102 spectrum in basic (pH = 10) water solution. (Main panel) Difference spectrum of Au102–KU-hybrid and Au102 (black), absorption spectrum of KU in water (green), and excitation spectrum of Au102–KU-hybrid (pH = 4.1) with detection at λ = 600 nm (red). (e) Normalized fluorescence intensity pH-dependence for Au102–KU-hybrid (red points) and aqueous KU solution (blue points). Red curve: Result of least squares fitting with using eqn (1) (for parameters, see text, direction of pH change from basic to acidic). Black curve: Ligand protonation behavior of Au102(p-MBA)44 Au102(p-MBA)44 from ref. 31. | ||
Cluster functionalization was confirmed using PAGE (Fig. 2b). A single band is observed for the reference Au102 (dashed black line) indicating monodisperse cluster size. For the hybrid sample we observe single main band, corresponding to Au102 cluster, and a distinctive band of a lower mobility species (dashed red line). Note that the band of the hybrid is different from the band of the non-covalently bound complex (Fig. 1e) implying a different type of interaction after covalent linking (For comparison, see ESI7†). The distinctive brown color of the Au102 hybrid and the similar PAGE running distance suggests that the gold core of the Au102 has remained intact during the covalent attachment of the KU. We performed transmission electron microscopy (TEM) for the hybrid sample (Fig. 2c; for additional images see ESI8†). The observed cluster size is similar to the bare Au102 cluster,3,20 confirming that covalent bonding of the KU dyes does not lead to change in the cluster size.
The UV-vis spectrum of the Au102–KU hybrid is shown in Fig. 2d (inset). The spectral features of KU are present in the Au102–KU hybrid spectra. This is most visible for the peak at ∼250 nm but also the three KU absorptions at 400–550 nm are observable. Similarly, presence of KU was evident in Raman spectra of dried Au102–KU samples (see ESI9†). A difference spectrum between the Au102–KU hybrid and Au102 is presented in Fig. 2d (main panel, black spectrum), where we have subtracted the cluster contribution from the hybrid spectrum (Abs(Au102–KU) − Abs(Au102)). The obtained KU spectrum in the hybrid is similar to the spectrum of free KU (green spectrum), but there are notable differences. The peak at ∼250 nm is broadened and the transitions at 400–550 nm are broadened and red-shifted by 8 nm indicating interaction between the dye and the cluster. We estimated the molar ratio between Au102 and KU in the hybrid based on the previously determined molar absorption coefficients of the constituents.27,34 The estimated ratio was 1 ± 1, which qualitatively agrees with approximate stoichiometry observed in analysis of PAGE images (Fig. 2b right panel). Presumably, the ratio could be changed by varying the reactant concentrations, but we have not studied this systematically.
Fig. 2e depicts the pH-dependence of the Au102–KU and KU fluorescence intensities. The bare KU-dye shows strong fluorescence throughout the studied pH range and the variation of intensity is less than 20%. The hybrid fluorescence on the other hand shows approximately 2 orders of magnitude difference in intensity between basic pH range and the lowest measured pH = 1.5. We verified that the fluorescence increase at low pH and quenching in basic conditions are reversible by cycling the pH between pH = 11 and pH = 4. We determined an approximate pKa = 2.37 and steepness factor k = 0.62 for the pH dependent intensity data by least squares fitting the points to a Henderson–Hasselbach equation
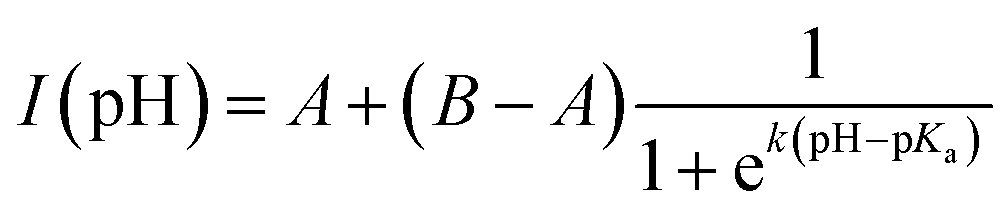 | (1) |
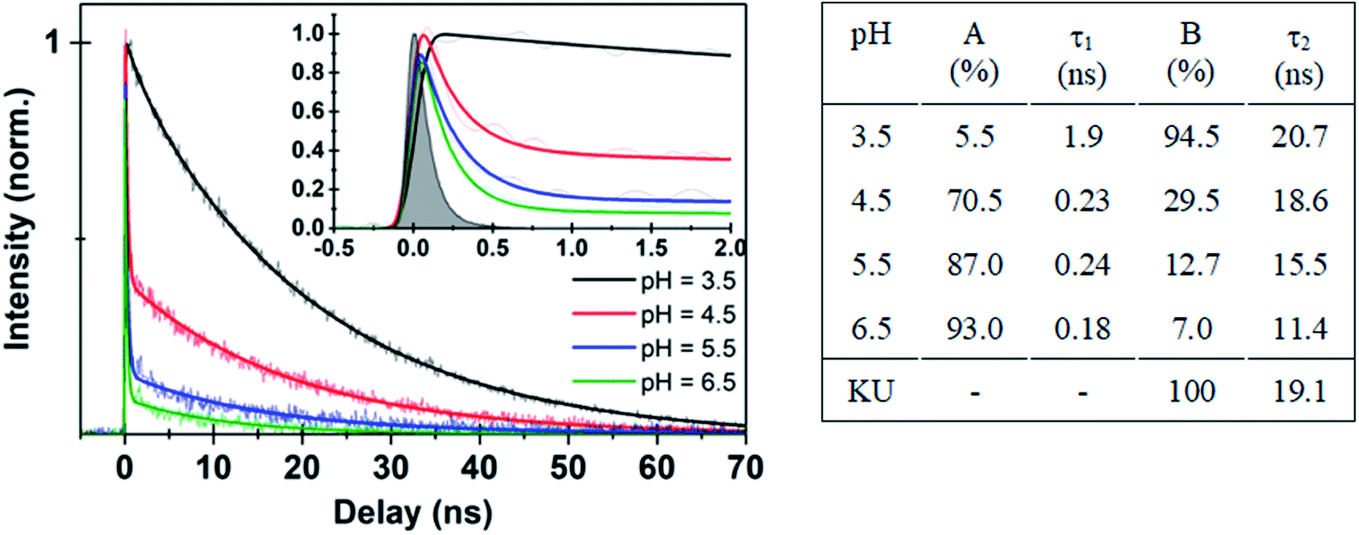 | ||
| Fig. 3 (Left panel) Time-resolved fluorescence data for Au102–KU hybrid at different pH values. Thick solid lines are deconvolution fits with two exponential functions (see ESI11†) to the experimental data points. Inset shows expanded view of the early time dynamics for the different pH values. (Right panel) Fitted parameters from different pH decay curves in comparison with KU dye. | ||
The interpretation of the fluorescence data suggests that the initially covalently bound complex dissociates at low pH. Otherwise, fluorescence would not be activated as quenching by energy transfer cannot be avoided due to the close proximity of the dye molecule (∼1 nm, see ESI5† for MD structures) to the gold nanocluster.36 The increase of fluorescence from the covalently bound hybrid at low pH can be explained by hydrolysis of the ester bond which is known to occur either at acidic or basic conditions.37 Effectively this leads to a transition of the system from a covalently bound hybrid to a non-covalently bound complex, where activation of the fluorescence is observed at low pH due to the spatial separation of the dye and the cluster. The dissociation process is also supported by excitation spectrum of the hybrid measured at pH = 4.1 (Fig. 2d, red spectrum), which has the same spectral shape as the free KU.
Further evidence for the dissociation of the hybrid was obtained from time-resolved fluorescence studies. Time traces of the bare KU dye and the initially covalently bound Au102–KU hybrid at high and low pH are shown in Fig. 3. The instrumental response is included for reference. The bare KU has a fluorescence lifetime of 19.1 ns in water (Fig. SI†10), in good agreement with literature value of 23.2 ns, measured at acetonitrile.27 The Au102–KU hybrid shows essentially the same lifetime as free KU at low pH (3.5). When pH is raised, two time-components are observed: a long time-component, similar to the free KU and a short, instrument-limited lifetime (<100 ps). The proportion of the short lifetime component increases at high pH. The fitted parameters are given in Fig. 3. The observation of two time-components from which the longer one is identical to the time constant of the free KU is in agreement with the model of quenching by complexation. The time constant for the complexed dye being less than 100 ps is also compatible with the predicted very fast energy transfer obtained by Förster resonance energy transfer calculations.
Live cell imaging using the Au102–KU complex and the hybrid
After establishing the fluorescent properties of the Au102–KU complex and the hybrid, we performed confocal fluorescence live cell imaging using different probe systems aiming at visualization of the acidification dynamics of cellular vesicles late endosomes and lysosomes along the endocytic route.25 The complex, hybrid, and KU dye control probes were introduced to cells by using a pulse chase experiment. One mM of the probes was internalized for 10 min at 37 °C in HeLa cells followed by quick 4 washes and a chase in the internalization medium at 37 °C. The probes were internalized into vesicles along the endocytic pathway, similar to any fluid-phase marker.25,38The covalently bound Au102–KU hybrid probe accumulated in vesicles with a perinuclear location by 2 hours as is expected for any fluid-phase marker entering low pH endosomes/lysosomes; see Fig. 4 (upper row). The fluorescence intensity of the vesicles was faint at 15 min post internalisation (p.i.) but increased in intensity with time. This observation suggests that a pH drop in the endosomes was linked with a release of the KU from the cluster and thereby led to the increase of fluorescence intensity. At 2 h p.i. the probe showed intense signal in perinuclear vesicles. This signal was even brighter after overnight incubation suggesting that accumulation into more acidic endosomes and lysosomes continued (Fig. 4, upper row, right).
 | ||
| Fig. 4 Confocal fluorescence microscopy images of HeLa cells treated with the cluster–dye hybrid (pH sensor) or the dye only for details of image analysis, see ESI12.† The hybrid was internalized for 10 min in the cells, quickly washed and then treated with internalization medium without the hybrid for 15 min, 2 h or overnight at 37 °C (upper row). As a control, HeLa cells were treated with the KU dye only (10 min internalization followed by further 40 min without the dye). pH dependence of the fluorescence was tested in a live-cell sample after overnight loading of Au102–KU hybrid and then adding bafilomycin A1 for 3 min. Bars, 20 μm. | ||
Further evidence for the pH dependence of the fluorescence signal was obtained by the addition of the inhibitor of the vacuolar ATPase, bafilomycin A1; see Fig. 4 (lower row) and Table SI12.1.† This drug neutralizes the pH of the endosomes very quickly by inhibiting the action of the proton pump across the endosomal membrane to pH = 7 and higher.25 After the pulse-chase of the probe for 2 h at 37 °C, 50 nM bafilomycin A1 was added and the visualization of the vesicles was continued. The intensity dropped very quickly by showing a dramatic loss already after 3 min and showing almost no intensity after 5 min (Fig. 4 (lower row)). The quantification of the intensity loss after 5 min from approximately 60–80 cells was statistically very significant (Table SI12.1†). The intensity dropped by more than 60% within 5 minutes.
In contrast, in a control experiment, in which the KU without the gold cluster conjugation was used, the fluorescence intensity was very low, it did not increase over time and the localization of the KU reference label looked similar at different time points. The signal coming from the cells with KU only is probably due to low affinity association with cellular membranes due to hydrophobic interactions. Additionally, the low intensity fluorescence signal did not change due to bafilomycin treatment for 5 min (Fig. 4, lower row, right and Table SI†12). Identical result was obtained with non-covalently bound Au102–KU complex probe (not shown). The fluorescence did not localize in any specific part of the cells and the intensity did not change significantly over time. This verifies that the KU did not alone enrich in acidic vesicles and was thus not prone to inhibition of the vacuolar ATPase in the membrane of the acidic vesicles.
Previously, also other tools for measuring pH in intracellular structures have been introduced. pH sensitive GFP variants has been developed to monitor pH changes in the locations where the GFP variant has been successfully targeted. McAnaney et al. introduced dual emission GFP variants, deGFPs that display green fluorescence for higher pH and blue fluorescence for lower pH values.39 Despite the sensitivity of these and other similar constructs, the drawback is the need to first express the molecule in cells, and then to successfully target the construct to the desired location. Typically, expressed protein constructs are widely distributed along the biosynthetic pathway, giving rise to fluorescence in several structures, such as endoplasmic reticulum, Golgi apparatus, and endosomes, depending on the targeting signals added to the construct. Here, the Au102–KU relies on a totally different idea: the molecule is internalized via vesicular uptake to cells where it travels further along the endocytic pathways like any fluid-phase marker. The intensity of the fluorescence is directly dependent on the acidity of the structures.
We have demonstrated a novel successful strategy to utilize covalent and electrostatic interactions between the atomically precise, water soluble gold nanoclusters Au102(pMBA)44 and the organic KU dye to create functional hybrid structures that can be used for intracellular pH imaging in live cells. Gold clusters play here a dual role both as nanocarriers and as regulators of the fluorescence intensity in the target. The cluster–dye hybrids are internalized into cells via endocytosis and accumulate inside endosomes, where the dye molecules are released via hydrolysis of the ester bond. Subsequent changes of the local pH influence the surface charge of the p-MBA ligand layer which in turn regulates the strength of the electrostatic attraction between the dye and the cluster modifying the extent of fluorescence quenching. In principle, other types of chemical transformation are possible, such as isomerization, but so far there is no evidence of such processes.
Several directions for further developments are envisaged. An absolute pH sensor may be developed by correlating the ratio of the two components of the fluorescence lifetimes of the Au102–KU hybrid to the degree of cluster–dye complexation and pH values. Fluorescent molecules that are sensitive to variations of the local ionic (Na+, Ca+, K+, …) concentrations may also be complexed to gold clusters by similar chemistry. As it has been already demonstrated that gold nanoclusters can be used for targeted binding to biomolecules and bionanoparticles creating site-specific labels for electron microscopy, multi-functional gold nanocluster–reporter complexes could be developed for complementary electron and fluorescence microscopy investigations of intracellular processes.
Conclusions
Covalent and non-covalent linking of a Au102 gold nanocluster and a KU dye was studied. Quenching of fluorescence of KU occurred by energy transfer upon complexation with Au102 nanocluster at high pH values. The degree of complexation depends on pH – the binding energy of KU to protonated form of Au102 is lower than to deprotonated form. The fluorescence intensity of KU changes by 2 orders of magnitude when going from low to high pH. A key finding of this work is that covalent linking between the Au102 and KU enables loading of the hybrid into living cells, which is otherwise not possible due to dissociation of the Au102–KU complex at physiological salt concentration. Once loaded to cells, the Au102–KU hybrid accumulates in endosomes where, upon acidification of them, the covalent ester bond is hydrolysed leading to recovery of the pH dependent reversible complexation of Au102 and KU. It was demonstrated that the Au102–KU hybrid enables imaging of acidification of endosomes in live cells. This work introduces a new strategy for developing intracellular biosensors from atomically precise gold nanoclusters by exploiting covalent linking of the nanocluster with a fluorescent dye and release of the dye inside the cells via acid-catalyzed hydrolysis. The presented concept is expected to be applicable to intracellular sensing of also other properties than pH, such as ion concentrations.Author contributions
E. H, T. L., and M. P. conceived the idea. E. H. designed, coordinated, and performed the experimental spectroscopy and analyzed the data and did the Förster energy transfer analysis. T. L. designed and carried out the synthesis and PAGE analysis of the covalent Au102–KU hybrid molecule. V. M. performed and analyzed the live cell experiments. E. P. performed and analysed MD simulations V. S. conducted PAGE experiments to evaluate the synthesis products and did fluorescence salt titration experiments of the non-covalently bound Au102(pMBA)44 and KU dye. K. S. performed electrophoresis experiments and analysis. A. A. did part of the spectroscopic experiments of pH dependent fluorescence of the Au102–KU hybrid. M. K. and L. L. did simulations (published separately) and theoretical considerations on energy transfer between Au102 and KU to help understand the quenching process. G. G. supervised the simulations and contributed to the interpretation of computational and experimental results. H. H. supervised computational work. M. P. Coordinated and supervised the work and wrote the initial manuscript with the assistance of the co-authors. M. P. and E. H. wrote the final draft of the manuscript. All the authors contributed in discussions and in refining the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Academy of Finland (Grants 292352, 294217, 290677, 257125 to H. H., G. G. and V. M.) and Jane and Aatos Erkko foundation (V. M.). Computations were performed on the JYU node of the FGCI national infrastructure. Pasi Myllyperkiö is thanked for his help with life-time analysis. T. L. thanks Jaakko Koivisto for fruitful discussions. Thomas Just Sørensen and Bo W. Larsen from the University of Copenhagen are thanked for kindly providing the KU dye.References
- T. Tsukuda, and H. Häkkinen, Protected Metal Clusters: from Fundamentals to ApplicationsElsevier, 2015 Search PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically precise colloidal metal nanoclusters and nanoparticles: fundamentals and opportunities, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Structure of a thiol monolayer-protected gold nanoparticle at 1.1 Å resolution, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- V. Marjomäki, et al., Site-specific targeting of enterovirus capsid by functionalized monodisperse gold nanoclusters, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1277–1281 CrossRef PubMed.
- M. Martikainen, et al., Hydrophobic pocket targeting probes for enteroviruses, Nanoscale, 2015, 7, 17457–17467 RSC.
- M. C. Stark, et al., Structural characterization of site-modulated nanocapsid with monodispersed gold clusters, Sci. Rep., 2017, 7, 17048 CrossRef PubMed.
- M. Azubel, et al., FGF21 trafficking in intact human cells revealed by cryo-electron tomography with gold nanoparticles, eLife, 2019, 8, e43146 CrossRef PubMed.
- X. Song, W. Zhu, X. Ge, R. Li, S. Li, X. Chen, J. Song, J. Xie, X. Chen and H. Yang, A New Class of NIR-II Gold Nanocluster-Based Protein Biolabels for In Vivo Tumor-Targeted Imaging, Angew. Chem., Int. Ed., 2021, 60, 1306 CrossRef CAS PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X.-D. Zhang, Atomic-Precision Gold Clusters for NIR-II Imaging, Adv. Mater., 2019, 31, 1901015 CrossRef CAS PubMed.
- Y. Xiao, Z. Wu, Q. Yao and J. Xie, Luminescent metal nanoclusters: Biosensing strategies and bioimaging applications, Aggregate, 2021, 2, 114–132 CrossRef.
- J. Zheng, J. T. Petty and R. M. Dickson, High quantum yield blue emission from water-soluble Au8 nanodots, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Highly fluorescent noble metal quantum dots, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Different sized luminescent gold nanoparticles, Nanoscale, 2012, 4, 4073–4083 RSC.
- K. L. Dimuthu, M. Weerawardene and C. M. Aikens, Theoretical insights into the origin of photoluminescence of Au25(SR)18-nanoparticles, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef PubMed.
- B. Varnholt, et al., Excited state interactions between the chiral Au38L24 cluster and covalently attached porphyrin, Phys. Chem. Chem. Phys., 2015, 17, 14788–14795 RSC.
- K. Pyo, et al., Unique energy transfer in fluorescein-conjugated Au22 nanoclusters leading to 160-fold pH-contrasting photoluminescence, J. Phys. Chem. Lett., 2018, 9, 5303–5310 CrossRef CAS PubMed.
- A. Fihey, F. Maurel and A. Perrier, Modeling the absorbance properties of a pyrene chromophore grafted onto a Au25 nanocluster: A TD-DFT study, J. Phys. Chem. C, 2014, 118, 4444–4453 CrossRef CAS.
- P. P. H. Cheng, et al., Dynamic and static quenching of fluorescence by 1–4 nm diameter gold monolayer-protected clusters, J. Phys. Chem. B, 2006, 110, 4637–4644 CrossRef CAS PubMed.
- A. Munoz-Losa, S. Vukovic, S. Corni and B. Mennucci, Nonplasmonic metal particles as excitation energy transfer acceptors: An unexpected efficiency revealed by quantum mechanics, J. Phys. Chem. C, 2009, 113, 16364–16370 CrossRef CAS.
- K. Salorinne, T. Lahtinen, S. Malola, J. Koivisto and H. Häkkinen, Solvation chemistry of water-soluble thiol-protected gold nanocluster Au102 from DOSY NMR spectroscopy and DFT calculations, Nanoscale, 2014, 6, 7823–7826 RSC.
- M. J. Abraham, et al., GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- E. Pohjolainen, X. Chen, S. Malola, G. Groenhof and H. Häkkinen, A unified AMBER-compatible molecular mechanics force field for thiolate-protected gold nanoclusters, J. Chem. Theory Comput., 2016, 12, 1342–1350 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general AMBER force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- N. Rintanen, et al., Promote α2β1 integrin turnover in non-recycling integrin pathway, Mol. Biol. Cell, 2012, 23, 448–463 CrossRef CAS PubMed.
- T. J. Sørensen, et al., Azadioxatriangulenium (ADOTA+): A long fluorescence lifetime fluorophore for large biomolecule binding assay, Methods Appl. Fluoresc., 2013, 1, 25001 CrossRef PubMed.
- E. Thyrhaug, T. J. Sørensen, I. Gryczynski, Z. Gryczynski and B. W. Laursen, Polarization and symmetry of electronic transitions in long fluorescence lifetime triangulenium dyes, J. Phys. Chem. A, 2013, 117, 2160–2168 CrossRef CAS PubMed.
- S. A. Bogh, M. Simmermacher, M. Westberg, M. Bregnhøj, M. Rosenberg, L. De Vico, M. Veiga, B. W. Laursen, P. R. Ogilby, S. P. A. Sauer and T. J. Sørensen, Azadioxatriangulenium and Diazaoxatriangulenium: Quantum Yields and Fundamental Photophysical Properties, ACS Omega, 2017, 2, 193–203 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, A unified view of ligand-protected gold clusters as superatom complexes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- S. Mustalahti, et al., Molecule-like photodynamics of Au102(pMBA)44 nanocluster, ACS Nano, 2015, 9, 2328–2335 CrossRef CAS PubMed.
- J. Koivisto, et al., Acid–base properties and surface charge distribution of the water-soluble Au102(pMBA)44 nanocluster, J. Phys. Chem. C, 2016, 120, 10041–10050 CrossRef CAS.
- S. Dileesh and K. R. Gopidas, Photoinduced electron transfer in azatriangulenium salts, J. Photochem. Photobiol., A, 2004, 162, 115–120 CrossRef CAS.
- M. Rosenberg, A. K. R. Junker, T. J. Sørensen and B. W. Laursen, Fluorescence pH Probes Based on Photoinduced Electron Transfer Quenching of Long Fluorescence Lifetime Triangulenium Dyes, ChemPhotoChem, 2019, 3, 233–242 CrossRef CAS.
- G. D. Scholes, Long-range resonance energy transfer in molecular systems, Annu. Rev. Phys. Chem., 2003, 54, 57–87 CrossRef CAS PubMed.
- E. Hulkko, et al., Electronic and vibrational signatures of the Au102(p-MBA)44 cluster, J. Am. Chem. Soc., 2011, 133, 3752–3755 CrossRef CAS PubMed.
- S. Bhowmick, S. Saini, V. B. Shenoy and B. Bagchi, Resonance energy transfer from a fluorescent dye to a metal nanoparticle, J. Chem. Phys., 2006, 125, 181102 CrossRef PubMed.
- O. Jogunola, T. Salmi, K. Eränen, J. Wärnå and J.-P. Mikkola, Rates and equilibria of ester hydrolysis: Combination of slow and rapid reactions, Chem. Eng. Process., 2011, 50, 665–674 CrossRef CAS.
- K. Karjalainen, et al., A raft-derived, pak1-dependent entry participates in a2b1 integrin-dependent sorting to caveosomes, Mol. Biol. Cell, 2008, 19, 2857–2869 CrossRef PubMed.
- T. B. McAnaney, E. S. Park, G. T. Hanson, S. J. Remington and S. G. Boxer, Green fluorescent protein variants as ratiometric dual emission pH sensors. 2. Excited-state dynamics, Biochemistry, 2002, 41, 15489–15494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00368b |
| This journal is © The Royal Society of Chemistry 2021 |