
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of organophosphorus compounds using H–P reagents derived from chiral alcohols†
Joseph D.
Gbubele
 and
Tomasz K.
Olszewski
*
and
Tomasz K.
Olszewski
*
Faculty of Chemistry, Wrocław University of Science and Technology, ul. Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: tomasz.olszewski@pwr.edu.pl
First published on 18th February 2021
Abstract
Chiral organophosphorus compounds, especially those containing C-stereogenic carbons in the proximity of the phosphorus atom, are known for their unique properties and have found wide applications that span from medicinal chemistry to enantioselective catalysis. However, the synthesis of such chiral molecules, especially with the precise control of stereochemistry at chiral carbon atoms, still remains a very challenging task. This review summarizes recent advances in the highly stereoselective formation of C- and, in some cases, also P-stereogenic organophosphorus compounds. The presented synthesis strategy is based on the use of H–P reagents bearing TADDOL, BINOL or a menthol moiety attached to the phosphorus atom and serving as a chiral auxiliary. Reactions of such chiral H–P species with different partners, e.g., alkenes, alkynes, imines, and carbonyl compounds, leading to structurally diverse chiral organophosphorus compounds with up to five chiral centers are comprehensively discussed. In each case, the stereochemical outcome of the reaction is influenced by the presence of the chiral alcohol used; therefore, the content of this review is compiled into sections with respect to the type of chiral alcohol attached to the phosphorus atom in the H–P species applied.
 Joseph D. Gbubele | Joseph D. Gbubele obtained his MS in 2019 in chemistry from the Wroclaw University of Science and Technology (WUST) under the Ignacy Lukasiewicz scholarship. He conducted his M.Sc. thesis at the Department of Bioorganic Chemistry, where he worked on the synthesis of aminophosphonic acids using cyclobutanone and its derivatives as substrates under the guidance of Prof. Paweł Kafarski. In October 2019, he started his Ph.D. studies at the Department of Physical and Quantum Chemistry of WUST. He is working on the development of the methodology for the diastereoselective preparation of structurally diverse substituted phosphonates and phosphonic acids under the direction of Dr Tomasz K. Olszewski. |
 Tomasz K. Olszewski | Tomasz K. Olszewski obtained his Ph.D. degree in 2006 from the Wroclaw University of Science and Technology (WUST), Poland (with Prof. Bogdan Boduszek) and the University of Montpellier, France (under the guidance of Prof. Claude Grison). After a postdoctoral period and working in industry, he returned to academia in 2012 and obtained habilitation in 2016. Currently, he holds the position of Associate Professor at WUST. His research interests are focused on the synthesis and applications of organophosphorus compounds and recently on catalysis. |
1. Introduction
Organophosphorus compounds due to their interesting physicochemical properties have found wide applications in many important areas of the chemical industry such as synthesis of utility chemicals, e.g., flame retardants,1 anticorrosive coatings and adhesives,2–4 ligands for catalysis,5–9 agrochemicals (e.g. insecticides, herbicides and fungicides),10–12 and finally pharmaceutically active compounds.13–18 Selected pharmaceutically important applications of chiral organophosphorus molecules include19–27 treatment of HIV and hepatitis B (Tenofovir),19 treatment of heart failure,20 antibiotics (Fosfamycin,21 Valinophos,22 Fosfazinomycin A23), renin24 and leucine aminopeptidase25 inhibitors, antiviral agents,26 and antibacterial and antifungal agents27 (Fig. 1).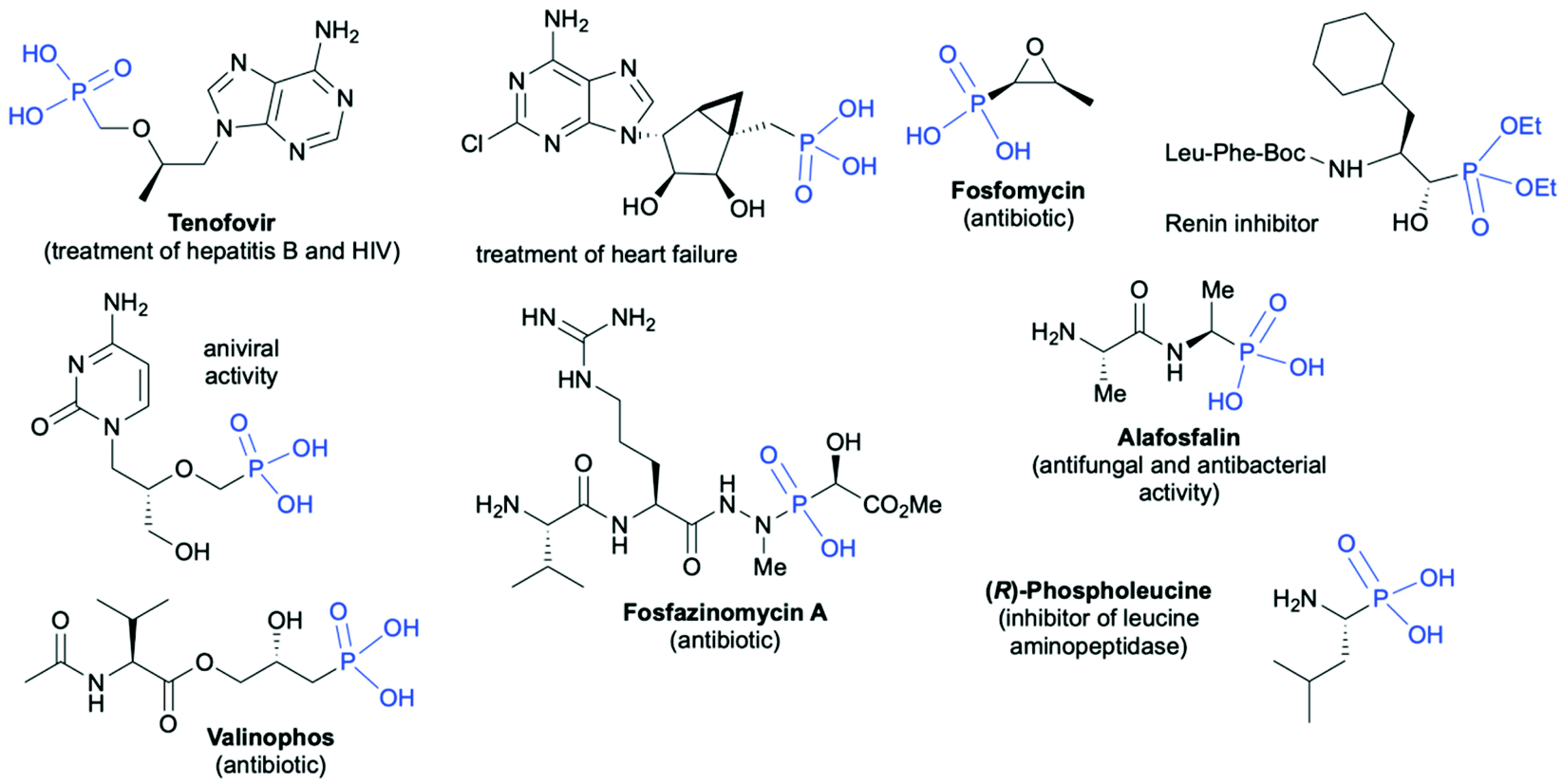 | ||
| Fig. 1 Selected examples of biologically active chiral organophosphorus compounds bearing C-stereogenic centers in the proximity of the phosphorus atom. | ||
The utility of organophosphorus compounds as biologically active molecules along with their applications as ligands for catalysis are both considered as most important and challenging since in serving to control biological and chemical processes it is very often essential to use chiral molecules with well-defined configuration, especially at the chiral carbon center attached to the phosphorus atom. Not surprisingly, owing to the importance of chiral organophosphorus compounds, several methods have emerged for their effective syntheses in an asymmetric fashion.28–31 Among the proposed strategies, the use of H–P species bearing a chiral auxiliary attached to the phosphorus atom represents an interesting approach in which asymmetric induction on a C atom is induced by the presence of a chiral auxiliary.32,33 The summarized literature shows that in the role of chiral auxiliaries readily available and non-expensive TADDOL, BINOL and menthol chiral alcohols are used (Fig. 2).
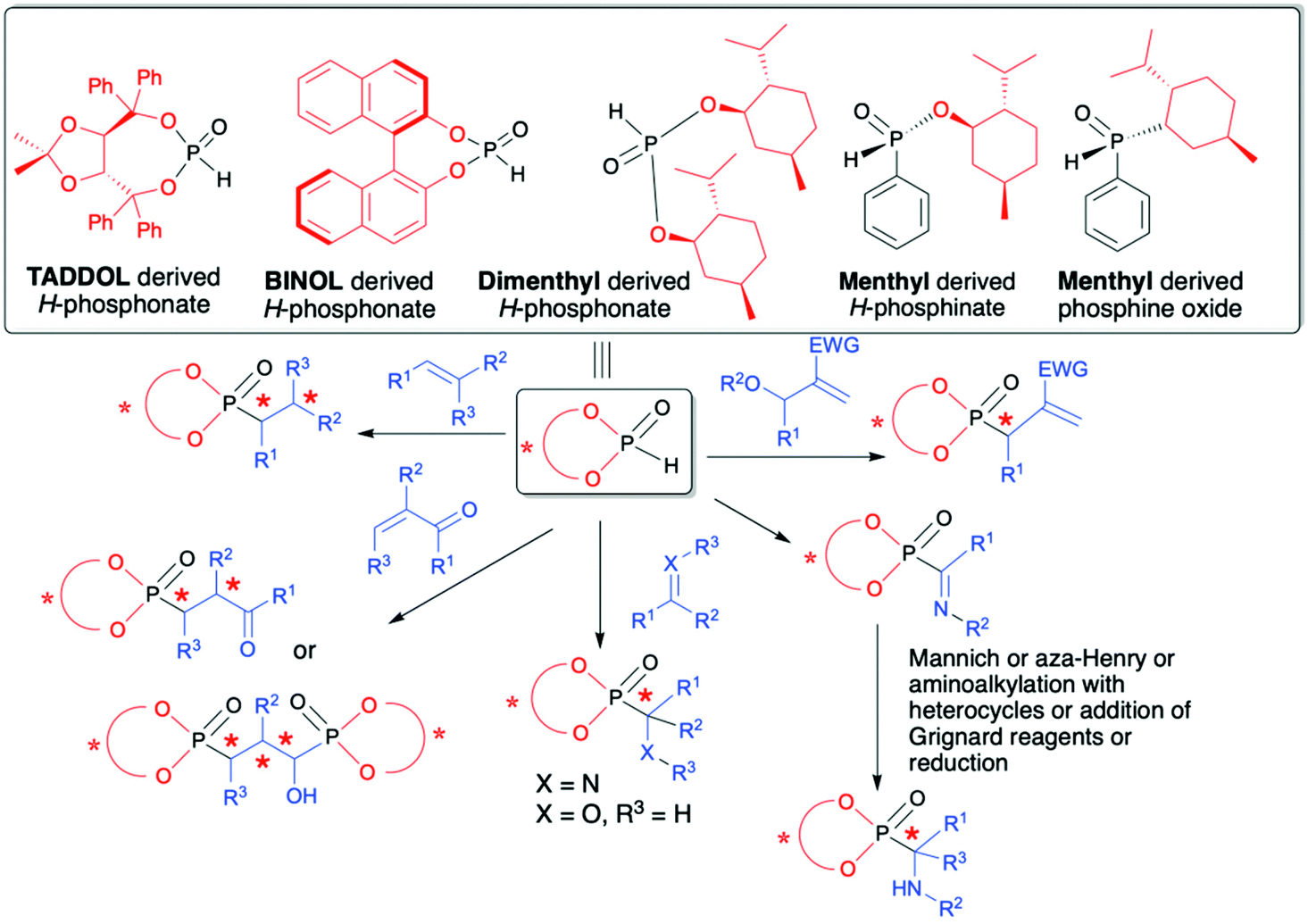 | ||
| Fig. 2 Commonly used chiral H–P species with chiral alcohols attached to the phosphorus atom and their application in the asymmetric synthesis of C-stereogenic organophosphorus compounds. | ||
Reactions of such chiral H–P species with different partners, e.g., alkenes, alkynes, imines and carbonyl compounds, provide convenient access to structurally diverse C-, and in some cases also P-, chiral organophosphorus compounds with up to five stereogenic centers. Additionally, the use of H–P species bearing a chiral auxiliary bound to the phosphorus atom can be considered as a useful and straightforward tool for monitoring the stereochemical outcome of the reaction by means of using 31P or/and 1H NMR spectroscopy since the addition of such chiral H–P species enables the formation of a pair of diastereomers in the product that are clearly distinguishable by NMR spectroscopy.
This review provides the reader with an in-depth overview of recent advances in the asymmetric synthesis of organophosphorus compounds containing C- and in some cases P-stereogenic centers with the use of chiral H–P species derived from TADDOL, BINOL or menthol, serving as chiral auxiliaries attached to the phosphorus atom. Special emphasis is placed on such important aspects as methods used for the determination of reaction stereoselectivity, determination of absolute configuration on the new stereogenic carbon atom and isolation of optically pure products. Since in each case the stereochemical outcome of the reaction is influenced by the presence of chiral alcohol used, therefore, the content of this review is compiled into sections with respect to the type of chiral alcohol attached to the phosphorus atom in H–P species used (Fig. 2).
2. Application of H–P species bearing chiral alcohol attached to the phosphorus atom
2.1. TADDOL-derived H-phosphonate (2)
TADDOLs and their derivatives have become an important source of chirality in organic synthesis,34–37 and TADDOL is considered one of the few privileged chiral ligands and catalysts.38 The synthesis of (S,S)- or (R,R)-4,5-bis(diphenylhydroxymethyl)-2,2-dimethyl-1,3-dioxalane (1) (TADDOL) has been known for decades39–41 and involves four-step transformation starting from affordable and commercially available tartaric acid dimethyl ester (either enantiomer) (Scheme 1). The H-phosphonate (R,R)-2 bearing an (R,R)-TADDOL chiral auxiliary bound to the phosphorus atom can be conveniently prepared, even on a multigram scale, from (R,R)-(1) in reaction with PCl3 followed by hydrolysis of the corresponding chloride (Scheme 1).40,42 The final (R,R)-(2) is an air and moisture stable white solid that can be stored for months without any precautions.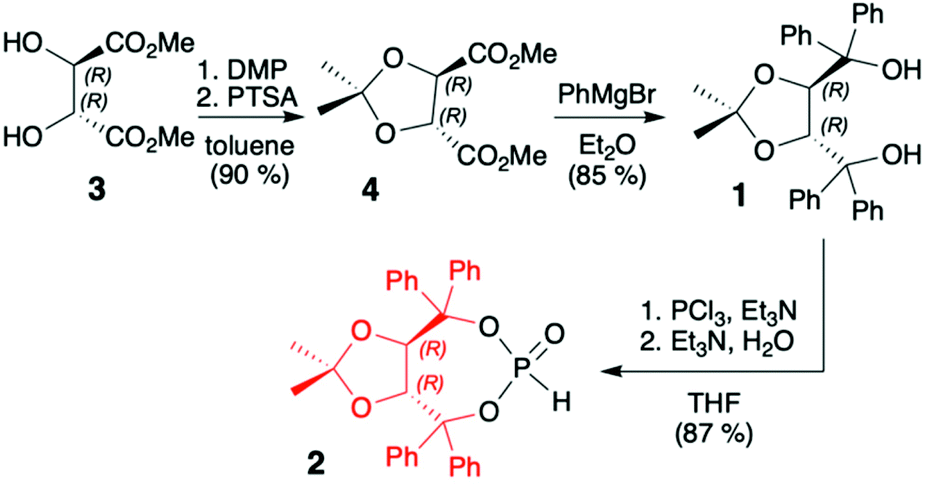 | ||
| Scheme 1 Synthesis of TADDOL H-phosphonate (2). DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C double bond.
Enders and co-workers reported a series of articles devoted to the use of enantiopure (R,R)-(2) in the asymmetric synthesis of organophosphorus compounds. In the seminal work, they used (R,R)-TADDOL H-phosphonate (2) in the successful asymmetric synthesis of β-nitrophosphonates (6) via phospha-Michael addition to aromatic nitroalkenes (Scheme 2).42 The developed protocol was based on the use of diethylzinc (Et2Zn), which in the reaction with (R,R)-2 formed a very reactive organozinc–phosphorus adduct43,44 that underwent smooth addition to nitroalkenes. During preliminary tests to achieve high diastereoselectivity of the reaction, low temperatures (−78 °C) were required, and under such conditions the organozinc–phosphorus adduct was found to be very insoluble.42 The solubility was improved by the addition of N,N,N′,N′-tetramethylenediamine (TMEDA) so that it was possible to perform the phospha-Michael addition at a low temperature and obtain the desired β-nitrophosphonates (6) with good yields (86–89%) and diastereoselectivities (de 84–96%) (Scheme 2).
C double bond.
Enders and co-workers reported a series of articles devoted to the use of enantiopure (R,R)-(2) in the asymmetric synthesis of organophosphorus compounds. In the seminal work, they used (R,R)-TADDOL H-phosphonate (2) in the successful asymmetric synthesis of β-nitrophosphonates (6) via phospha-Michael addition to aromatic nitroalkenes (Scheme 2).42 The developed protocol was based on the use of diethylzinc (Et2Zn), which in the reaction with (R,R)-2 formed a very reactive organozinc–phosphorus adduct43,44 that underwent smooth addition to nitroalkenes. During preliminary tests to achieve high diastereoselectivity of the reaction, low temperatures (−78 °C) were required, and under such conditions the organozinc–phosphorus adduct was found to be very insoluble.42 The solubility was improved by the addition of N,N,N′,N′-tetramethylenediamine (TMEDA) so that it was possible to perform the phospha-Michael addition at a low temperature and obtain the desired β-nitrophosphonates (6) with good yields (86–89%) and diastereoselectivities (de 84–96%) (Scheme 2).
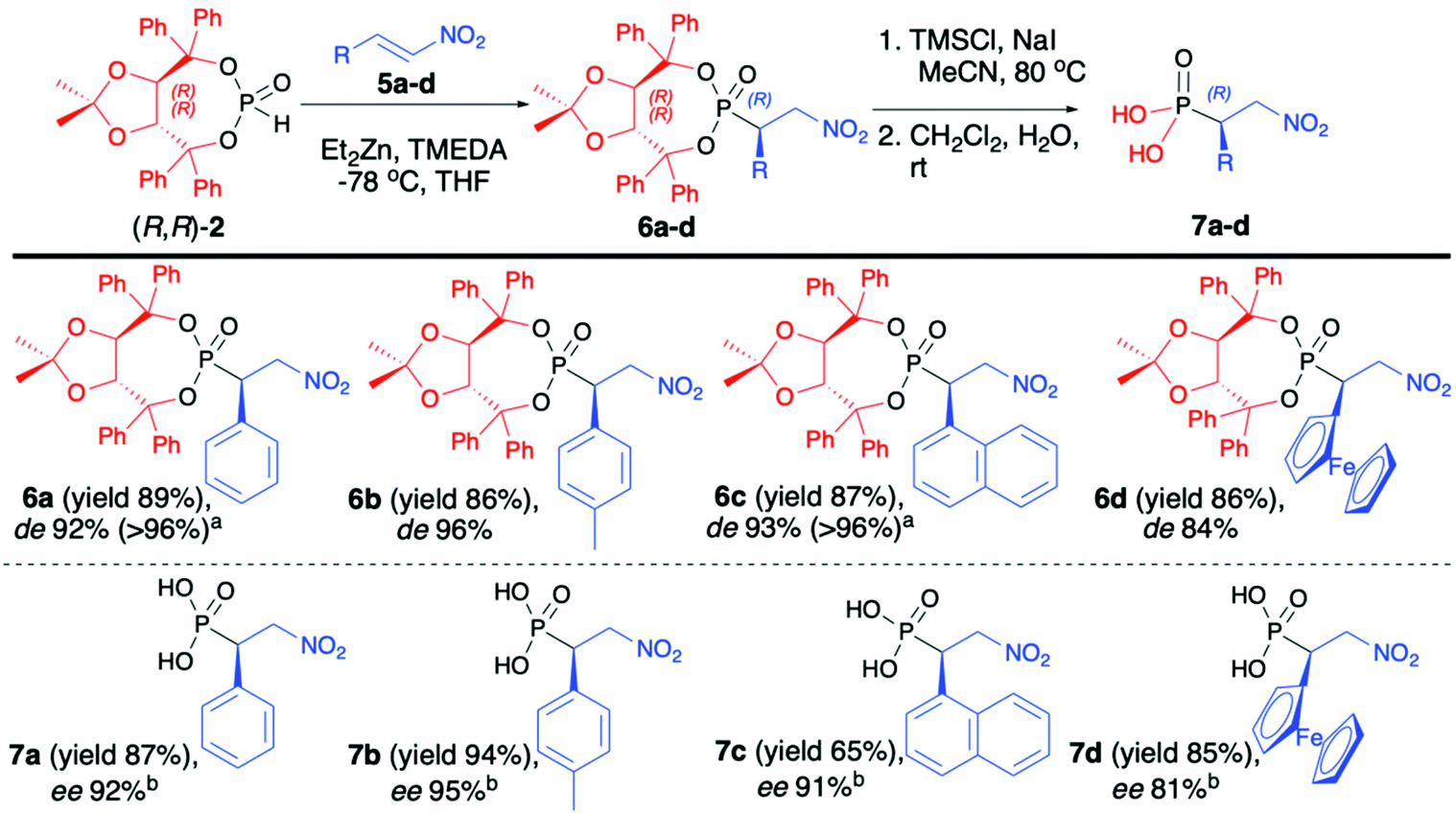 | ||
Scheme 2 Phospha-Michael addition of (R,R)-TADDOL H-phosphonate (2) to aromatic nitroalkenes-selected examples. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) de values after epimer separation by chromatography. bee determined by HPLC over a chiral stationary phase of the methyl ester derivatives. de values after epimer separation by chromatography. bee determined by HPLC over a chiral stationary phase of the methyl ester derivatives. | ||
The conjugate additions were also tested with other bases such as n-butyllithium, sodium or potassium hydride, and n-butyllithium in the presence of copper salts or dibutylmagnesium but resulted in a much lower diasteroselectivity.45 The authors postulated, based on the single-crystal X-ray analysis of the obtained β-nitrophosphonates (6), that the newly formed C-stereogenic center in the major epimer had R configuration. It is noteworthy that high asymmetric induction of the reaction was not observed in the case of aliphatic nitroalkenes. Subsequent removal of the chiral auxiliary bound to the phosphorus atom under racemization free and mild conditions (TMSCl and NaI) led to optically pure β-nitrophosphonic acids (7) (ee 81–95%) with good yields (65–94%) (Scheme 2).42
As a continuation of their work, Enders et al. reported on diastereoselective phospha-Michael addition of enantiopure (R,R)-(2) to α,β-unsaturated malonates (8) under heterogeneous conditions in the presence of Fe2O3/KOH as a solid base, leading to desired β-substituted β-phosphono malonates (9) with good yields (64–75%), excellent diastereoselectivity (de >99) and S configuration at the new stereogenic carbon center (Scheme 3).46 Use of aliphatic malonates resulted in the formation of the desired addition products with high yields albeit with low diastereoselectivity (de 15–30%). Subsequent removal of the chiral auxiliary bound to the phosphorus atom led to very polar acids that were not isolated but directly esterified with CH2N2 to corresponding methyl esters (10) (Scheme 3).46 Several parameters had influenced the diastereoselectivity and yield of the conjugate addition reaction. The authors reported that the presence of a metal oxide, as a solid support, was crucial for the successful outcome of the reaction. Fe2O3 was responsible for the activation of the P–H bond in (R,R)-TADDOL H-phosphonate (2) towards deprotonation with a base, such as KOH.
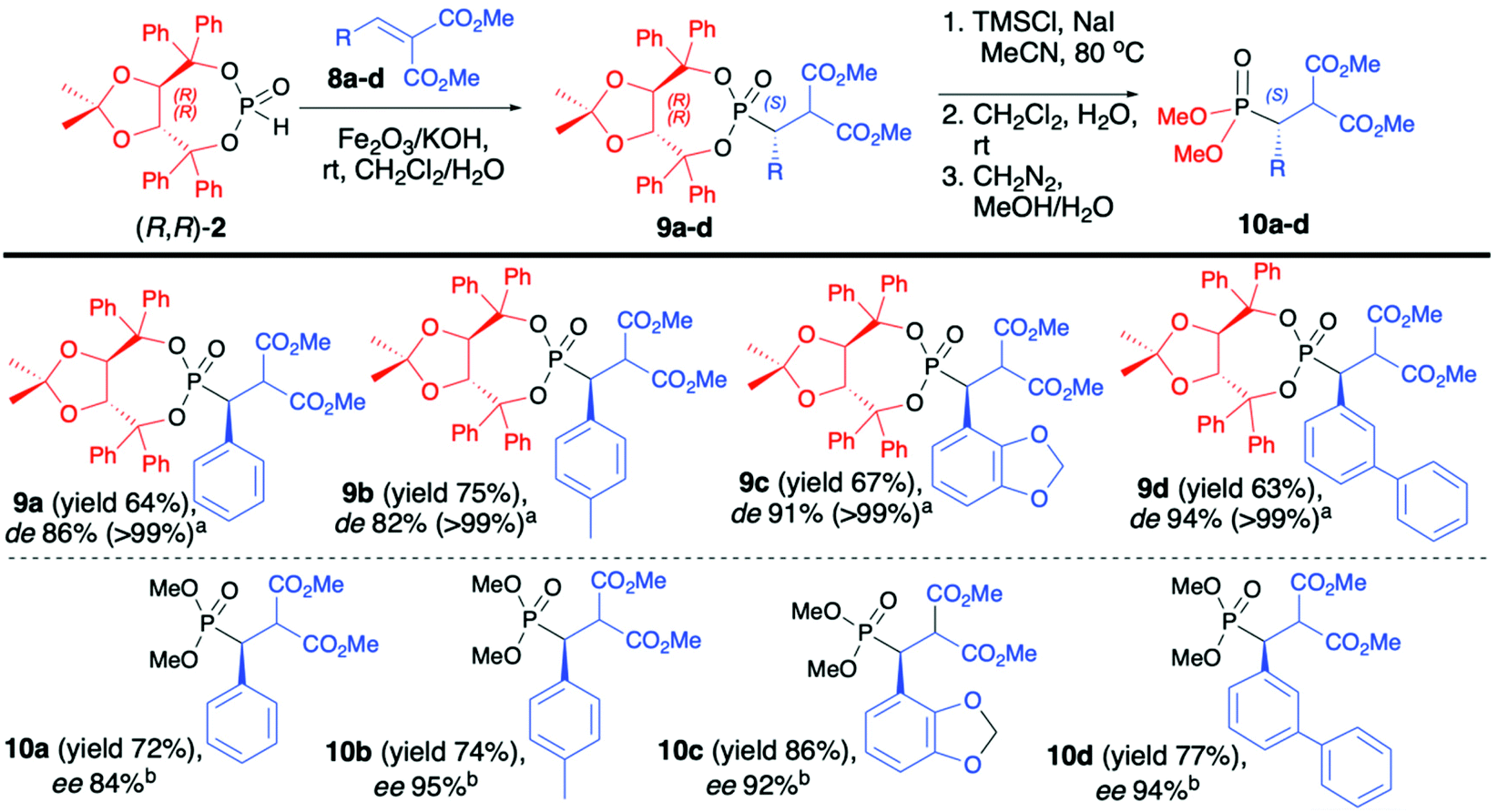 | ||
| Scheme 3 Phospha-Michael addition of (R,R)-TADDOL H-phosphonate (2) to malonates-selected examples. ade values after epimer separation by chromatography. bee determined by HPLC over a chiral stationary phase. | ||
No reaction was observed when only KOH was used. The diastereoselectivity of the reaction was strongly dependent on the molar ratio of the base and H-phosphonate bearing an (R,R)-TADDOL chiral auxiliary bound to the phosphorus atom (a 1:2.5 molar ratio of (R,R)-(2)/KOH being the optimal combination) and the method of pre-treatment of the solid base (drying at high vacuum for 72 h at 140 °C were found to be the optimal conditions). Likewise, the choice of the solvent was important as the best diastereoselectivity was obtained when CH2Cl2 was utilized, and the use of oxygen-containing solvents such as MeOH, THF, acetone or Et2O led to a substantial decrease in the de. Surprisingly, when the reaction was performed in CH2Cl2 in the presence of traces of water, the diastereoselectivity of the addition was proved to be time-dependent, increasing with longer reaction times. As an explanation, the authors suggested that the reaction product undergoes retro-Michael addition. This establishes an equilibrium, which leads to the virtually complete conversion of the starting materials into the major diastereoisomers.45
:5) (Scheme 4).47 Importantly, in each case, the pure major diastereomer was isolated by means of simple column chromatography. The use of other protecting groups of the iminic nitrogen such as benzyl and para-toluenesulfonyl or different bases for metalation of H-phosphonate such as LDA or only ZnEt2 led to desired products albeit with significantly lower diastereoselectivities (dr 75:25 in the best case). The success of the developed methodology was attributed to the presence of a bulky H-phosphonate bearing an (R,R)-TADDOL chiral auxiliary bound to the phosphorus atom, a large diphenylphosphinyl substituent on the iminic nitrogen and its influence on the stereoselective nucleophilic addition as well as electronic activation of the imine and a combination of those factors with the use of Et2Zn. The latter formed a very reactive organozinc–phosphorus adduct which requires the presence of TMEDA to increase the solubility of such adduct ensuring high yields of the reaction.42–45 At the end of the reaction sequence, pure major diastereomers of α-aminophosphonates (12a,d) were subjected to simultaneous removal of the chiral auxiliary bound to the phosphorus atom and diphenylphosphinyl group under acidic conditions, which led to enantiomerically pure (R)-α-aminophosphonic acids (13a,d) with good yields (82% and 78%, respectively) (Scheme 4).47
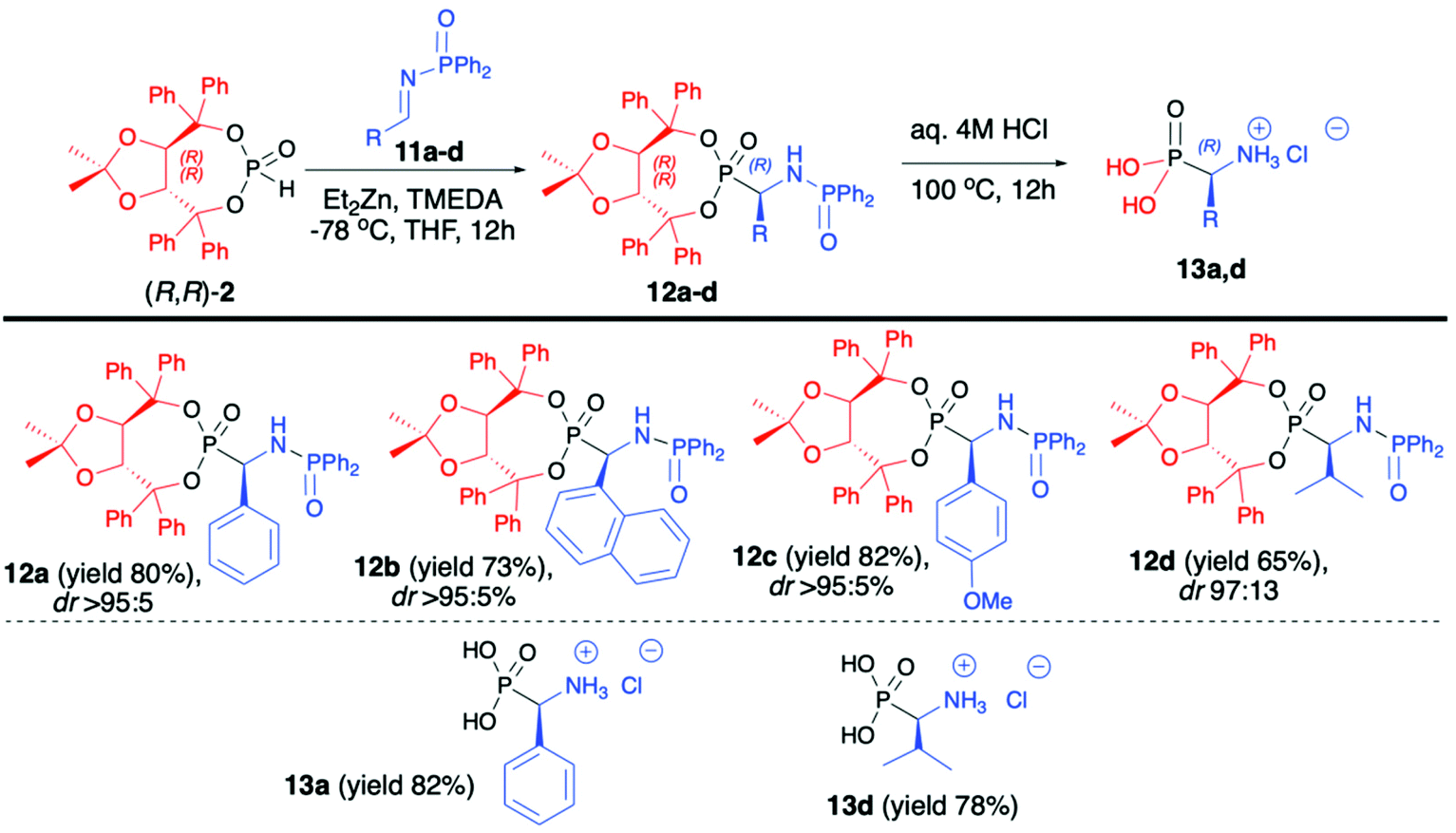 | ||
| Scheme 4 Aza-Pudovik addition of (R,R)-TADDOL H-phosphonate (2) to N-diphenylphosphinyl aldimines (11) – selected examples. dr values are established based on 31P NMR of the crude reaction mixture. | ||
In continuation of our interest in the asymmetric synthesis of α-aminophosphonates, we have developed an improved protocol for the highly diastereoselective hydrophosphonylation of imines with (R,R)-TADDOL H-phosphonate (2) based on the use of enantiomerically pure (S)-N-tert-butylsulfinyl imines (14a–d) at room temperature and with the use of K2CO3 as a base (Scheme 5).48 The desired α-aminophosphonates (15a–d) were obtained in good yields (80–87%) and excellent diastereoselectivities (dr >95:5 in the majority of cases); consequently, pure major diastereomers could be easily isolated by simple crystallization. Subsequent removal of the chiral auxiliary bound to the phosphorus atom and a tert-butylsulfiny protecting group led to enantiomerically pure (R)-α-aminophosphonic acids (16a–d) in good yields (72–92%). It is worth noting that the use of the opposite enantiomer of the imine, namely, (R)-N-tert-butylsulfiny imine led to a significant decrease in the reaction diastereoselectivity (dr 22:78). A broad substrate scope, mild and simple reaction conditions, high yields and diastereoselectivity and simple isolation of major diastereoisomer can be considered as the main advantages of this protocol (Scheme 5).48
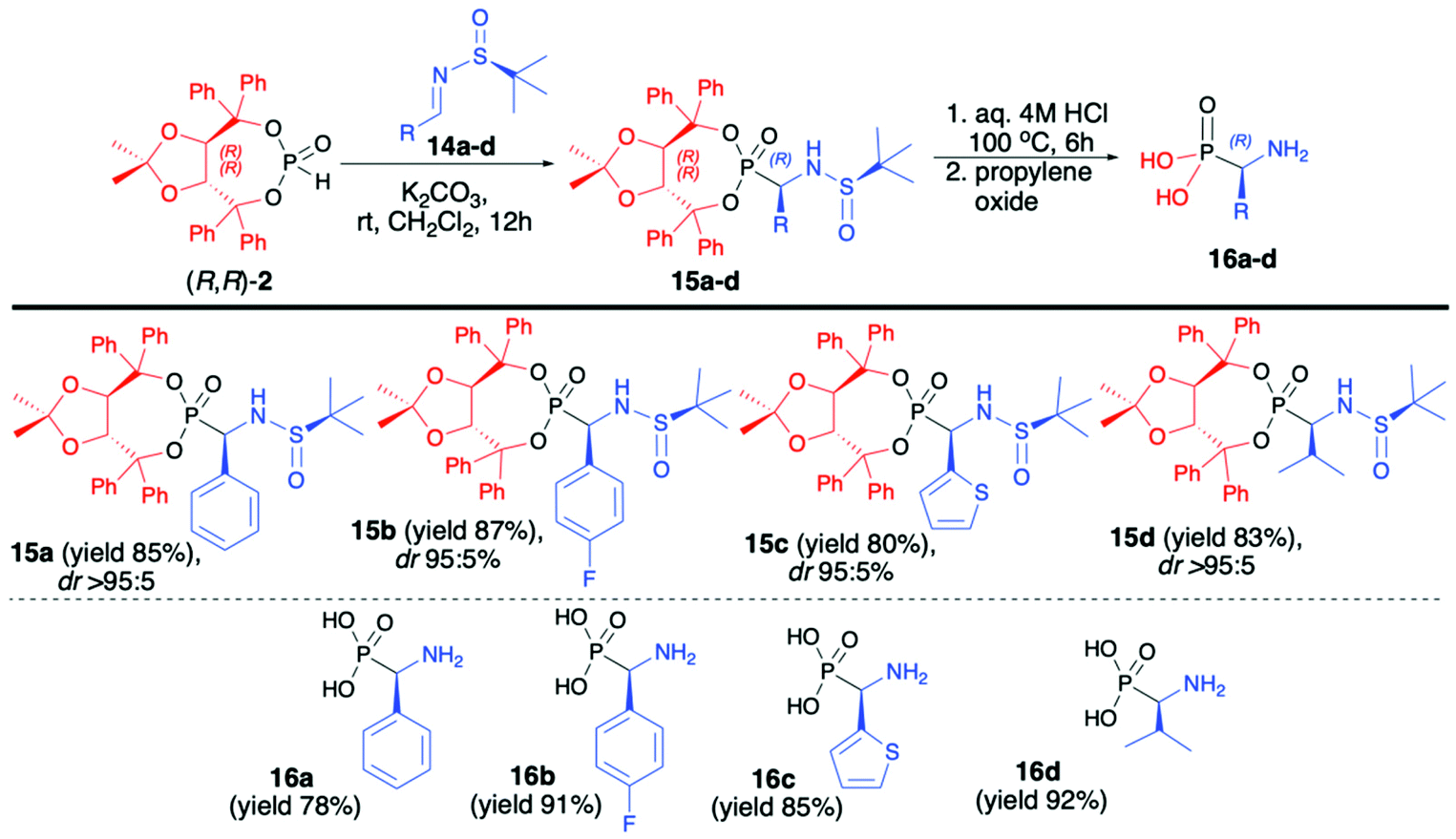 | ||
| Scheme 5 Aza-Pudovik addition of (R,R)-TADDOL H-phosphonate (2) to (S)-N-tert-butylsulfinylimines (14) – selected examples. dr values are established based on 31P NMR of the crude reaction mixture. | ||
Vicario et al. used (R,R)-TADDOL H-phosphonate (2) for hydrophosphonylation of imine 17 and obtained the corresponding racemic α-aminophosphonate 18 (yield 93%, dr 77:23) that after chlorination with an excess of trichloroisocyanuric acid (TCCA) followed by the treatment with an excess of poly-(4-vinylpyridine) (Poly-Py) gave α-ketiminophosphonate 19 (Scheme 6).49 The latter was subjected to nucleophilic addition of Grignard reagents at −80 °C. Although no diastereoselectivity was observed when a bulky 2-naphthylmagnesium bromide was added and the desired tetrasubstituted α-aminophosphonate 20b (yield 80%, dr 55:45) was formed, excellent diastereoselectivity was obtained when a smaller methylmagnesium bromide was used. The formed tetrasubstituted α-aminophosphonate 20a was obtained in 81% yield and dr 94:6, allowing for easy isolation of the major diastereoisomer by simple crystallization from Et2O (Scheme 6).
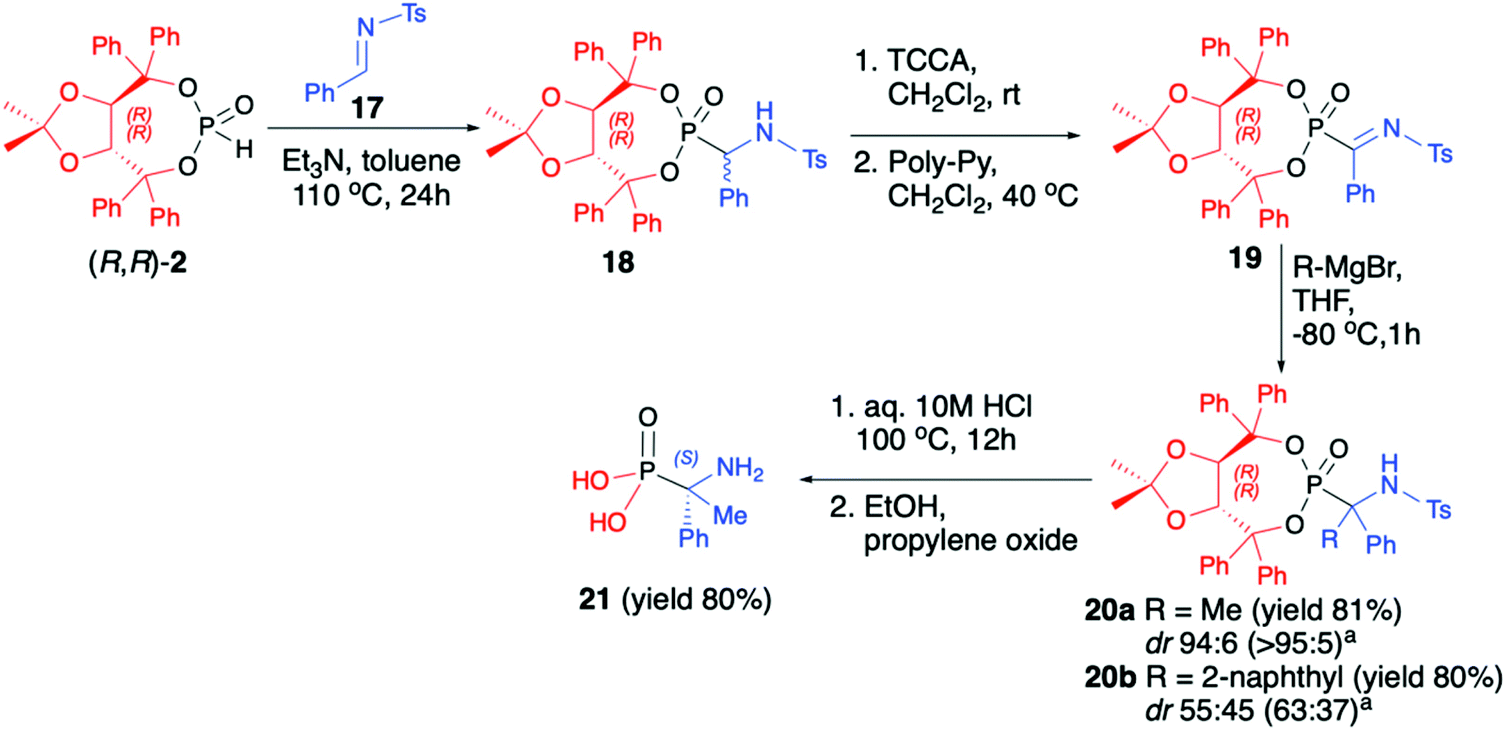 | ||
| Scheme 6 Preparation of tetrasubstituted α-aminophosphonates from trisubstituted α-aminophosphonates with the use of enantiopure (R,R)-2. adr values after chromatographic purification. Ts – toluenesulfonyl. | ||
Finally, the major diastereoisomer was subjected to simultaneous removal of the chiral auxiliary bound to the phosphorus atom and deprotection of the nitrogen atom under acidic conditions and led to the desired tetrasubstituted α-aminophosphonic acid 21 in 80% yield and as a pure S enantiomer (Scheme 6).
Recently, we have reported on the hydrophosphonylation of enantiomerically pure (R,R)-hexahydroquinoxalinone (3-oxo-2,5-diazabicyclo[4.4.0]dec-4-ene) (22) with (R,R)-2 in the presence of Et3N in toluene at 80 °C (Scheme 7).50 The desired bicyclic α-aminophosphonate 23 was isolated in a pure form with acceptable yield (72%) albeit with low diastereoselectivity (dr 60:40). The DFT calculations performed for the reaction of imine 22 with dimethyl H-phosphonate suggested that the energies of the two formed epimers of the corresponding α-aminophosphonate were essentially the same, which agreed with the observed experimental diastereomeric ratio (dr ca. 1:1).
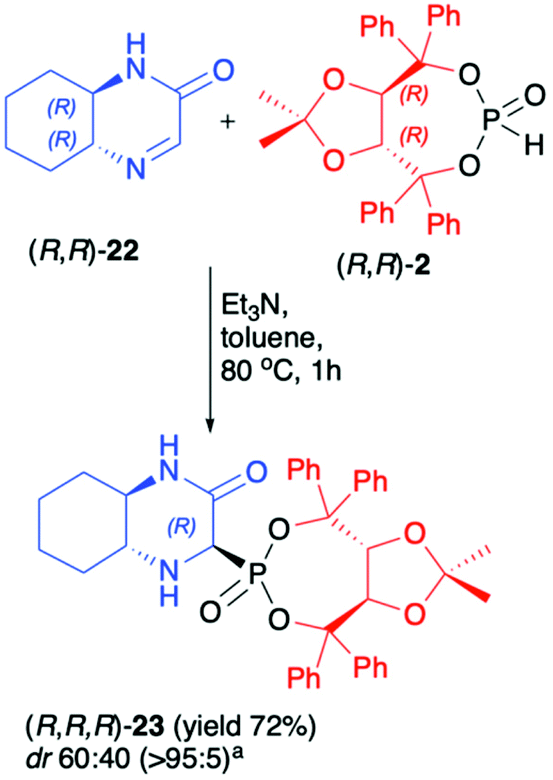 | ||
| Scheme 7 Asymmetric synthesis of hexahydroquinoxalin-2(1H)-one-derived α-aminophosphonate (23). a major epimer isolated by means of column chromatography. | ||
Therefore, it seemed likely that the two epimers readily interconvert under the reaction conditions and this precluded their formation with high diastereoselectivity even when chiral (R,R)-2 was used. Due to the presence of the (R,R)-TADDOL chiral auxiliary bound to the phosphorus atom in α-aminophosphonate 23, however, a pure major diastereomer could be easily separated by column chromatography. The absolute configuration at the newly formed stereogenic carbon in the major diastereoisomer of 23 was unambiguously assigned as R based on the X-ray analysis. Unfortunately, we have observed that in the case of pure diastereoisomer of 23, fast epimerisation takes place during its storage in solution. We attributed this behaviour to the structure of the bicyclic α-aminophoshonate where the presence of both carbonyl and phosphonate groups renders the proton attached to the stereogenic α-C prone to dissociation, which led to epimerisation.
After examining the utility of (R,R)-2 in the hydrophosphonylation of imines and asymmetric synthesis of α-aminophosphonates, we turned our attention towards the preparation of α-hydroxyphosphonates and the utility of aldehydes as substrates in the hydrophosphonylation reaction (Scheme 8).51 Carbonyl compounds are more challenging substrates for the asymmetric hydrophosphonylation since, unlike in the case of imines where different stereodirecting groups can be placed on the iminic nitrogen atom, there is no possibility of introducing such functionalities on simple aldehydes. Therefore, the use of H–P nucleophile bearing chiral auxiliary bound to the phosphorus atom and careful optimization of reaction conditions were the key to success in the preparation of α-hydroxyphosphonates in an asymmetric fashion. In that aspect, we have developed a diastereoselective and general method for the hydrophosphonylation of aldehydes with the use of (R,R)-TADDOL H-phosphonate (2) (Scheme 8).51 In order to obtain high diastereoselectivity, reactions were performed at −78 °C. Moreover, the choice of a base had a crucial effect on the reaction diastereoselectivity.
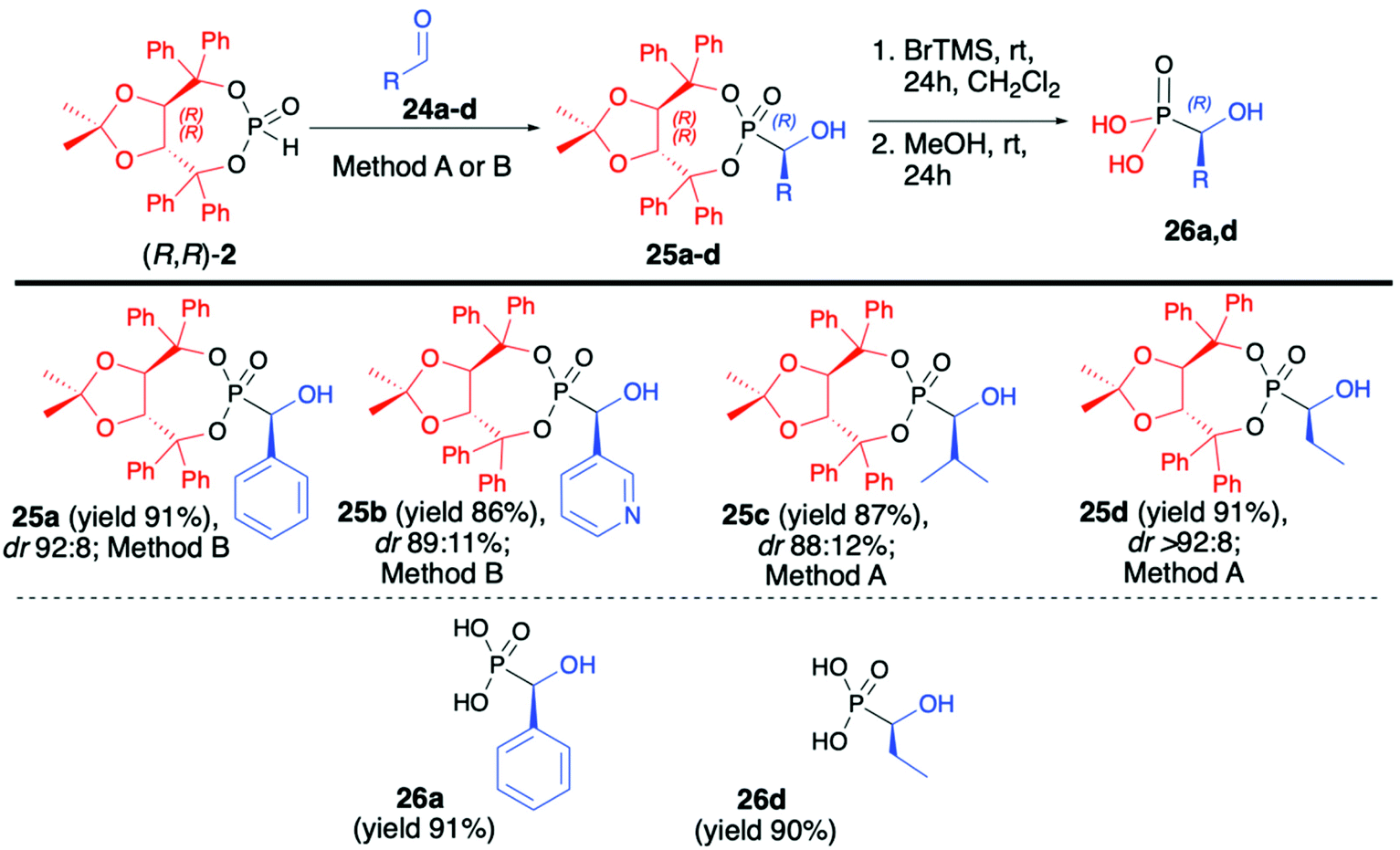 | ||
| Scheme 8 Pudovik reaction between enantiopure (R,R)-(2) and aldehydes (24a–d) – selected examples. Method A: Et2Zn, TMEDA, −78 °C, THF, 12 h; Method B: LDA, −78 °C, THF, 12 h. BrTMS – bromotrimethylsilane. | ||
In the case of aromatic and heteroaromatic aldehydes, the use of Et2Zn and TMEDA gave the best results, whereas application of LDA was found to be beneficial in the case of aliphatic aldehydes. In both cases, corresponding α-hydroxy-phosphonates were obtained with high asymmetric induction (up to dr >92:8) and in good yields (up to 91%) (Scheme 8). In each case, isolation of the pure major diastereoisomer was possible by simple crystallization from Et2O. Subsequent removal of the chiral auxiliary under mild reaction conditions gave enantiomerically pure (R)-α-hydroxyphosphonic acids (26a,d) in good yields (91 and 90%, respectively).
We have also tested more challenging 2-azanorbornane-derived aldehyde 27 for the hydrophosphonylation with (R,R)-2 (Scheme 9).52
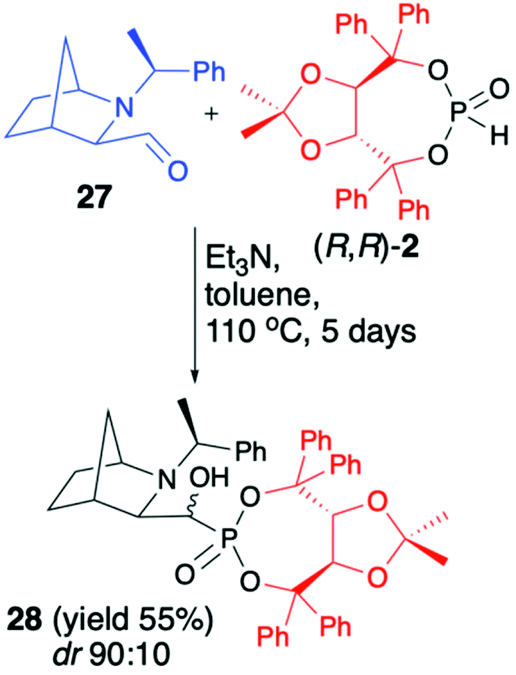 | ||
| Scheme 9 Diastereoselective preparation of 2-azanorbornane-derived α-hydroxyphosphonate 28. | ||
We have quickly realized, however, that configurationally stable, rigid, bicyclic aldehyde 27 is not prone to hydrophosphonylation. After heating of aldehyde 27 with (R,R)-2 in the presence of Et3N in toluene at 110 °C for 5 days, the desired α-hydroxyphosphonate 28 was formed with good diastereoselectivity (dr 90:10), albeit with low conversion (50%), which was attributed to thermal instability of (R,R)-2 that decomposed to (R,R)-TADDOL (1).
2.2. Application of BINOL H-phosphonate (31) and its derivatives as H–P species bearing a chiral auxiliary bound to the phosphorus atom
1,1′-Binaphthyl-2,2′-diol (BINOL) is considered one of the most representative axially chiral C2 symmetric molecules in organic chemistry.53–56 The steric hindrances between 2,2′-hydroxyls and between 8,8′-hydrogens restrict the free rotation of the two 2-naphthol units around the 1,1′-bond, which orient the chirality of BINOL (Scheme 10). Importantly, the rigid structure and the C2 symmetry of the chiral binaphthyl molecules play an important role in chiral induction, and just like TADDOL, BINOL is also considered one of the few privileged chiral ligands.38 The preparation of BINOL H-phosphonate (31) is known for almost three decades and is based on the classical addition of PCl3 in the presence of Et3N to the appropriate diol 29 and followed by hydrolysis of the corresponding chloride 30 by the addition of H2O and Et3N (Scheme 10).57–59 Replacements of H2O/Et3N, for the hydrolysis of chloride in the second reaction step, with tert-butanol or formic acid were also reported in the literature as ways to obtain 31 with higher yield.60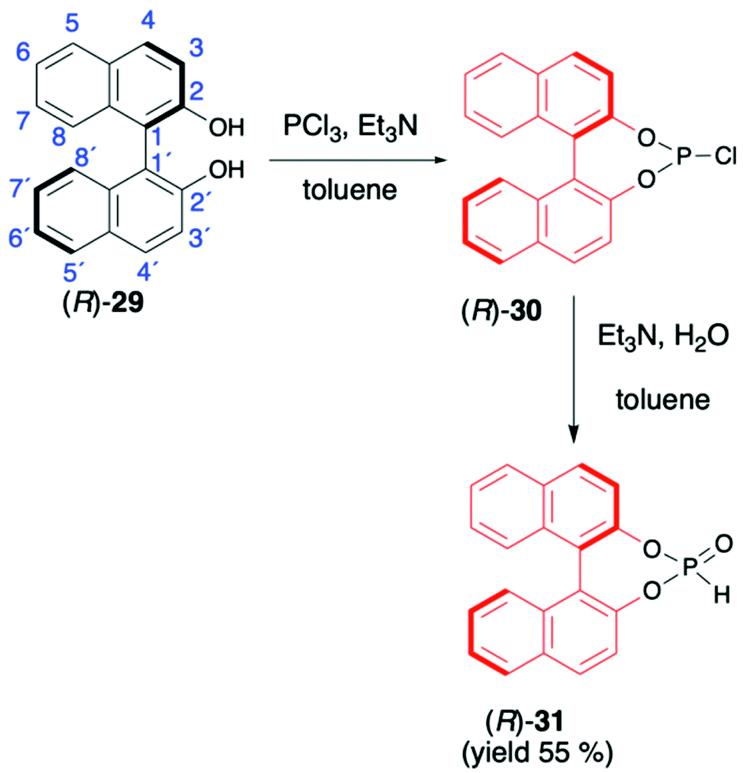 | ||
| Scheme 10 Synthesis of (R)-BINOL H-phosphonate (31). | ||
BINOL-derived H-phosphonate 31 is a white solid that is stable once in pure form. Reports can be found in the literature, however, where authors state that 31 decomposes to the starting BINOL when purified by column chromatography; hence, crystallization should be used as a purification method.58,59 Additionally, we have observed during our work on hydrophosphonylation of imines that fast transesterification of BINOL H-phosphonate 31 and corresponding aminophosphonates occurs when the reaction is carried out in alcoholic solvents.47 This behaviour was assigned to the ease of cleavage of endocyclic P–O bonds in BINOL H-phosphonate due to the twisted structure of BINOL auxiliary and the low stability of the 7-membered ring, including four sp2 carbons.
N and CC double bonds.
Martens and co-workers reported the first highly diastereoselective hydrophosphonylation of 3-thiazoline-derived imines 32a–e using racemic (rac)-BINOL-derived H-phosphonate (31) in the presence of BF3·OEt2, leading to the corresponding 4-thiazolidynylphosphonates 33a–e (Scheme 11).61,62 Derivatives of thiazole, thiazoline and thiazolidine are known for their important biological activity; therefore, synthesis of such compounds in an asymmetric fashion is of high importance.63–66 The desired aminophosphonates 33a–e were obtained with very good diastereoselectivity (up to dr >95:5) albeit with moderate yields (30–68%). The steric hindrance of the substituents R2 in imines 32 had a crucial impact on the diastereoselectivity of the hydrophosphonylation and the larger the substituent, the better was the diastereoselectivity of the reaction observed (Scheme 11). In the case of 4-thiazolidynylphosphonates 33a and c, pure major diastereoisomers were isolated by column chromatography and the X-ray analysis of crystals prepared from those compounds revelated that both the BINOL H-phosphonate part and the newly formed asymmetric carbon atom had R configuration. Unfortunately, no attempts to remove the chiral auxiliary bound to the phosphorus atom or to perform any further transformation of the obtained products were presented.
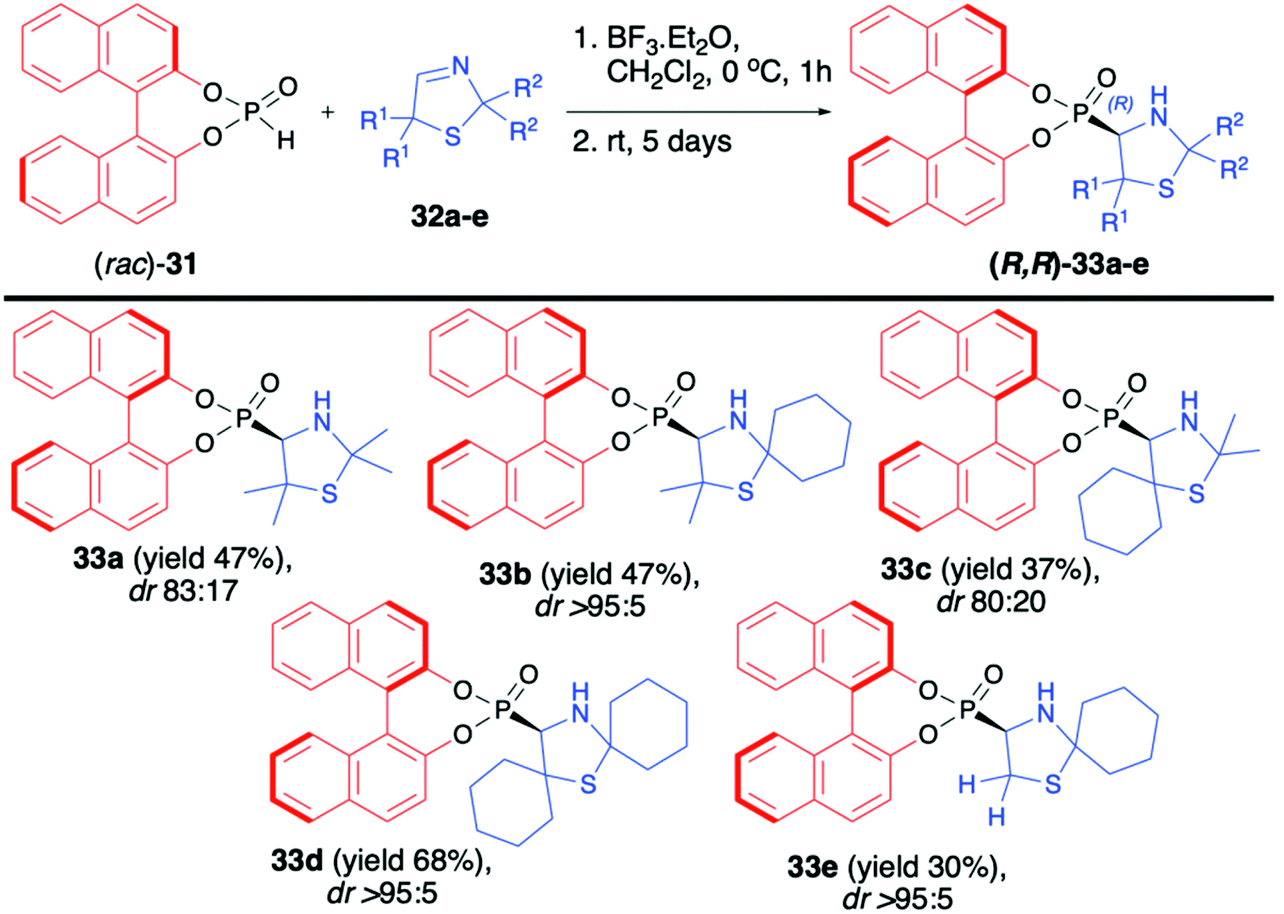 | ||
| Scheme 11 Diastereoselective hydrophosphonylation of 3-thiazolines (32) with (rac)-BINOL H-phosphonate (31). | ||
An interesting application of (rac)-BINOL H-phosphonate (31) was described by Wu et al., who investigated Cu-catalysed reductive coupling of N-tosylhydrazones with H-phosphine oxides (Scheme 12).67 When (rac)-31 was used in the presence of CuCl2 (20 mol%) and K2CO3 in dioxane at 110 °C, product 35 with a new C(sp3)–P bond was obtained in 66% yield.
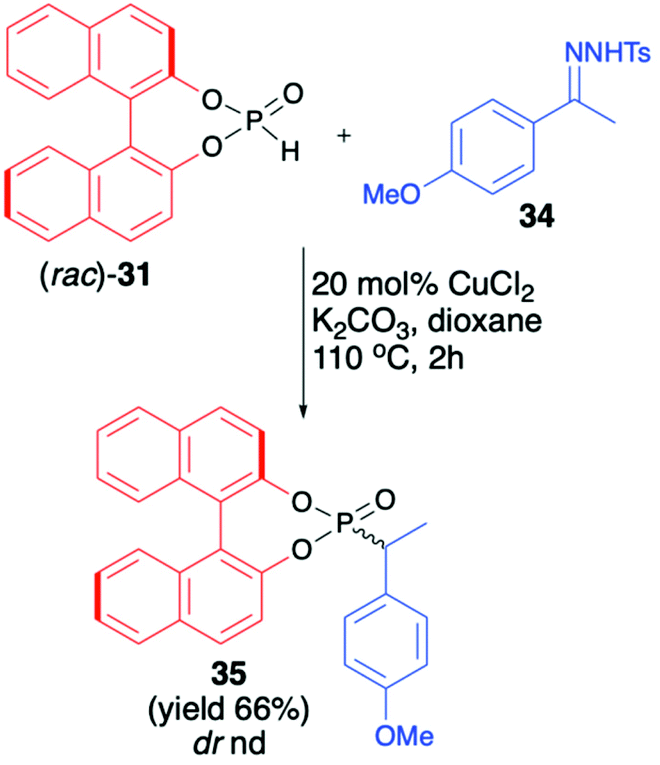 | ||
| Scheme 12 C(sp3)–P bond formation via Cu-catalyzed reductive coupling of N-tosylhydrazone 34 with BINOL-derived H-phosphonate (31). nd – not determined. | ||
Unfortunately, the authors neither examined the diastereoselectivity of the reaction nor attempted to remove the chiral auxiliary from the reaction product (Scheme 12).67 A recent example of the copper-catalysed synthesis of organophosphorus compounds with the use of (R)-BINOL H-phosphonate (31) was described by Mahesh and Anand.68 The authors elaborated an elegant protocol for the preparation of indolizine-based phosphonates based on Cu-catalysed 5-endo-dig-cyclization of 2-(2-enynyl)pyridines followed by remote hydrophosphonylation. The reaction of pyridine derivative 36 with (R)-31 in the presence of CuI (10 mol%) in dichloroethane at 70 °C during 8 h produced the desired indolizine-derived phosphonate in good yield (70%) albeit with low diastereoselectivity (dr 1:1.2) (Scheme 13).
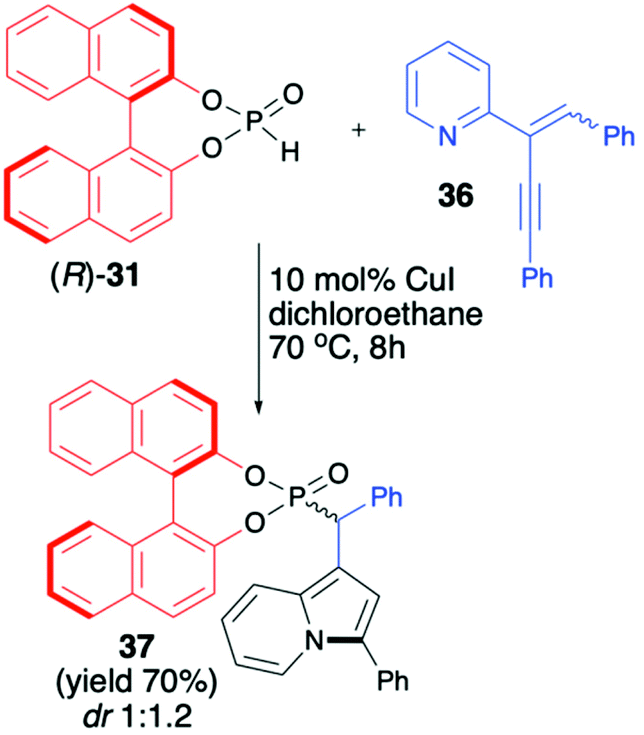 | ||
| Scheme 13 Cu-catalysed hydrophosphonylation of 2-(2-enyl)pyridine 36 with (R)-31. | ||
The authors, however, did not perform any optimisation of the reaction conditions towards improving diastereoselectivity, attempts to separate the diastereomers or further removal of the chiral auxiliary bound to the phosphorus atom.
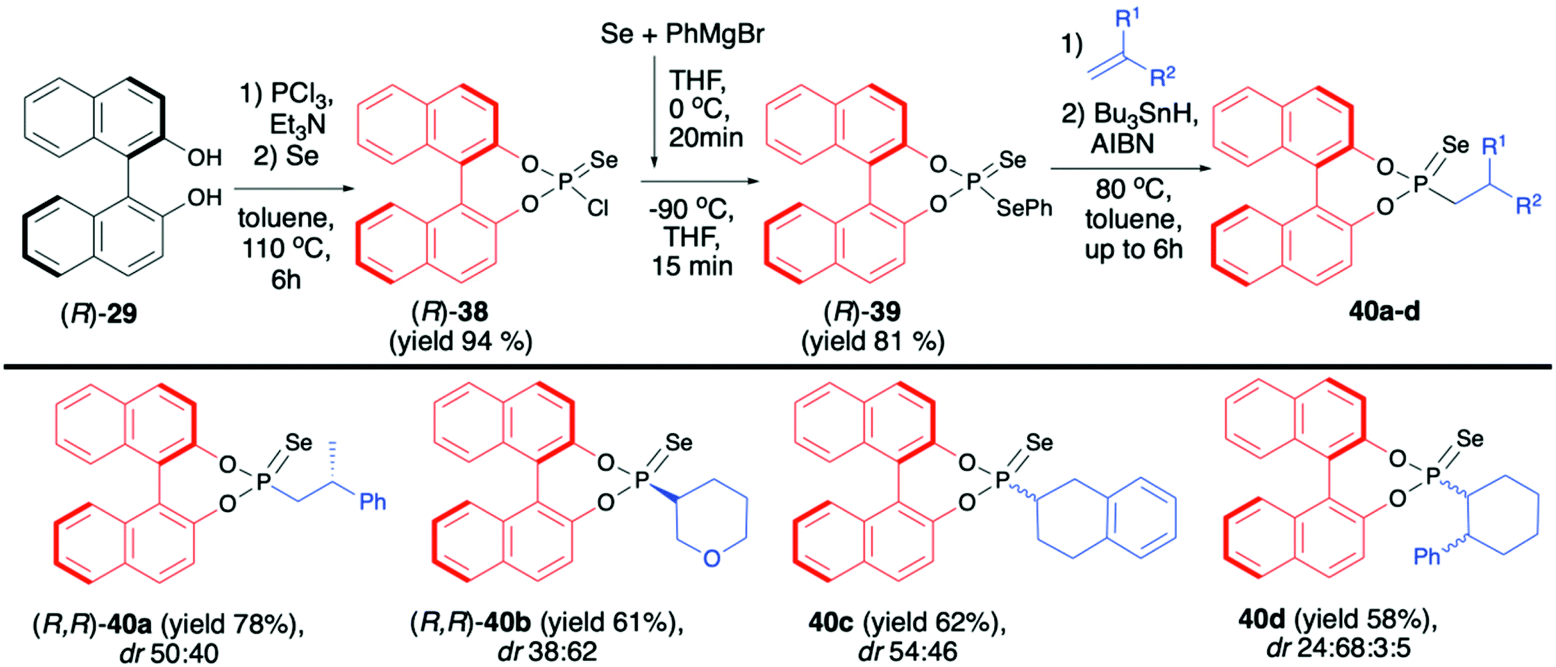 | ||
| Scheme 14 Hydrophosphonylation of alkenes with (R)-39 – selected examples. dr is calculated based on 31P NMR of the crude reaction mixture. For 40a and 40b, the absolute configuration is given for the major diastereomer based on X-ray analysis. Bu3SnH – tributyltin hydride; AIBN – azobisisobutyronitrile. | ||
(R)-38 was subsequently transformed into phosphonodiselenoic acid ester (R)-39 and used by the Murai group for the hydrophosphonylation of alkenes (Scheme 14).70
The reaction proceeded under radical conditions and in an anti-Markovnikov fashion. Several alkenes were reacted with (R)-39 in the presence of Bu3SnH and AIBN leading to the desired phosphorylated products 40 with good yields (up to 78%) and moderate diastereoselectivities (dr up to 38:62) (Scheme 14).70 Importantly, the diastereoselectivity could be improved by the introduction of bulky substituents in the 3,3′-positions of the binaphthyl group, e.g., an analogue of 40c with triisopropylsilyl groups in the 3,3′-positions of BINOL part was obtained with excellent dr 92:8 and good yield (67%).70 In the majority of cases, the obtained diastereoisomers could be separated by simple column chromatography. In the case of pure diastereoisomers of 40a and 40b, the absolute configuration of the stereogenic carbon, established by means of X-ray analysis, was found to be R. Finally, to demonstrate their utility as useful building blocks, phosphonoselenoic acid esters 40 were transformed into corresponding phosphonic acid esters 41 and phosphonite boranes 43 using classical conditions (Scheme 15). It is worth mentioning that in the case of transformation of pure diastereoisomer of 40a (dr >99:1) into its oxygen analogue 41a, no racemisation was observed.
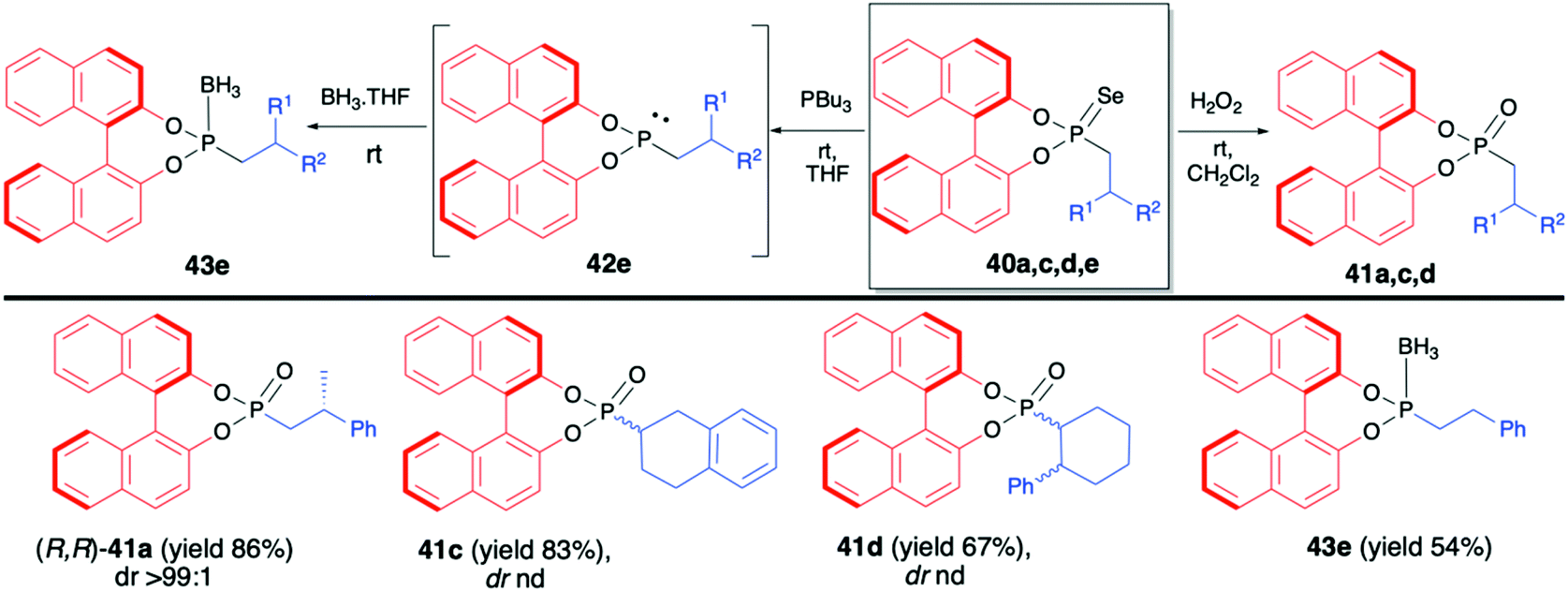 | ||
| Scheme 15 Further transformations of phosphonoselenoic acid esters 40 – selected examples. nd – not determined. | ||
In continuation of their interest in the use of BINOL-derived phosphonoselenoyl chloride (38), Murai et al. reported on the reaction of (S)-38 with Grignard reagents.71 The use of chiral Grignard reagents (in the racemic form) such as sec-butylmagnesium bromide (44a), sec-decylmagnesium bromide (44b) and 2-ethylhexylmagnesium bromide (45) led to desired products 46a,b and 47 as mixtures of diastereoisomers albeit with no diastereoselectivity (Scheme 16). The diastereomers could however be easily distinguished by 31P NMR and separated by simple chromatography or crystallization. In the case of 46a, the absolute configuration of the stereogenic carbon was found to be S, as determined by X-ray analysis (Scheme 16).71
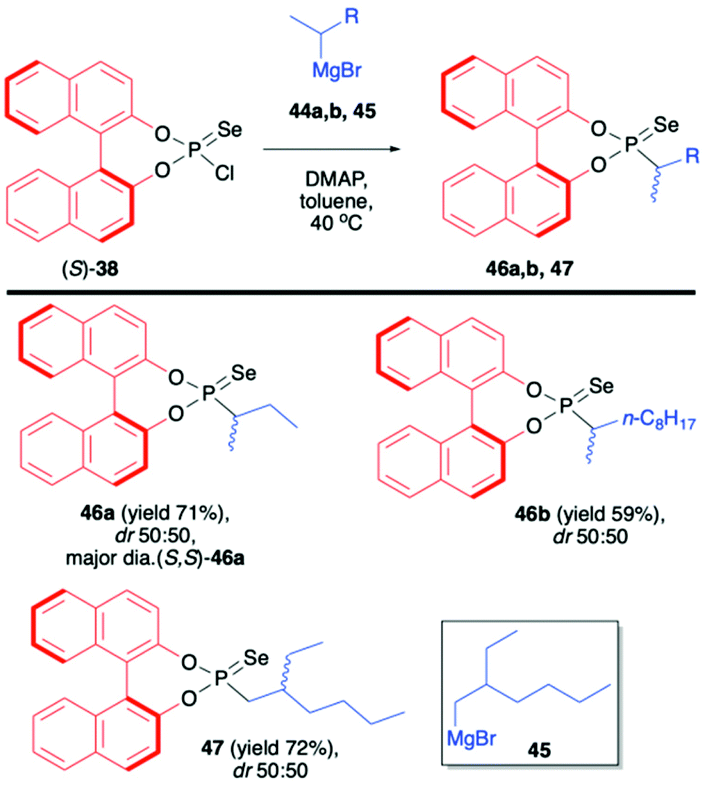 | ||
| Scheme 16 Addition of chiral Grignard reagents to (S)-38. dr is calculated based on 31P NMR of the crude reaction mixture. The absolute configuration is given for the major diastereomer based on X-ray analysis. DMAP – 4-dimethylaminopyridine. | ||
Recently, Murai and co-workers reported on the use of (S)-BINOL-derived phosphonoselenoates 48 and phosphonates 50 in diastereoselective sequential deprotonation–alkylation reaction leading to new organophosphorus compounds with tri- and tetrasubstituted carbon centers adjacent to the phosphorus atom, 49 and 51, respectively (Schemes 17 and 18).72
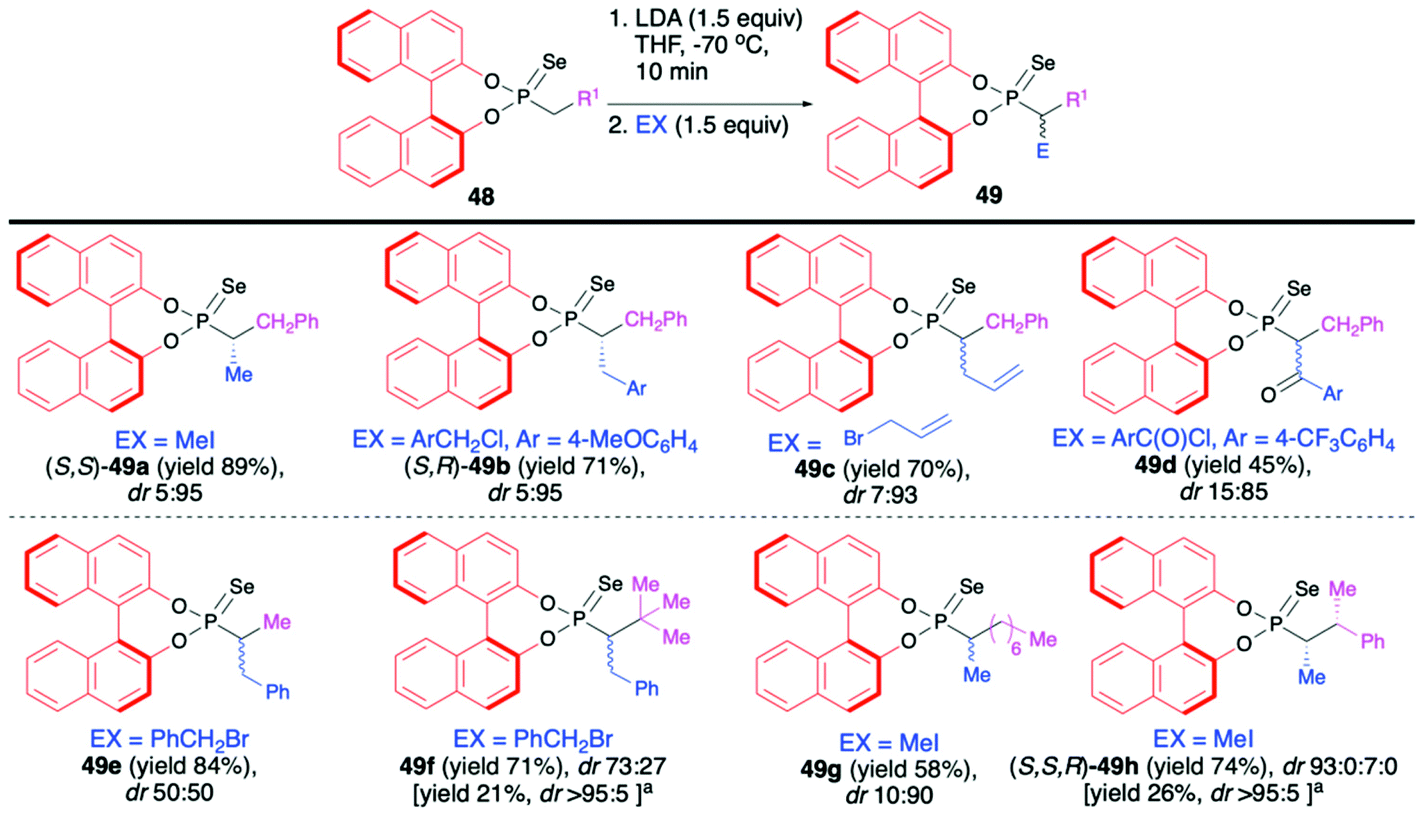 | ||
| Scheme 17 Sequential deprotonation–alkylation of different BINOL-derived phosphonoselenoates 48 with a variety of halides – selected examples. dr is calculated based on 31P NMR of the crude reaction mixture. The absolute configuration is given for the major diastereomer based on X-ray analysis. aYield and dr after recrystallization. | ||
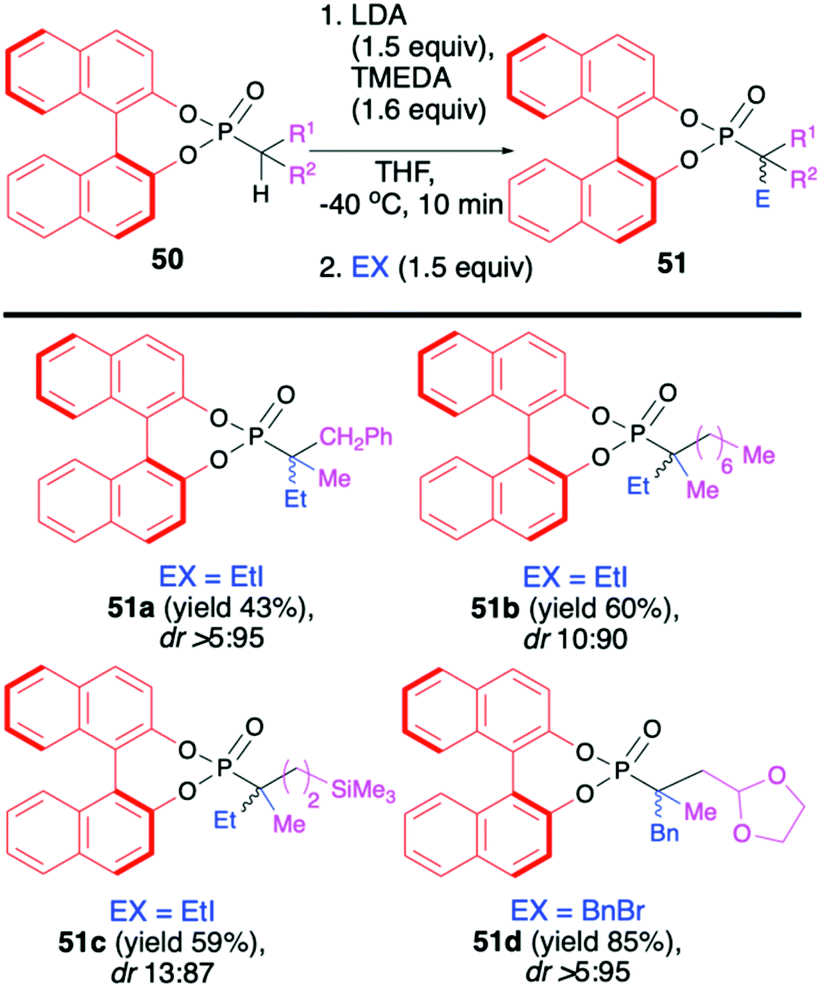 | ||
| Scheme 18 Reaction of different BINOL-derived phosphonates 50 with halides and diastereoselective construction of chiral quaternary carbon centre attached to the P-atom. dr is calculated based on 31P NMR of the crude reaction mixture. | ||
In the case of phosphonoselenoates 48, the deprotonation proceeded smoothly with LDA at −70 °C in THF and subsequent addition of the appropriate halides to the in situ generated carbanion resulted in the formation of desired trisubstituted alkylated products 49 with good yields (up to 89%) and high diastereoselectivities (dr up to 5:95) (Scheme 17). In the case of products 49f and 49h, simple recrystallization of the crude product resulted in an improvement of the original diastereoselectivity. Due to the presence of BINOL unit in each case, the diastereomers formed were easily discriminated by means of 31P NMR.72
The absolute configuration of the new carbon centers was unambiguously assigned by means of X-ray analysis in the case of products 49a, 49b and 49f (Scheme 17). The authors postulated that the BINOL part exerts a strong influence on the stereochemical fate of the carbon atom connected to the phosphorus.
To explain the stereochemical outcome of the deprotonation–alkylation reaction, the authors assumed that the electrophile approaches the racemic carbon center, generated during the deprotonation, from the sterically unhindered site. Unfavourable nonbonding interaction between the incoming electrophile and a binaphthyl group of the lithiated phosphonoselenoate could be present.72
In turn, the formation of the tetrasubstituted carbon centers adjacent to the phosphorus atom was possible exclusively with the use of trisubstituted phosphonates 50 (analogous phosphono-selenoates did not undergo deprotonation–alkylation reaction) (Scheme 18). Deprotonation was also carried out with the use of LDA, albeit in the presence of TMEDA and at −40 °C in THF in order to obtain the highest yield of the products (up to 85%) and diastereoselectivity (dr up to >5:95) possible.72 Finally, to demonstrate the utility of the obtained (S)-BINOL-derived alkylated phosphonoselenoates, the authors performed derivatization and removal of the chiral auxiliary bound to the phosphorus atom for phosphonoselenoate 49h (dr 86:8:6:0) as a model substrate (Scheme 19).72 Dialkylation–deselenation of 49h (dr 86:8:6:0) with n-BuLi led to the desired chiral phosphine 52 (dr 90:10) suitable for applications as a ligand in, e.g., catalysis. Transformation of phosphonoselenoate 49h to the corresponding phosphonate 53 by reaction with H2O2 followed by alcoholysis with EtONa afforded the desired diethyl phosphonate 54 with moderate yield (41%) and dr of 88:12. Importantly in both cases, the authors state that the BINOL chiral auxiliary could be recovered, which is an additional advantage of this useful chiral auxiliary continuously used with success by the Murai group.73,74
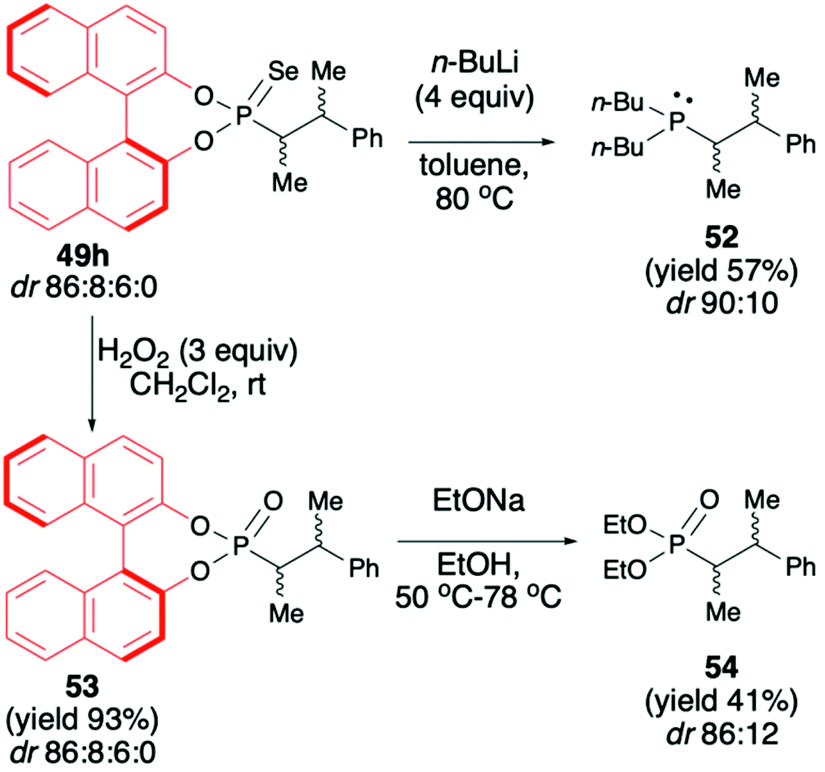 | ||
| Scheme 19 Derivatization of BINOL-derived phosphonate 49h with the recovery of the chiral auxiliary in each case. dr-calculated based on 31P NMR of the crude reaction mixture. | ||
2.3. Application of menthyl-derived H–P species in the stereoselective preparation of organophosphorus compounds
(1R,2S,5R)-2-Isopropyl-5-methylcyclohexanol, [(−)-menthol], is perhaps one of the most readily available, naturally occurring and inexpensive chiral building blocks very often applied in the preparation of chiral H-phosphonates, phosphinates and phosphine oxides subsequently used as H–P species bearing chiral auxiliary bound to the phosphorus atom in the asymmetric preparation of organophosphorus compounds, C- and also P-chiral.75,76 The simplest case is represented by (−)-O,O-di-(1R,2S,5R)-menthyl H-phosphonate (56) that can be conveniently obtained by the reaction of (−)-menthol with phosphorus trichloride and subsequent hydrolysis of dimenthyl chlorophosphite (55) formed (Scheme 20, reaction (1)).77 In turn, the preparation of (−)-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59), although much more problematic, is also quite well described in the literature, and today convenient synthetic protocols are available for the preparation of diastereomerically pure (RP)-(59) even on a multi-gram scale (Scheme 20, reactions (2a) and (2b)). The leading protocol is based on the reaction of PhPCl2 (57), pyridine and (−)-menthol followed by repeated recrystallization of the mixture of diastereoisomers at low temperature and was pioneered by Emmick and Letsinger78 followed by Mislow et al.79 and recently improved by Han et al. (Scheme 20, reaction (2a)).76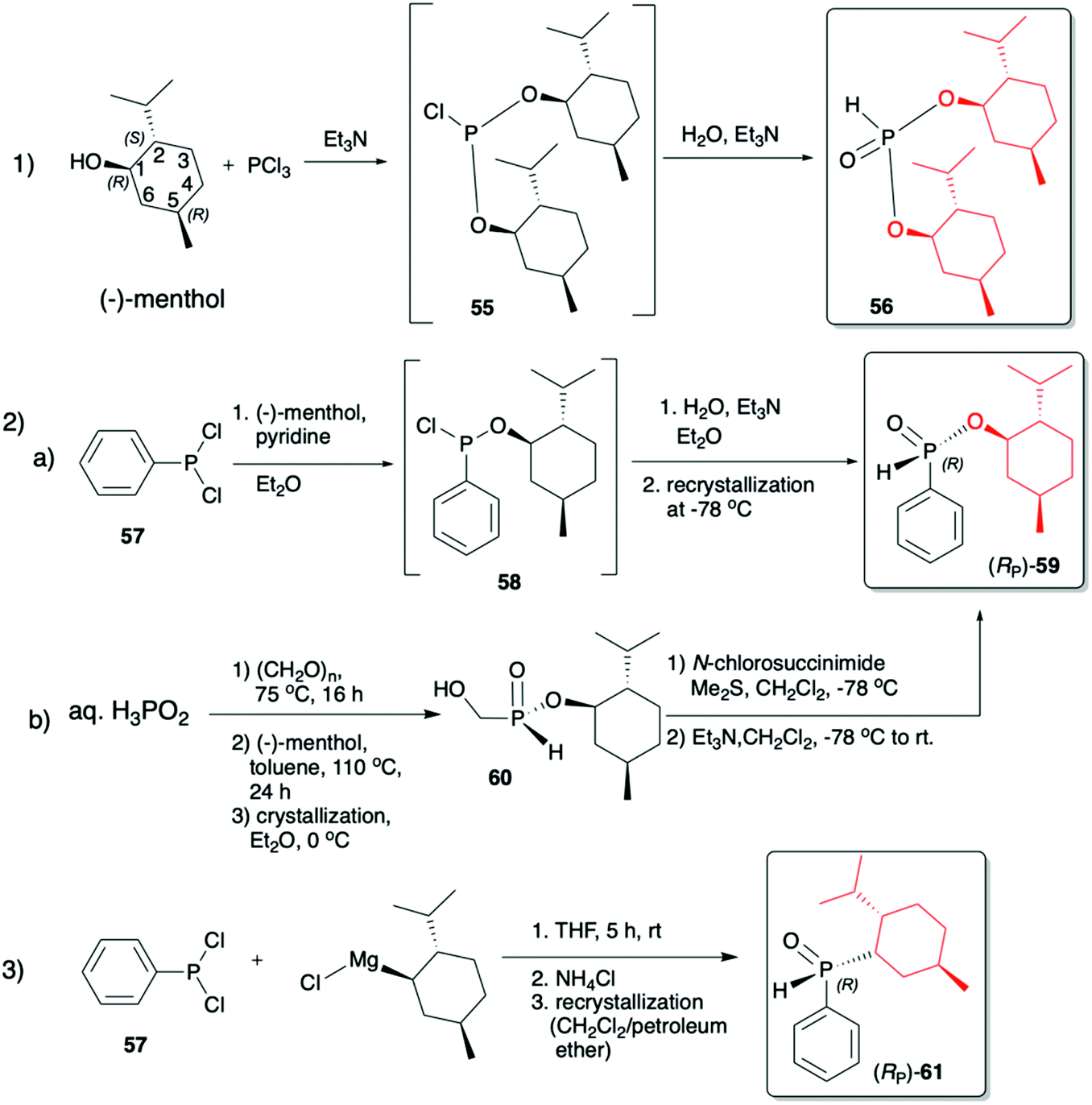 | ||
| Scheme 20 Syntheses of (−)-O,O-di-(1R,2S,5R)-menthyl-H-phosphonate (56), (−)-O-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59) and (−)-(1R,2S,5R)-menthyl phenylphosphine oxide (RP)-(61). | ||
A recent alternative method for the preparation of (RP)-(59) was reported quite recently by Montchamp (Scheme 20, reaction (2b)).80,81 The synthesis is based on the use of (hydroxymethyl)-H-phosphinate (SP)-(60) easily accessible by the reaction of aqueous hypophosphorous acid with paraformaldehyde, followed by azeotropic distillation with menthol in toluene and subsequent crystallization at 0 °C to afford (SP)-(60) (10% yield, de >99%). Subsequent stereoselective cleavage of the hydroxymethyl moiety using the Corey–Kim oxidation to unmask the P(O)H moiety leads to the desired (RP)-(59) in 91% yield and de >99% (Scheme 20, reaction (2b)). Finally, (−)-menthol can also be used to prepare chiral phosphine oxide (RP)-(61). The reported procedure is based on the reaction between (−)-menthyl-magnesium chloride and dichloro(phenyl)phosphine (57) followed by recrystallization of the crude product to yield diastereomerically pure (RP)-(61) in 34% yield (Scheme 20, reaction (3)).82
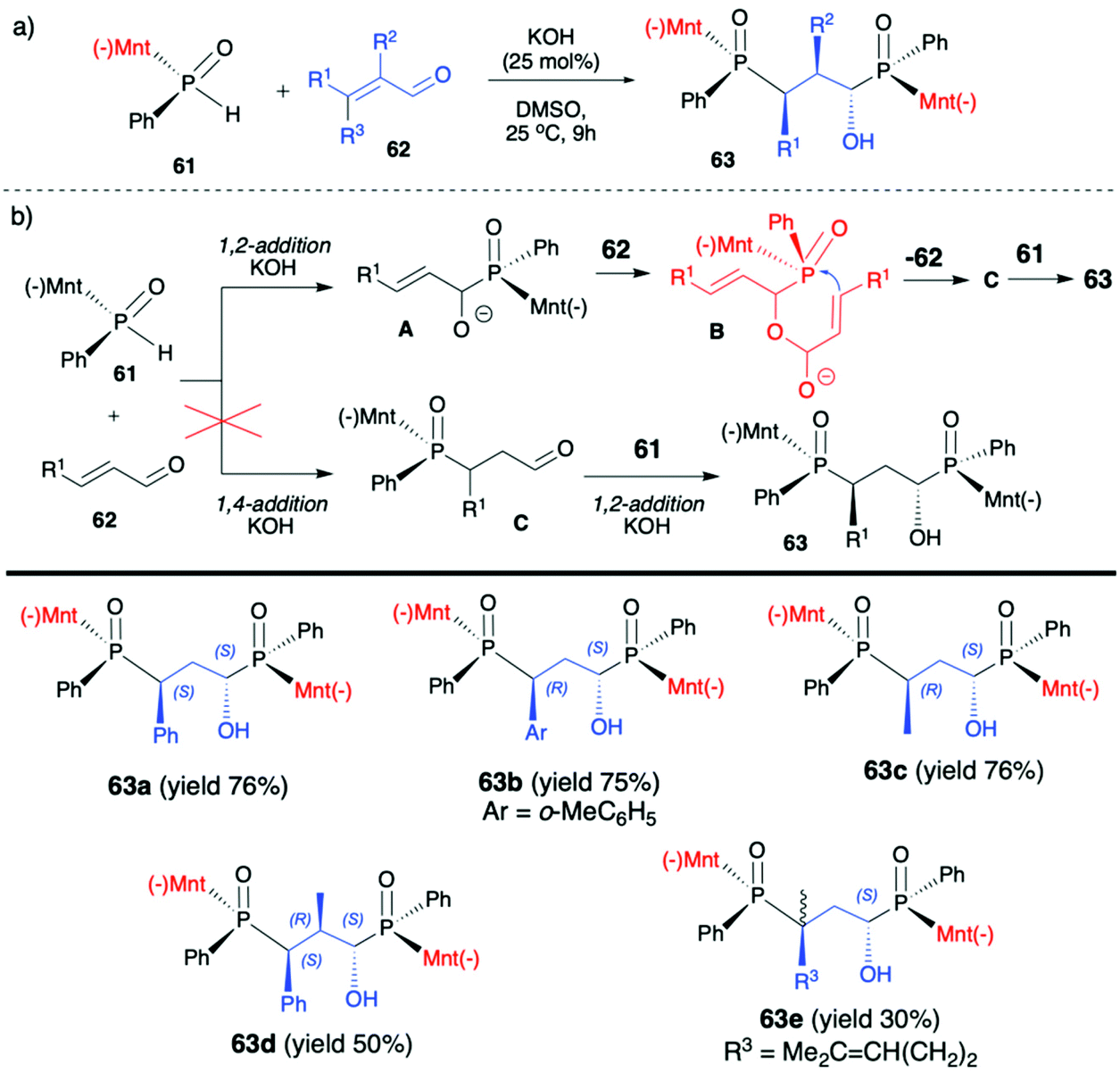 | ||
| Scheme 21 (a) Reaction of vinyl aldehydes 62 with (−)-(1R,2S,5R)-menthyl phenylphosphine oxide (RP)-(61) leading to P,C-stereogenic 1,3-bisphosphinylpropanes 63 – selected examples. (b) Postulated reaction mechanism. (−)Mnt = 1R,2S,5R-menthyl. | ||
Later on, the same authors focused on the addition of (RP)-(61) to unsaturated ketones 64 and developed a procedure leading stereoselectively to β-phosphino ketones 65 (Scheme 22).84
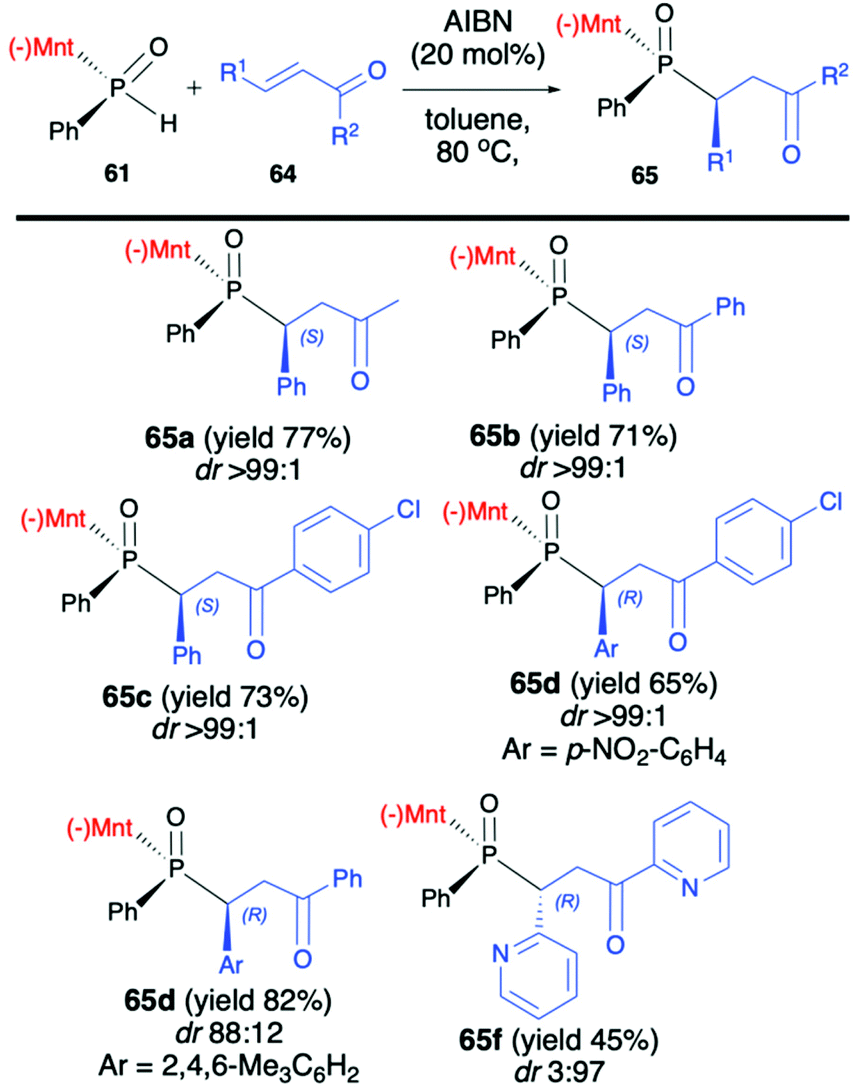 | ||
| Scheme 22 Preparation of β-phosphino ketones via the addition of chiral (RP)-61 to vinyl ketones 64 – selected examples. Yields and dr are given for isolated products. AIBN = azobisisobutyronitrile. (−)Mnt = 1R,2S,5R-menthyl. | ||
The methodology was based on the gentle heating of reagents in toluene at 80 °C in the presence of a catalytic amount of AIBN (20 mol%), which was found to accelerate the reaction and slightly increase the dr. It is worth mentioning that the excellent stereoselectivity of the reaction (dr >99:1 in most cases) enabled easy isolation of pure major diastereomers by simple recrystallization of the crude products from hexane. Recently, Zhao et al. also presented results on the addition of (RP)-(61) and its derivatives to a triple bond in diethyl acetylenedicarboxylate and ethyl propiolate leading stereoselectively to corresponding bis-phosphine derivatives (up to dr >99 and yield up to 99%) when the reaction was conducted at 100 °C in decane.85
Another application of (RP)-(61) and also (−)-O-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59) in the reaction with alkenes was reported by Teck-Peng Loh et al. and was based on the use of allylic carbonates 66 as substrates in the presence of a catalytic amount of quinine (20 mol%) in toluene at room temperature (Scheme 23).86 Reactions in general proceeded with high diastereoselectivity, and major diastereomers could be easily separated by flash chromatography. The absolute configuration of the products was established by X-ray crystal analysis. The utility of the obtained P,C-stereogenic allylic compounds 68 was demonstrated as an example by subsequent reaction with (RP)-(61) in the presence of K2CO3 in DMSO at room temperature yielding the corresponding 1,3-bisphosphinylpropane as a mixture of diastereomers (73% yield, dr 57:43) from which the major diastereomer could be separated by flash chromatography. Later on, the same authors attempted to use (RP)-(59) in the barium-catalysed C–OH/P–H dehydrative cross-coupling; however, their attempts were unsuccessful and (RP)-(59) did not react under the optimized conditions in contrary to the diaryl phosphine oxides tested.87
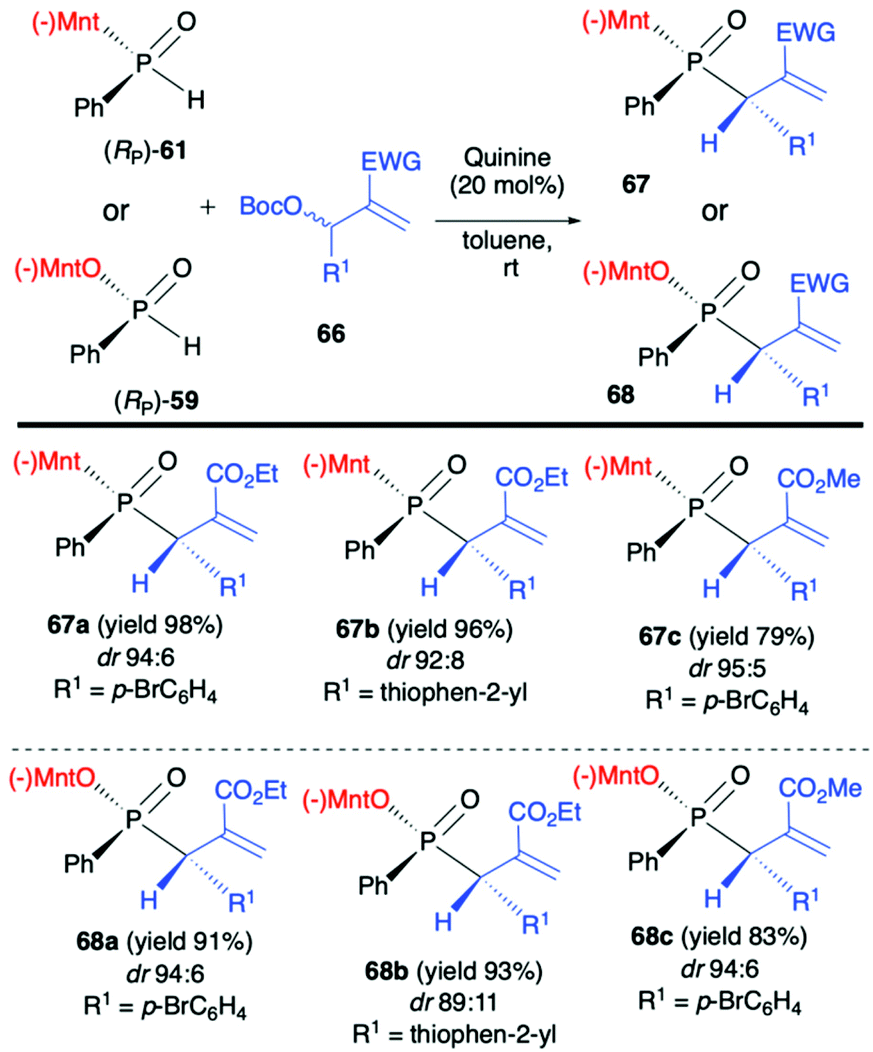 | ||
| Scheme 23 Diastereoselective P–C bond formation between allylic carbonates 66 and chiral (RP)-(59) and (RP)-(61) in the presence of quinine – selected examples. Yields are given for isolated products. dr is determined by 31P NMR of the crude reaction mixture. (−)Mnt = 1R,2S,5R-menthyl. | ||
:1), (+)-74a (dr 1.7:1), (+)-74b (dr 1.3:1) and (+)-75 (dr 1.6:1), respectively, as an additional proof for the utility of a dimenthoxyphosphonyl group as a good stereodirecting chiral auxiliary attached to the phosphorus atom (Scheme 24).
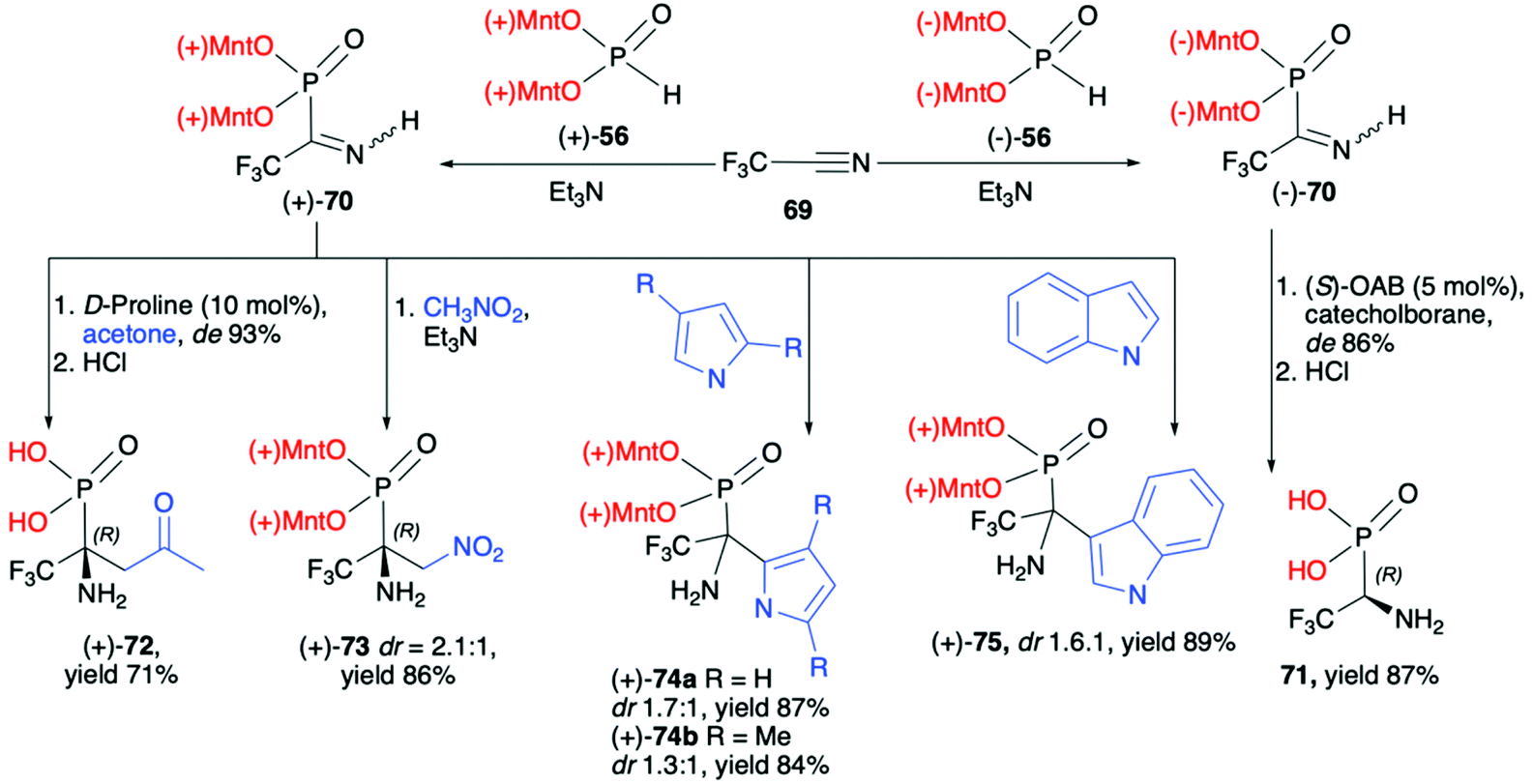 | ||
| Scheme 24 Reactions of optically active O,O-dimenthyl α-iminotrifluoroethylphosphonates (+)-70 and (−)-70. Yields are given for isolated products. (S)-OAB = (S)-1-methyl-3,3-diphenyl-tetrahydro pyrrolo[1,2c][1,3,2]oxazaborole. (−)Mnt = 1R,2S,5R-menthyl; (+)Mnt = 1S,2R,5S-menthyl. | ||
As an example of double asymmetric addition, the use of optically pure (−)- and (+)-lithium salts of O,O-di-(1R,2S,5R)-menthyl H-phosphonate (76) by the hydrophosphonylation of pure enantiomers of both chiral N-(p-tolylsulfinyl)-benzaldimines (+)- and (−)-77a,b at −78 °C, leading to the corresponding α-aminophosphonates 78 with high diastereoselectivity (dr up to 100:0) was reported by Łyżwa (Scheme 25).90 The enantiomers of (−)-76a and (+)-(S)-77a as well as of (+)-76b and (−)-(R)-77b were assigned as the matched pairs of isomers, whereas the enantiomers (−)-76a and (−)-(R)-77b or (+)-76b and (+)-(S)-77a were assigned as the mismatch pairs. In each case, the pure major diastereoisomer was isolated by means of chromatography. The absolute configuration of the stereogenic carbon was assigned after hydrolysis of the pure diastereomers of α-aminophosphonates 78 to the corresponding α-aminophosphonic acids 16a with (R)- or (S)-configuration (Scheme 25).
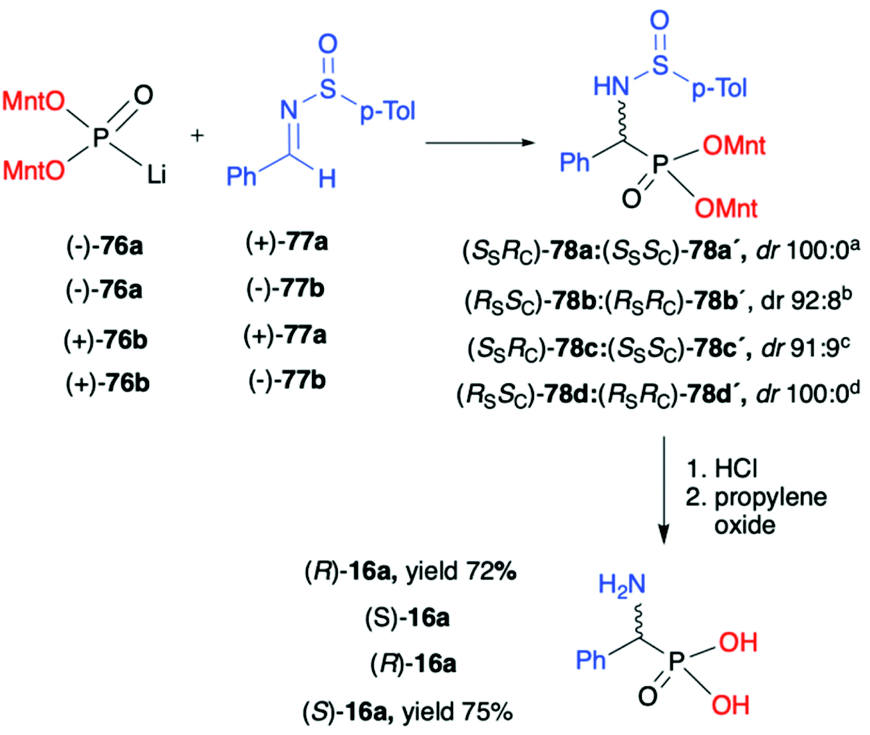 | ||
| Scheme 25 Asymmetric addition of lithium salts of chiral H-dimenthyl phosphonates (+)-76 and (−)-76 to chiral sulfinylimines (+)-77a and (−)-77b. aYield of major diastereomer 88%, byield of major diastereomer 77%, cyield of major diastereomer 75%, dyield of major diastereomer 86%. (−)Mnt = 1R,2S,5R-menthyl; (+)Mnt = 1S,2R,5S-menthyl. | ||
In turn, the pure enantiomer of P-chiral (−)-O-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59) was used by Miao et al. in a successful and diastereoselective Lewis acid catalysed phospha-Mannich addition to O-pivaloylated N-galactosylimines 80, obtained from 2,3,4,6-tetra-O-pivaloyl-β-D-galactopyranosyl-amine 79 and aldehydes (Scheme 26).91 Under the optimized reaction conditions, good to excellent yields (up to 91%) and diastereoselectivity (dr up to >20:1) were obtained for aromatic and substituted aromatic imines 80. However, in the case of heterocyclic and aliphatic substituents, yields and diastereoselectivities were lower or no reaction was observed. Furthermore, the absolute configuration at the phosphorus atom was determined unambiguously by X-ray analysis showing that the phospha-Mannich reaction took place with the retention of the configuration at phosphorus. The structure showed that the relative configuration of the β-N-glycoside-α-aminophosphinates 81 main product could be assigned as (SC,SP). Finally, the removal of the O-pivaloylated N-galactosyl substituent from 81a under acidic conditions resulted in the formation of a pure enantiomer of the corresponding α-aminophosphinate 82 (yield 86%) without racemization. In turn, Zhao et al. reported on the use of (RP)-(59) phospha-Mannich reaction with pure enantiomers of (R)-1-phenylethanimines 82, derived from various substituted aromatic and also aliphatic aldehydes, leading to the corresponding α-aminophosphinates 83 with high diastereoselectivity (dr >99:1) (Scheme 27).92 Based on the X-ray analysis, the absolute configuration of the stereogenic carbon in the major diastereomers was assigned as S, and also S at the phosphorus, since the reaction occurred with retention of configuration at phosphorus. Importantly, the use of (S)-1-phenylethanimine or non-chiral imines resulted in low diastereoselectivity (dr 50:50) of the reaction with (RP)-(59). The authors suggest that the chirality on the stereogenic carbon atom was induced by the chiral aldimine rather than the chiral phosphorus. The (RP)-(59) exhibited matched effect with (R)-1-phenylethanimines and mismatched effect with (S)-1-phenylethanimines. The same authors also used (−)-(1R,2S,5R)-menthyl phenylphosphine oxide (RP)-(61) as a chiral auxiliary in the phospha-Mannich reaction with chiral and non-chiral imines (Scheme 28).93 The reactions with (RP)-(61) and (R)-1-phenylethanimines and non-chiral imines resulted in the formation of the desired α-aminophosphinates 85 with moderate yields and diastereoselectivities (dr up to 77:23). Noteworthily, in each case, diastereomers could be easily separated by preparative TLC. Yields and diastereoselectivities could be improved when the reaction of (RP)-(61) was performed with (S)-1-phenylethanimines (Scheme 28). The much better diastereoselectivity was assigned to the matched induction between (S)-1-phenylethanimines and (RP)-(61). Additionally, the authors stated that the chirality was induced mainly by the stereogenic phosphorus atom. The absolute configuration in the dominant stereoisomers of the reaction products was assigned as RP,RC (Scheme 28).
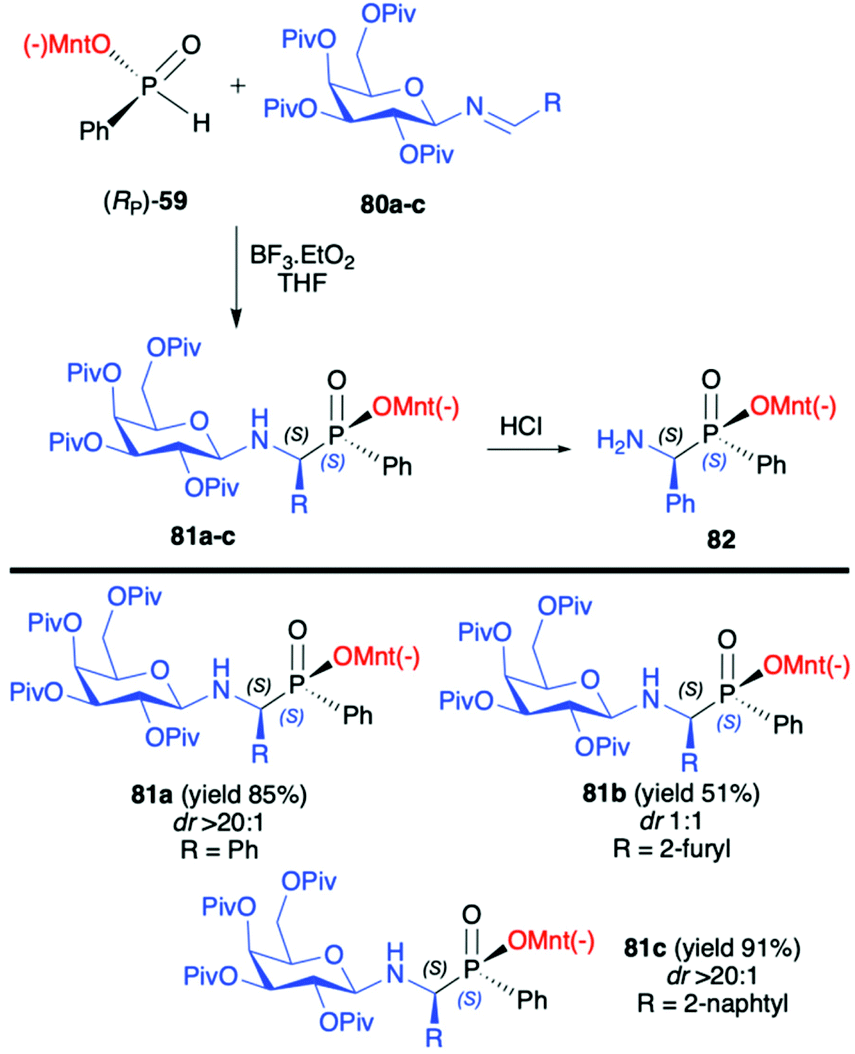 | ||
| Scheme 26 Diastereoselective phospha-Mannich reaction of (RP)-(59) with aldimies 80a–c leading to α-aminophosphinates 81a–c-selected examples. Yields are given for isolated products. dr is determined by 31P NMR of the crude reaction mixture. (−)Mnt = 1R,2S,5R-menthyl. | ||
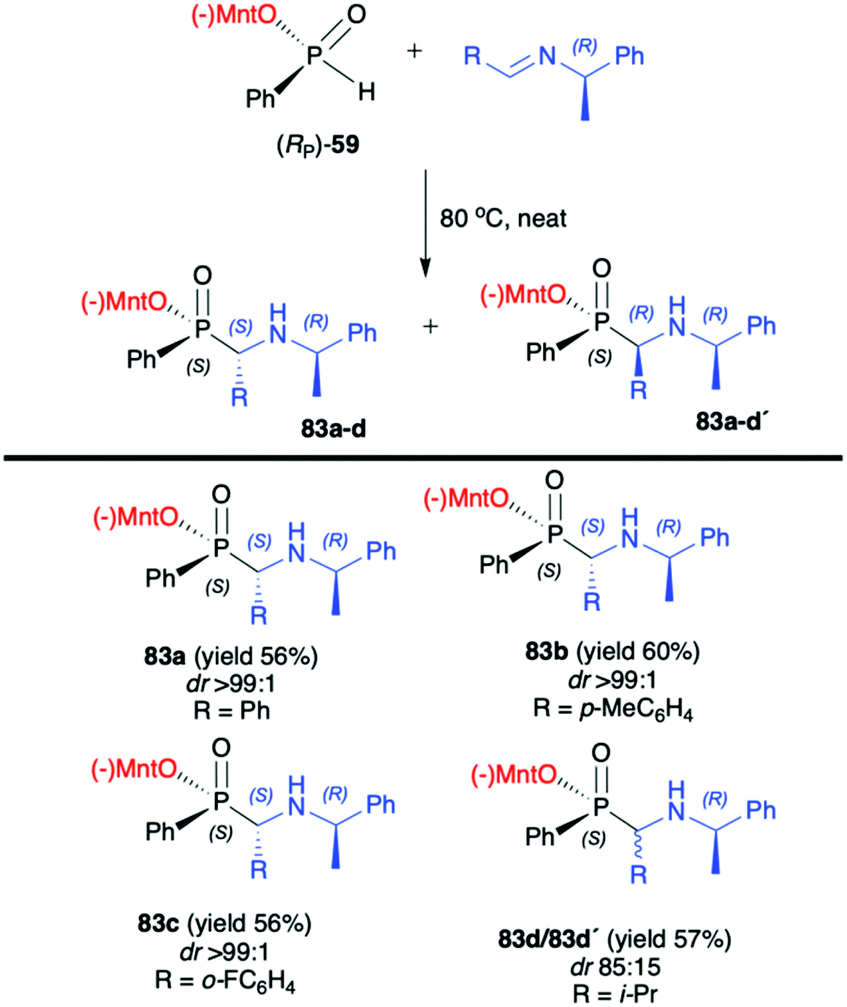 | ||
| Scheme 27 Reaction of (RP)-(59) with enantiomerically pure (R)-1-phenylethanimines-selected examples. Yields and dr are given for isolated products after recrystallization. (−)Mnt = 1R,2S,5R-menthyl. | ||
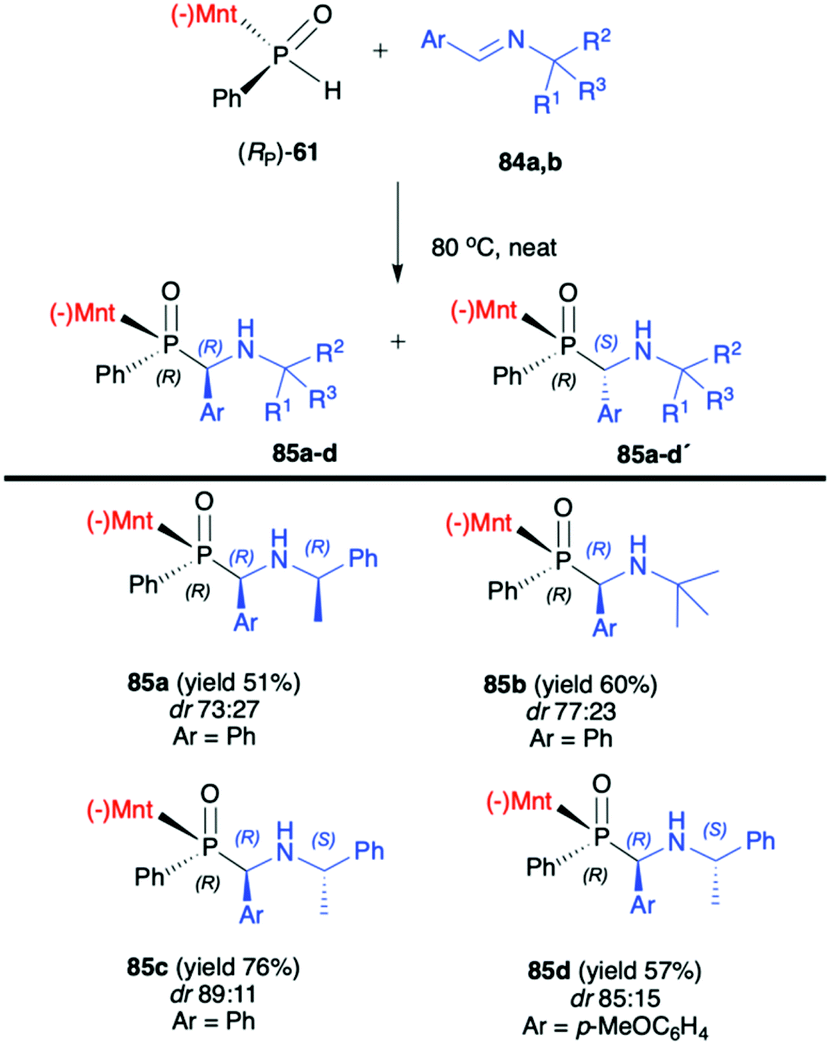 | ||
| Scheme 28 Reaction of (RP)-(61) with imine – selected examples. dr is determined by 31P NMR of the crude reaction mixture. Yields are given for isolated products. (−)Mnt = 1R,2S,5R-menthyl. | ||
Finally, Zhao et al. in a recent publication described the use of (RP)-(61) in the Atherton–Todd coupling reaction with ammonia solution to obtain (SP)-(−)-menthyl phenylphosphinamide (86) in 94% yield and dr >99:1. Subsequently, 86 was condensed with aromatic aldehydes in the presence of TiCl4/Et3N to obtain the corresponding N-phosphonyl imines 87. Finally, the imines were subjected to the 1,2-addition of the Grignard reagents to obtain P-chiral phosphinamides 88 (Scheme 29).94 The desired products were obtained with good yields albeit with moderate diastereoselectivity.
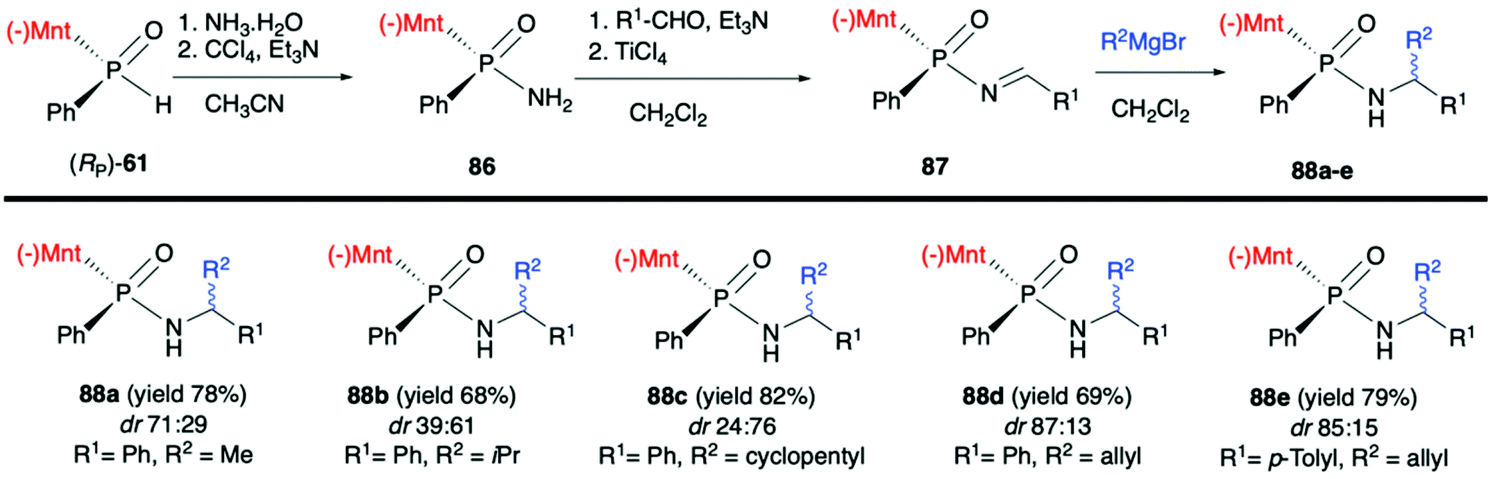 | ||
| Scheme 29 Application of (RP)-(61) in the synthesis of imines 87 and their reaction with Grignard reagents – selected examples. dr is determined by 31P NMR. Yield is given for an isolated mixture of diastereomers. (−)Mnt = 1R,2S,5R-menthyl. | ||
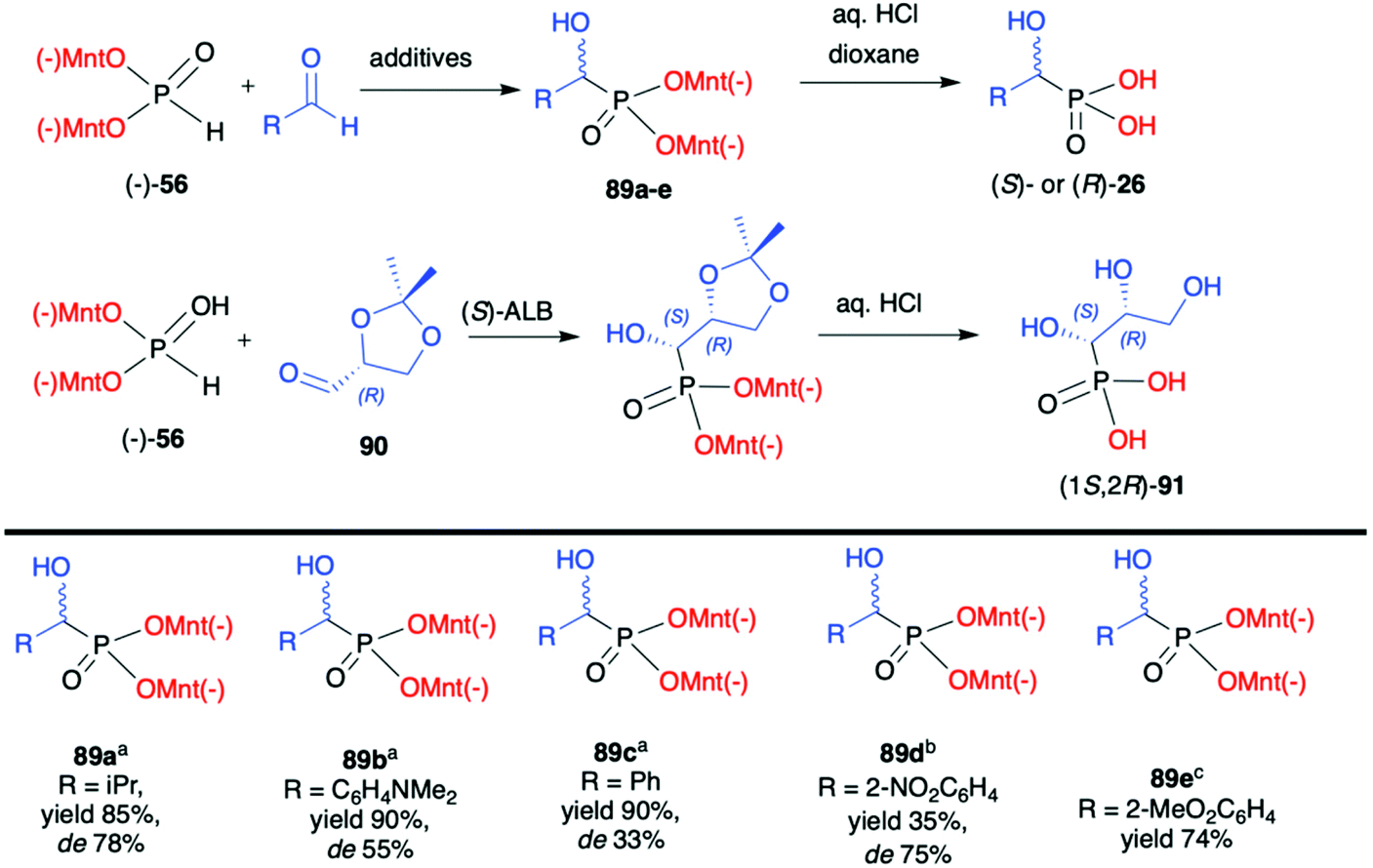 | ||
| Scheme 30 Hydrophosphonylation of aldehydes with (−)-56. In the case of products 89a–c, dr was established based on 31P NMR spectra of the crude product and pure major diastereomer was separated by crystallization. aAs base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was used. bCatalytic amount of cinchonidine was used. cCatalytic amount of quinine was used. (S)-ALB = (S)-aluminium lithium bis(binaphthoxide). (−)Mnt = 1R,2S,5R-menthyl. | ||
Later on, Kolodiazhnyi et al. demonstrated that diastereoselectivity of the reaction involving aromatic aldehydes and (−)-56 could be improved by the addition of a catalytic amount of cinchonine alkaloids and its structure influenced the stereochemical direction of the reaction.96 For instance, in the case of 2-nitrobenzaldehyde and the use of cinchonidine, the desired (R)-α-hydroxyphosphonate 89d was obtained with de 75%. In turn, the application of quinine gave rise to (S)-α-hydroxyphosphonate 89d. Importantly, the use of cinchonine alkaloids in a similar reaction but with non-chiral dimethyl H-phosphonate resulted in poor enantioselectivity (ee ≤33%) clearly demonstrating the stereodirecting properties and utility of the menthyl-derived H-phosphonate (−)-56.
Finally, the same authors also used 2,3-D-isopropylidene-(R)-glyceraldehyde (90) as chiral aldehyde in the reaction with (−)-56. After extensive experimentation, it was established that the best diastereoselectivity (de 85%) could be obtained with the use of (S)-aluminium lithium bis(binaphthoxide) as a catalyst in THF at room temperature. Noteworthily, diastereomers could be easily separated by crystallization and obtained in a pure form and subsequently hydrolysed to enantiomerically pure hydroxyphosphonic acid (1S,2R)-91 (Scheme 30).97
Kolodiazhnyi et al. also demonstrated that racemic α-hydroxyphosphonates bearing dimenthyl auxiliary, prepared in the reaction of aldehydes with (−)-56 catalysed by DBU, could be used to obtain the corresponding α-ketophosphonates 92a–c (by oxidation with the use of pyridinium dichromate/trimethylchlorosilane) that were subsequently reduced in diastereoselective fashion to optically pure α-hydroxyphosphonates 89a,c,f (Scheme 31).98,99 Similar strategy was also applied to β-ketophosphonate 93. The latter was prepared in a three-step one-pot procedure from dimenthyl methylphosphonate by the reaction with butyllithium to form the corresponding carbanion that was further reacted with cuprous bromide to form cuprous derivative that was finally reacted with acyl chlorides to produce the appropriate β-ketophosphonates. The authors after testing the easily available chiral molecules demonstrated that the best reducing agent was adduct (R)-95 obtained from the reaction of (R,R)-tartaric acid with sodium borohydride. The stereochemistry of the reduction of α- and β-ketophosphonates with (R)-95 depended on the absolute configuration of tartaric acid. Thus, the reduction of α- and β-ketophosphonates with (R)-95 yielded α- and β-hydroxyphosphonates with S configuration, whereas the reduction of α- and β-ketophosphonates with (S)-95 resulted in the formation of (R)-α- and β-hydroxyphosphonates. Therefore, asymmetric inductions of chiral (1R,2S,5R)-menthyl groups and (R,R)-tartaric acid acted in one direction (matched effect), increasing the diastereofacial selectivity of the reagents, whereas asymmetric inductions of (1R,2S,5R)-menthyl groups and (S,S)-tartaric acid were mismatched and acted in opposite directions decreasing the resulting stereoselectivity.
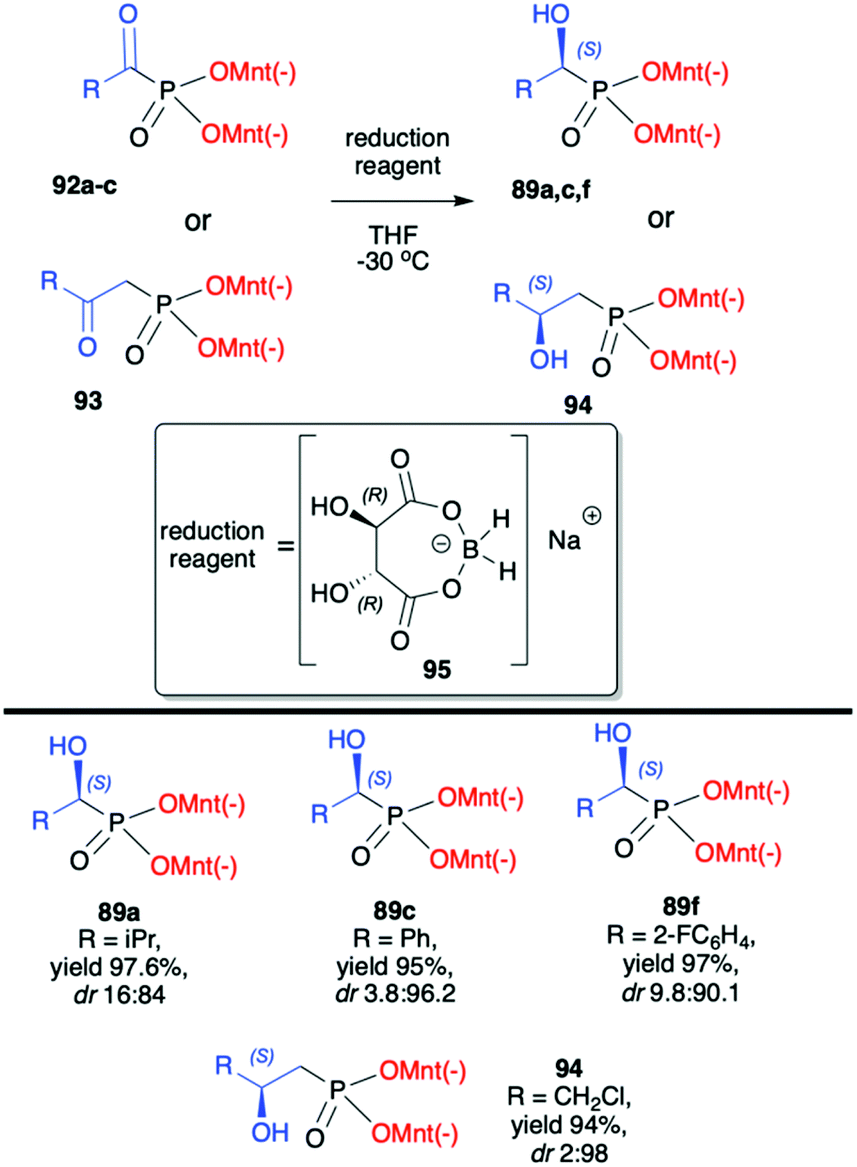 | ||
| Scheme 31 Diastereoselective reduction of α- and β-ketophosphonates leading to α- and β-hydroxyphosphonates – selected examples. (−)Mnt = 1R,2S,5R-menthyl. | ||
Li-Biao Han et al. examined the use of (−)-O-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59) in diastereoselective rhodium- or iridium-catalysed hydrophosphonylation of aldehydes (Scheme 32).100 During optimization of the structure of the catalyst, the [RhHCl2(PPh3)3] obtained by the treatment of Wilkinson's catalysts [RhCl(PPh3)3] with diluted HCl was found to give the best diastereoselectivity of the addition (up to dr 95:5). As an alternative, a series of iridium catalysts were tested, and [IrCl(cod)2] with PPh3 was found to give comparable results (up to dr 97:3). The pure major diastereomer could be obtained by simple crystallization, and the X-ray analysis of the crystals revealed R configuration on the hydroxy carbon and S configuration on the phosphorus. The authors postulated that the reaction most probably proceeds by transition-metal-catalyzed and optically pure menthyl-derived H-phosphinate-directed mechanism.
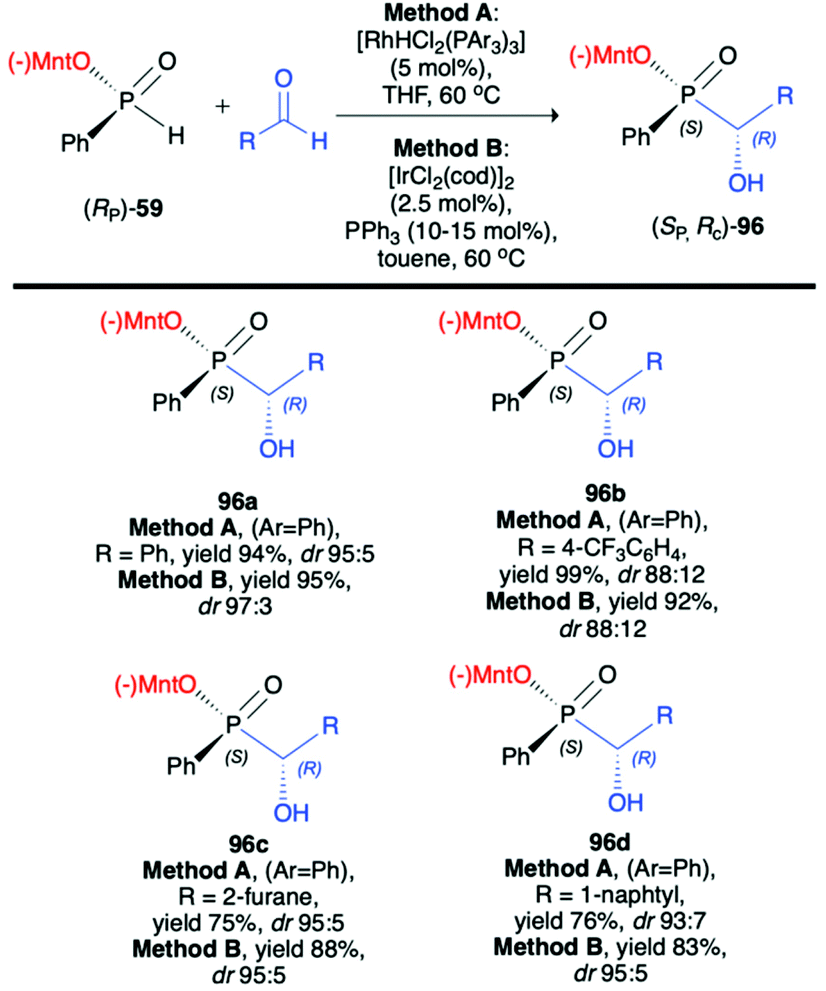 | ||
| Scheme 32 Rhodium or iridium catalysed hydrophosphonylation of aldehydes with (RP)-(59) leading to P- and C-stereogenic α-hydroxyphosphinates 96 – selected examples; dr is established based on 31P NMR spectra of the crude product. cod – 1,5-cyclooctadiene. (−)Mnt = 1R,2S,5R-menthyl. | ||
Very recently, Chang-Qui Zhao and Qiang Li et al. presented an interesting application of (−)-(1R,2S,5R)-menthyl phenylphosphine oxide (RP)-(61) in the reaction with alkenylaldehydes 97 to form α-phosphinyl propargyl alcohols 98, which were subsequently converted to P,axial-stereogenic allenyl phosphine oxides 99 in the presence of pyridine and Ph2PCl (Scheme 33).101 Although the addition of (RP)-(61) to aldehydes 97 did not proceed with high diastereoselectivity in all cases, the presence of chiral auxiliary bound to the phosphorus atom group allowed for easy separation of the pure diastereomers from the reaction mixture. The subsequent reaction of pure diastereoisomers 98 with Ph2PCl in the presence of pyridine led dominantly to (SP,SA)-99 (e.g. dr 99:1 for 99a) in the case of (RP,RC)-98, whereas (RP,SC)-98′ also gave (RP,SA)-99 as the major product but with lower stereoselectivity (e.g. dr 81:19 for 99a). The authors claimed that such results could be explained by the fact that axial chirality was mainly controlled by stereogenic phosphorus.101 Finally, Li-Biao Han et al. examined also the utility of (−)-(1R,2S,5R)-menthyl phenylphosphine oxide (RP)-(61) and (−)-O-(1R,2S,5R)-menthyl phenyl-H-phosphinate (RP)-(59) in the reaction with aldehydes and ketones under simple base catalysed conditions (Scheme 34).102,103 In the case of ketones, the best results were obtained with the use of K2CO3 as a base in DMSO at room temperature.
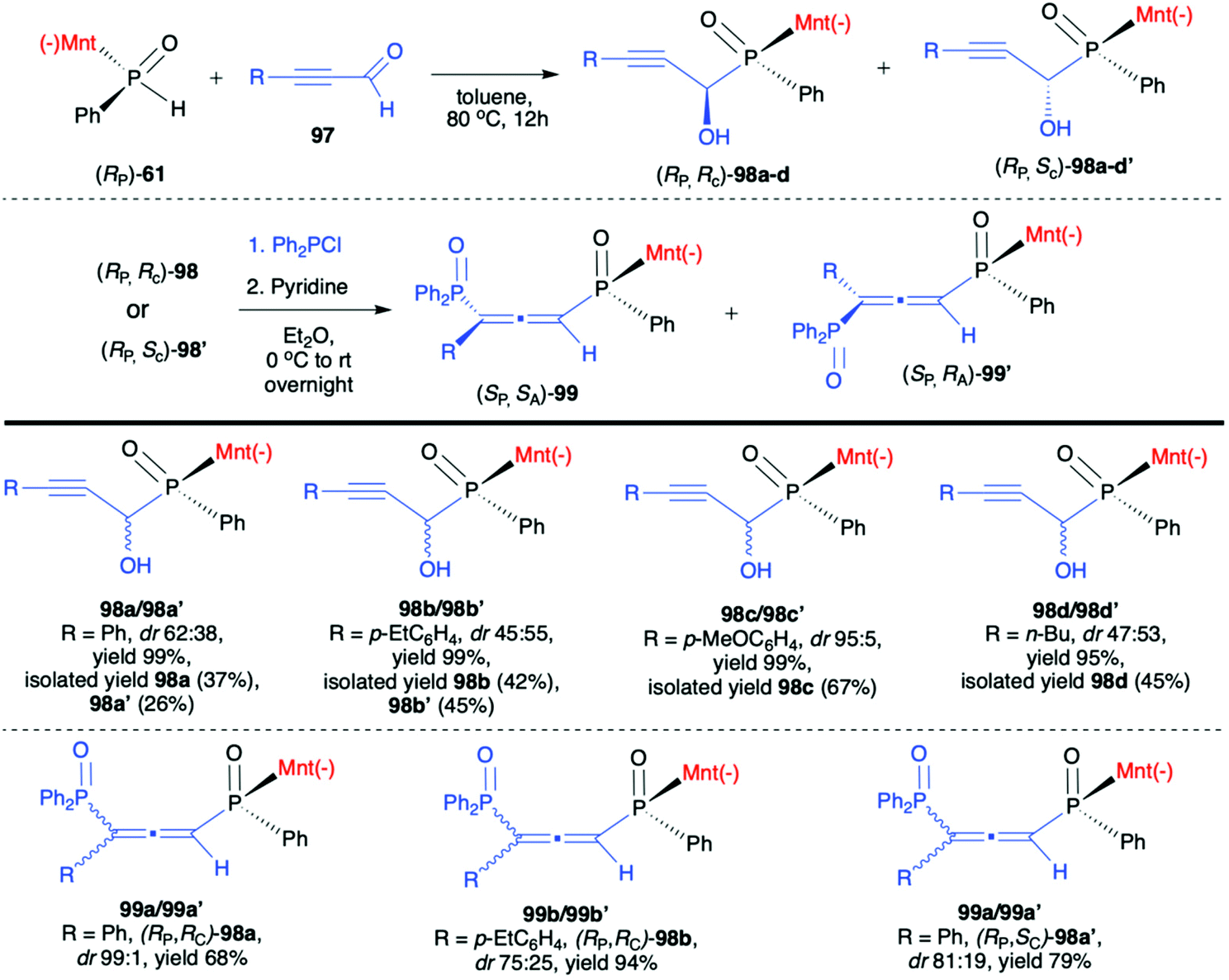 | ||
| Scheme 33 Stereoselective preparation of P,axial-stereogenic allenyl bisphosphine oxides from optically pure α-hydroxyphosphinates 98via chirality transfer – selected examples. dr is established based on 31P NMR spectra of the crude product. Diastereoisomers could be easily separated and obtained in pure form by means of crystallization or chromatography. (−)Mnt = 1R,2S,5R-menthyl. | ||
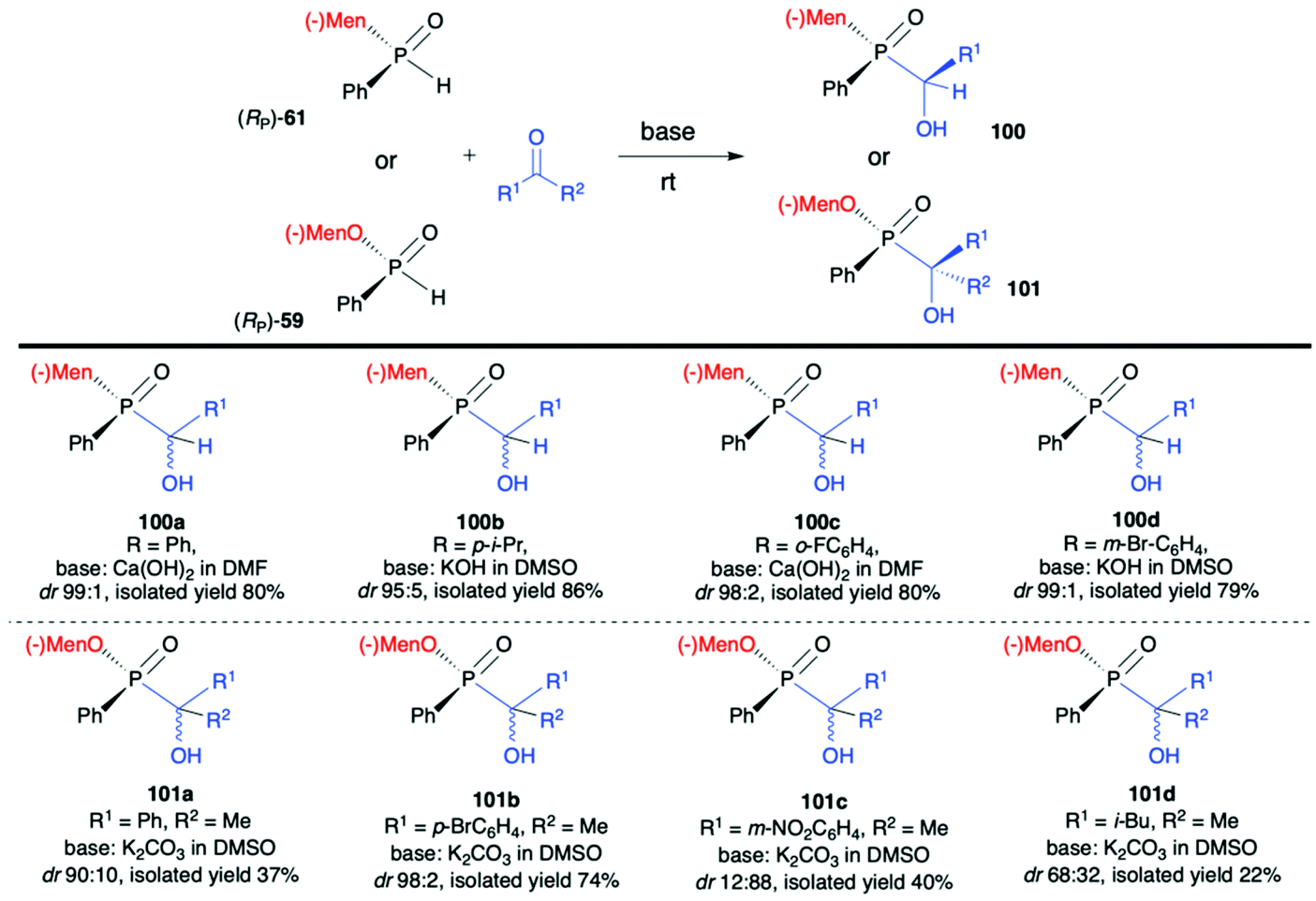 | ||
| Scheme 34 Nucleophilic addition of (RP)-(61) and (RP)-(59) to aldehydes and ketones – selected examples. dr is established based on 31P NMR spectra of the crude product. Diastereoisomers could be easily separated and obtained in pure form by means of crystallization or chromatography. (−)Mnt = 1R,2S,5R-menthyl. | ||
The aromatic and p-substituted aromatic ketones gave the corresponding quaternary α-hydroxyphosphinates 101 in good, isolated yields and diastereoselectivity (dr ≥90:10, with predominantly R configuration of the asymmetric C atom and with retention of configuration at P atom in the major diastereomer); in turn less satisfactory results were obtained with ortho or meta substituted aromatic ketones. In the case of the latter, even reversed selectivity was observed (Scheme 34, 101c). The use of aliphatic ketones gave low yields and diastereoselectivity. In turn, for aldehydes, the use of Ca(OH)2 in DMF or KOH in DMSO was found to give the best results. The desired α-hydroxyphosphine oxides 100 were obtained in good yields and stereoselectivity practically regardless of the structure of the aldehyde used (dr ≥95:5, with predominantly S configuration of the asymmetric C atom and R configuration of the P atom in the major diastereomer). Examining the progress of the reactions over time and improvement of the diastereoselectivity with time, the authors reasoned that both additions of (RP)-(61) to aldehydes and (RP)-(59) to ketones were reversible, and the thermodynamic stabilities of diastereomers formed were different.
3. Conclusions
We have demonstrated that, derived from chiral alcohols, optically pure H-phosphonates, phosphinates and phosphine oxides as H–P species bearing a chiral auxiliary attached to the phosphorus atom can be considered as very effective tools for the highly diastereoselective preparation of a variety of structurally diverse chiral organophosphorus compounds containing C- and, in some cases, also P-stereogenic centers (Table 1). The most commonly used chiral alcohols are TADDOL, BINOL and menthol, as inexpensive and easily available building blocks. Their subsequent transformation into the optically pure H–P reagents can be carried out without any synthesis challenges. The TADDOL, BINOL and dimenthyl-derived H-phosphonates are used in cases where the creation of a new C-stereogenic center in the product is desired. In turn, application of pure enantiomers of menthyl-derived H-phosphinates and phosphine oxides is recommended if both C- and P-stereogenic products are to be obtained. The presented examples clearly demonstrate that in each case the use of such H–P reagents bearing chiral auxiliary attached to the phosphorus atom not only has a significant influence on the asymmetric induction and highly diastereoselective formation of new C-stereogenic centers but also is beneficial for isolating optically pure single stereoisomers of the reaction product simply by means of crystallization or chromatography. An additional advantage of the use of such chiral H–P reagents is the possibility of fast and reliable controlling of the stereochemical outcome of the reaction by means of 31P or/and 1H NMR spectroscopy since the pairs of diastereomers formed in the product are clearly distinguishable by NMR. Because of the important benefits originating from the use of TADDOL, BINOL and menthol derived from optically pure H-phosphonates, phosphinates and phosphine oxides as H–P species bearing a chiral auxiliary attached to the phosphorus atom, we are confident that many more applications of these useful chiral H–P reagents will appear in the near future and will serve as a driving force for further developments in the exciting field of synthesis and applications of chiral organophosphorus compounds.| Advantages and disadvantages | H–P species bearing chiral auxiliary attached to the phosphorus atom | Possible applications | Ref. |
|---|---|---|---|
| + Easily prepared from tartaric acid esters (4 steps) |
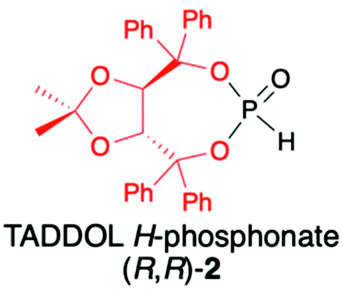
|
1. Stereoselective synthesis of C-stereogenic organophosphorus compounds | |
| + Chiral part easily cleaved with aq. HCl or BrTMS | 2. Highly diastereoselective addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (de ≥90%) C bond (de ≥90%) |
42, 45 and 46 | |
| − In solution undergoes decomposition to TADDOL when heated above 80 °C | 3. Highly diastereoselective addition to CN (chiral and non-chiral imines) and CO bond (aldehydes and ketones) (de ≥90%) |
47–52 | |
| + Easily obtained from BINOL and PCl3 (2 steps) |
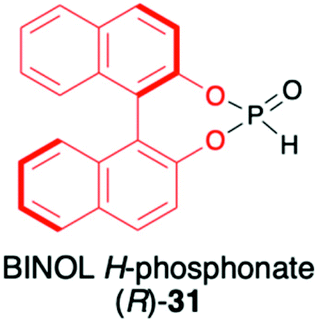
|
1. Stereoselective synthesis of C-stereogenic organophosphorus compounds | |
| + Chiral part cleaved with nBuLi or EtONa and BINOL recovered | 2. Addition to cyclic imines with good dr (95:5) but moderate yields (≤50%) |
61 and 62 | |
| − Decomposition to BINOL when purified by column chromatography | 3. Catalytic addition to hydrazones and CC bonds with low dr (1:1.2) |
67 and 68 | |
| 4. Diastereoselective reactions of Se derivative of (R)-31 with Grignard reagents and halides | 70–72 | ||
| 5. Good stereodirecting properties | |||
| + Easily prepared from menthol and PCl3 (2 steps) |
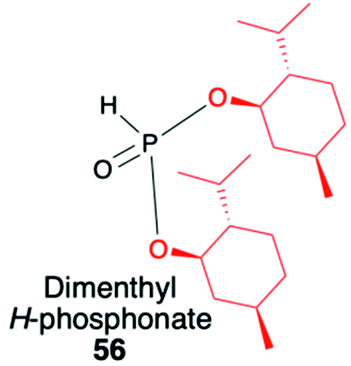
|
1. Stereoselective synthesis of C-stereogenic organophosphorus compounds | |
| + Chiral part easily cleaved with aq. HCl | 2. Highly diastereoselective addition to chiral imines (dr ≥92:8) |
90 | |
| 3. Moderate diastereoselectivty in addition to CO bond (chiral and non-chiral aldehydes) (de ≤85%) |
95–99 | ||
| 4. Moderate stereodirecting properties | 88, 98 and 99 | ||
| + Relatively easily prepared and reproducible protocols are described in the literature |
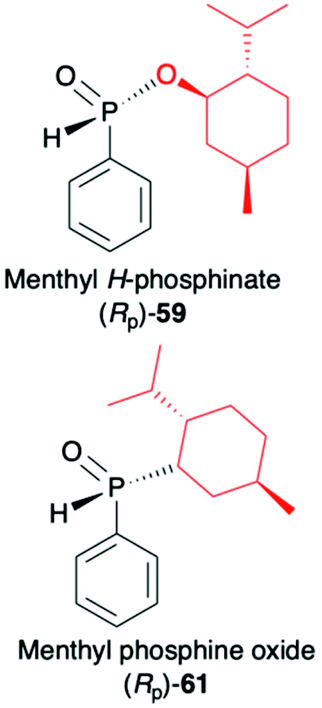
|
1. Stereoselective synthesis of C- and P-stereogenic organophosphorus compounds | |
| − Due to the chirality on the P-atom, synthesis is more challenging than in the case of other H–P species bearing chiral auxiliary attached to the phosphorus, (R,R)-2, (R)-31 and 56 | 2. Highly diastereoselective addition to alkenes and alkynes (dr ≥90:10) |
83–85 and 101 | |
| 3. Highly diastereoselective addition to imines | 91–93 | ||
| 4. Highly diastereoselective addition to CO bond (de ≥90%) |
100, 102 and 103 | ||
| 5. Good stereodirecting properties | 94 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding from the Polish Ministry of Science and Education for the Wrocław University of Science and Technology.Notes and references
- S. Wendels, T. Chavez, M. Bonnet, K. A. Salmeia and S. Gaan, Materials, 2017, 10, 784 CrossRef PubMed.
- M. Wehbi, A. Mehdi, C. Negrell, G. David, A. Alaaeddine and B. Ameduri, ACS Appl. Mater. Interfaces, 2020, 12, 38 CrossRef CAS PubMed.
- Ch. Verma, D. K. Verma, E. E. Ebenso and M. A. Quraishi, Heteroat. Chem., 2018, 29, e21437 CrossRef.
- G. David and C. Negrell-Guirao, Complexation with Metals: Anticorrosion Phosphorus-Containing Polymer Coatings, in Phosphorus-Based Polymers: From Synthesis to Applications, ed. S. Monge and G. David, The Royal Society of Chemistry, Oxford, U.K., 2014, pp. 210–224 Search PubMed.
- A. Cabre, A. Riera and X. Verdaguer, Acc. Chem. Res., 2020, 53, 676 CrossRef CAS PubMed.
- B. T. Ingoglia, C. C. Wagen and S. L. Buchwald, Tetrahedron, 2019, 75, 4199 CrossRef CAS PubMed.
- H. Guo, Y. Ch. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765 RSC.
- M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC.
- E. Dann and A. McLeod, Pest Manage. Sci., 2020 DOI:10.1002/ps.6156.
- Ch. Zhou, X. Luo, N. Chen, L. Zhang and J. Gao, J. Agric. Food Chem., 2020, 68, 3344 CrossRef CAS PubMed.
- R. H. Hall, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 258 CrossRef CAS.
- M. M. Abdou, Tetrahedron, 2020, 76, 131251 CrossRef CAS.
- J. B. Rodriguez and C. Gallo-Rodriguez, ChemMedChem, 2019, 14, 190 CAS.
- G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed.
- Ch. M. Sevrain, M. Berchel, H. Couthon and P.-A. Jaffres, Beilstein J. Org. Chem., 2017, 13, 2186 CrossRef CAS PubMed.
- S. Demkowicz, J. Rachon, M. Dasko and W. Kozak, RSC Adv., 2016, 6, 7101 RSC.
- Ch. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981 CrossRef CAS PubMed.
- B. L. Robbins, R. V. Srinivas, C. Kim, N. Bischofberger and A. Fridland, Antimicrob. Agents Chemother., 1998, 42, 612 CrossRef CAS PubMed.
- T. S. Kumar, S.-Y. Zhou, B. V. Joshi, R. Balasubramanian, T. Yang, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2010, 53, 2562 CrossRef CAS PubMed.
- K. Grif, M. P. Dierich, K. Pfaller, P. A. Miglioli and F. Allerberger, J. Antimicrob. Chemother., 2001, 48, 209 CrossRef CAS PubMed.
- K.-S. Ju, J. Gao, J. R. Doroghazi, K.-K. A. Wang, C. J. Thibodeaux, S. Li, E. Metzger, J. Fudala, J. Su, J. K. Zhang, J. Lee, J. P. Cioni, B. S. Evans, R. Hirota, D. P. Labeda, W. A. van der Donk and W. W. Metcalf, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12175 CrossRef CAS PubMed.
- K.-K. A. Wang, T. L. Ng, P. Wang, Z. Huang, E. P. Balskus and W. A. van der Donk, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- D. V. Patel, K. Rielly-Gauvin, D. E. Ryono, C. A. Free, W. L. Rogers, S. A. Smith, J. M. DeForrest, R. S. Oehl and E. W. Petrillo, J. Med. Chem., 1995, 38, 4557 CrossRef CAS PubMed.
- P. P. Giannousis and P. A. Bartlett, J. Med. Chem., 1987, 30, 1603 CrossRef CAS PubMed.
- E. De Clercq, Med. Res. Rev., 2011, 31, 118 CrossRef CAS PubMed.
- E. C. L. Marrs, L. Varadi, A. F. Bedernjak, K. M. Day, M. Gray, A. L. Jones, S. P. Cummings, R. J. Anderson and J. D. Perry, Molecules, 2020, 25, 1445 CrossRef CAS PubMed.
- L. Chen, X.-Y. Liu and Y.-X. Zou, Adv. Synth. Catal., 2020, 362, 1724 CrossRef CAS.
- A. Maestro, E. Martinez de Marigorta, F. Palacios and J. Vicario, Asian J. Org. Chem., 2020, 9, 538 CrossRef CAS.
- L. Chen, Synthesis, 2018, 50, 440 CrossRef CAS.
- M. Ordonez, J. L. Viveros-Ceballos, C. Cativiela and F. J. Sayago, Tetrahedron, 2015, 71, 1745 CrossRef CAS.
- G. Diaz-Muñoz, I. L. Miranda, S. K. Sartori, D. C. de Rezende and M. Alves Nogueira Diaz, Chirality, 2019, 31, 776 CrossRef PubMed.
- O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 1998, 9, 1279 CrossRef CAS.
- J. Pedroni and N. Cramer, Chem. Commun., 2015, 51, 17647 RSC.
- H. W. Lam, Synthesis, 2011, 2011 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2008, 64, 10279 CrossRef CAS.
- D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92 CrossRef CAS.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- D. Seebach and A. K. Beck, Helv. Chim. Acta, 1987, 70, 954 CrossRef CAS.
- Y.-X. Wang, S.-L. Qi, Y.-X. Luan, X.-W. Han, S. Wang, H. Chen and M. Ye, J. Am. Chem. Soc., 2018, 140, 5360 CrossRef CAS PubMed.
- D. Dailler, R. Rocaboy and O. Baudoin, Angew. Chem., Int. Ed., 2017, 56, 7218 CrossRef CAS.
- D. Enders, L. Tedeschi and J. W. Bats, Angew. Chem., Int. Ed., 2000, 39, 4605 CrossRef CAS.
- J. G. Noltes, Recl. Trav. Chim. Pays-Bas, 1965, 84, 782 CrossRef CAS.
- J. Boersma and J. G. Noltes, Recl. Trav. Chim. Pays-Bas, 1973, 92, 229 CrossRef CAS.
- D. Enders, L. Tedeschi and D. Forster, Synthesis, 2006, 1447 CrossRef CAS.
- L. Tedeschi and D. Enders, Org. Lett., 2001, 3, 3515 CrossRef CAS PubMed.
- F. Palacios, T. K. Olszewski and J. Vicario, Org. Biomol. Chem., 2010, 8, 4255 RSC.
- T. K. Olszewski and M. Majewski, Tetrahedron: Asymmetry, 2015, 26, 846 CrossRef CAS.
- J. Vicario, P. Ortiz and F. Palacios, Eur. J. Org. Chem., 2013, 7095 CrossRef CAS.
- J. Iwanejko, A. Brol, B. Szyja, M. Daszkiewicz, E. Wojaczynska and T. K. Olszewski, Tetrahedron, 2019, 75, 1431 CrossRef CAS.
- T. K. Olszewski, Tetrahedron: Asymmetry, 2015, 26, 393 CrossRef CAS.
- T. K. Olszewski, E. Wojaczynska, R. Wieczorek and J. Bakowicz, Tetrahedron: Asymmetry, 2015, 26, 601 CrossRef CAS.
- S. Yu and L. Pu, Tetrahedron, 2015, 71, 745 CrossRef CAS.
- J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS.
- J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed.
- L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS PubMed.
- N. Greene and T. P. Kee, Synth. Commun., 1993, 23, 1651 CrossRef CAS.
- X. Linghu, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070 CrossRef.
- Y.-X. Wang, S.-L. Qi, Y.-X. Luan, X.-W. Han, S. Wang, H. Chen and M. Ye, J. Am. Chem. Soc., 2018, 140, 5360 CrossRef CAS PubMed.
- M. T. Reetz, T. Sell and R. Goddard, Chimia, 2003, 57, 290 CrossRef CAS.
- I. Schlemminger, A. Lutzen, A. Willecke, W. Maison, R. Koch, W. Saak and J. Martens, Tetrahedron Lett., 2000, 41, 7285 CrossRef CAS.
- I. Schlemminger, A. Willecke, W. Maison, R. Koch, A. Lutzen and J. Martens, J. Chem. Soc., Perkin Trans. 1, 2001, 2804 RSC.
- S. Othman and M. Kozurkova, Curr. Med. Chem., 2018, 25, 1968 CrossRef CAS PubMed.
- G. Turan-Zitouni, L. Yurttas, A. Tabbi, G. A. Ciftci, H. E. Temel and Z. A. Kaplancikli, Molecules, 2018, 23, 135 CrossRef PubMed.
- A. Ahmad, A. Ahmad, R. Sudhakar, H. Varshney, N. Subbarao, S. Ansari, A. Rauf and A. U. Khan, J. Biomol. Struct. Dyn., 2017, 35, 3412 CrossRef CAS.
- D. Havrylyuk, O. Roman and R. Lesyk, Eur. J. Med. Chem., 2016, 113, 145 CrossRef CAS.
- L. Wu, X. Zhang, Q.-Q. Chen and A.-K. Zhou, Org. Biomol. Chem., 2012, 10, 7859 RSC.
- S. Mahesh and R. V. Anand, Eur. J. Org. Chem., 2017, 2698 CrossRef CAS.
- T. Murai, D. Matsuoka and K. Morishita, J. Am. Chem. Soc., 2006, 128, 4584 CrossRef CAS.
- T. Murai, Y. Maekawa, M. Monzaki, T. Ando and T. Maruyama, Chem. Commun., 2013, 49, 9675 RSC.
- T. Murai, Y. Maekawa, Y. Hirai, K. Kuwabara and M. Minoura, RSC Adv., 2016, 6, 15180 RSC.
- Y. Maekawa, T. Maruyama and T. Murai, Org. Lett., 2016, 18, 5264 CrossRef CAS PubMed.
- K. Kuwabara, Y. Maekawa, M. Minoura, T. Maruyama and T. Murai, J. Org. Chem., 2020, 85, 14446–14455 CrossRef CAS PubMed.
- K. Kuwabara, Y. Maekawa and T. Murai, Tetrahedron, 2020, 76, 131152 CrossRef CAS.
- S. Lemouzy, L. Giordano, D. Hérault and Ge. Buono, Eur. J. Org. Chem., 2020, 3351 CrossRef CAS.
- T. Chen and L.-B. Han, Synlett, 2015, 26, 1153 CrossRef CAS.
- P. Bałczewski, A. Szadowiak, A. Bodzioch, T. Białas, W. M. Wieczorek and M. Szyrej, J. Organomet. Chem., 2007, 692, 997 CrossRef.
- T. L. Emmick and R. L. Letsinger, J. Am. Chem. Soc., 1968, 90, 3459 CrossRef CAS.
- W. B. Farnham, R. K. Murray Jr. and K. Mislow, J. Am. Chem. Soc., 1970, 92, 5809 CrossRef CAS.
- O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS PubMed.
- O. Berger and J.-L. Montchamp, Org. Biomol. Chem., 2016, 14, 7552 RSC.
- J.-P. Wang, S.-Z. Nie, Z.-Y. Zhou, J.-J. Ye, J.-H. Wen and Ch.-Q. Zhao, J. Org. Chem., 2016, 81, 7644 CrossRef CAS PubMed.
- H. Zhang, Y.-M. Sun, Y. Zhao, Z.-Y. Zhou, J.-P. Wang, N. Xin, S.-Z. Nie, Ch.-Q. Zhao and L.-B. Han, Org. Lett., 2015, 17, 142 CrossRef CAS PubMed.
- J.-P. Wang, S.-Z. Nie, Z.-Y. Zhou, J.-J. Ye, J.-H. Wen and Ch.-Q. Zhao, J. Org. Chem., 2016, 81, 7644 CrossRef CAS PubMed.
- B.-X. Yan, H.-X. Zheng, J.-J. Ye, M.-R. Qiu, Y. Zhang, Ch.-Q. Zhao and Q. Li, Asian J. Org. Chem., 2020, 9, 566 CrossRef CAS.
- P. Xie, L. Guo, L. Xu and T.-P. Loh, Chem. – Asian J., 2016, 11, 1353 CrossRef CAS PubMed.
- X. Wo, P. Xie, W. Fu, C. Gao, Y. Liu, Z. Sun and T.-P. Loh, Chem. Commun., 2018, 54, 11132 RSC.
- Y. V. Rassukana, Y. Y. Khomutnyk, A. D. Synytsya and P. P. Onys'ko, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 718 CrossRef CAS.
- Y. V. Rassukana, O. V. Stanko and P. P. Onys'ko, J. Fluorine Chem., 2019, 219, 123 CrossRef CAS.
- P. Łyżwa, Heteroat. Chem., 2014, 25, 15 CrossRef.
- S. Liu, Y. Li, Z. Yin, Q. Yu and Z. Miao, J. Org. Chem., 2017, 82, 2481 CrossRef CAS PubMed.
- M. Yang, H. Xu, Z.-Y. Zhou, H. Zhang, L.-J. Liu, Y.-M. Sun, S.-Z. Nie and C.-Q. Zhao, Tetrahedron: Asymmetry, 2016, 27, 815 CrossRef CAS.
- Z.-Y. Zhou, H. Zhang, L. Yao, J.-H. Wen, S.-Z. Nie and Ch.-Q. Zhao, Chirality, 2016, 28, 132 CrossRef CAS PubMed.
- T.-T. Ding, Ch.-Ch. Qin, D.-D. Gao, X.-J. Li, H. Yan, R.-M. Liu, Ch.-Q. Zhao and S.-Z. Nie, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 102 CrossRef CAS.
- O. I. Kolodiazhnyi, E. V. Grishkun, S. Sheiko, O. Demchuk, H. Thoennessen, P. G. Jones and R. Schmutzler, Tetrahedron: Asymmetry, 1998, 9, 1645 CrossRef CAS.
- A. O. Kolodyazhnaya, V. P. Kukhar and O. I. Kolodyazhnyi, Russ. J. Gen. Chem., 2008, 78, 2043 CrossRef CAS.
- A. O. Kolodiazhna, V. P. Kukhar, A. N. Chernega and O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2004, 15, 1961 CrossRef CAS.
- V. V. Nesterov and O. I. Kolodiazhnyi, Tetrahedron, 2007, 63, 6720 CrossRef CAS.
- I. Guliaiko, V. Nesterov, S. Sheiko, O. I. Kolodiazhnyi, M. Freytag, P. G. Jones and R. Schmutzler, Heteroat. Chem., 2008, 19, 133 CrossRef CAS.
- Q. Li, T. Chen, Q. Xu and L.-B. Han, Chem. – Eur. J., 2016, 22, 6213 CrossRef CAS PubMed.
- M.-R. Qiu, H.-X. Zheng, J.-J. Ye, B.-X. Yan, Ch.-Q. Zhao and Q. Li, Org. Biomol. Chem., 2020, 18, 3017 RSC.
- H. Zhang, Y.-M. Sun, L. Yao, S.-Y. Ji, Ch.-Q. Zhao and L.-B. Han, Chem. – Asian J., 2014, 9, 1329 CrossRef CAS PubMed.
- Y.-M. Sun, N. Xin, Z.-Y. Xu, L.-J. Liu, F.-J. Meng, H. Zhang, B.-C. Fu, Q.-J. Liang, H.-X. Zheng, L.-J. Sun, Ch.-Q. Zhao and L.-B. Han, Org. Biomol. Chem., 2014, 12, 9457 RSC.
Footnote |
| † Dedicated to Prof. Claude Grison on the occasion of her birthday. |
| This journal is © The Royal Society of Chemistry 2021 |