DOI:
10.1039/D0OB02151B
(Review Article)
Org. Biomol. Chem., 2021,
19, 2847-2855
Mutual kinetic resolution: probing enantiorecognition phenomena and screening for kinetic resolution with racemic reagents†
Received
26th October 2020
, Accepted 1st March 2021
First published on 15th March 2021
Abstract
Enantiorecognition between a racemic reagent and a racemic substrate can be a valuable process in organic synthesis. This review highlights representative examples of this phenomenon and the use of mutual kinetic resolution as a method for screening of kinetic and/or parallel kinetic resolutions.
 Stephen G. Davies | Steve Davies is the fifth Waynflete Professor of Chemistry at the University of Oxford. He has been the recipient of numerous prizes, including the following Royal Society of Chemistry awards: Hickinbottom Fellowship, Corday Morgan Medal and Prize, Award for Organometallic Chemistry, Bader Award, Tilden Lectureship, Award for Stereochemistry, and Perkin Prize for Organic Chemistry. He has published more than 600 papers and has research interests ranging from organometallic chemistry, asymmetric synthesis and natural product chemistry to medicinal chemistry and drug discovery. In 2014, he was elected Dr Honoris Causa, University of Salamanca (Spain). |
 Ai M. Fletcher | Ai Fletcher obtained a B. En. from Keio University, Japan, then moved to the U.K. where she pursued a Ph.D. at Imperial College London under supervision of Professor Chris Braddock. Since completing her Ph.D. in 2004, she has explored a range of chemistry as a post-doctoral researcher at the University of Regensburg (Professor Oliver Reiser), and at the University of Bath (Professor Michael Willis), she joined the group of Professor Steve Davies in Oxford in 2007, where she has been involved with the development of asymmetric synthetic methodology and its application to the total synthesis of natural products. |
 Paul M. Roberts | Paul Roberts graduated with an M.Chem. from Jesus College, Oxford, in 2000, which was followed by a D.Phil. with Professor Steve Davies in the area of the asymmetric synthesis of piperidine alkaloids employing a ring closing metathesis approach. In 2005, he took up a post-doctoral position with Professor Davies at Oxford, where his research interests centre upon natural product synthesis and the development of new stereoselective methodologies, for example to effect the chemo- and stereoselective functionalisation of allylic amines with a range of electrophilic reagents. |
 James E. Thomson | Jim Thomson studied chemistry at the University of Oxford where he gained an M.Chem. (2003) and then D.Phil. (2007), working with Professor Steve Davies in the area of β-amino acid organocatalysis. He then took up a post-doctoral position with Professor Davies, as a Junior Research Fellow, and in 2010 was appointed to a Research Fellowship in association with St Catherine's College, Oxford. His research interests centre upon the development of novel asymmetric transformations and the total synthesis of natural products. |
Introduction
Horeau, Kagan and Vigneron first reported an example of double asymmetric induction in 19681 and Masamune et al. defined the concept of double asymmetric induction in both racemic and enantiopure substrate systems.2 Mutual kinetic resolution (MKR) was mentioned by Heathcock and co-workers in 1981:3,4 “mutual kinetic resolution is expected in reactions between two chiral racemic compounds and that the magnitude of the effect should be proportional to the inherent diastereoselectivity shown by each compound in its reaction with achiral reaction partners”. In their report, Heathcock and co-workers showed that treatment of the racemic ketone 1 with LDA followed by addition of racemic aldehyde 2 and TMEDA gave aldol adduct 3 as a single diastereoisomeric product. This process involves formation of two new stereogenic centres which could thus afford up to eight possible diastereoisomeric products. The stereochemical outcome of this reaction was explained by two factors: (1) the relative stereochemistries of the two newly formed stereogenic centres are governed by the well-documented preferential stereoselective addition of the corresponding chelated (Z)-enolate derived from treatment of 1 with LDA, and an aldehyde; and (2) the (Z,R)-enantiomer of the racemic enolate 4 reacts with the (R)-enantiomer of the racemic aldehyde 2 faster than it does with the (S)-enantiomer of the racemic aldehyde 2; similarly and exactly complementary, the (Z,S)-enantiomer of the racemic enolate, ent-4, reacts faster with the (S)-enantiomer of the racemic aldehyde 2 than it does with the (R)-enantiomer of the racemic aldehyde 2, thus leading to the same diastereoisomer of the product in racemic form (Scheme 1).
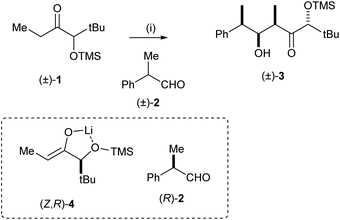 |
| | Scheme 1 Reagents and conditions: (i) LDA, THF, −70 °C, 2 h then TMEDA, (±)-2, 20 min. | |
Since Heathcock's report, several mutual kinetic resolutions have appeared in the literature. When high levels of enantiorecognition occur, it is as a result of the fact that one of the enantiomers of the racemic reagent reacts significantly faster with one of the enantiomers of the racemic substrate than with the other. However, the levels of enantiorecognition between two racemic reactants are not easily predictable and the majority of the reports have been one-off examples of reagent enhanced doubly diastereoselective reactions. We came to realise that this process could be useful, and have employed mutual kinetic resolution as a reliable method to evaluate the potential for efficient kinetic and/or parallel kinetic resolution protocols5 by quantifying levels of enantiorecognition using the stereoselectivity factor E.6 In this review, we will describe representative examples of mutual kinetic resolutions reported in the literature since Heathcock's seminal work, and examples where mutual kinetic resolution could be employed in screening while evaluating kinetic and/or parallel kinetic resolution for the analogous system of enantiopure reagents.
Mutual kinetic resolution: chiral recognition between racemic reactants
Several examples of mutual kinetic resolutions resulting in formation of one major (racemic) diastereoisomeric product have been reported in the literature;7 some of those will be presented in this section.
Total synthesis of erythromycin A
Several examples of mutual kinetic resolution have been reported in the area of target synthesis where a racemic reagent and a racemic substrate produced a major diastereoisomeric product with high diastereoselectivity. For example, in Woodward's posthumous report of the total synthesis of erythromycin A,8 erythronolide A seco acid 8, a key precursor for the synthesis, was prepared using an aldol reaction between racemic aldehyde 5 and the racemic enolate derived from ketone 6. The ketone 6 was treated with MeLi followed by addition of aldehyde 5, and then subsequent oxidation gave diketone 7 as a major diastereoisomeric product (83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 dr). In the corresponding reaction of enantiopure reagents 5 and 6, diketone 7 was obtained in 76% yield as a single diastereoisomer (Scheme 2). Diketone 7 was elaborated to erythnolide A seco acid 8.
:17 dr). In the corresponding reaction of enantiopure reagents 5 and 6, diketone 7 was obtained in 76% yield as a single diastereoisomer (Scheme 2). Diketone 7 was elaborated to erythnolide A seco acid 8.
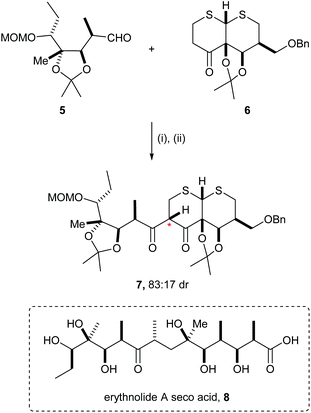 |
| | Scheme 2 Reagents and conditions: (i) MeLi, THF, −50 °C; (ii) TFAA, DMSO, CH2Cl2, −78 °C. | |
Mutual kinetic resolution of an α-chiral aldehyde and a chiral silylenol ether in an aldol reaction
Yamamoto and co-workers reported the highly efficient mutual kinetic resolution of racemic aldehyde 2 and racemic cyclic silylenol ether 9.9 Racemic 9 was treated with racemic 2 in the presence of TiCl4, which gave furanone 10 in 62% yield and very high diastereoisomeric purity (Scheme 3).10 The high diastereoselectivity in this reaction was rationalised by the combination of the preferential diastereofacial selectivity upon the addition of the chiral silyl enol ether 9 and preferential diastereofacial addition to aldehyde 2 in accordance with Cram's rule.11
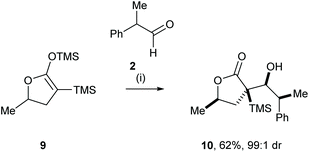 |
| | Scheme 3 Reagents and conditions: (i) TiCl4, CH2Cl2, −78 °C, 1 h. | |
Doubly diastereoselective reaction: nucleophilic addition of a chiral organolithium reagent to a chiral ketone
Johnson and Meanwell reported the total synthesis of the ginseng sesquiterpene (−)-β-panasinsene 14 employing the high degree of enantiorecognition between chiral organolithium reagent 12 and chiral ketone 11 as the key step in their synthesis.12 Due to the low efficiency when ketone 11 was exposed to Wittig olefination conditions, the authors demonstrated an alternative methylenation procedure employing a resolution exploiting sulfoximine chemistry. Addition of racemic (N-methylphenylsulfonimidoyl)methyllithium 12 to racemic ketone 11 gave 13 as essentially a single diastereoisomeric product. Treatment of the resulting adduct 13 with aluminium amalgam and AcOH gave (±)-β-panasinsene 14 in 67% yield from 11 (Scheme 4). This finding indicated that (+)-(S)-sulfoxiimine reagent 12 should react faster with the (−)-enantiomer of ketone 11, thus an enantioselective synthesis of (−)-β-panasinsene 14 was subsequently devised and successfully executed.
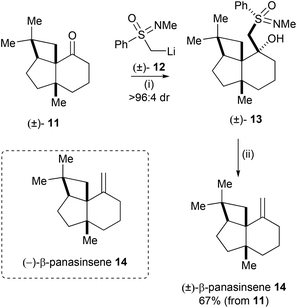 |
| | Scheme 4 Reagents and conditions: (i) (±)-12, −78 °C; (ii) aluminium amalgam, AcOH, THF, H2O. | |
Mutual kinetic resolution of oxiranyl carbaldimine by base-induced heterochiral dimerization
Wurthwein and co-workers reported an interesting case of mutual kinetic resolution in an aza-Darzens type reaction in their stereoselective synthesis of a highly functionalised aziridine.13 Treatment of racemic oxiranyl carbaldimine 15 with LDA and KOtBu gave dimerised aziridine 18 in 53% yield. The unexpected formation of 18 was explained by the deprotonation of the α-proton of epoxide 15 followed by nucleophilic attack of the resultant carbanion 16 to the enantiomeric oxiranyl carbaldimine 15 to give intermediate 17. The authors proposed regioselective nucleophilic ring-opening of the epoxide via a well-defined six-membered transition state and subsequent base induced second epoxide ring-opening gave aziridine 18 in 53% yield with high diastereoselectivity (Scheme 5). The high enantiorecognition in this reaction was demonstrated by the control experiment where analogous treatment of enantioenriched starting epoxide (S,S)-15 (86% ee) gave about 10 different products. The lack of the other enantiomer [i.e., the “matched” reactant14 pairing for (S,S)-15] in this process suppressed the formation of the dimerised aziridine product.
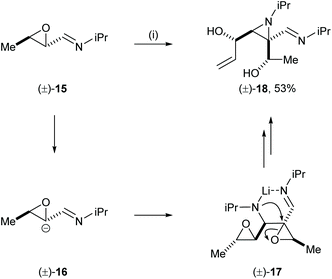 |
| | Scheme 5 Reagents and conditions: (i) LDA, KOtBu, THF, −78 °C to rt, 1 h. | |
Mutual kinetic resolution in the Michael addition reaction between a chiral lithium amide reagent and a chiral iron crotonoyl complex
Iron acyl complexes incorporating the chiral iron auxiliary [(C5H5)Fe(CO)(PPh3)] were originally developed for the highly diastereoselective functionalisation of the corresponding enolates and have been employed widely in stereoselective synthesis.15 We reported a high degree of chiral recognition in the Michael addition reaction between chiral lithium amides such as racemic lithium N-(3,4-dimethoxybenzyl)-N-(α-methylbenzyl)amide (RS)-20 and the chiral iron crotonyl complex (RS)-19.16 Treatment of iron crotonyl complex (RS)-19 with 2.0 equiv. of lithium amide (RS)-20 gave a 94:3:3 mixture of (RSFe,RS,αRS)-21, (RSFe,RS,αSR)-22 and (RSFe,SR,αRS)-23 in 82% combined yield (Scheme 6). This high degree of mutual kinetic resolution could be translated to a 15:1 rate difference between the “matched” [(R)-19 and (S)-20] and the “mismatched” [(S)-19 and (S)-20] pairs of reagents, or their antipodal pairs. This observation formed the basis for the development of secondary lithium amides derived from α-methybenzylamine and its derivatives as highly useful “chiral ammonia equivalents” in asymmetric synthesis.17
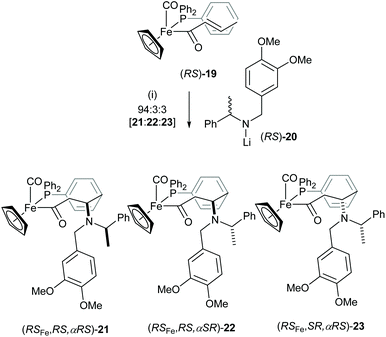 |
| | Scheme 6 Reagents and conditions: (i) THF, −78 °C, 2 h. | |
Stereoselective alkylation of succinoyl iron complexes via a mutual kinetic resolution strategy
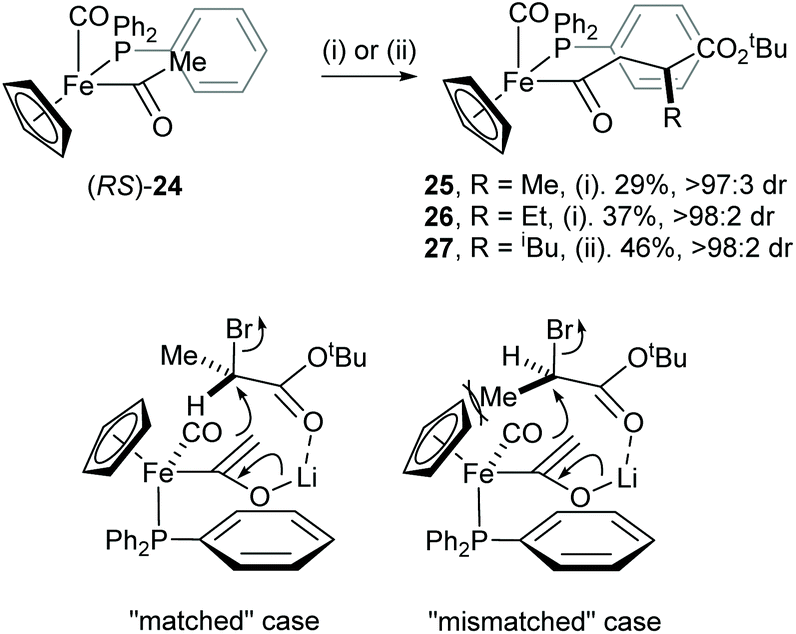
An efficient mutual kinetic resolution process was also found in the alkylation reaction between the iron acetyl enolate and α-bromoesters.18 Treatment of racemic iron acyl complex (RS)-24 with BuLi followed by addition of racemic RCHBrCO2tBu (R = Me or Et) or racemic iPrCH(OTf)CO2tBu gave racemic complexes 25–27 with excellent diastereoselectivity (>97:3 dr). The stereochemical outcome of this reaction was rationalised by a steric interaction of the α-methyl group within the electrophile and the cyclopentadienyl ligand of the iron complex 24, which occurs in the “mismatched” case (Scheme 7).
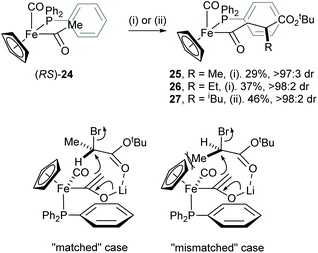 |
| | Scheme 7 Reagents and conditions: (i) BuLi, THF, −78 °C then (RS)-RCHBrCO2tBu, −78 °C, 10 min; (ii) BuLi, THF, −78 °C then (RS)-RCHOTfCO2tBu, −78 °C, 10 min. | |
Stereoselective acylation of heterocyclic amines via a mutual kinetic resolution strategy
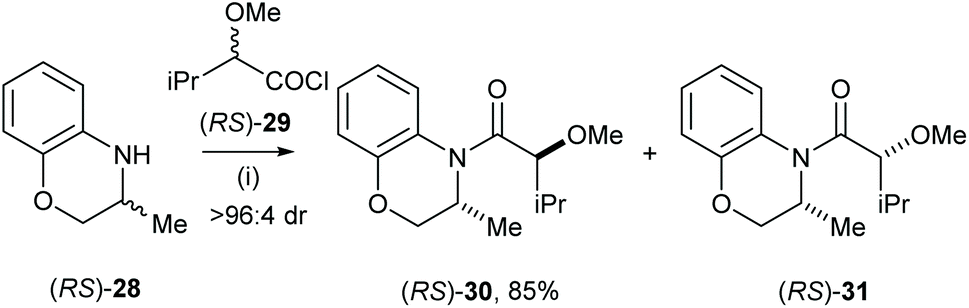
Very recently Vakarov and co-workers studied stereoselective acylation of racemic heterocylic amines and racemic 2-alkoxyacychloride.19 For example, racemic amine (RS)-28 was treated with racemic acylchloride (RS)-29 in toluene for 6 h, which gave the major diastereoisomeric product (RS)-30 with high selectivity (>96:4 dr) and isolated in 85% yield (Scheme 8). Acylation of the analogues system were also extensively studied.20,21
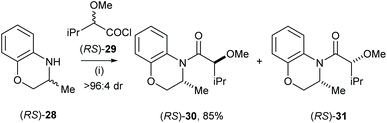 |
| | Scheme 8 Reagents and conditions: (i) (RS)-29, toluene, 20 °C, 6 h. | |
Mutual kinetic resolution as a screening method for kinetic and parallel kinetic resolutions
Mutual kinetic resolution can be a key initial evaluation method to assess the level of the enantiorecognition of the reagents. The levels of enantiorecognition are quantified by the stereoselectivity factor E,5 which is determined from the product distribution using 1H NMR spectroscopic analysis (i.e., ratio of the major diastereoisomeric product to the minor diastereoisomeric product). In this approach, the effects of mass action are eliminated, allowing the magnitude of the stereoselectivity factor E to be obtained independent of the reaction conversion.22,23 If high enantiorecognition is seen (i.e., high E value, typically >10) between the reacting reagents, efficient kinetic and parallel kinetic resolution can be expected.
Asymmetric syntheses of substituted cispentacin derivatives
The cyclic β-amino acid, 2-aminocyclopentane carboxylic acid (cispentacin) and its derivatives have attracted considerable attention in synthetic and biological chemistry due to their secondary structural properties, biological activities,24 and use as an efficient organocatalyst.25,26 We have enhanced the structural diversity of substituted cispentacin derivatives available by employing kinetic resolutions or parallel kinetic resolution strategies.27–29 Conjugate addition of racemic lithium amide reagent (RS)-32 to a range of 5-substituted methyl (RS)-cyclopent-1-ene-carboxylates 33–35, bearing an α-branched 5-substituent, gave major diastereoisomeric products 36–38 in good yield (>80%) and with high diastereoselectivities (>94:6 dr) (Scheme 9).
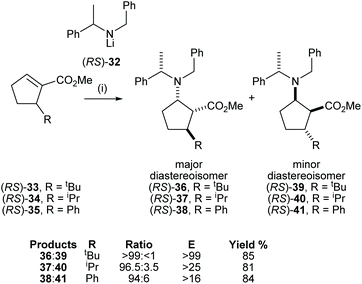 |
| | Scheme 9 Reagents and conditions: (i) (RS)-32, THF, −78 °C, 2 h. | |
High E values (>16 in each case) obtained in these mutual kinetic resolutions suggested that efficient kinetic resolutions could be developed in this system for the asymmetric synthesis of the corresponding enantiopure substituted cispentacin derivatives.29a Treatment of enantiopure lithium amide (S)-32 (0.8 equiv.) with (RS)-33 (1.0 equiv.) gave β-amino ester 36 in 94:6 dr and 42% isolated yield. The resolved α,β-unsaturated ester (S)-33 was also isolated in 41% yield and >99:1 er. β-Amino ester 36 was then elaborated to the corresponding β-amino acid 42 in 65% yield (over 2 steps) and 99:1 dr (Scheme 10).
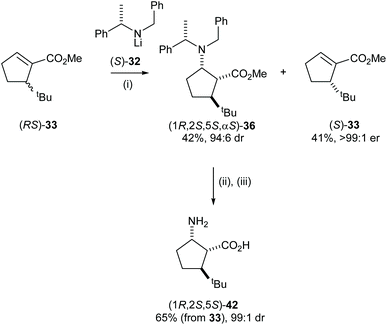 |
| | Scheme 10 Reagents and conditions: (i) (S)-32(0.8 equiv.), THF, −78 °C, 2 h; (ii) Pd(OH)2/C, MeOH, H2 (5 atm), rt; (iii) 20% HCl (aq), then Dowex 50Q-X8. | |
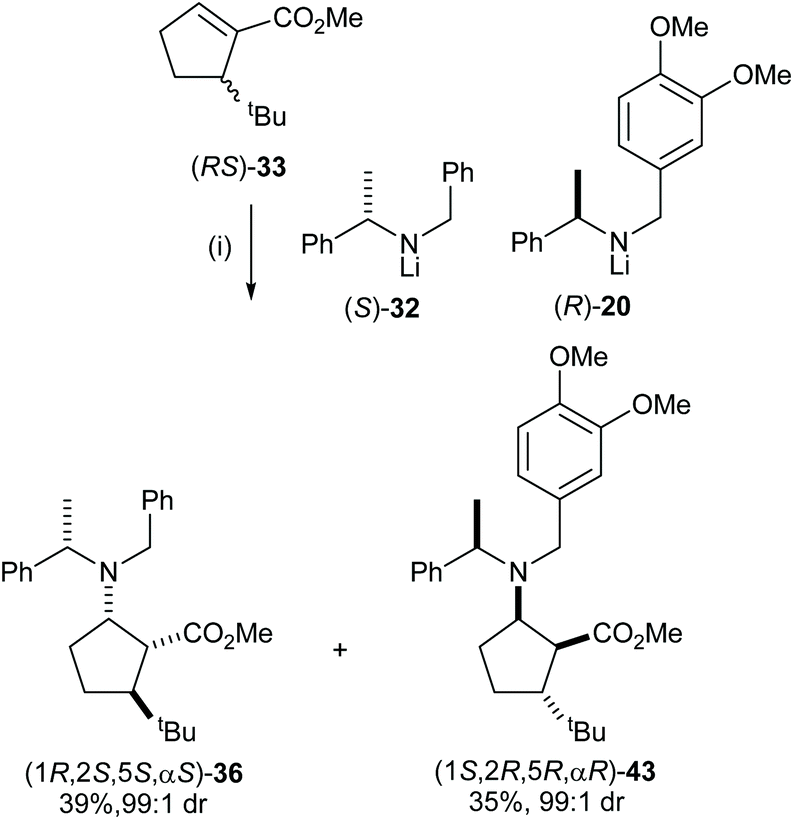
After developing a successful kinetic resolution for these conjugate addition reactions, we have shown that a 50:50 mixture of lithium (S)-N-benzyl-N-(α-methylbenzyl)amide 32 and lithium (R)-N-(3,4-dimethoxybenzyl)-N-(α-methylbenzyl)amide 20 could act as an optimal pseudoenantiomeric mixture for the corresponding parallel kinetic resolution. Conjugate addition of lithium N-benzyl-N-(α-methylbenzyl)amide 32 and lithium N-(3,4-dimethoxybenzyl)-N-(α-methylbenzyl)amide 20 to a series of α,β-unsaturated esters proceeded at similar rates28 and the high polarity of N-(3,4-dimethoxybenzyl)-N-(α-methylbenzyl)amino groups compared to N-benzyl-N-(α-methylbenzyl)amino groups facilitates easy separation of the pseudoenantiomeric β-amino ester products by flash column chromatography. A range of enantiomerically pure 5-substituted cispentacin derivatives were synthesised via a parallel kinetic resolution (PKR) strategy employing a pseudoenantiomeric mixture of lithium amides (S)-32 and (R)-20.29a For example, addition of a 50:50 mixture of (S)-32 and (R)-20 to α,β-unsaturated ester (RS)-33 gave a 50:50 mixture of β-amino esters 36 and 43 which were isolated as essentially single diastereoisomers in 39% and 35% yield, respectively, upon purification (Scheme 11).
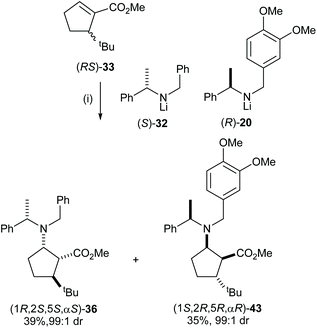 |
| | Scheme 11 Reagents and conditions: (i) (S)-32 (1.5 equiv.), (R)-20 (1.5 equiv.), THF, −78 °C, 2 h. | |
This strategy was also applied in kinetic27a,c,28 and parallel kinetic30 resolutions for the synthesis of enantiopure 3-alkyl-substituted cispentacin derivatives, and 6-alkyl-substituted cishexacin derivatives.31
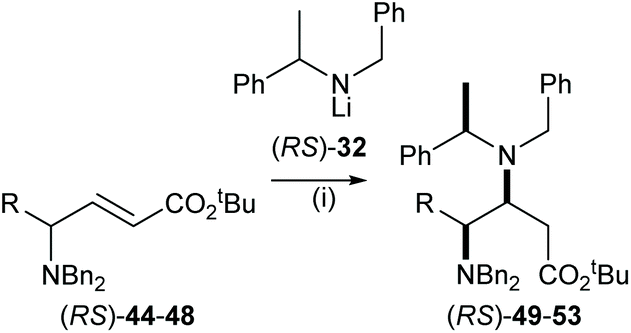
Mutual kinetic resolution of acyclic γ-amino-α,β-unsaturated esters
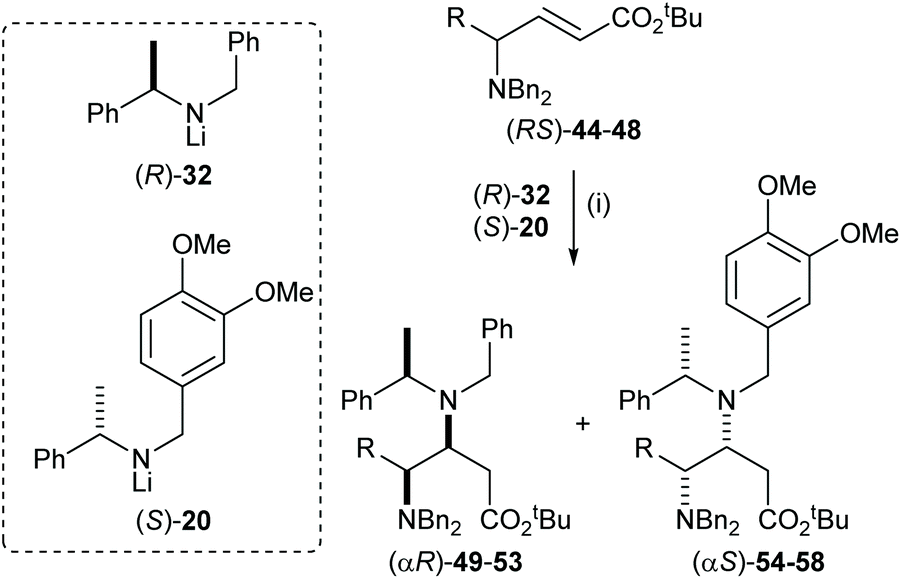
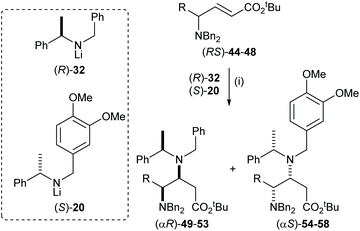
The highly efficient mutual kinetic resolution upon conjugate addition of chiral lithium amide reagents to chiral Michael acceptors was also observed in acyclic systems. Treatment of a range of acyclic γ-amino esters (RS)-44–48 with 1.6 equiv. of (RS)-32 gave γ,β-diamino esters 49–53 in >95:5 dr (Table 1). These results allowed us to predict that (RS)-44–48 were viable substrates for the corresponding parallel kinetic resolution protocol.
Table 1 Reagents and conditions: (i) (RS)-32 (1.6 equiv.), THF, −78 °C, 2 h
The parallel kinetic resolution of α,β-unsaturated esters (RS)-44–48 using a 50:50 pseudoenantiomeric mixture of lithium amides (R)-32 and (S)-20 gave the corresponding (αR)-products 49–53 and (αS)-products 54–58 in 31–42% yield and 40–43% yield, respectively, as a single diastereoisomer in each case (Table 2).32
Table 2 Reagents and conditions: (i) (S)-20 (2.0 equiv.), (R)-32 (2.0 equiv.), THF, −78 °C, 2 h
|
|
| |
R |
(αR) product yield % |
(αS) product yield % |
|
44
|
Me |
49, 40 |
54, 43 |
|
45
|
Et |
50, 31 |
55, 40 |
|
46
|
Bn |
51, 42 |
56, 43 |
|
47
|
iBu |
52, 39 |
57, 43 |
|
48
|
CH2OBn |
53, 37 |
58, 40 |
Resolution of 2-phenyl propanoic acid derivatives using pseudo enantiomeric oxazolidine-2-ones
Eames and co-workers reported the diastereoselective coupling of 2-phenylpropanoate and lithiated oxazolidine-2-ones, including a computational analysis of the potential pathway.33 This strategy enabled efficient resolution of 2-phenyl propanoic acid derivatives. For example, treatment of racemic 4-phenyloxazolidin-2-one 59 with nBuLi followed by the addition of racemic (RS)-60 gave syn-61 in 70% yield and 97:3 dr (Scheme 12).
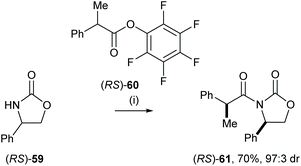 |
| | Scheme 12 Reagents and conditions: (i) nBuLi, THF, −78 °C. | |
The high levels of mutual kinetic resolution observed in this process allowed its application in a parallel kinetic resolution. Under the optimised conditions, racemic 2-methylphenyl propanoate (RS)-60 was treated with pseudoenantiomeric lithiated oxazolidine-2-one of (R)-59 and lithiated oxazolidine-2-thione of (S)-6234 to give enantiomerically pure syn-61 in 51% yield and 96:4 dr and syn-63 in 24% and 98:2 dr. Hydrolysis of enantiomerically pure 61 with LiOH in the presence of H2O2 gave (S)-64 in 90% yield and >98:2 er (Scheme 13).
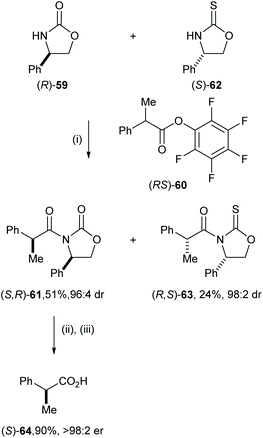 |
| | Scheme 13 Reagents and conditions: (i) nBuLi, THF, −78 °C, then (RS)-60 (2.0 equiv.), 2 h; (ii) LiOH, H2O2, H2O, THF, rt, 12 h; (iii) 3.0 M aq HCl. | |
Asymmetric syntheses of absouline
The levels of enantiorecognition (as quantified by the stereoselectivity factor, E) may also determine which approach (i.e., kinetic/parallel kinetic resolution or doubly diastereoselective reaction) would be more suitable for asymmetric preparation in target synthesis. We initially envisaged an enantiodivergent synthesis of both of the enantiomers of absouline 69via a parallel kinetic resolution approach. The level of enantiorecognition between chiral α,β-unsaturated ester (RS)-65 and the chiral lithium amide reagent 32 was initially evaluated by MKR, so that the selectivity factor could be directly quantified. Conjugate addition of racemic 32 to α,β-unsaturated ester (RS)-65 gave an 84:16 mixture of (3RS,4SR,αSR)-66 and (3RS,4RS,αSR)-67, which were isolated in 63% and 8% yield, respectively. This product ratio is consistent with E ≈ 5, indicating at best a modest level of enantiorecognition in this process, which suggested that α,β-unsaturated ester (RS)-65 is not a suitable substrate for the parallel kinetic resolution protocol. Thus, an approach based upon the application of double asymmetric induction was investigated. Enantiopure α,β-unsaturated ester (S)-65 was prepared from (S)-proline, and doubly diastereoselective conjugate addition of (S)-32 to α,β-unsaturated ester (S)-65 gave 66 in 74% yield as a single diastereoisomer. β-Amino ester 66 was elaborated to (−)-absouline 69 in four steps: hydrogenolytic N-debenzylation of 66 followed by cyclisation under acidic conditions gave 68·HCl in quantitative yield. Reduction of 68·HCl with excess DIBAL-H followed by DCC mediated coupling with (E)-p-methoxycinnamic acid to give (−)-absouline 69 in 49% yield over two steps (Scheme 14).35 By employing mutual kinetic resolution, evaluation of the level of enantiorecognition was possible without preparation of the corresponding enantiopure substrate and reagent, which led to an efficient determination of the synthetic strategy to employ, a doubly diastereoselective reaction in this case.
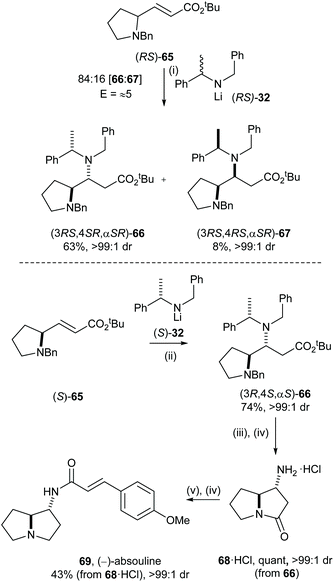 |
| | Scheme 14 Reagents and conditions: (i) (RS)-32, THF, −78 °C, 2 h; (ii) (S)-32, THF, −78 °C, 2 h; (iii) H2 (5 atm), Pd(OH)2/C (50% w/w), HCl (1.25 M in MeOH), rt, 48 h; (iv) 3.0 M aq HCl, 90 °C, 18 h; (v) DIBAL-H (1.0 M in THF), THF, 0 °C then rt, 18 h; (vi) trans-p-methoxycinnamic acid, DCC, DMAP, CH2Cl2, 0 °C then rt, 3 h. | |
Conclusions
In conclusion, this review presents several important cases of mutual kinetic resolution (the chiral recognition phenomena between two racemic reactants) that have appeared in the literature. We have identified this process as a powerful tool to screen for efficient kinetic and/or parallel kinetic resolution protocols by simple determination of the selectivity factor E, which can be calculated from the product distribution of major and minor diastereoisomers. In the future, the authors hope that use of mutual kinetic resolution to determine the selectivity factor E will become a more widely recognised approach in the synthetic community, which should allow an expedient evaluation of the levels of enantiorecognition that may be expected in a particular scenario; in turn it is hoped that access to this information will allow future synthetic endeavours to be planned with a greater degree of certainty of success.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- A. Horeau, H.-B. Kagan and J. P. Vigneron, Bull. Soc. Chim. Fr., 1968, 3795 CAS.
-
S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1 Search PubMed.
- C. H. Heathcock, M. C. Pirrung, J. Lampe, C. T. Buse and S. D. Young, J. Org. Chem., 1981, 46, 2290 CrossRef CAS.
- C. H. Heathcock, M. C. Pirrung, C. T. Buse, J. P. Hagen, S. D. Young and J. E. Sohn, J. Am. Chem. Soc., 1979, 101, 7077 CrossRef CAS.
- Some representative reviews for parallel kinetic resolutions, see:
(a) J. Eames, Angew. Chem., Int. Ed., 2000, 39, 885 CrossRef CAS;
(b) J. R. Dehli and V. Gotor, Chem. Soc. Rev., 2002, 31, 365 RSC.
- A. Horeau, Tetrahedron, 1975, 31, 1307 CrossRef CAS.
- T. Berkenbusch, A. C. Laungani, R. Brückner and M. Keller, Tetrahedron Lett., 2004, 45, 9517 CrossRef CAS.
- R. B. Woodward, E. Logusch, K. P. Nambiar, K. Sakan, D. E. Ward, B.-W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card, C. H. Chen, R. B. Chenevert, A. Fliri, K. Frobel, H.-J. Gals, D. G. Garratt, K. Hayakawa, W. Heggie, D. P. Hesson, D. Hoppe, I. Hoppe, J. A. Hyatt, D. Ikeda, P. A. Jacobi, K. S. Kim, Y. Kobuke, K. Kojima, K. Krowicki, V. J. Lee, T. Leutert, S. Malchenko, J. Martens, R. S. Matthews, B. S. Ong, J. B. Press, T. V. Rajan Babu, G. Rousseau, M. Sauter, M. Suzuki, K. Tatsuta, L. M. Tolbert, E. A. Truesdale, I. Uchida, Y. Ueda, T. Uyehara, A. T. Vasella, W. C. Vladuchick, P. A. Wade, R. M. Williams and H. N.-C. Won, J. Am. Chem. Soc., 1981, 103, 3210 CrossRef.
- Y. Tomo and K. Yamamoto, Tetrahedron Lett., 1985, 26, 1061 CrossRef CAS.
- Other examples of mutual kinetic resolution in aldol reactions have also been reported in the literature; for example, see: D. E. Ward and A. Kazemeini, J. Org. Chem., 2012, 77, 10789 CrossRef CAS PubMed.
- A. Mengel and O. Reiser, Chem. Rev., 1999, 99, 1191 CrossRef CAS.
- C. R. Johnson and N. A. Meanwell, J. Am. Chem. Soc., 1981, 103, 7667 CrossRef CAS.
- D. Alickmann, R. Fröhlich and E.-U. Wurthwein, Org. Lett., 2001, 3, 1527 CrossRef CAS PubMed.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1 CrossRef.
- For example, see:
(a) N. Aktogu, H. Felkin and S. G. Davies, J. Chem. Soc., Chem. Commun., 1982, 1303 RSC;
(b) K. Broadley and S. G. Davies, Tetrahedron Lett., 1984, 25, 1743 CrossRef CAS;
(c) S. G. Davies, I. M. Dordor, J. C. Walker and P. Warner, Tetrahedron Lett., 1984, 25, 2709 CrossRef CAS;
(d) S. G. Davies, J. I. Seeman and I. H. Williams, Tetrahedron Lett., 1986, 27, 619 CrossRef CAS;
(e) S. G. Davies, Pure Appl. Chem., 1988, 60, 13 CAS;
(f) S. G. Davies, D. Middlemiss, A. Naylor and M. Wills, Tetrahedron Lett., 1989, 30, 2971 CrossRef CAS;
(g) S. G. Davies, R. Polywka and P. Warner, Tetrahedron, 1990, 46, 4847 CrossRef CAS;
(h) G. J. Bodwell, S. G. Davies and S. C. Preston, J. Organomet. Chem., 1991, 402, C56 CrossRef CAS;
(i) G. J. Bodwell, S. G. Davies and A. A. Mortlock, Tetrahedron, 1991, 47, 10077 CrossRef CAS;
(j) S. G. Davies and G. L. Gravatt, J. Chem. Soc., Chem. Commun., 1988, 780 RSC;
(k) G. J. Bodwell and S. G. Davies, Tetrahedron: Asymmetry, 1991, 2, 1075 CrossRef CAS;
(l) S. L. Brown, S. G. Davies, D. F. Foster, J. I. Seeman and P. Warner, Tetrahedron Lett., 1986, 27, 623 CrossRef CAS;
(m) S. G. Davies, R. J. C. Easton, A. Gonzalez, S. C. Preston, K. H. Sutton and J. C. Walker, Tetrahedron, 1986, 42, 3987 CrossRef CAS;
(n) S. G. Davies, R. J. C. Easton, K. H. Sutton, J. C. Walker and R. H. Jones, J. Chem. Soc., Perkin Trans. 1, 1987, 489 RSC;
(o) S. G. Davies, D. Middlemiss, A. Naylor and M. Wills, Tetrahedron Lett., 1989, 30, 587 CrossRef CAS;
(p) S. G. Davies, D. Middlemiss, A. Naylor and M. Wills, J. Chem. Soc., Chem. Commun., 1990, 797 RSC;
(q) S. G. Davies, H. M. Kellie and R. Polywka, Tetrahedron: Asymmetry, 1994, 5, 2563 CrossRef CAS.
- S. G. Davies and O. Ichihara, J. Chem. Soc., Chem. Commun., 1990, 1554 RSC.
-
(a) S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2017, 28, 1842 CrossRef CAS;
(b) S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2012, 23, 1111 CrossRef CAS;
(c) S. G. Davies, P. D. Price and A. D. Smith, Tetrahedron: Asymmetry, 2005, 16, 2833 CrossRef CAS.
- G. Bashiardes, G. J. Bodwell, S. P. Collingwood, J. F. Costello, S. G. Davies, A. M. Fletcher, A. Ganer, S. C. Preston and J. E. Thomson, Tetrahedron, 2014, 70, 8938 CrossRef CAS.
- S. A. Vakarov, D. A. Gruzdev, L. S. Sadretdinova, M. I. Kodess, A. A. Tumashov, E. B. Gorbunov, G. L. Levit and V. P. Krasnov, Chem. Heterocycl. Compd., 2018, 54, 437 CrossRef CAS.
- S. A. Vakarov, M. A. Korolyova, D. A. Gruzdev, M. G. Pervova, G. L. Levit and V. P. Krasnov, Russ. Chem. Bull., 2019, 68, 1257 CrossRef CAS.
- M. A. Korolyova, S. A. Vakarov, D. N. Kozhevnikov, D. A. Gruzdev, G. L. Levit and V. P. Krasnov, Eur. J. Org. Chem., 2018, 4577 CrossRef CAS.
- H. B. Kagan, Adv. Synth. Catal., 2001, 343 Search PubMed.
- D. W. Johnson Jr. and D. A. Singleton, J. Am. Chem. Soc., 1999, 121, 9307 CrossRef.
- F. Fülöp, Chem. Rev., 2001, 2181 CrossRef PubMed.
- S. G. Davies, R. L. Sheppard, A. D. Smith and J. E. Thomson, Chem. Commun., 2005, 3802 RSC.
- S. G. Davies, A. J. Russell, R. L. Sheppard, A. D. Smith and J. E. Thomson, Org. Biomol. Chem., 2007, 5, 3190 RSC.
- For the kinetic resolution of 3-alkyl-cyclopent-1-ene-carboxylates, see:
(a) S. Bailey, S. G. Davies, A. D. Smith and J. M. Withey, Chem. Commun., 2002, 2910 RSC;
(b) M. E. Bunnage, A. M. Chippindale, S. G. Davies, R. M. Parkin, A. D. Smith and J. M. Withey, Org. Biomol. Chem., 2003, 1, 3698 RSC;
(c) M. E. Bunnage, S. G. Davies, R. M. Parkin, P. M. Roberts, A. D. Smith and J. M. Withey, Org. Biomol. Chem., 2004, 2, 3337 RSC.
- S. G. Davies, A. C. Garner, M. J. C. Long, A. D. Smith, M. J. Sweet and J. M. Withey, Org. Biomol. Chem., 2004, 2, 3355 RSC.
- For the kinetic and parallel kinetic resolution of 5-alkyl-cyclopent-1-ene-carboxylates, see:
(a) S. G. Davies, D. Díez, M. M. El Hammouni, A. C. Garner, N. M. Garrido, M. J. C. Long, R. M. Morrison, A. D. Smith, M. J. Sweet and J. M. Withey, Chem. Commun., 2003, 2410 RSC;
(b) S. G. Davies, A. C. Garner, M. J. C. Long, R. M. Morrison, P. M. Roberts, E. D. Savory, A. D. Smith, M. J. Sweet and J. M. Withey, Org. Biomol. Chem., 2005, 3, 2762 RSC.
- Y. Aye, S. G. Davies, A. C. Garner, P. M. Roberts, A. D. Smith and J. E. Thomson, Org. Biomol. Chem., 2008, 6, 2195 RSC.
- S. G. Davies, M. J. Durbin, S. J. S. Hartman, A. Matsuno, P. M. Roberts, A. J. Russell, A. D. Smith, J. E. Thomson and S. M. Toms, Tetrahedron: Asymmetry, 2008, 19, 2870 CrossRef CAS.
- S. G. Davies, J. A. Lee, P. M. Roberts, J. E. Thomson and J. Yin, Org. Lett., 2012, 14, 218 CrossRef CAS PubMed.
- N. Al Shaye, D. M. Benoit, S. Chavda, E. Coulbeck, M. Dingjan, J. Eames and Y. Yohannes, Tetrahedron: Asymmetry, 2011, 22, 413 CrossRef.
- The authors showed that lithiated oxazolidine-2-one of 59 reacted at least four times faster than the lithiated oxazolidine-2-thione of 62.
- S. G. Davies, A. M. Fletcher, C. Lebée, P. M. Roberts, J. E. Thomson and J. Yin, Tetrahedron, 2013, 69, 1369 CrossRef CAS.
Footnote |
| † This article is part of the special issue dedicated to The Horeau Principle. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ai M.
Fletcher
,
Paul M.
Roberts
and
James E.
Thomson
*,
Ai M.
Fletcher
,
Paul M.
Roberts
and
James E.
Thomson