
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational evolution following the sequential molecular dehydrogenation of PMDI on a Cu(111) surface†
Lacheng
Liu
 ab,
Alexander
Timmer
ab,
Elena
Kolodzeiski
ab,
Hong-Ying
Gao
*abd,
Harry
Mönig
ab,
Henning
Klaasen
c,
Xiangzhi
Meng
abe,
Jindong
Ren
abf,
Armido
Studer
c,
Saeed
Amirjalayer
*ab and
Harald
Fuchs
*ab
ab,
Alexander
Timmer
ab,
Elena
Kolodzeiski
ab,
Hong-Ying
Gao
*abd,
Harry
Mönig
ab,
Henning
Klaasen
c,
Xiangzhi
Meng
abe,
Jindong
Ren
abf,
Armido
Studer
c,
Saeed
Amirjalayer
*ab and
Harald
Fuchs
*ab
aPhysikalisches Institut, Westfälische Wilhelms-Universität, Wilhelm-Klemm-Straße 10, 48149 Münster, Germany. E-mail: samir_01@uni-muenster.de; fuchsh@uni-muenster.de
bCenter for Nanotechnology (CeNTech), Heisenbergstraße 11, 48149 Münster, Germany
cOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany
dSchool of Chemical Engineering and Technology, Tianjin University, 300072 Tianjin, China. E-mail: gaohongying@tju.edu.cn
eInstitut für Experimentelle und Angewandte Physik, Christian-Albrechts-Universität zu Kiel, Leibnizstraße 19, 24118 Kiel, Germany
fCAS Key Laboratory of Nanophotonic Materials and Devices, CAS Key Laboratory of Standardization and Measurement for Nanotechnology, National Center for Nanoscience and Technology, Beijing 100190, P. R. China
First published on 8th October 2021
Abstract
Molecular spatial conformational evolution following the corresponding chemical reaction pathway at surfaces is important to understand and optimize chemical processes. Combining experimental and theoretical methods, the sequential N–H and C–H dehydrogenation of pyromellitic diimide (PMDI) on a Cu(111) surface are reported. STM experiments and atomistic modeling allow structural analysis at each well-defined reaction step. First, exclusively the aromatic N–H dehydrogenation of the imide group is observed. Subsequently, the C–H group at the benzene core of PMDI gets activated leading to a dehydrogenation reaction forming metalorganic species where Cu adatoms pronouncedly protruding from the surface are coordinated by one or two PMDI ligands at the surface. All reactions of PMDI induce conformational changes at the surface as confirmed by STM imaging and DFT simulations. Such conformational evolution in sequential N–H and C–H activation provides a detailed insight to understand molecular dehydrogenation processes at surfaces.
Introduction
On-surface synthesis is a promising approach to prepare covalently bonded nanostructures having potential application for nanomaterials and nano-devices. So far, several reaction types such as Ullmann coupling,1–4 Glaser coupling,5,6 carbene coupling,7,8 cycloaddition,9–11 and polymerization including carboxylic acid derivatives12,13 have been shown to be suitable processes in on-surface chemistry. Among the reported reaction types, the Ullmann reaction is the most popular approach for synthesis of cycloarene,14 graphene nanoribbons,15–18 biphenylene sheet,19 and other one-dimensional polymers,20–24 or two-dimensional networks25–27 sequentially by carbon halide bond activation followed by C–C coupling. In addition, by dehalogenative homocoupling direct formation of polymers connected via C–C double28 and triple bonds29 has been achieved as well. However, a limiting factor of on-surface Ullmann reactions is halogen contamination at the surface. Along these lines, on-surface dehydrogenation reactions avoid this kind of contamination, since only hydrogen gas is formed as the by-product. Therefore, surface assisted dehydrogenation reactions via C–H activation,30–35 N–H activation,30,36–39 O–H activation,12,40,41 and Si–H activation42 have attracted great attention. The hierarchical dehydrogenative coupling of amine groups and aromatic C–H bonds has been reported.30 Inspired by these works, we studied the chemo selective stepwise dehydrogenation of imide N–H and aromatic C–H groups within an aromatic π system. It is expected that the molecular spatial conformation at the surface will be dominated by the corresponding dehydrogenation states. The molecular conformational evolution based on its dehydrogenation state is also important to understand the pathway of multiple dehydrogenation reactions. Considering the dehydrogenation of arenes bearing multiple C–H bonds, regioselectivity is a challenge. An effective approach is to select a precursor limited to few reactive C–H bonds. For instance, s-indacene-1,3,5,7(2H,6H)-tetrone (INDO4)43 has only two potential positions for C–H activation, reducing the complexity of the regioselectivity problem. For the present work, we chose pyromellitic diimide (PMDI, 97%, Sigma-Aldrich) containing two types of potentially activated X–H bonds (X = C, N) for selective X–H activation studies and sequential dehydrogenation of PMDI on the Cu(111) surface.As shown in Scheme 1, PMDI consists of a benzene core that is conjugated by two annulated imide functionalities, overall bearing four carbonyl groups at the 1,3,5,7-positions, two N–H groups at the 2,6-positions and two C–H groups at the 4,8-positions. The different chemical nature of the two X–H bonds (Scheme 1, N–H highlighted in blue, and C–H highlighted in red) provides the opportunity to monitor the dehydrogenation reaction. The bonding energy of the N–H in five-membered heteroatomic amine (such as pyrrole, indole, and carbazole, around 88 kcal mol−1) is significantly lower than the bonding energy of C–H in atomic benzene (around 113 kcal mol−1), which allows the stepwise activation of N–H and C–H on the metal surface.44 Furthermore, a previous study by L. Chi and co-workers showed that removal of an H atom from aniline has a significantly lower barrier than the dehydrogenation of the adjacent C atom on the Cu(111) surface (compare 1.04 eV to 1.67 eV), resulting in a stepwise dehydrogenation of the 4,4′′-diamino-p-terphenyl (DATP) via N–H and C–H activations, respectively.30 Thus, stepwise reaction via N–H and C–H activations of PMDI is expected. The conformational evolution of the PMDI molecules during the dehydrogenation is affected by two types of interaction, the intermolecular interaction between adjacent PMDI molecules within the self-assembled monolayer and the molecule substrate interaction. When PMDI is deposited on a Cu(111) surface, the molecules arrange in a self-assembled structure, which is stabilized by hydrogen bonds between neighboring molecules (see Fig. S1†). Over time, selective thermal N–H bond activation occurs at or even below room temperature (RT). As a result, the hydrogen will secede, and the nitrogen forms a coordination bond to the Cu surface. Complete scission of the hydrogen atoms from the N–H groups is achieved upon annealing the sample to 405 K (PMDI′ in Scheme 1). Upon further increasing the annealing temperature to 485 K, the PMDI′–Cu complex with one Cu adatom is formed. All these reaction states are accompanied by their corresponding geometries at surface, which help to understand the stepwise dehydrogenation mechanism. The stepwise dehydrogenations of aromatic N–H and C–H groups in PMDI was studied by scanning tunnelling microscopy (STM), combined with X-ray photoelectron spectroscopy (XPS) under ultra-high vacuum (UHV) condition. The experimental results were further supported by density functional theory (DFT).
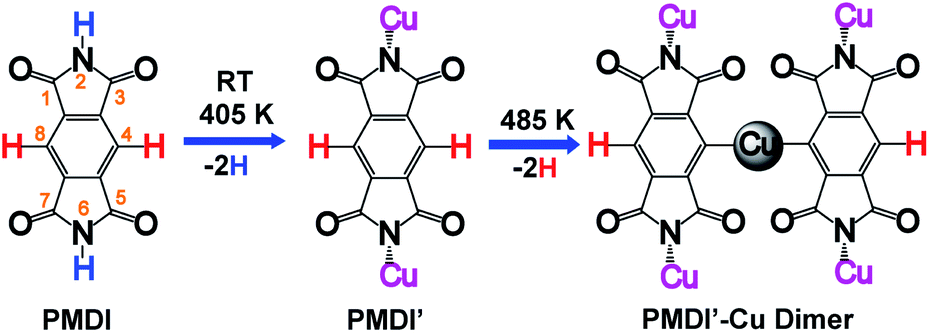 | ||
| Scheme 1 Illustration of the sequential on-surface dehydrogenation of PMDI on Cu(111) surface. | ||
Results and discussion
PMDI was deposited by sublimation from a quartz crucible in UHV at 463 K onto a Cu(111) surface at RT and cooled to 78 K for STM analysis. As shown in Fig. 1a, a self-assembled structure of PMDI is observed with a unit cell a = 0.89 ± 0.02 nm, b = 0.68 ± 0.02 nm, θ = 70 ± 1° (inset Fig. 1a). To get insight into the molecule-substrate interaction, a single PMDI molecule was optimized on a Cu(111) surface at the DFT level (Fig. 1c). The calculation showed that the PMDI only slightly bended upon adsorption on the surface. To understand the molecule–molecule interaction, additional calculations were performed in the gas phase, which shows the intermolecular interaction of the PMDI molecules is hydrogen bonding between the carbonyl group and the N–H group (Fig. S1†). Notably, some molecules appear with a lower contrast (highlighted by a green circles in Fig. 1a, also see Fig. S2†) compared with the molecules in the hydrogen bonded network which was not observed neither on Au(111) nor on Ag(111) surfaces (see in Fig. S3†). To understand this, PMDI was deposited on a cold Cu(111) surface by means of cold deposition (detailed in the method section). STM images (low coverage in Fig. 1b, high coverage in Fig. S4†) show the molecules with lower contrast (highlighted by a green arrow) as compared to the intact PMDI (highlighted by a white arrow). This might be caused by N–H bond activation of some of the PMDI molecules on the Cu(111) surface at RT or below. As a result, some of the hydrogen atoms split from the N–H groups leading to the interaction of the nitrogen with the Cu(111) substrate. A possible structure model of the PMDI after N–H activation (PMDI′) was optimized by DFT on a Cu(111) surface (Fig. 1d). It shows a stronger interaction with the substrate compared with the intact PMDI molecule, since the nitrogen atoms interact stronger with the Cu atoms at the surface resulting in a pronounced bending of the molecules.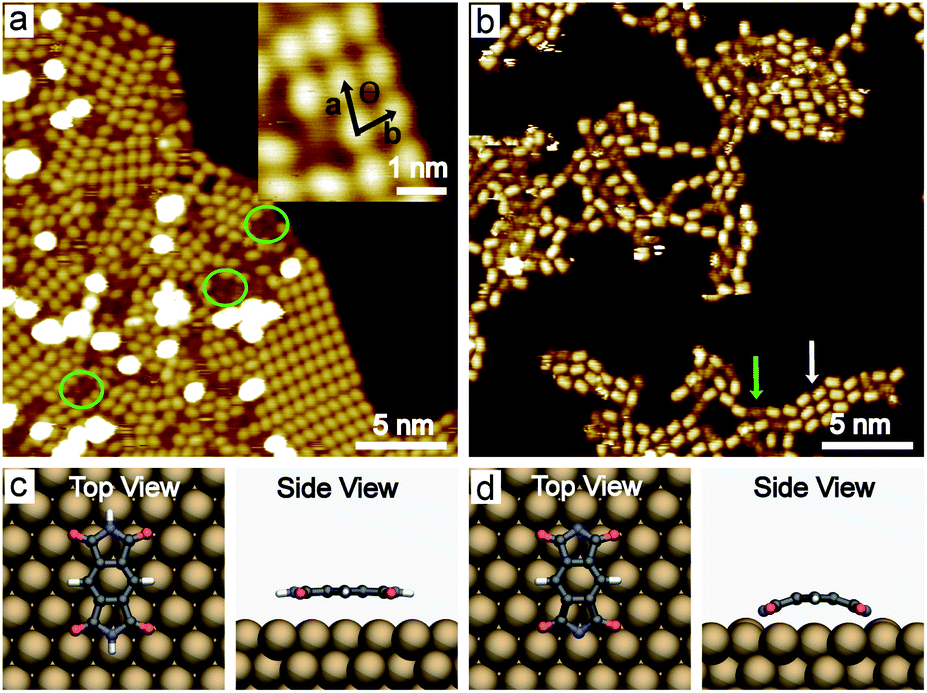 | ||
| Fig. 1 STM images of PMDI deposition on Cu(111) surfaces. (a) STM image of PMDI molecules as deposited on Cu(111) surface at RT, inset: zoomed-in image (−1 V, 10 pA). (b) STM image of PMDI on Cu(111) surface by cold deposition (−1 V, 50 pA). (c) Ball-and-stick optimized DFT model of intact PMDI on Cu(111) surface, top view (top) and side view (bottom). Black, blue, red, and white balls stand for C, N, O and H atoms, respectively. (d) Ball-and-stick model of the optimized structure of the PMDI′ on Cu(111) surface, top view (top) and side view (bottom). | ||
After annealing to 405 K, a Moire pattern structure was observed on Cu(111) surface in the overview STM image (Fig. 2a). The Cu(111) surface and step edges were sculpted by the molecules. It is worth noting that the molecules exhibit two different topographic features in the STM images at −0.2 V and 1 V (Fig. 2b and c) due to two electronic states of the molecules on Cu(111). Both experimental STM topographic features fit very well with the theoretical DFT model of PMDI′. The former exhibits more contrast at the backbone and the later exhibits more contrast at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. It indicates that after annealing to 405 K most of the molecules were transferred to PMDI′. Thus, the N–H activation of PMDI was completed at this temperature. The molecule–molecule interaction largely dominated by N–H⋯O hydrogen bonding is reduced and the substrate–molecule interaction between the Cu-surface and the PMDI molecules becomes the main leading force. Newly formed molecular bonds between adjacent PMDI molecules such as N–Cu–N or N–N bonds are suppressed by the reaction conditions. Forming such bonds, a migration of the PMDI molecules would be necessary to adjust the molecular positions and orientations respectively. Provided by the assembly after deposition originating from the intermolecular hydrogen-bond network the molecular degrees of freedom for such translational and rotational motions are restricted which hinders the formation of intermolecular couplings. Moreover, no such diffusion or migration inside the close-packed assembly was observed by STM in our experiments. The N–Cu–N or N–N structure is, therefore, unlikely to be formed in this case. The strong interaction between the N and Cu atom hinders the formation of the N–N bond, as described before.30,45
O groups. It indicates that after annealing to 405 K most of the molecules were transferred to PMDI′. Thus, the N–H activation of PMDI was completed at this temperature. The molecule–molecule interaction largely dominated by N–H⋯O hydrogen bonding is reduced and the substrate–molecule interaction between the Cu-surface and the PMDI molecules becomes the main leading force. Newly formed molecular bonds between adjacent PMDI molecules such as N–Cu–N or N–N bonds are suppressed by the reaction conditions. Forming such bonds, a migration of the PMDI molecules would be necessary to adjust the molecular positions and orientations respectively. Provided by the assembly after deposition originating from the intermolecular hydrogen-bond network the molecular degrees of freedom for such translational and rotational motions are restricted which hinders the formation of intermolecular couplings. Moreover, no such diffusion or migration inside the close-packed assembly was observed by STM in our experiments. The N–Cu–N or N–N structure is, therefore, unlikely to be formed in this case. The strong interaction between the N and Cu atom hinders the formation of the N–N bond, as described before.30,45
 | ||
| Fig. 2 STM images of PMDI on Cu(111) surface after annealing to 405 K. (a) Overview STM image (0.5 V, 10 pA), (b and c) zoomed-in STM images of the same area obtained at different scanning bias ((b) −0.2 V, 100 pA, the inset; (c) 1 V, 100 pA). The inset simulation STM images of PMDI′ in (b) and (c) was obtained based on the optimized DFT model shown in Fig. 1d at −0.2 V and 1 V, respectively. | ||
Increasing the annealing temperature to 485 K, spots with remarkable higher contrast compared to the dehydrogenated molecules were observed connecting the molecules in chains, as shown in Fig. 3a. The zoomed-in image (Fig. 3b) shows the bright spot mainly links with one or two molecules, taking the nearby located individual PMDI′ molecule as the reference. The bright spot is suggested to be Cu adatom on the Cu(111) surface and coordinated with one or two PMDI′ molecules via C–Cu coordination30 to form PMDI′–Cu dimer (highlighted by a dashed rectangle in green) and PMDI′–Cu monomer (highlighted by a dashed rectangle in white). It is deduced that C–H of PMDI′ was activated after annealing to 485 K. The PMDI′–Cu dimers form a linear structure following regular shift (left or right side) in the same plane with the neighbouring dimers and individual PMDI′ molecules. It is supposed that the driving force to form linear structure should be C–H⋯O hydrogen bonding between the dimers, or between the PMDI′–Cu dimer and PMDI′ (see Fig. S5†). Therefore, only one of the C–H bonds in the PMDI′ forms the C–Cu coordination, while the other is activated. The optimized DFT model of a PMDI′-Cu dimer on Cu(111) surface was proposed as shown in Fig. 3c, where a Cu adatom is linking two PMDI′ via C–Cu coordination at the carbon of the central benzene group. Notably, the side view image shows two CO groups of the PMDI′ at the C–Cu side are obviously closer to the surface compared to those at the C–H side. The schematic structure of PMDI′-Cu monomer on Cu(111) surface was shown in Fig. S6† where a Cu adatom is linking the PMDI′ molecule via C–Cu coordination. After further annealing to higher temperature (505 K) more PMDI′-Cu dimers were observed (Fig. S7†).
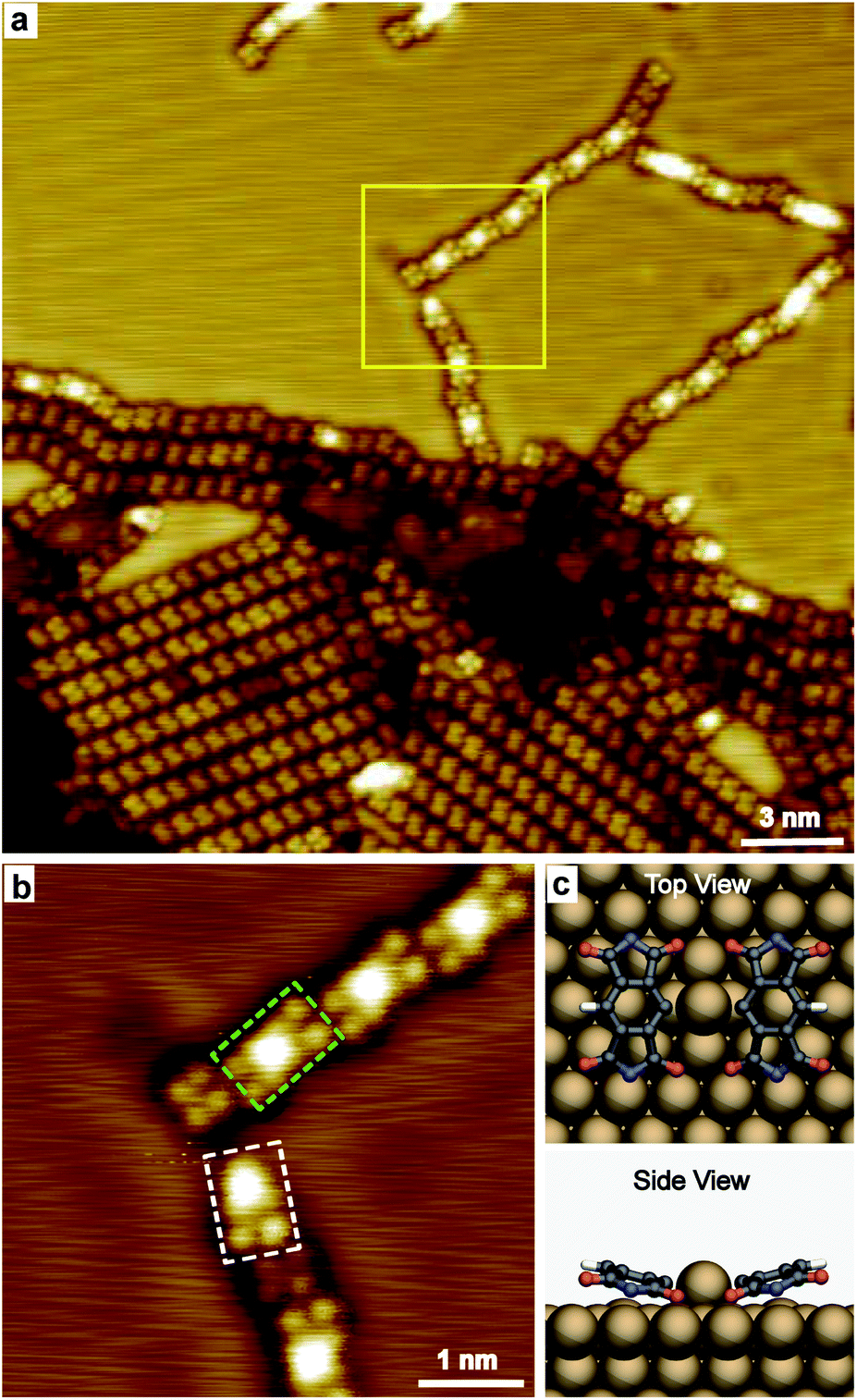 | ||
| Fig. 3 STM images of PMDI on Cu(111) after annealing to 485 K. (a) Overview STM image (0.3 V, 30 pA). (b) Zoomed-in STM image at the area marked by yellow square in (a) (0.3 V, 50 pA). A PMDI′-Cu dimer and a PMDI′-Cu monomer was highlighted by rectangle in green and in white, respectively. (c) Ball-and-stick optimized DFT model of a PMDI′-Cu dimer on Cu(111) surface, top view (top) and side view (bottom). | ||
To further elucidate and confirm the sequential dehydrogenation reaction of PMDI on Cu(111), we performed XPS measurements on the different sample states. As shown in Fig. 4a, two clear peaks are already observed (at 400.6 eV and 398.8 eV), when PMDI molecules are deposited on Cu(111) at RT. In accordance with the previous studies,46,47 we assign the peak at higher binding energy (BE) to the intact N–H and the one at lower BE to the nitrogen bound to the Cu surface (N–Cu). This confirms that N–H activation in PMDI is already possible at RT on Cu(111). After annealing to 405 K, the imide nitrogen peak disappears indicating that complete dehydrogenation of N–H occurred. After annealing to 485 K, a peak shift of the imide nitrogen can be observed to the lower BE side to 398.4 eV, due to the molecular conformational changes in the following C–H reactions (Fig. 3). The corresponding O 1s spectrum is depicted in Fig. 4b. The spectrum of PMDI deposited at RT can be fitted with a single peak at Eb[CO] (O 1s) = 531.9 eV which is consistent with the single oxygen species of the molecule. After thermal annealing to 405 K, there is no significant variation in the O 1s spectrum. Further thermal annealing to 485 K, induces a significant broadening of the oxygen peak suggesting the appearance of an additional oxygen component. The position of the initial peak is changed to Eb[CO (up)] (O 1s) = 531.8 eV and a second component appears at Eb[CO (down)] (O 1s) = 531.0 eV. A possible explanation for the observation of the second component could be an effect due to the reduction of the vertical distance between the oxygen species and the substrate.48–50 This finding would be congruent with the STM and DFT data describing the system after annealing to 485 K. In case of the PMDI′–Cu dimer (Fig. 3c), the tilted geometry that pulling down two oxygen atoms at the C–Cu side to the surface are significantly closer to the surface compared to the PMDI′. The STM data (Fig. 3a) shows that after annealing to 485 K, a mixture of these states exists on the surface, which explains the existence of two peaks in the corresponding XPS data. The corresponding C 1s spectrum (in Fig. S8†) further confirms the complete dehydrogenation of the N–H groups after annealing to 405 K. The peak at the low BE side (Eb = 284.4 eV) of the main carbon peak is commonly considered as a strong indication for the formation of organometallic bonds.51–53 After annealing to 485 K, the obvious broadening, and the shift towards lower BE of the C 1s peak confirm the formation of a significant amount of C–Cu bonds.
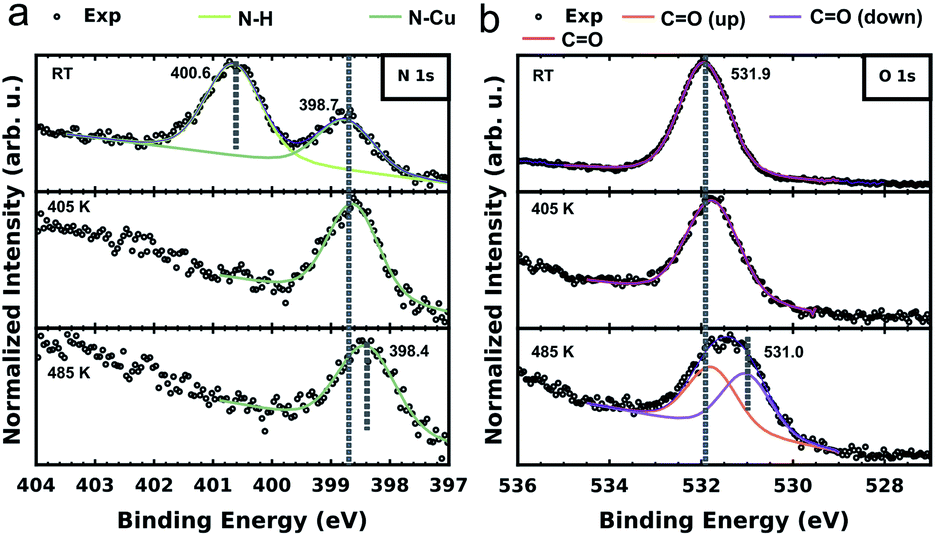 | ||
| Fig. 4 X-ray photoelectron spectra and curve-fitting of the N 1s (a) and O 1s (b) core levels of PMDI on Cu(111). From top to bottom the data in each tile corresponds to the as deposited state at room temperature, first annealing step to 405 K and the final annealing step to 485 K, respectively. All sample states were checked by STM before the XPS measurement. | ||
Conclusions
In summary, we have presented a dehydrogenation study of PMDI on a Cu(111) surface via sequential N–H and C–H activation. The N–H activation of PMDI partially occurs at RT and even lower temperature, and is completed after annealing to 405 K. The C–H activation occurs after annealing to 485 K, leading to PMDI′–Cu coordination with Cu adatoms. Importantly, the stepwise dehydrogenation processes are accompanied by the geometric change of PMDI molecules. It has been shown that the conformational evolution of PMDI molecules during dehydrogenation depends strongly on both the intermolecular interaction between adjacent molecules which establish a pre-ordering after deposition and the intermolecular interaction between molecules and substrate, which gains importance with continuing dehydrogenation. This study provides an important contribution for understanding the reaction pathway and mechanism of such conformational evolution processes.Experimental methods
The STM measurements were performed with an ultrahigh-vacuum (UHV) low-temperature STM system (Omicron). The base pressure was ∼2 × 10−11 mbar. The single crystal Au(111), Ag(111) and Cu(111) surfaces were cleaned with several cycles of Ar+ sputtering and high temperature annealing. The molecules were thermally evaporated from quartz crucibles onto the metal surfaces. The sublimation temperature of PMDI was at 190 °C. For cold deposition here means the substrate was cooled to 78 K and subsequently transferred to the deposition position in the preparation chamber. After deposition, the crystal was transferred back to the STM chamber. The whole process was finished within five minutes and the highest temperature of the sample was lower than room temperature. Thermal annealing of the samples which lasted 30 minutes was carried out in the preparation chamber and monitored with an infrared thermometer.Author contributions
L. Liu, X. Meng, and J. Ren performed the STM experiments. A. Timmer, L. Liu, and H. Mönig performed XPS experiments. E. Kolodzeiski and S. Amirjalayer performed theoretical studies. H. Klaasen and A. Studer provided chemical support in the data analysis and discussion. H.-Y. Gao, S. Amirjalayer, and H. Fuchs supervised the project. L. Liu prepared the manuscript supervised by H.-Y. Gao, S. Amirjalayer, and H. Fuchs. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (TRR 61, and SFB 858, FU 299/19, AM 460/2-1, GA 2430/1-1, MO 2345/4-1) for financial support.References
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed
.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS PubMed
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456–4458 RSC
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed
- H.-Y. Gao, H. Wagner, D. Zhong, J.-H. Franke, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2013, 52, 4024–4028 CrossRef CAS PubMed
- M. Matena, T. Riehm, M. Stöhr, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 2414–2417 CrossRef CAS PubMed
- L. Liu, H. Klaasen, A. Timmer, H.-Y. Gao, D. Barton, H. Mönig, J. Neugebauer, H. Fuchs and A. Studer, J. Am. Chem. Soc., 2018, 140, 6000–6005 CrossRef CAS PubMed
- F. Bebensee, C. Bombis, S.-R. Vadapoo, J. R. Cramer, F. Besenbacher, K. V. Gothelf and T. R. Linderoth, J. Am. Chem. Soc., 2013, 135, 2136–2139 CrossRef CAS PubMed
- O. Díaz Arado, H. Mönig, H. Wagner, J.-H. Franke, G. Langewisch, P. A. Held, A. Studer and H. Fuchs, ACS Nano, 2013, 7, 8509–8515 CrossRef PubMed
- H. Zhou, J. Liu, S. Du, L. Zhang, G. Li, Y. Zhang, B. Z. Tang and H.-J. Gao, J. Am. Chem. Soc., 2014, 136, 5567–5570 CrossRef CAS PubMed
- H.-Y. Gao, P. A. Held, M. Knor, C. Mück-Lichtenfeld, J. Neugebauer, A. Studer and H. Fuchs, J. Am. Chem. Soc., 2014, 136, 9658–9663 CrossRef CAS PubMed
- P. A. Held, H.-Y. Gao, L. Liu, C. Mück-Lichtenfeld, A. Timmer, H. Mönig, D. Barton, J. Neugebauer, H. Fuchs and A. Studer, Angew. Chem., Int. Ed., 2016, 55, 9777–9782 CrossRef CAS PubMed
- Q. Fan, D. Martin-Jimenez, S. Werner, D. Ebeling, T. Koehler, T. Vollgraff, J. Sundermeyer, W. Hieringer, A. Schirmeisen and J. M. Gottfried, J. Am. Chem. Soc., 2020, 142, 894–899 CrossRef CAS PubMed
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed
- H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Kalashnyk, K. Mouhat, J. Oh, J. Jung, Y. Xie, E. Salomon, T. Angot, F. Dumur, D. Gigmes and S. Clair, Nat. Commun., 2017, 8, 14735 CrossRef PubMed
- N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Müllen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef PubMed
- M. Kolmer, A. K. Steiner, I. Izydorczyk, W. Ko, M. Engelund, M. Szymonski, A.-P. Li and K. Amsharov, Science, 2020, 369, 571–575 CrossRef CAS PubMed
- Q. Fan, L. Yan, M. W. Tripp, O. Krejčí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth and J. M. Gottfried, Science, 2021, 372, 852–856 CrossRef CAS PubMed
- J. A. Lipton-Duffin, O. Ivasenko, D. F. Perepichka and F. Rosei, Small, 2009, 5, 592–597 CrossRef CAS PubMed
- J. A. Lipton-Duffin, J. A. Miwa, M. Kondratenko, F. Cicoira, B. G. Sumpter, V. Meunier, D. F. Perepichka and F. Rosei, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11200–11204 CrossRef CAS PubMed
- T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 135, 3576–3582 CrossRef CAS PubMed
- Q. Sun, L. Cai, H. Ma, C. Yuan and W. Xu, ACS Nano, 2016, 10, 7023–7030 CrossRef CAS PubMed
- Q. Fan, D. Martin-Jimenez, D. Ebeling, C. K. Krug, L. Brechmann, C. Kohlmeyer, G. Hilt, W. Hieringer, A. Schirmeisen and J. M. Gottfried, J. Am. Chem. Soc., 2019, 141, 17713–17720 CrossRef CAS PubMed
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489 CrossRef CAS PubMed
- S. Schlogl, T. Sirtl, J. Eichhorn, W. M. Heckl and M. Lackinger, Chem. Commun., 2011, 47, 12355 RSC
- C. Rogers, C. Chen, Z. Pedramrazi, A. A. Omrani, H. Z. Tsai, H. S. Jung, S. Lin, M. F. Crommie and F. R. Fischer, Angew. Chem., Int. Ed., 2015, 54, 15143–15146 CrossRef CAS PubMed
- P. H. Jacobse, A. van den Hoogenband, M. E. Moret, R. J. M. Klein Gebbink and I. Swart, Angew. Chem., Int. Ed., 2016, 55, 13052–13055 CrossRef CAS PubMed
- Q. Sun, B. V. Tran, L. Cai, H. Ma, X. Yu, C. Yuan, M. Stöhr and W. Xu, Angew. Chem., Int. Ed., 2017, 56, 12165–12169 CrossRef CAS PubMed
- Q. Sun, X. Yu, M. Bao, M. Liu, J. Pan, Z. Zha, L. Cai, H. Ma, C. Yuan, X. Qiu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 4035–4038 CrossRef CAS PubMed
- Q. Li, B. Yang, J. Björk, Q. Zhong, H. Ju, J. Zhang, N. Cao, Z. Shi, H. Zhang, D. Ebeling, A. Schirmeisen, J. Zhu and L. Chi, J. Am. Chem. Soc., 2018, 140, 6076–6082 CrossRef CAS PubMed
- D. Zhong, J. H. Franke, S. K. Podiyanachri, T. Blömker, H. Zhang, G. Kehr, G. Erker, H. Fuchs and L. Chi, Science, 2011, 334, 213–216 CrossRef CAS PubMed
- H.-Y. Gao, D. Zhong, H. Mönig, H. Wagner, P. A. Held, A. Timmer, A. Studer and H. Fuchs, J. Phys. Chem. C, 2014, 118, 6272–6277 CrossRef CAS
- Q. Sun, L. Cai, Y. Ding, L. Xie, C. Zhang, Q. Tan and W. Xu, Angew. Chem., Int. Ed., 2015, 54, 4549–4552 CrossRef CAS PubMed
- C. Wang, Q. Jin, C. Shu, X. Hua, Y. Long and P. Liu, Chem. Commun., 2017, 53, 6347–6350 RSC
- Q. Fan, S. Werner, J. Tschakert, D. Ebeling, A. Schirmeisen, G. Hilt, W. Hieringer and J. M. Gottfried, J. Am. Chem. Soc., 2018, 140, 7526–7532 CrossRef CAS PubMed
- M. Matena, J. Björk, M. Wahl, T.-L. Lee, J. Zegenhagen, L. H. Gade, T. A. Jung, M. Persson and M. Stöhr, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125408 CrossRef
- A. Basagni, L. Colazzo, F. Sedona, M. Di Marino, T. Carofiglio, E. Lubian, D. Forrer, A. Vittadini, M. Casarin, A. Verdini, A. Cossaro, L. Floreano and M. Sambi, Chem. Eur. J., 2014, 20, 14296–14304 CrossRef CAS PubMed
- X. Meng, H. Klaasen, L. Viergutz, B. S. Lammers, M. C. Witteler, H. Mönig, S. Amirjalayer, L. Liu, J. Neugebauer, H.-Y. Gao, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2021, 60, 1458–1464 CrossRef CAS PubMed
- M. Knor, H.-Y. Gao, S. Amirjalayer, A. Studer, H. Gao, S. Du and H. Fuchs, Chem. Commun., 2015, 51, 10854–10857 RSC
- S. Fischer, A. C. Papageorgiou, J. A. Lloyd, S. C Oh, K. Diller, F. Allegretti, F. Klappenberger, A. P. Seitsonen, J. Reichert and J. V. Barth, ACS Nano, 2014, 8, 207–215 CrossRef CAS PubMed
- L. Liu, H. Klaasen, M. C. Witteler, B. S. Lammers, A. Timmer, H. Kong, H. Mönig, H.-Y. Gao, J. Neugebauer, H. Fuchs and A. Studer, Nat. Chem., 2021, 13, 350–357 CrossRef CAS PubMed
- F. Bebensee, K. Svane, C. Bombis, F. Masini, S. Klyatskaya, F. Besenbacher, M. Ruben, B. Hammer and T. R. Linderoth, Angew. Chem., Int. Ed., 2014, 53, 12955–12959 CrossRef CAS PubMed
- C. Barckholtz, T. A. Barckholtz and C. M. Hadad, J. Am. Chem. Soc., 1999, 121, 491–500 CrossRef CAS
- M. Knor, H.-Y. Gao, S. Amirjalayer, A. Studer, H. Gao, S. Du and H. Fuchs, Chem. Commun., 2015, 51, 10854–10857 RSC
- P. Borghetti, G. D. Santo, C. Castellarin-Cudia, M. Fanetti, L. Sangaletti, E. Magnano, F. Bondino and A. Goldoni, J. Chem. Phys., 2013, 138, 144702 CrossRef PubMed
- J. P. Macquet, M. M. Millard and T. Theophanides, J. Am. Chem. Soc., 1978, 100, 4741–4746 CrossRef CAS
- A. Franco-Cañellas, Q. Wang, K. Broch, D. A. Duncan, P. K. Thakur, L. Liu, S. Kera, A. Gerlach, S. Duhm and F. Schreiber, Phys. Rev. Mater., 2017, 1, 013001 CrossRef
- G. Heimel, S. Duhm, I. Salzmann, A. Gerlach, A. Strozecka, J. Niederhausen, C. Bürker, T. Hosokai, I. Fernandez-Torrente, G. Schulze, S. Winkler, A. Wilke, R. Schlesinger, J. Frisch, B. Bröker, A. Vollmer, B. Detlefs, J. Pflaum, S. Kera, K. J. Franke, N. Ueno, J. I. Pascual, F. Schreiber and N. Koch, Nat. Chem., 2013, 5, 187–194 CrossRef CAS PubMed
- B. Stadtmüller, N. Haag, J. Seidel, G. van Straaten, M. Franke, C. Kumpf, M. Cinchetti and M. Aeschlimann, Phys. Rev. B, 2016, 94, 235436 CrossRef
- M. D. Giovannantonio, M. E. Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. F. Revurat, A. Cossaro, A. Verdini, D. F. Perepichka, F. Rosei and G. Contini, ACS Nano, 2013, 7, 8190–8198 CrossRef PubMed
- R. Gutzler, L. Cardenas, J. Lipton-Duffin, M. E. Garah, L. E. Dinca, C. E. Szakacs, C. Fu, M. Gallagher, M. Vondracek, M. Rybachuk, D. F. Perepichka and F. Rosei, Nanoscale, 2014, 6, 2660–2668 RSC
- X. Zhou, F. Bebensee, Q. Shen, R. Bebensee, F. Cheng, Y. He, H. Su, W. Chen, G. Q. Xu, F. Besenbacher, T. R. Linderoth and K. Wu, Mater. Chem. Front., 2017, 1, 119–127 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00590a |
| This journal is © The Royal Society of Chemistry 2021 |