
Monomeric vanadium oxide: a very efficient species for promoting aerobic oxidative dehydrogenation of N-heterocycles†
Zhenbing
Xie
ab,
Bingfeng
Chen
 *a,
Lirong
Zheng
c,
Fangfang
Peng
a,
Huizhen
Liu
*ab and
Buxing
Han
*ab
*a,
Lirong
Zheng
c,
Fangfang
Peng
a,
Huizhen
Liu
*ab and
Buxing
Han
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloidal and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: chenbf@iccas.ac.cn; liuhz@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing, 100049, P. R. China
cInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 25th November 2020
Abstract
Monomeric active species are very interesting in heterogeneous catalysis. In this work, we proposed a method to prepare VOx–NbOy@C catalysts, which involve the one-pot hydrothermal synthesis of inorganic/organic hybrid materials containing V/Nb followed by thermal treatment under a reducing atmosphere. The prepared catalysts were characterized using different techniques, such as high-angle annular dark-field scanning transmission electron microscopy and X-ray absorption fine structure spectroscopy. It was shown that monomeric VOx species were dispersed homogeneously in the catalysts. The VOx–NbOy@C catalysts displayed high performance in the aerobic oxidative dehydrogenation of N-heterocycles to aromatic heterocycles. It was demonstrated that the selectivity of reaction over the catalyst with a very small amount of V (0.07 wt%) was much higher than that over the NbOy@C, and the catalyst also exhibited excellent stability in the reaction. The detailed study indicated that monomeric VO2 species were the most effective for promoting the reaction.
Introduction
Vanadium metal is very attractive in catalysts because it is an earth-abundant metallic element and has a low toxicity compared to most other heavy metals. Vanadium-based catalysts have been widely used in chemical, petroleum and environmental industries, such as industrial production of H2SO4,1 maleic anhydride2 and phthalic anhydride,3 and selective catalytic reduction (SCR) of harmful NOx.4,5 Vanadium-based catalysts, such as vanadium silicate zeolites6 and divanadium-substituted phospho-tungstate,7 have been reported in the catalytic hydroxylation of arenes to phenols. Moreover, supported vanadium oxides catalysts have also been used in the catalytic dehydrogenation of light alkanes.8 The catalytic performances of supported vanadium oxide catalysts are closely correlated with the dispersion state of active sites, in which the reported vanadium species exist as monomeric VOx species, polymeric surface VO4 sites or V2O5 nanoparticles.9 It has been reported that the isolated VOx species exhibited higher catalytic activity and selectivity in the catalytic dehydrogenation of propane compared with their oligomerized counterparts.10 Conventional methods like impregnation and anchoring always result in a broad variety of VxOy species. However, other catalyst synthesis methods, such as flame spray pyrolysis11 and atomic layer deposition,12 were restricted by the complicated procedures and high cost of volatile molecular metal precursors. Hence, developing highly dispersed vanadium-based catalysts using a feasible procedure is highly desirable for their applications.During the last decade, much attention has been paid to the study of single-atom catalysts (SAC), which exhibit distinctive performance and achieve the highest utilization efficiency of metal atoms.13 Recently, Farha and co-workers reported the synthesis of a single-atom-based organo-vanadium species incorporated in MOFs, which could catalyse the selective oxidation of alcohol.14 However, the poor thermal stability of MOF materials hinders their application. Owing to the high surface free energy of SAC,15 the atomic dispersed metal sites are generally stabilized by the N atoms with an empty orbital on the surface of nitrogen doped carbon materials,16 or defects of metal oxides,17 metal sulfides,18 metal nitrides19 and metal–organic frameworks (MOFs).20
The catalytic dehydrogenation of N-heterocycles is referred to as an important route of synthesis of the corresponding aromatic heterocycles, which are common intermediates in pharmaceutical and biological molecules.21 Several supported metal catalysts, such as Rh,22 Co,23 Fe24 or Ni25 catalysts, have been reported in the non-oxidative dehydrogenation of N-heterocycles. Although homogeneous oxovanadium (V) catalysts have been applied in the oxidative dehydrogenation of N-heterocycles,26 heterogeneous catalysts are preferred owing to their heterogeneity and easy separation from the reaction mixture. Although V2O5 has been used to catalyse the dehydro-aromatization, the catalyst dosage was greater than the stoichiometric value.27 Niobium-based catalytic materials have been used in several selective oxidations and acid-catalysed reactions.28 Although supported V2O5/Nb2O5 catalysts have been reported in the oxidative dehydrogenation of ethane and propane,29,30 mononuclear vanadium catalysts have not been reported previously for the oxidative dehydrogenation of N-heterocycles.
Herein, we developed a novel method for the fabrication of VOx–NbOy@C catalysts, in which monomeric VOx species were dispersed uniformly in the catalytic materials. Owing to the excellent coordination ability of tartaric acid, the natural carboxylic acid was used to control the dispersion of the vanadium species, which is different from the conventional methods used for vanadium-based catalysts. In this route, the inorganic/organic hybrid materials containing V/Nb were initially prepared via the one-pot hydrothermal synthesis. Then the materials were made to undergo thermal treatment under a reducing atmosphere to obtain VOx–NbOy@C-T catalysts, in which T represents the temperature of heat-treatment. It was found that the catalyst was very efficient and stable for catalyzing the oxidative dehydrogenation of N-heterocycles (Scheme 1).
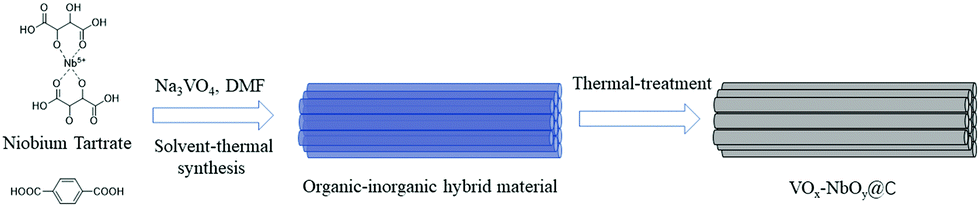 | ||
| Scheme 1 The schematic illustration of the preparation procedure for VOx–NbOy@C materials. | ||
Experimental
Materials
Niobium pentaoxide (99.9%), trisodium tetraoxovanadate dodecahydrate (Na3VO4·12H2O, 99%), HF (≥40.0%), ammonium hydroxide (25–28%), L-(+)-tartaric acid (99.5%), N,N-dimethylformamide (DMF, AR), methanol (AR), dimethyl sulfoxide (DMSO, AR), dioxane (AR), t-BuOH (AR), and CH3CN (AR) were purchased from Sinopharm Chemical Reagent. Benzonitrile (AR), chlorobenzene (AR) and 1,3,5-trimethylbenzene (AR) were provided by Acros. Terephthalic acid (99%), 1,2,3,4-tetrahydroquinoline (99%), 7-nitro-1,2,3,4-tetra-hydroquinoline (98%), 6-methyl-1,2,3,4-tetrahydro-quinoline (98%), 6-bromo-1,2,3,4-tetrahydro-quinoline (98%), indoline (99%), 6-nitroindoline (98%), 1,2,3,4-tetrahydro-isoquinoline (98%), and 1,2,3,4-tetrahydro-quinoxaline (98%) were obtained from Alfa Aesar.Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:2 (v/v), 80 mL). The obtained white precipitate of Nb2O5·nH2O was filtered and washed with 5% ammonia solution to remove fluoride ions. Finally, the solid was added into the aqueous tartaric acid solution (320 mL, 0.25 mol L−1). After stirring for a few minutes, aqueous niobium tartrate solution was obtained.
:H2O = 1:2 (v/v), 80 mL). The obtained white precipitate of Nb2O5·nH2O was filtered and washed with 5% ammonia solution to remove fluoride ions. Finally, the solid was added into the aqueous tartaric acid solution (320 mL, 0.25 mol L−1). After stirring for a few minutes, aqueous niobium tartrate solution was obtained.
Terephthalic acid was used as a ligand for preparing the organic–inorganic hybrid material, analogous to the metal–organic frameworks. DMF was used to enhance the solubility of terephthalic acid in the aqueous solution. After sonication for 1.0 h, the clear solution was transferred into hydrothermal reaction vessels. After solvothermal treatment at 220 °C for 8 h, the hybrid material was obtained. In order to remove residual terephthalic acid, the obtained white powder was made to undergo Soxhlet extraction with MeOH and was washed with DMF. After being dried overnight at 383 K under vacuum, NbOy@C was obtained by thermal treatment at 800 °C under H2/Ar (H2: 10%, v/v).
Catalytic reaction
Oxidative dehydrogenation of N-heterocycles was performed in a 12 mL stainless autoclave equipped with a pressure gauge, a magnetic stirrer, and an electric temperature controller. The catalyst (50.0 mg), the solvent (2.0 mL) and N-containing compounds (0.5 mmol) were introduced into the reactor. The sealed autoclave was flushed with O2 three times, and pressurized with O2 to 0.5 MPa. After the designated temperature was reached, the reaction mixture was stirred at a rate of 800 rpm to start the reaction. At the end of the reaction, the autoclave was cooled to ambient temperature in water and slowly depressurized. Conversion and product composition were analyzed by GC and GC-MS. Chlorobenzene was used as the internal standard. GC was performed on a GC-2014 (SHIMADZU) equipped with a high-temperature capillary column (MXT-1, 30 m, 0.25 mm I.D.) and an FID detector. GC-MS was performed on a GCT Premier/Waters instrument equipped with a capillary column (DB-5MS/J&W Scientific, 30 m, 0.25 mm I.D.). For NMR analysis, the crude reaction mixture was washed with H2O several times to remove DMSO and the aqueous phase was extracted with CH2Cl2. After drying with Na2SO4 and evaporating under vacuum, the crude products were purified by column chromatography [hexane/ethyl acetate] to get the corresponding product yields.Catalyst characterization
XRD patterns were obtained on a Rigaku D/Max-2500 X-ray diffractometer using Cu Kα radiation. The elemental analysis was performed using a PerkinElmer Optima 2000 OV inductively coupled plasma-optical emission spectroscope (ICP-OES). Transmission electron microscopy (TEM) was performed on a JEOL 2010 TEM equipped with an attachment for local energy dispersion analysis (EDX). The accelerating voltage was 200 kV, and the spot size was 1 nm. X-Ray photoelectron spectra (XPS) were recorded on a Thermo Scientific ESCA Lab 250 Xi using 200 W mono-chromated Al Kα radiation (1486.6 eV). The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon. Nitrogen adsorption–desorption analysis was carried out at 77 K on a NOVA 2200e adsorption analyser. Before the adsorption analysis, the samples were degassed under vacuum at 300 °C in the port of the adsorption analyser. 1H NMR and 13C NMR spectra were recorded using a Bruker Ultra-Shield 400 MHz spectrometer (1H = 400 MHz), with CDCl3 as the solvent unless otherwise indicated. Tetramethyl-silane (TMS) was used as an internal standard. Raman spectra were recorded on a LabRAM HR Raman microscope equipped with a 532 nm laser source and a 100× objective lens. The sample powders were sandwiched between two glass slides before the test.The X-ray absorption spectra (XAS) including the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) of the samples at the V K-edge were collected at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF), China. The electron storage ring of BSRF was operated at 23 keV. The spectra were recorded in the solid-state detectors for the V K-edge by using an Si(311) double-crystal monochromator, respectively. The Athena software package was used to analyse the data. Theoretical XANES spectra were calculated by using the FEFF8.2 code.
Results and discussion
Firstly, the niobium-based inorganic/organic hybrid materials were synthesized using a facile hydrothermal process using niobium tartrate, Na3VO4 and terephthalic acid with DMF as solvent. After Soxhlet extraction with MeOH to remove residual terephthalic acid, the hybrid materials were subjected to high temperature under a H2/Ar (H2: 10%, v/v) atmosphere, and the obtained VOx–NbOy@C material had a rod-like morphology (Fig. 1a–c). In the absence of Na3VO4, similar nanorod-structured materials (simplified as NbOE@C) were also synthesized (Fig. 1d and Fig. S1, ESI†). The excessive tartrates provided the coordination environments for complexing the vanadium and niobium precursors during the hydrothermal procedure. Owing to the excellent coordination ability of tartaric acid, this natural carboxylic acid was helpful for uniform dispersion of VOx and NbOy species in the hybrid materials. | ||
| Fig. 1 SEM images of the VOx–NbOy@C-800 material (a)–(c) and the NbOy@C-800 material (d). | ||
The Raman spectra of VOx–NbOy@C showed only two bands corresponding to the G (1580 cm−1) and D (1360 cm−1) bands, which are the characteristic peaks of carbon materials,32 but no peak derived from VOx species was detected (Fig. S2, ESI†). The carbonaceous species, derived from the carbonization of the residual terephthalic acid and tartrate, acted as a support for monomeric VOx species and NbOy oxides. Besides, they also provided good porosity of the catalysts. ICP analysis revealed that the content of V in VOx–NbOy@C-800 was 0.07 wt%. As shown in the high-resolution TEM images (Fig. 2b and c), the lattice space of the VOx–NbOy@C-800 nanorods is 0.342 nm, corresponding to the [400] planes of NbO2.33 Before thermal treatment, both NbOy@C and VOx–NbOy@C showed a pseudo-hexagonal Nb2O5 phase (JCPDS no. 28-0317) (Fig. S3, ESI†). After thermal-treatment at 600 °C under a reducing atmosphere, both NbO2 and Nb2O5 phases were detected in VOx–NbOy@C (Fig. 2B). Further increasing the temperature resulted in transition from the Nb2O5 phase to the tetragonal NbO2 phase, which was confirmed by XRD analysis. However, no crystal phases related to vanadium species were detected by X-ray diffraction analysis (Fig. S4, ESI†), which indicated that either no crystal vanadium oxide phase was formed in the VOx–NbOy@C catalysts or the amount of vanadium oxides was too low to be detected using the XRD method. Moreover, high-angle annular dark-field scan TEM (HAADF-STEM) images and corresponding elemental mapping of VOx–NbOy@C-800 showed the homogeneous distribution of V, Nb, O and C elements throughout the materials (Fig. 2A).
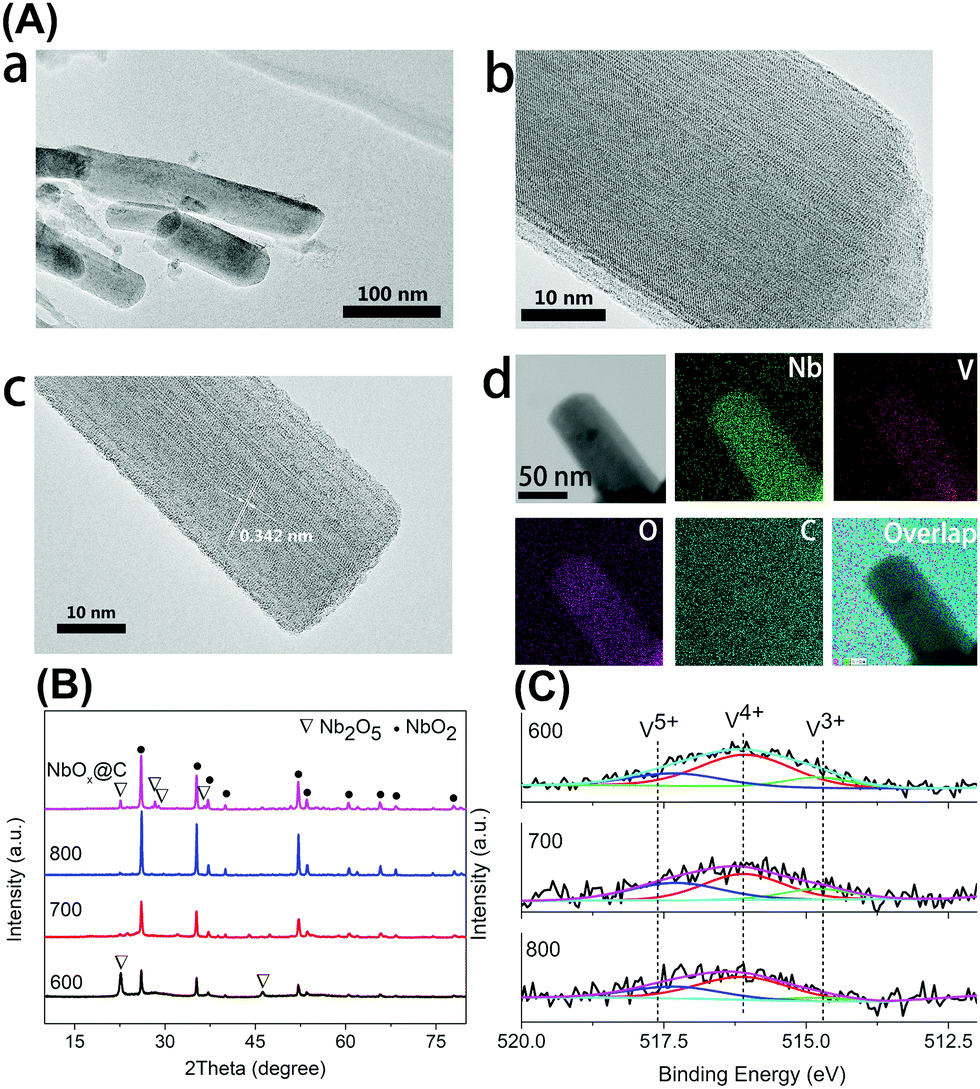 | ||
| Fig. 2 The characterization of the VOx–NbOy@C catalyst: (A) TEM images of VOx–NbOy@C-800: (a) low-resolution TEM images; (b) and (c) high-resolution TEM images; (d) EDS mapping; (B) PXRD patterns of NbOy@C and VOx–NbOy@C undergoing thermal treatment at different temperatures; (C) XPS curve-fitting of the V 2p3/2 photoelectronic peak of VOx–NbOy@C catalysts after thermal treatment at different temperatures. | ||
X-Ray photoelectron spectroscopy (XPS) was performed to characterize the surface compositions of catalytic materials. The high-resolution of the V 2p3/2 spectra can be deconvoluted into three peaks located at 517.6 eV, 516.1 eV and 514.7 eV (Fig. 2C), which can be assigned to the V5+, V4+ and V3+ species, respectively.34 The V 2p3/2 spectra also demonstrate that the main composition of VOx–NbOy@C was the V4+ species. The Nb 3d spectra showed three peaks at the binding energies of 207.4 eV (3d5/2), 206.8 eV (3d5/2) and 205.8 eV (3d5/2) (Fig. S5, ESI†), which can be ascribed to Nb5+, Nb4+ and Nb3+ species, respectively.35 Although the content of low valence Nb species increased with the thermal-treatment temperature, the Nb5+ species existed as the main component in these materials (Table S1, ESI†). From N2 adsorption–desorption isotherms (Fig. S6, ESI†), the BET surface area of the VOx–NbOy@C-800 was found to be 59.9 m2 g−1, and the BJH average pore diameter was centred at 3.9 nm.
The chemical oxidation state and local coordinate structures of the V species in the VOx–NbOy@C were further studied by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy at the V K-edge. As shown in the XANES spectra (Fig. 3a), VOx–NbOy@C-600 showed a strong pre-edge peak comparable to that of VO2, which can be assigned to the so-called 1s–3d transition originated mainly from the mixing of 2p orbitals of the oxygen anions with 3d orbitals of the vanadium atoms.36 After thermal treatment at higher temperatures, the intensity of the pre-edge peak decreased, and the binding energy at adsorption edge shifted to lower energy (Fig. 3a and Fig. S7, ESI†). These results indicate that the vanadium species was reduced to lower valence under high temperature treatment.37 The position of the white line peak of VOx–NbOy@C was close to that of VO2, indicating that the average valence state of V in the hybrid was approximately V4+. Moreover, the position of the white line peak of these catalysts shifted to low energy with increasing thermal-treatment temperature (Fig. S7, ESI†), suggesting that the valence state of V species decreased in this procedure. From the FT-EXAFS spectra (Fig. 3b), the peak located at around 1.6 Å was ascribed to the first V–O coordination shell.38 Furthermore, the scattering peak due to the presence of the neighbouring V atoms (V–O–V) in the as-synthesized VOx–NbOy@C was hardly detected compared with V2O5 or VO2.39 These results demonstrate that the vanadium species in the VOx–NbOy@C catalysts existed in the isolated monomeric VO4 state with tetrahedral coordination.
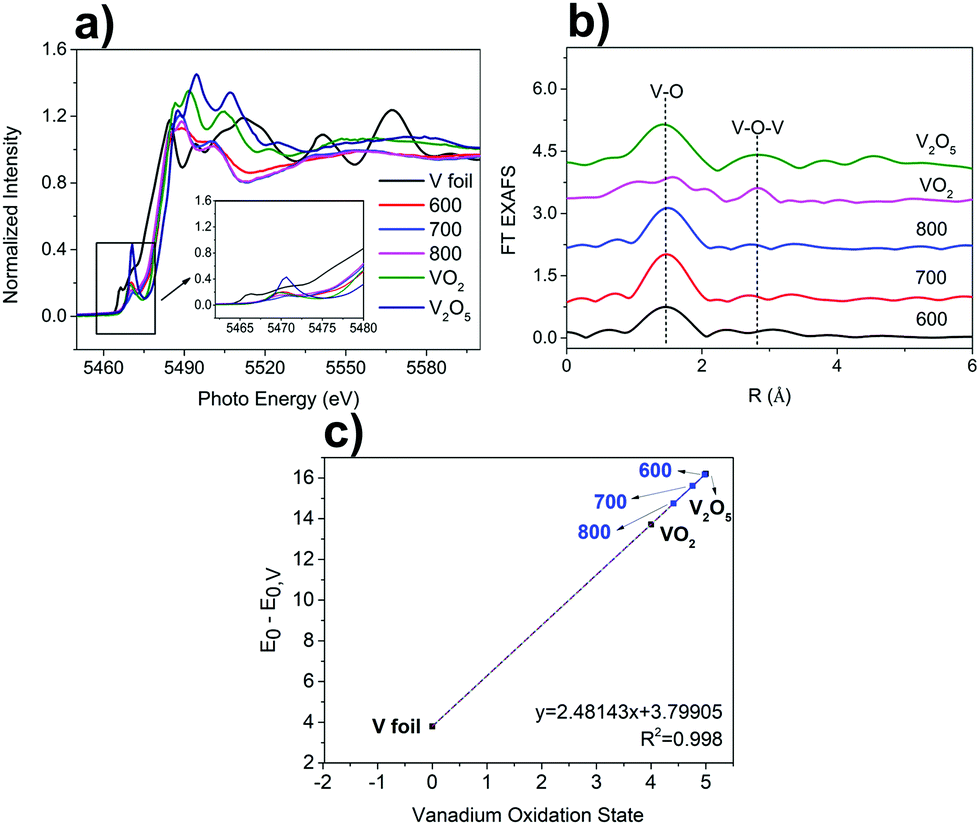 | ||
| Fig. 3 (a) V K-edge XANES spectra (the inset shows a magnified view of the pre-edge peaks) and (b) Fourier transformed V K-edge EXAFS spectra of the VOx–NbOy@C catalysts and the reference compounds; (c) the plot of the relative edge position to E0 (5465 eV) vs. the formal vanadium oxidation state, with the measurements for the VOx–NbOy@C samples indicated on the calibration curve. | ||
The catalytic performance of the VOx–NbOy@C was evaluated in the dehydrogenation of 1,2,3,4-tetrahydroquinoline. The commercial vanadium oxides showed low catalytic selectivity in this reaction (Table 1, entries 1 and 2). The nanorod-like niobium-based catalyst NbOy@C gave 56.0% yield of quinoline. Compared with the NbOy@C catalysts, incorporation of the vanadium species improved catalytic selectivity effectively, indicating a synergistic effect between vanadium oxide and niobium oxide. It is well known that the reducibility of vanadium in the redox mechanism is stronger than niobium.40 The catalytic performance of the VOx–NbOy@C catalyst was significantly influenced by the thermal-treatment temperature. VOx–NbOy@C-600 showed medium catalytic selectivity of the product. Quinoline yields increased with increasing thermal-treatment temperature (Table 1, enties 4–6). VOx–NbOy@C-800 achieved the highest catalytic performance with 92.5% yield of quinoline at full conversion of the substrate. As shown in Fig. 3c, the formal vanadium oxidation states deduced from the calibration curve decreased with increasing thermal-treatment temperature.41 The increased proportion of low valent V species (Fig. 3c) may be the main reason for the improved selectivity of the catalyst treated at higher temperature. A small amount of quinolinone was detected owing to the over-oxidation reaction.42 It has been reported that the V4+ species in the mesoporous vanadia–titania nanoparticle (VTN) catalysts played a crucial role in the oxidative dehydrogenation of ethyl lactate to ethyl pyruvate.43
| Entrya | Catalysts | Conv.b (%) | Sel.c (%) |
|---|---|---|---|
| a Reaction conditions: 1,2,3,4-tetrahydroquinoline (0.5 mmol), catalyst (50 mg), DMSO (1.5 mL), H2O (0.5 mL), O2 (0.5 MPa), 120 °C/16 h. b The yields were obtained by GC using chlorobenzene as the internal standard. c The other product was quinolinone. | |||
| 1 | V2O5 | 99 | 59.3 |
| 2 | VO2 | 99 | 60.7 |
| 3 | NbOy@C-800 | 99 | 56.0 |
| 4 | VOx–NbOy@C-600 | 99 | 61.0 |
| 5 | VOx–NbOy@C-700 | 99 | 79.2 |
| 6 | VOx–NbOy@C-800 | 99 | 92.5 |
Reaction conditions were further optimized using VOx–NbOy@C-800, and the results are listed in the Tables S2 and S3 (ESI†). Solvents play a crucial role in this reaction. Organic solvents with weak polarity or moderate polarity, such as 1,3,5-trimethylbenzene, benzotrifluoride and dioxane, gave poor results. When polar solvents were used, quinoline was obtained in medium or good yield. These results could be related to the improved adsorption of the nonpolar reactant in the presence of polar solvents. The DMSO/H2O (v/v: 3/1) mixed solvent gave the best results. Furthermore, the scope of the reaction was explored under the optimized reaction conditions (Table 2). Tetrahydroquinoline derivatives bearing electron-donating groups (Table 2, entry 2) or electron-withdrawing groups (Table 2, entries 3 and 4) were catalytically transformed into the corresponding aromatic heterocycles in good to excellent yields. Apart from the tetrahydroquinoline derivatives, oxidative dehydrogenation of 1,2,3,4-tetrahydro-isoquinoline (Table 2, entry 5) and 1,2,3,4-tetrahydro-quinoxaline (Table 2, entry 8) also gave a high yield of the corresponding products. A moderate yield of indole was obtained in the case of indoline (Table 2, entry 6). While heterocycles were substituted with one strong electron withdrawing group like the NO2 group, the desired yield of products could be obtained by increasing the oxygen pressure (Table 2, entries 3 and 7). These results indicate that electron-deficient aromatic rings retard adsorption or coordination of the substrates with the VOx–NbOy@C catalyst. It has been reported that electron-deficient groups usually impair the electron density of the aromatic ring which may lead to more difficult coordination of the substrates with the catalyst.44 The yield of the reaction catalyzed by our V-based catalyst was among the highest reported in the literature (listed in the Table S4, ESI†), although the content of V in the catalyst was very low (0.07%), indicating that the monomeric VOx species were very efficient for promoting the reaction.
| Ent. | Substrates | Products | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: N-heterocycles (0.5 mmol), catalyst (50 mg), DMSO (1.5 mL), H2O (0.5 mL), O2 (0.5 MPa), 120 °C/16 h. b Yields are for the isolated product. c O2 (1.0 MPa). | |||
| 1 |
|
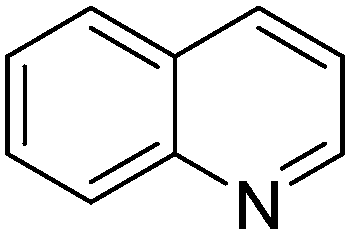
|
92.5 |
| 2 |
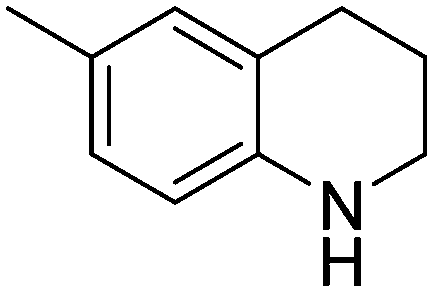
|
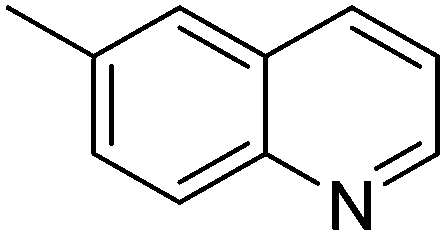
|
93.6 |
| 3 |
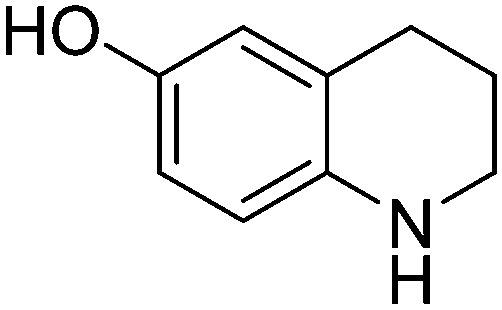
|
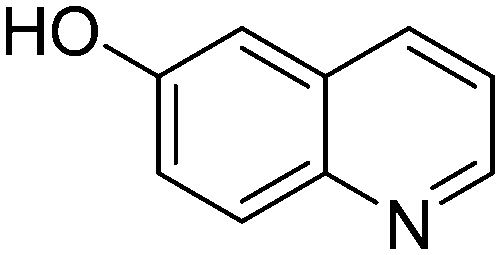
|
65.9 |
| 4 |
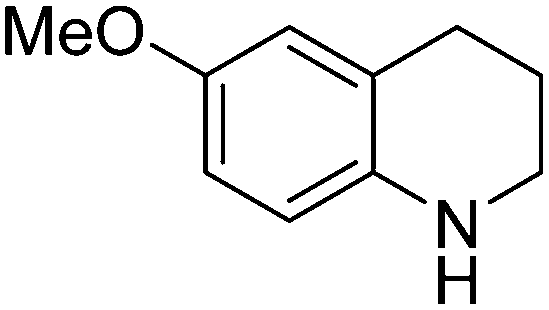
|
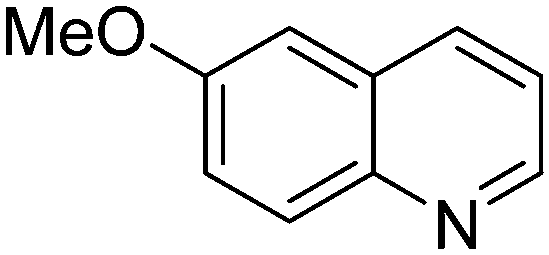
|
53.9 |
| 5c |
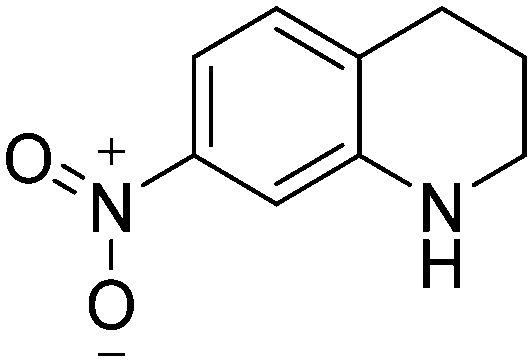
|
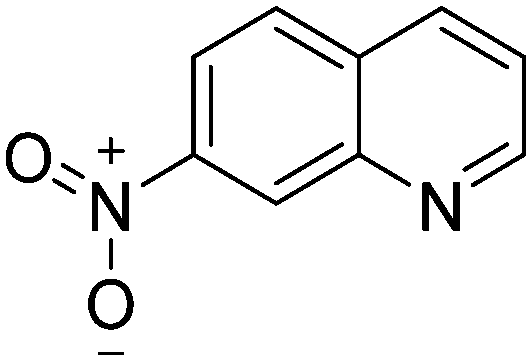
|
87.3 |
| 6 |
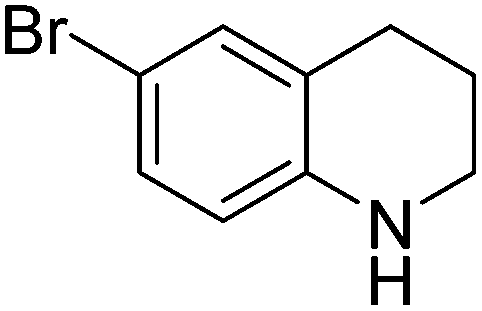
|
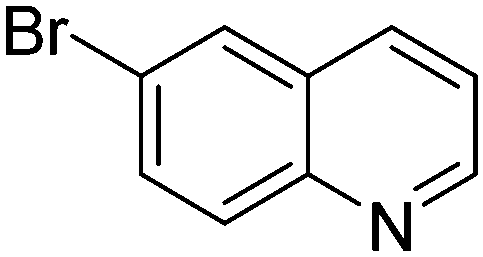
|
78.8 |
| 7 |
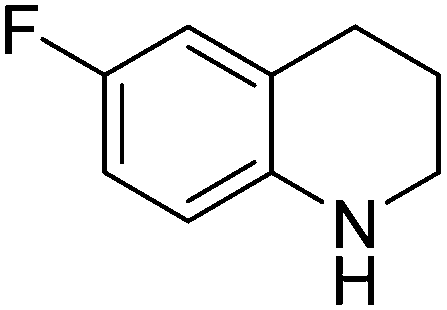
|
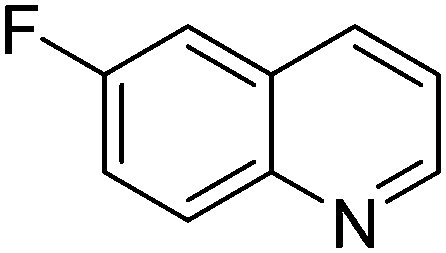
|
61.9 |
| 8 |
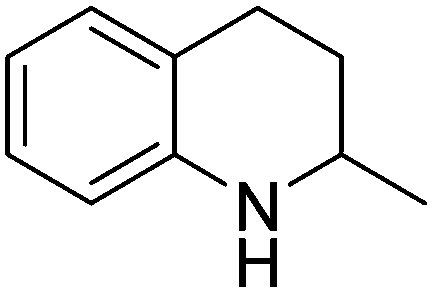
|
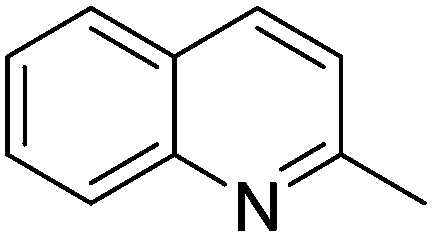
|
74.2 |
| 9 |
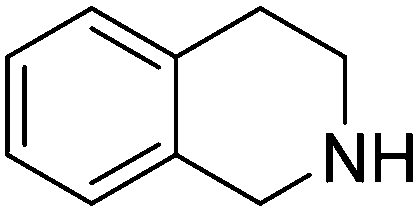
|
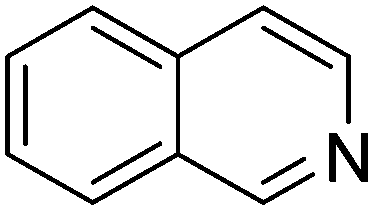
|
89.8 |
| 10 |
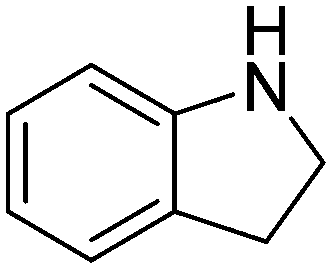
|
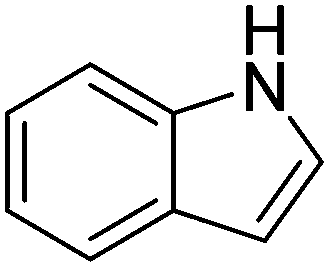
|
62.1 |
| 11c |
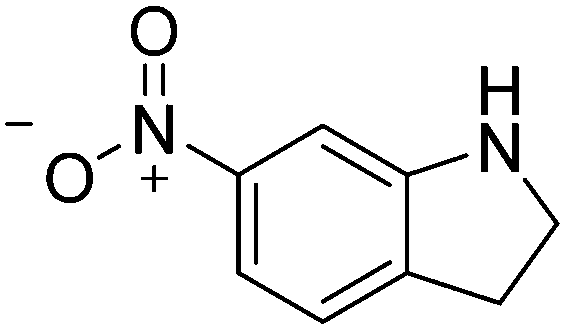
|
|
86.4 |
| 12 |
|
|
79.9 |
The recyclability of the VOx–NbOy@C catalyst was further investigated in the oxidative dehydrogenation of 1,2,3,4-tetra-hydroquinoline. The used catalyst was recovered from the reaction mixture via simple centrifugation and reused in the next cycle after rinsing with DMSO. As shown in Fig. 4, the catalytic performance did not change notably after reusing four times, indicating an excellent catalytic stability of the catalyst. As shown in the XRD analysis of the reused VOx–NbOy@C catalyst (Fig. S8, ESI†), there is no significant change in the crystal phase structure of the reused catalyst compared with the fresh one. Moreover, the chemical composition of the catalyst, detected by XPS analysis, did not change obviously after reusing four times (Fig. S9, ESI†). Therefore, the VOx–NbOy@C catalyst displayed good recyclability in the reaction.
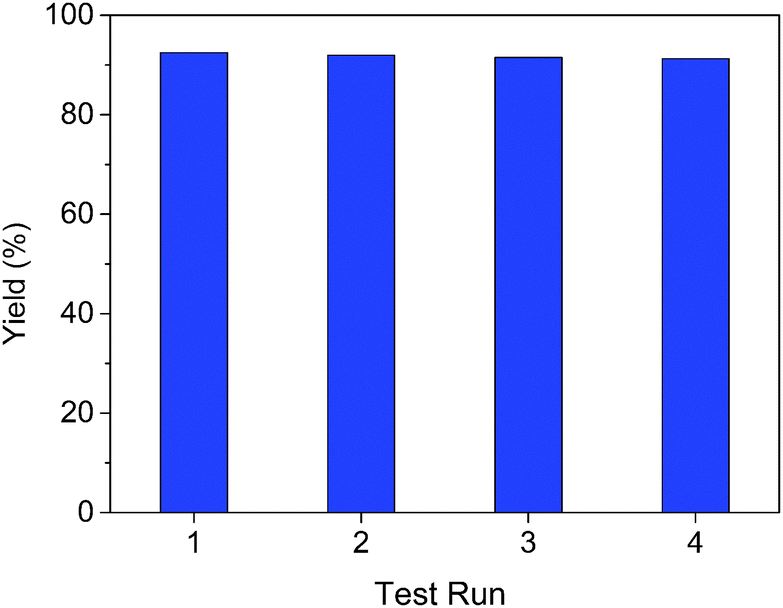 | ||
| Fig. 4 Recyclability of VOx–NbOy@C in the oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline. | ||
Conclusions
In summary, the VOx–NbOy@C catalytic materials, in which the monomeric VOx species were dispersed uniformly in the catalysts, was synthesized using a one-pot hydrothermal synthesis of the V/Nb inorganic/organic hybrid materials using the vanadium precursor, niobium tartrate and terephthalic acid followed by thermal treatment under a reducing atmosphere. The VOx–NbOy@C catalyst displayed excellent performance in the oxidative dehydrogenation of N-heterocycles. The monomeric VOx species and the NbOy in the composite catalytic materials showed a synergistic effect for promoting the reaction. The catalyst also exhibited good catalytic stability. We believe that the method used to prepare catalysts with monomeric VOx species can also be used to design other effective catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017YFA0403003, 2017YFA0403103), the National Natural Science Foundation of China (21871277 and 21403248), and the Recruitment Program of Global Youth Experts of China, and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- M. Mazidi, R. M. Behbahani and A. Fazeli, Appl. Catal., B, 2017, 209, 190–202 CrossRef CAS.
- M. J. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2013, 135, 4600–4603 CrossRef CAS PubMed.
- T. Lange, S. Heinrich, C. Liebner, H. Hieronymus and E. Klemma, Chem. Eng. Sci., 2012, 69, 440–448 CrossRef CAS.
- A. Marberger, D. Ferri, M. Elsener and O. Krocher, Angew. Chem., Int. Ed., 2016, 55, 11989–11994 CrossRef CAS PubMed.
- M. H. Zhu, J. K. Lai, U. Tumuluri, Z. L. Wu and I. E. Wachs, J. Am. Chem. Soc., 2017, 139, 15624–15627 CrossRef CAS PubMed.
- Y. Zhou, Z. P. Ma, J. J. Tang, N. Yan, Y. H. Du, S. B. Xi, K. Wang, W. Zhang, H. M. Wen and J. Wang, Nat. Commun., 2018, 9, 2931 CrossRef.
- K. Kamata, T. Yamaura and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7275–7278 CrossRef CAS PubMed.
- (a) J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed; (b) X. Rozanska, R. Fortrie and J. Sauer, J. Am. Chem. Soc., 2014, 136, 7751–7761 CrossRef CAS PubMed; (c) Z. J. Zhao, T. F. Wu, C. Y. Xiong, G. D. Sun, R. T. Mu, L. Zeng and J. L. Gong, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS PubMed; (d) U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- (a) S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haessner, Y. Saih, E. Abou-Hamad, I. Llorens, J. L. Hazemann, K. Kohler, V. D’Elia and J. M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS; (b) U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- B. Schimmoeller, Y. J. Jiang, S. E. Pratsinis and A. Baiker, J. Catal., 2010, 274, 64–75 CrossRef CAS.
- B. Mitra, I. E. Wachs and G. Deo, J. Catal., 2006, 240, 151–159 CrossRef CAS.
- (a) B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed; (b) E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. H. Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed; (c) P. X. Liu, Y. Zhao, R. X. Qin, S. G. Mo, G. X. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. D. Zang, B. H. Wu, G. Fu and N. F. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed; (d) L. Nie, D. H. Mei, H. F. Xiong, B. Peng, Z. B. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed; (e) Y. J. Chen, S. F. Ji, W. M. Sun, W. X. Chen, J. C. Dong, J. F. Wen, J. Zhang, Z. Li, L. R. Zheng, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2018, 140, 7407–7410 CrossRef CAS PubMed; (f) B. Zhang, H. Asakura, J. Zhang, J. G. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- X. J. Wang, X. Zhang, P. Li, K. Otake, Y. X. Cui, J. F. Lyu, M. D. Krzyaniak, Y. Y. Zhang, Z. Y. Li, J. Liu, C. T. Buru, T. Islamoglu, M. R. Wasielewski, Z. Li and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 8306–8314 CAS.
- X. F. Yang, A. Q. Wang, B. T. Qiao, J. Li, J. Y. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- (a) T. Zhang, D. Zhang, X. H. Han, T. Dong, X. W. Guo, C. S. Song, R. Si, W. Liu, Y. F. Liu and Z. K. Zhao, J. Am. Chem. Soc., 2018, 140, 16936–16940 CrossRef CAS PubMed; (b) S. B. Tian, Z. Y. Wang, W. B. Gong, W. X. Chen, Q. C. Feng, Q. Xu, C. Chen, C. Chen, Q. Peng, L. Gu, H. J. Zhao, P. Hu, D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2018, 140, 11161–11164 CrossRef CAS PubMed.
- (a) T. B. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Y. Zhang, J. H. Huang, H. Sun, M. Haruta, A. Q. Wang, B. T. Qiao, J. Li and T. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed; (b) J. W. Wan, W. X. Chen, C. Y. Jia, L. R. Zheng, J. C. Dong, X. S. Zheng, Y. Wang, W. S. Yan, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- G. L. Liu, A. W. Robertson, M. M. J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- L. Tsetseris, N. Kalfagiannis, S. Logothetidis and S. T. Pantelides, Phys. Rev. Lett., 2007, 99, 125503 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- (a) V. Sridharan, P. A. Suryavanshi and J. C. Menendez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) D. V. Jawale, E. Gravel, N. Shah, V. Dauvois, H. Y. Li, I. N. N. Namboothiri and E. Doris, Chem. – Eur. J., 2015, 21, 7039–7042 CrossRef CAS PubMed; (b) C. Deraedt, R. Ye, W. T. Ralston, F. D. Toste and G. K. Somorjai, J. Am. Chem. Soc., 2017, 139, 18084–18092 CrossRef CAS PubMed.
- (a) Y. H. Han, Z. Y. Wang, R. R. Xu, W. Zhang, W. X. Chen, L. R. Zheng, J. Zhang, J. Luo, K. L. Wu, Y. Q. Zhu, C. Chen, Q. Peng, Q. Liu, P. Hu, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 11262–11266 CrossRef CAS PubMed; (b) G. Jaiswal, M. Subaramanian, M. K. Sahoo and E. Balaraman, ChemCatChem, 2019, 11, 2449–2457 CrossRef CAS.
- G. Jaiswal, V. G. Landge, D. Jagadeesan and E. Balaraman, Nat. Commun., 2017, 8, 2147 CrossRef PubMed.
- P. Ryabchuk, A. Agapova, C. Kreyenschulte, H. Lund, H. Junge, K. Junge and M. Beller, Chem. Commun., 2019, 55, 4969–4972 RSC.
- N. Zumbragel, M. Sako, S. Takizawa, H. Sasai and H. Groger, Org. Lett., 2018, 20, 4723–4727 CrossRef PubMed.
- M. Karki, H. C. Araujo and J. Magolan, Synlett, 2013, 1675–1678 CAS.
- (a) K. Tanabe and S. Okazaki, Appl. Catal., A, 1995, 133, 191–218 CrossRef CAS; (b) I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS PubMed; (c) P. Carniti, A. Gervasini, C. Tiozzo and M. Guidotti, ACS Catal., 2014, 4, 469–479 CrossRef CAS.
- (a) A. Qiao, V. N. Kalevaru, J. Radnik, A. S. Kumar, N. Lingaiah, P. S. S. Prasad and A. Martin, Catal. Commun., 2013, 30, 45–50 CrossRef CAS; (b) M. Sarzi-Amade, S. Morselli, P. Moggi, A. Maione, P. Ruiz and M. Devillers, Appl. Catal., A, 2005, 284, 11–20 CrossRef CAS; (c) N. Ballarini, F. Cavania, C. Cortelli, C. Giunchi, P. Nobili, F. Trifirò, R. Catani and U. Cornaro, Catal. Today, 2003, 78, 353–364 CrossRef CAS.
- (a) M. Ziolek, A. Lewandowska, B. Grzybowska and A. Klisinska, React. Kinet. Catal. Lett., 2003, 80, 199–206 CrossRef CAS; (b) B. Kilos, M. Aouine, I. Nowak, M. Ziolek and J. C. Volta, J. Catal., 2004, 224, 314–325 CrossRef CAS; (c) R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS.
- A. Sarkar and P. Pramanik, Microporous Mesoporous Mater., 2009, 117, 580–585 CrossRef CAS.
- (a) J. S. Xu, J. X. Zhu, X. F. Yang, S. W. Cao, J. G. Yu, M. Shalom and M. Antonietti, Adv. Mater., 2016, 28, 6727 CrossRef CAS; (b) W. Kicinski and A. Dziura, Carbon, 2014, 75, 56–67 CrossRef CAS.
- J. Sun, W. Sun, L. Du, C. Du, Y. Gao and G. Yin, Fuel Cells, 2018, 18, 360–368 CrossRef CAS.
- (a) W. Q. Wu, K. L. Ding, J. Liu, T. Drake, P. Stair and E. Weitz, J. Phys. Chem. C, 2017, 121, 26794–26805 CrossRef CAS; (b) Y. L. Meng, T. Wan, S. Chen, Y. J. Zhao, X. B. Ma and J. L. Gong, Appl. Catal., B, 2014, 160, 161–172 CrossRef; (c) V. V. Kaichev, G. Y. Popova, Y. A. Chesalov, A. A. Saraev, D. Y. Zemlyanov, S. A. Beloshapkin, A. Knop-Gericke, R. Schlögl, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2014, 311, 59–70 CrossRef CAS.
- (a) E. N. Alvar, B. Zhou and S. H. Eichhorn, J. Mater. Chem. A, 2016, 4, 6540–6552 RSC; (b) C. W. Lin, A. Posadas, T. Hadamek and A. A. Demkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 035110 CrossRef.
- C. J. Patridge, L. Whittaker, B. Ravel and S. Banerjee, J. Phys. Chem. C, 2012, 116, 3728–3736 CrossRef CAS.
- T. Mitsudome, K. Miyagawa, Z. Maeno, T. Mizugaki, K. Jitsukawa, J. Yamasaki, Y. Kitagawa and K. Kaneda, Angew. Chem., Int. Ed., 2017, 56, 9381–9385 CrossRef CAS.
- Y. F. Wu, L. L. Fan, Q. H. Liu, S. Chen, W. F. Huang, F. H. Chen, G. M. Liao, C. W. Zou and Z. Y. Wu, Sci. Rep., 2015, 5, 9328 CrossRef CAS.
- K. Kaneda, T. Hara, N. Hashimoto, T. Mitsudome, T. Mizugaki and K. Jitsukawa, Catal. Today, 2010, 152, 93–98 CrossRef CAS.
- M. Ziolek, Catal. Today, 2003, 78, 47–64 CrossRef CAS.
- C. J. Patridge, C. Jaye, T. A. Abtew, B. Ravel, D. A. Fischer, A. C. Marschilok, P. H. Zhang, K. J. Takeuchi, E. S. Takeuchi and S. Banerjee, J. Phys. Chem. C, 2011, 115, 14437–14447 CrossRef CAS.
- X. J. Cui, Y. H. Li, S. Bachmann, M. Scalone, A. E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS PubMed.
- W. Zhang, G. Innocenti, P. Oulego, V. Gitis, H. H. Wu, B. Ensing, F. Cavani, G. Rothenberg and N. R. Shiju, ACS Catal., 2018, 8, 2365–2374 CrossRef CAS.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04708b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |