DOI:
10.1039/D1OB01535D
(Review Article)
Org. Biomol. Chem., 2021,
19, 9138-9147
Total synthesis of bioactive tetracyclic norditerpene dilactones
Received
5th August 2021
, Accepted 10th September 2021
First published on 10th September 2021
Abstract
Tetracyclic norditerpene dilactones are an important class of terpenoids that have been isolated from both terrestrial and marine sources, typically from Podocarpus plants and from filamentous fungi. This class of molecules shares a common 6/6/6/5 tetracyclic ring skeleton, which possesses a densely oxygenated carbon framework and contiguous stereocenters. What's more challenging for synthetic chemists are the consecutive sp2-hybridized carbon centers, which exacerbates the strain/rigidity of the whole molecule. In addition, many of these molecules display promising biological activities, such as antitumor, insecticidal, anti-feedant, allelopathic, and antibiotic activities. The unique structures and interesting biological profiles of norditerpene dilactones have attracted considerable attention from synthetic chemists. Herein we summarize the synthetic efforts with respect to tetranorditerpene dilactones.
1. Introduction
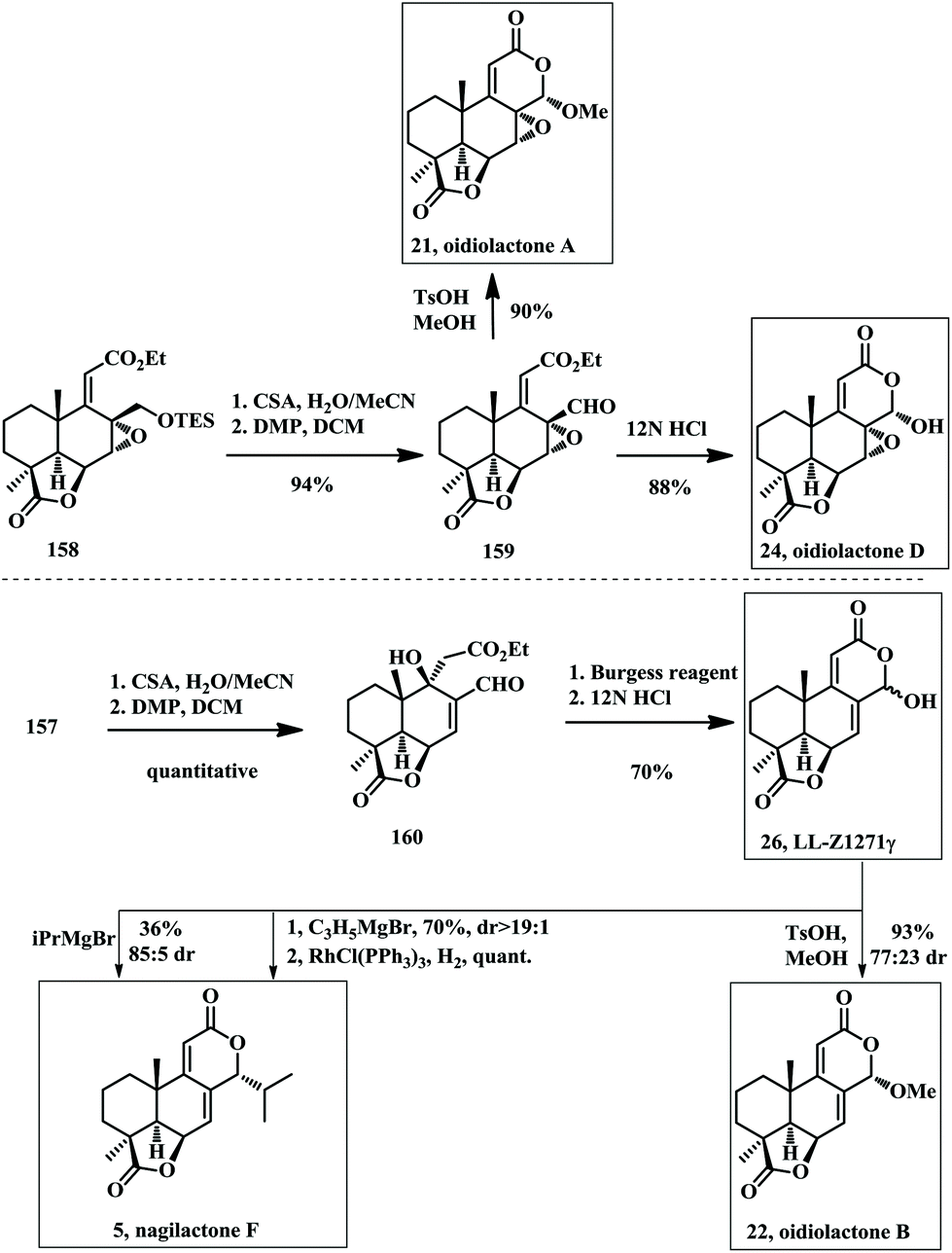
Terpenes are a very important class of natural products with dazzling structural diversity and huge numbers. In addition, they display a variety of biological activities, such as antitumor,1 antifungal,2 antifeedant,3 insecticidal4 and herbicidal5 properties and have complex structures and various oxidation states. Norditerpenoid dilactones belong to the diterpene family but with truncated carbon numbers even up to four carbons, which render them tetranorditerpenes.5c The norditerpenoids have been isolated from various sources including plants of the genera Podocarpus and Chamaecyparis as well as from fungal species derived from plants, peanut molds, and soil samples. Among these, most nor- and bisnorditerpene dilactones are usually discovered from plants while tetranorditerpene dilactones are generally isolated from fungus-derived secondary metabolites. With respect to this antifungal activity, it is worth emphasizing that a few of these compounds, e.g. oidiolactone A (21), oidiolactone B (22), and oidiolactone D (24), have shown promising inhibitory effects against Candida albicans (IC50 = 50, 26 and >220 nM) and the dimorphic fungus Histoplasma capsulatum, which causes one of the most severe mycoses.6 In 2001, it was suggested that oidiolactone B (22), a fungal metabolite from Oidiodendron griseum, might be used as an anti-inflammatory drug lead in the treatment of IL-1 (interleukin-1) and TNF (tumor necrosis factor)-mediated diseases.7 Wentilactone A (27) was originally isolated from Aspergillus wentii of mouldy peanuts in 1980 by J. W. Dorner and co-workers8 and a marine-derived endophytic fungus, Aspergillus wentii EN-48, was isolated in 2012 by Wang and co-workers.9 This tetranorditerpene dilactone exerted a significant inhibitory effect on the lung carcinoma cell lines NCI-H460 and NCI-H446 without markedly inhibiting the proliferation of the normal HUVEC cells.10 While wentilactone B (28) was isolated along with wentilactone A by Wang's group, it was shown to trigger apoptosis and inhibit metastasis in HCC cell lines.11 We have summarized all the reported natural products that belong to this norditerpene family (Fig. 1). Structurally speaking, this family can be categorized into nor- and bisnorditerpene dilactones and tetranorditerpene dilactones based on their carbon numbers.
 |
| | Fig. 1 Representative norditerpene dilactone natural products. | |
Both categories of these norditerpene dilactones share a 6/6/6/5 polyfused ABCD-ring system with two lactones. This family of natural products contain at least four contiguous stereogenic centers, two of which are all-carbon quaternary stereocenters. Within the BC ring junction, there are up to five contiguous sp2 hybridized carbon centers, which greatly restrain the conformational versatility and may also be the structural foundation for their remarkable biological activity. This family of natural products is more synthetically challenging because they all contain the characteristic γ- and δ-lactone rings, which make these molecules sensitive to both basic and acidic conditions. While sharing many similarities, they do differ from one another, mainly in the oxidation states of the A ring and C ring. Up to now, more than 70 norditerpenoid dilactones have been isolated, and there are many reports on the activity of these compounds. However, there are no comprehensive reviews summarizing the total synthetic studies towards these compounds. We herein summarize all the synthetic studies on them in a chronological way and focus on their strategies in order to provide a clear scenario of how these natural products are assembled.
2. Discussion
In this section, we will focus on the details of chemical synthetic studies of tetranorditerpene dilactones. To the best of our knowledge, there has been no review concentrating on the total synthesis of this class of natural products, despite the fact that many total syntheses have been reported in recent years. They have already become indispensable components in the vast terpenoid natural products. Their unique biological profile and pharmaceutical potential have made efficient preparation of these natural products highly desirable and urgent. A summary of retrosynthetic analysis from different groups is displayed in Fig. 2, featuring an annulation sequence. We will be summarizing all the examples in a chronological way in order to provide a comprehensive summary on this topic from an objective perspective as best as we can.
 |
| | Fig. 2 Summary of retrosynthetic analysis. | |
2.1 Semi-synthesis of oidiolactone B (aka. LL-Z12771α) by Adinolfi and coworkers in 1972
LL-Z1271α (22), which is also known as oidiolactone B, was isolated from the fermentation products of the Acrostalagmnu species by Ellestad and co-workers in 1969.12 An interesting observation was that a small amount of LL-Z1271γ (26) was isolated alongside. The following medicinal research revealed that LL-Z1271α (22) displayed not only anti-bacterial activity both in vitro and in vivo, but also selective inhibitory activity on IL-1β (IC = 0.049 nm).7
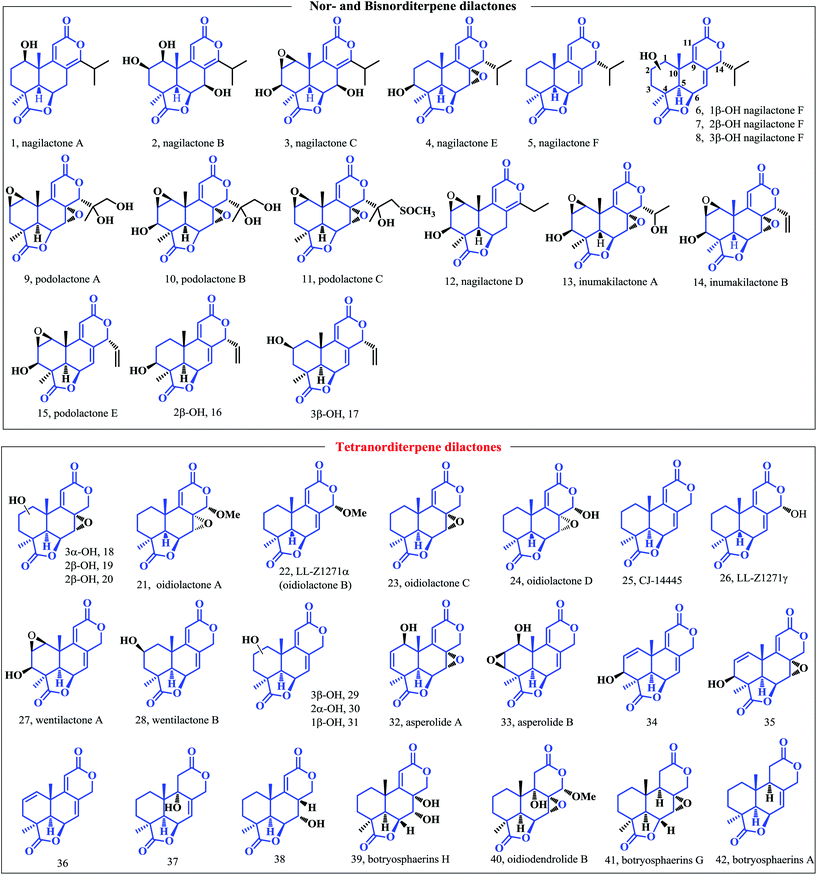
In 1972, a semi-synthesis of oidiolactone (LL-Z1271α, 22) was accomplished by M. Adinolfi and co-workers (Scheme 1) starting from the (+)-ketolactone IV,13 which according to the authors was “easily available by degradation of marrubin”.14 Bromination of 43 in glacial acetic acid gave the bromoketone 44, which underwent elimination by treatment with DBU smoothly affording the unsaturated ketone intermediate 45. When 45 was exposed to lithium ethoxy-acetylide in THF, the 1,2-addition product of ethynyl carbinol 46 was obtained in 90% yield. The conjugated ester of 47 was obtained by the treatment of 46 under the conditions of catalytic aqueous H2SO4 and ethanol. The reaction proceeded through hydrolysis of ethyl acetylide forming intermediate 46-II, followed by elimination yielding compound 47. The SeO2 mediated Riley oxidation and subsequent acetal formation/lactonization cascade afforded the desired product lactol 48 and the less oxidized by-product C14-deoxyl lactone in 53% and 18% yield, respectively. In this synthesis, the author used the degradation of the natural product marrubin as the starting material, which is astonishingly expensive (>$60 per mg from Sigma Aldrich). The desired natural product LL-Z1271α (22) can be obtained by less than 10 steps of chemical transformations from marrubin. However, the synthesis efficiency to obtain oidiolactone B is low because esterification and methylation of C14 resulted in more by-products, which has left more room for improvement in future research.
 |
| | Scheme 1 Semi-synthesis of oidiolactone B (LL-Z1271α) by Adinolfi and coworkers. | |
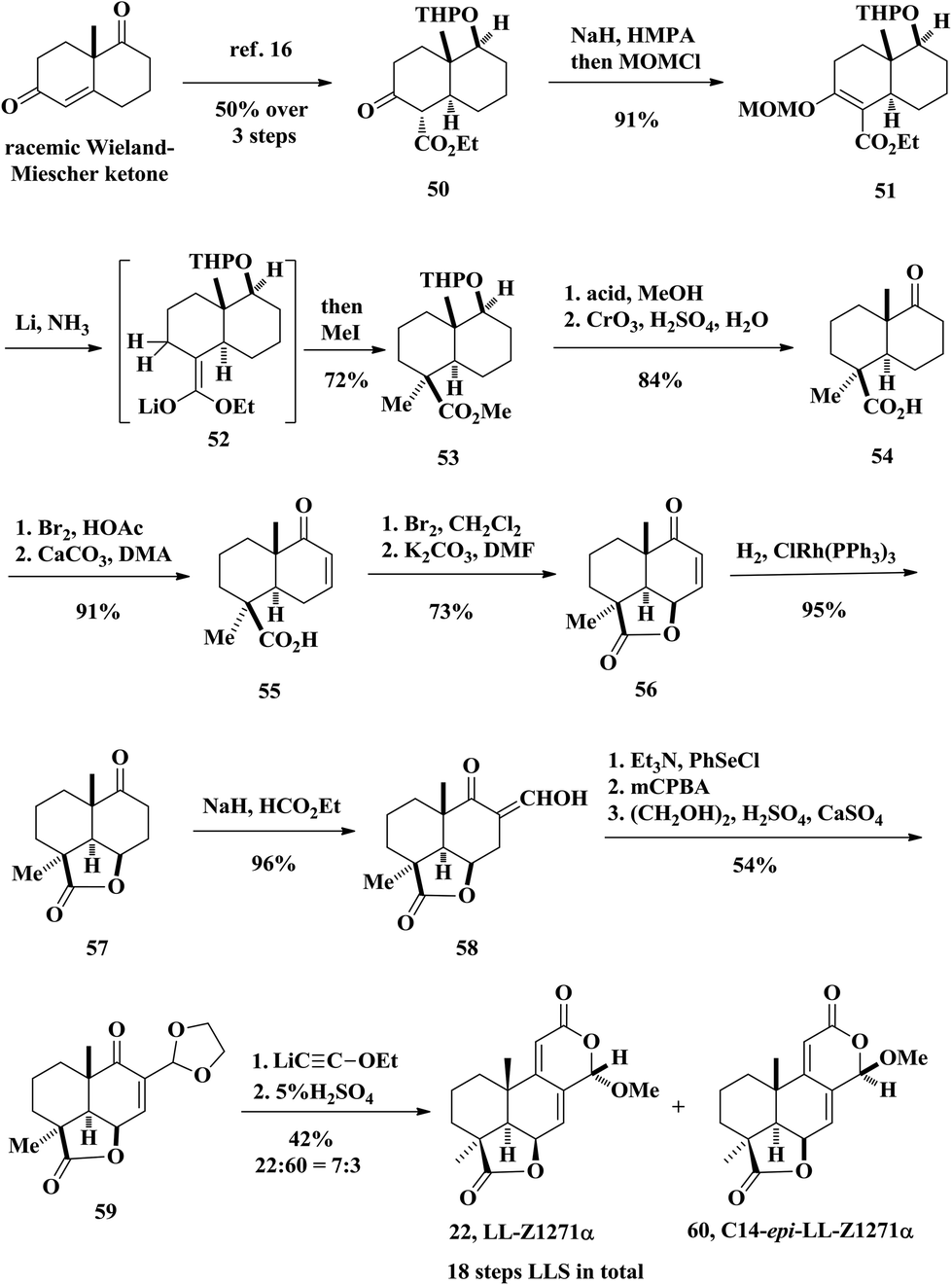
2.2 Total synthesis of oidiolactone B by Welch in 1976
In 1976, Welch and co-workers reported the first stereoselective total synthesis of (±)-LL-Z1271α (rac-22) from the well-known racemic Wieland–Miescher ketone (Scheme 2).15 The intermediate 50 was obtained through a literature procedure in three steps with an overall 50% yield.16 Ketone αC–H bonds were deprotonated under NaH and the resulting enolate was trapped by MOMCl, followed by the reductive-elimination reaction developed by Coates and Shaw, affording 53.17 This reaction sequence is unique because it proceeded through deoxygenation and concomitant stereoselective methylation in a single step. The authors did try introducing a methyl group based on β-keto ester 50, but obtained only the undesired diastereoisomer of the methylation product. The removal of the tetrahydropyranyl protecting group from 53 was realized by acid-catalyzed hydrolysis followed by oxidation of the secondary alcohol using Jones’ reagent to afford the keto ester 54. Bromination of 54 with bromine and then dehydrohalogenation of the crude bromoketone gave rise to enone ester 55. Then the γ-lactone 56 was formed by allylic bromination of enone acid 55 followed by SN2′ displacement of the resulting bromide with the carboxylic acid anion at close proximity. Hydrogenation of the conjugated olefin with Wilkinson's catalyst gave the saturated ketone 57. Lactone 57 when allowed to react with sodium hydride in the presence of ethyl formate produced hydroxymethylene ketone 58. Grieco elimination was used to convert hydroxymethylene ketone 58 to an α,β-unsaturated keto aldehyde followed by protection of the aldehyde to produce 59. Finally, the synthesis was completed by the aforementioned Meyer–Schuster rearrangement of enone acetal 59. As demonstrated by the previous work, the desired natural product LL-Z1127α was obtained in 29% yield and the C14-epimer 60 was obtained in 13% yield. The total synthesis of oidiolactone B (LL-Z1127α, 22) was achieved in 18 linear steps.
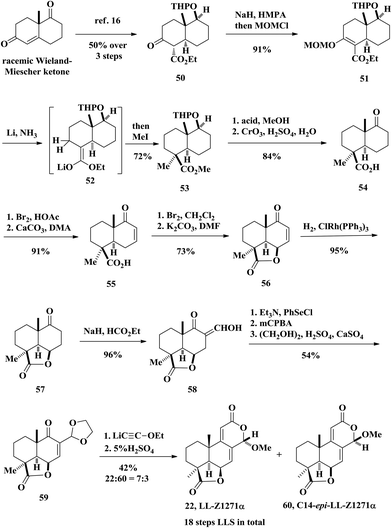 |
| | Scheme 2 Total synthesis of oidiolactone B (LL-Z1271α) by Welch. | |
2.3 Semi-synthesis of nagilactone F by Hayashi
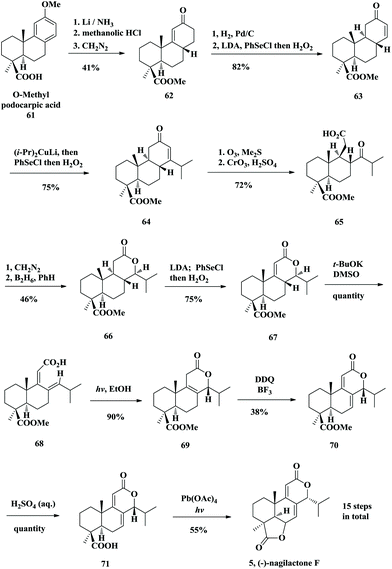
Nagilactone F (5) is a member of the class of biologically active norditerpenoid dilactones, which was isolated from Podocarpus plants. An asymmetric semi-synthesis of nagilactone F was reported by Hayashi and co-coworkers in 1982 (Scheme 3).18 Their synthesis commenced with a known natural product named (+)-podocarpic acid (61) and took 15 synthetic steps with 1.2% overall yield to obtain (−)-nagilactone F (5). The key step involved a photochemical cyclization of a dienoic acid, in which the desired stereochemistry at C14 was furnished selectively.
 |
| | Scheme 3 Semi-synthesis of nagilactone F by Hayashi in 1982. | |
Firstly, Birch reduction followed by acid treatment of the naturally derived O-methyl podocarpic acid produced 62. Conversion of 62 to the desired Δ13,14-enone 63 by hydrogenation and following Grieco elimination was achieved with an overall 82% yield. The obtaining enone 63 was treated with a lithium diisopropylcuprate-dimethyl sulfide complex. The desired 1,4-conjugate addition successfully introduced the isopropyl group and the resulting enolate intermediate was directly trapped by phenylselenyl chloride. The 14-isopropyl-13-selenylated ketone was obtained, which was converted to the enone 64 by Grieco elimination. Ozonolysis of unsaturated ketone 64 and subsequent Jones oxidation led to carboxylic acid 65. The tricyclic lactone 66 was formed through esterification and then an intramolecular lactonization took place upon reduction of the ketone group with borane. Unfortunately, the more stable equatorial 14β-isopropyl group resulted in compound 66, primarily due to the more stable chair-conformation of the C ring. However, the stereochemistry of the C14-isopropyl group was different from that of the natural nagilactone F. Therefore, t-BuOK mediated hydrolysis of the lactone afforded a dienoic acid intermediate, which was subjected to radiation in ethanol with a medium-pressure mercury lamp at 0 °C. A 6π conrotatory pericyclic reaction took place under a UV lamp and resulted in the exclusive formation of the tricyclic lactone 69 as a single diastereoisomer. The exclusive formation of the desired isomer 69 in the photoreaction could be rationalized by the favorable rotation of the C8–C14 bond during the lactonization to such a direction that the large isopropyl group avoids the nonbonded interaction with the angular methyl group at C10, in a concerted 6π-electrocyclic reaction involving the carbonyl system, although a radical cyclization of a photochemically produced biradical at C14 and the carbonyl oxygen cannot be ruled out. The dienolide 70 was obtained through oxidation by DDQ. Upon hydrolysis under acidic conditions, compound 71 was obtained in almost quantitative yield. Nagilactone F was successfully obtained by oxidation with lead tetraacetate in benzene under UV irradiation. The reaction was thought to proceed through allylic oxidation by Pb(OAc)4 and the resulting allyl cation was trapped by the carboxylic acid group to form the D-ring lactone moiety.
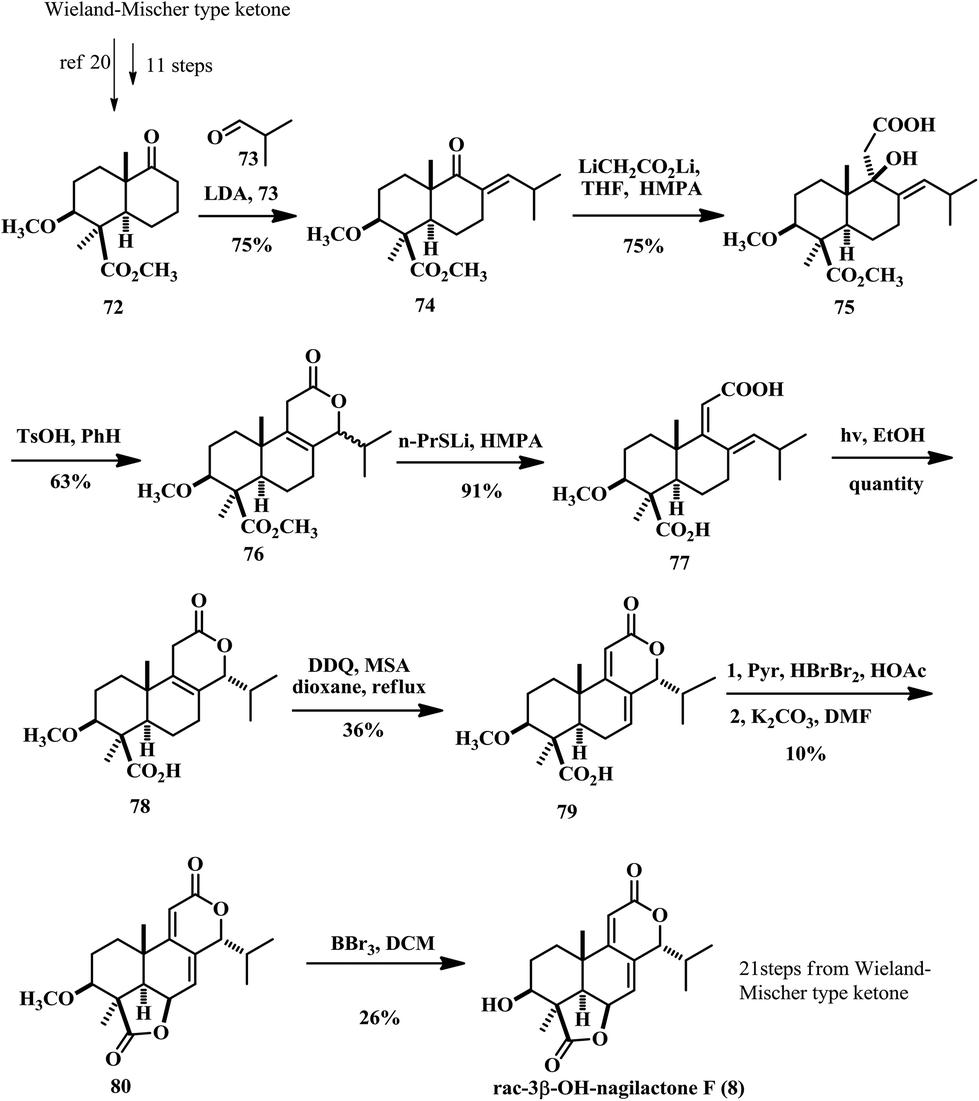
2.4 Total synthesis of 3β-OH nagilactone F by Groot
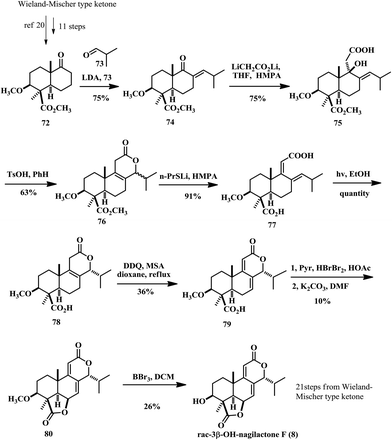
In 1986, Groot and co-worker accomplished the total synthesis of A ring functionalized tetranorditerpene dilactone, 3β-OH nagilactone F (8) (Scheme 4).19 They used the 3β-methoxy keto ester 72 as the starting compound for the synthesis, which itself took about 11 steps to prepare from the Wieland–Miescher type ketone.2072 was subjected to aldol condensation with isobutyraldehyde 73 and subsequent dehydration led to the formation of enone 74. 1,2-Addition of dilithioacetate afforded tertiary alcohol 75 in 75% yield. The cyclization of the 1,2-adduct 75 led to the expected δ-lactones 76 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :l mixture of the α- and β-C14 epimers. To obtain the single diastereoisomer of α-C14 lactone 78, they treated a mixture of the epimers with lithium n-propyl mercaptide in HMPA to afford the diene dicarboxylic acid 77. This is a similar intermediate to that reported by Hayashi in Scheme 3. They employed the same UV irradiation method to achieve stereoselective ring closure to the tricyclic acid 78 in quantitative yield. Then oxidation of 78 was also inspired by Hayashi's work, with DDQ resulting in the formation of dienolides 79 in only 36% yield. The authors mentioned that they used lead tetraacetate to forge the γ-lactone ring but failed. So they had to take a detour by selective bromination of the tri-substituted olefin due to its electronic properties, followed by an E2-type elimination promoted by K2CO3. The resulting allylic bromide underwent a SN2′ reaction with the carboxylic acid group to close the γ-lactone of 79. The overall efficiency of this cascade reaction is very low (10% over two steps). Finally, demethylation of 80 was achieved with boron tribromide and the racemic (±)-3β-hydroxynagilactone F (8) was obtained in 26% yield, indicating poor chemoselectivity during the deprotection reaction and instability of this compound under acidic conditions.
:l mixture of the α- and β-C14 epimers. To obtain the single diastereoisomer of α-C14 lactone 78, they treated a mixture of the epimers with lithium n-propyl mercaptide in HMPA to afford the diene dicarboxylic acid 77. This is a similar intermediate to that reported by Hayashi in Scheme 3. They employed the same UV irradiation method to achieve stereoselective ring closure to the tricyclic acid 78 in quantitative yield. Then oxidation of 78 was also inspired by Hayashi's work, with DDQ resulting in the formation of dienolides 79 in only 36% yield. The authors mentioned that they used lead tetraacetate to forge the γ-lactone ring but failed. So they had to take a detour by selective bromination of the tri-substituted olefin due to its electronic properties, followed by an E2-type elimination promoted by K2CO3. The resulting allylic bromide underwent a SN2′ reaction with the carboxylic acid group to close the γ-lactone of 79. The overall efficiency of this cascade reaction is very low (10% over two steps). Finally, demethylation of 80 was achieved with boron tribromide and the racemic (±)-3β-hydroxynagilactone F (8) was obtained in 26% yield, indicating poor chemoselectivity during the deprotection reaction and instability of this compound under acidic conditions.
 |
| | Scheme 4 Total synthesis of 3β-OH-nagilactone F by Groot in 1986. | |
2.5 Total synthesis of (−)-nagilactone F by Burke
In 1983, Burke and co-workers reported their strategy constructing a tricyclic model system of the nagilactones through an intramolecular Diels–Alder (IMDA) reaction with different kinds of the trimethylsilyl-substituted butadiene units and various electronically tuned dienophilic olefins (Scheme 5).21 They found that increasing the steric demand in the transition state was disadvantageous, for example with 82a and 82b, 83a and 83b, which either required higher temperature as in the case of 82a (less than 30% yield), 82b (52% yield) and 83a (less than 20% yield), or failed to react as in the case of 83b. Noticing the γ-lactone subunit present in nagilactone F, they designed and used acrylate substrates 84a and 84b. However, they proved to be rather sensitive precursors to thermolysis or Lewis acid catalysis because only elimination and/or polymerization were observed but no desired cycloaddition. Then they also failed to construct the tricyclic model system with the dimethylfulvene acrylate generator 85. Finally they prepared the fumarates 90 and 91, which increased the reactivity by introducing the cis-β-carbonyl ester. The desired IMDA reaction took place in good yield at 90 °C. However, the IMDA reaction proceeded via an exo transition state with respect to the lactone carbonyl, which afforded products 92 and 93 bearing a different diastereochemistry compared to the natural nagilactones.
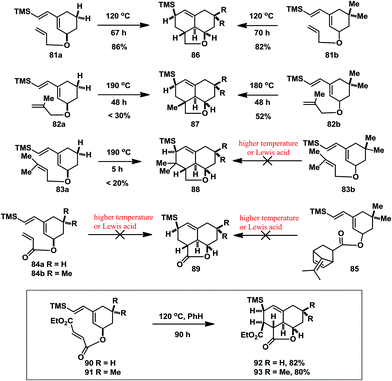 |
| | Scheme 5 Model studies towards the tricyclic core structures of nagilactones by Burke in 1983. | |
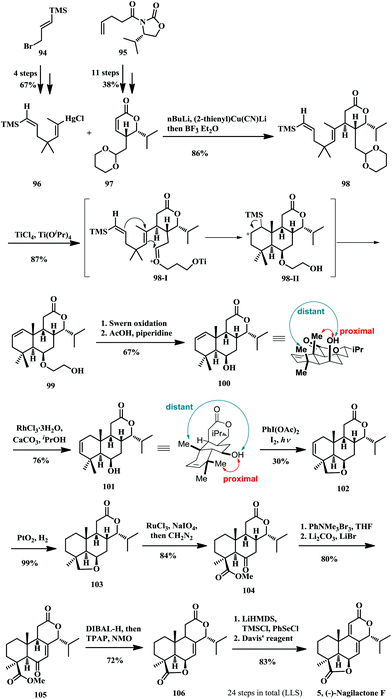
In 1994, Burke and co-workers successfully accomplished the asymmetric total synthesis of (−)-nagilactone F (Scheme 6) starting from the vinylmercurial derivative 9622 and lactone 97,23 which was prepared according to the literature procedures.24 The key step of this process was the cationic polyene cyclization promoted by mixed TiCl4 and Ti(OiPr)4 as Lewis acids from the compound 98, which was obtained via conjugate addition of the mixed higher order cuprate 96 to chiral pentenolide 97. The cyclization was postulated to proceed under Prins-like cyclization through the chair transition state and led to excellent diastereoselectivity. The desired product 99 was obtained with a good yield as a single diastereoisomer, whose structure was confirmed by X-ray crystallographic analysis. The ethoxyl group was removed through Swern oxidation and subsequent oxy retro-Michael addition provided the secondary alcohol 100. Then they aimed to construct the lactonic D-ring of nagilactones through a regioselective intramolecular remote functionalization involving a C6 alkoxy radical, but they found that it was easier for the C10 methyl group in 100 to form a carbon radical for intramolecular hydrogen atom-transfer. What's more, they measured the distance between the hydroxyl group and the C10 methyl group and found that it was smaller than that to the C4 methyl group by the computational method. From the 3D drawing of compound 100, it is clear that the angular C4 methyl group is in close proximity to the hydroxyl group compared to the C10 methyl group. A conformation adjustment was achieved by the hydrated RhCl3 mediated olefin isomerization. The migrated olefin rendered compound 101 adopting a new half-chair/chair conformation, which put the angular methyl group and hydroxyl group both at the equatorial position. This moved the C4 angular methyl group far away from the hydroxyl group while leaving the C4 methyl group in proximity to the OH group. When compound 101 was subjected to the photochemical conditions developed by Suárez and coworkers,25 the desired tetrahydrofuran product 102 and its by-product were formed in a ratio of 1:1.4. Although compound 102 could only be obtained in 30% yield, the conformation controlled hypoiodite mediated C–H abstraction is very promising. The olefin in the A-ring was reduced by hydrogenation catalysed by PtO2 in quantitative yield to afford the tetracyclic compound 103, which was oxidized by RuCl3/NaIO4 mediated ether oxidation to yield the keto ester 104. Bromination and elimination provided enone 105 in 80% overall yield, which was diastereoselectively reduced to tetraol with excess DIBAL-H. Without purification, the mixture was treated with TPAP and NMO to re-construct the C- and D-ring lactone moieties to furnish product 106 in decent yield (72%). Finally, a Grieco elimination procedure was applied to generated the double bond in the C-ring to provide (−)-nagilactone F. The synthesis proceeded through 24 steps (LLS) and the first asymmetric total synthesis of (−)-nagilactone F (5) was accomplished.
 |
| | Scheme 6 Total synthesis of nagilactone F by Burke in 1994. | |
2.6 Total synthesis of LL-Z1271α, LL-Z1271γ, and nagilactone F by Barrero
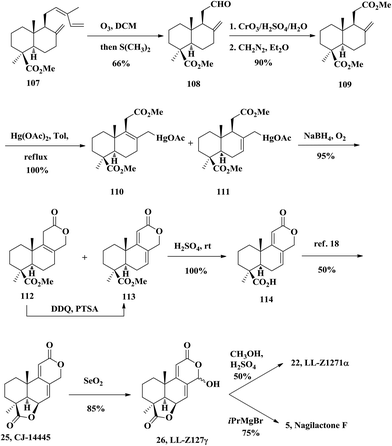
The Barrero group carried out a series of synthetic efforts towards this family of products from 1995 to 2002. In 1995, they accomplished the semi-total synthesis of LL-Z1271α (22), LL-Z1271γ (26), and nagilactone F (5) (Scheme 7), using the methyl ester of communic acid 107 as the starting material.26 The diester 109 was obtained through careful ozonolysis (∼60% conversion of 107) of the communic acid methyl ester 107. The resulting aldehyde 108 was oxidized with the Jones reagent and then esterified with diazomethane. The treatment of diester 109 with mercuric acetate led to hydroxy-mercuration to a pair of organomercurials 110 and 111. The mixture of organomercurials 110 and 111 were reduced with NaBH4 and the resulting borane compound underwent an oxidation in the presence of O2 giving lactone 112 and dienolide 113. The former can be swiftly converted to the desired dienolide 113 upon exposure to DDQ oxidation. The compound 113 was converted to free carboxylic acid 114via hydrolysis. Using Hayashi's conditions,18 the CJ-14445 (25) lactone ring was constructed successfully. The reaction of 25 with selenium dioxide gave LL-Z1271γ (26), which serves as a ready precursor to access other congeners. LL-Z1271γ (26) was treated with methanol/sulphuric acid giving as the main product LL-Z1271α (22) in 50% yield; they treated 26 with isopropylmagnesium bromide and obtained the natural product (−)-nagilactone F (5).
 |
| | Scheme 7 Total synthesis of LL-Z1271α, LL-Z1271γ and nagilactone F by Barrero in 1995. | |
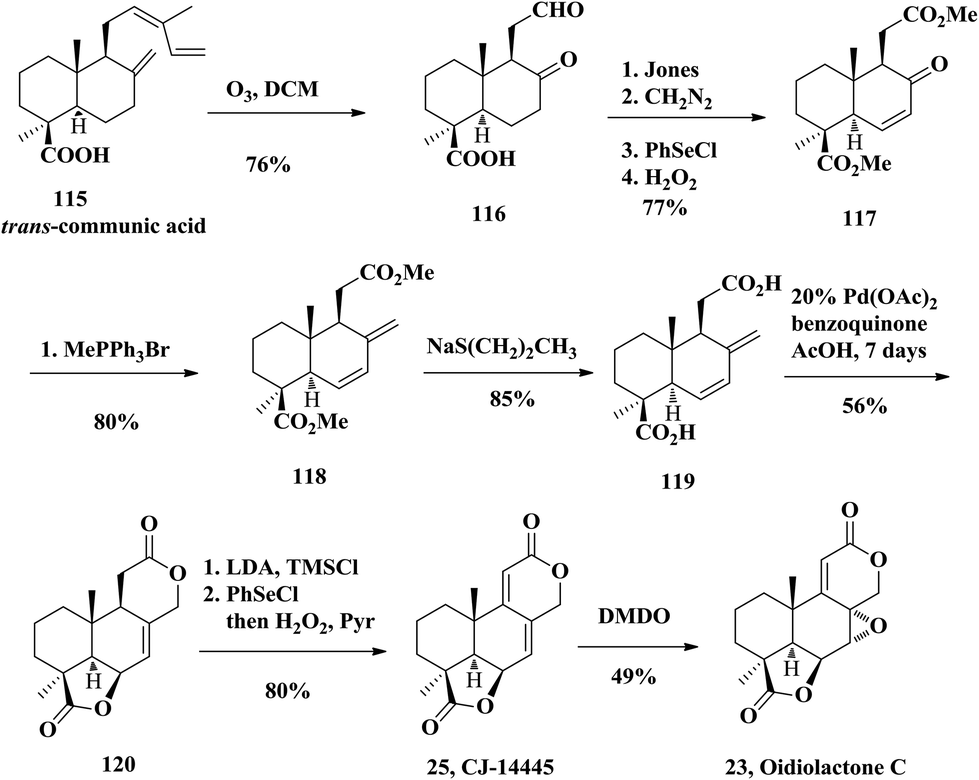
2.7 Total synthesis of oidiolactone C by Barrero
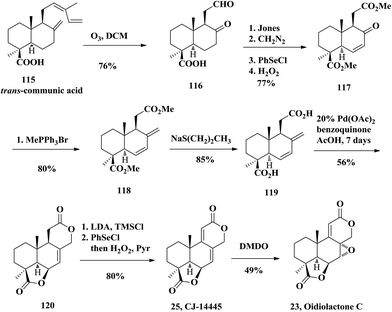
Oidiolactone C (23) was isolated from filamentous fungi, Oidiodendron truncatum, by Kawai and co-workers in 1999.28 They also investigated the antifungal activity of oidiolactone C, suggesting promising results against Candida albicans and other related species. In 2002, Barrero and co-workers firstly accomplished the synthesis of the antifungal oidiolactone C (Scheme 8) starting again from diterpenic trans-communic acid (115) with an overall yield of 11.7%.27 The key step in their synthetic process consists of a new bislactonization reaction catalyzed by Pd(II), which gives rise to the podolactone-type tetracyclic skeleton from a norlabdadienedioic acid 115. The communic acid was treated with ozone in DCM to obtain keto aldehyde 116. Oxidation of 116 with Jones reagent and esterification with diazomethane, followed by Grieco elimination, provided the conjugated ketone 117. They converted 117 to diacid 119 through Wittig reaction, followed by ester decomposition using sodium propanethiolate. Then the diacid 119 was treated with 20 mol% Pd(OAc)2 and p-benzoquinone to obtain the key intermediate dilactone 120. This reaction was proposed to undergo Wacker-type γ-lactone formation upon disubstituted olefin activation by Pd(OAc)2. The resulting allyl-Pd(II) intermediate could be trapped by the upper carboxyl group to form the δ-lactone. Towards this end, the bislactonization was accomplished. Finally, Grieco elimination furnished the natural product CJ-14445 (25) and subsequently epoxidation with DMDO provided oidiolactone C (23) in 49% yield diastereoselectively.
 |
| | Scheme 8 Total synthesis of oidiolactone C by Barreo in 2002. | |
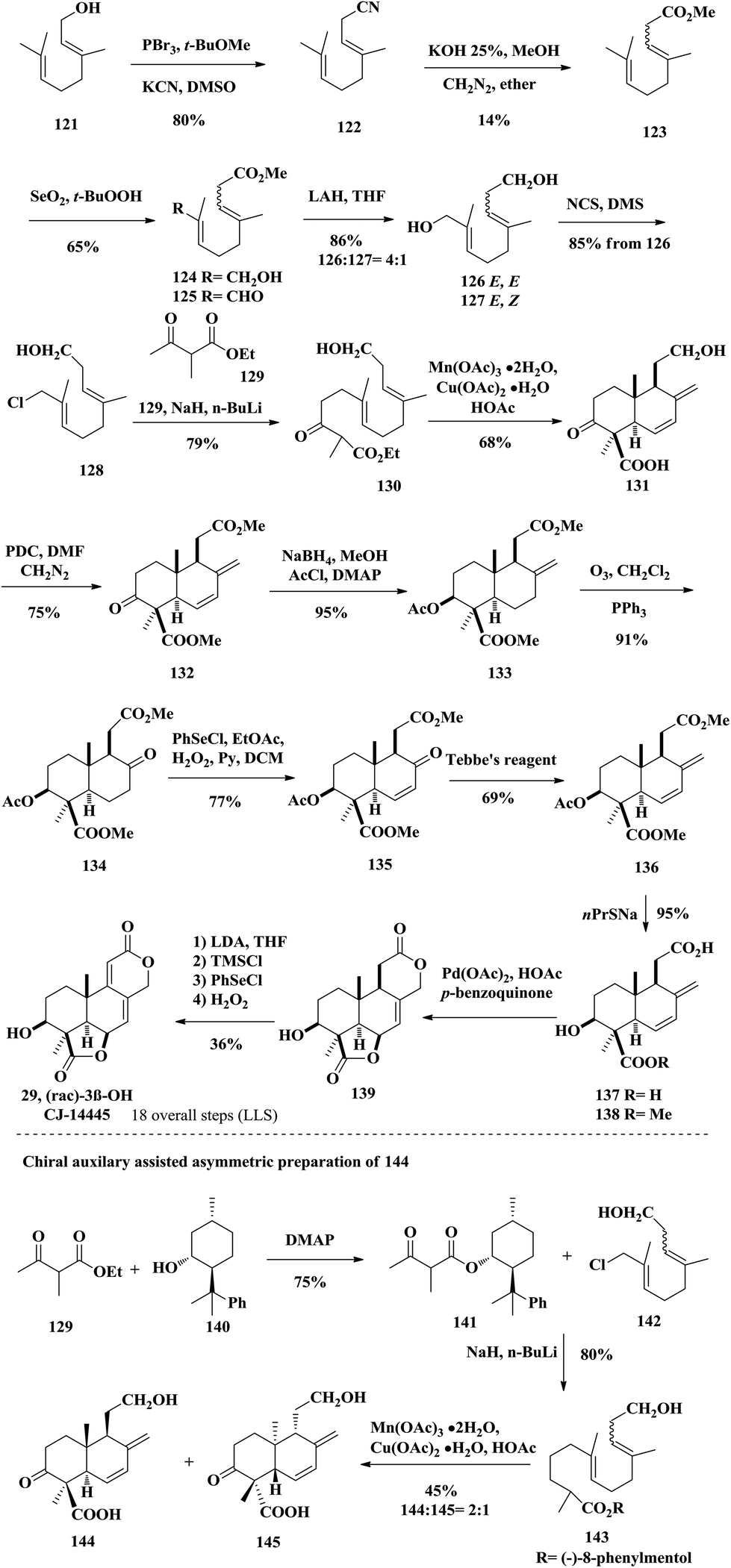
2.8 Synthetic studies towards wentilactone B by Barrero
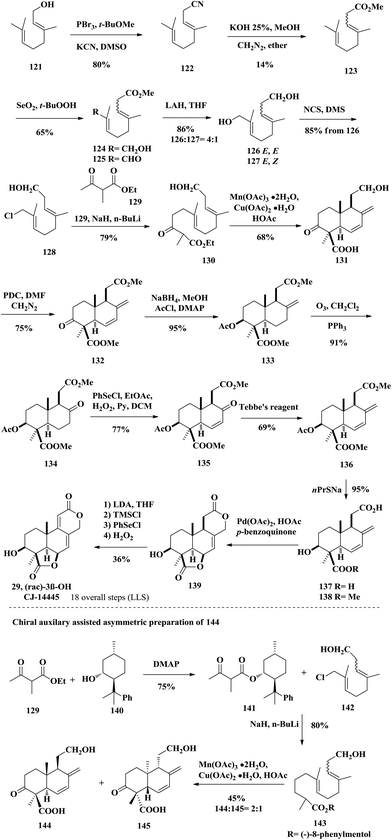
In the same year of 2002, the Barrero group reported the synthesis of A-ring functionalized 3β-CJ-14445 (29) and speculated the structure of wentilactone B (28) based on their synthesis (Scheme 9); however, they did not report their synthesized wentilactone B.29 The process started from commercially available geraniol 121, which was treated with PBr3 and KCN to produce 122. Hydrolysis of the cyano group and esterification with CH2N2 furnished 123 as a mixture of geometrical olefin isomers. Oxidation adjustment through Riley oxidation and LiAlH4 reduction of compound 125 provided diols 126 and 127 as E/Z isomers with the desired isomer 126 as the major product. Regioselective chlorination of the E,E isomer 126 with NCS afforded chloride 128, which was treated with β-ketoester to afford the radical cyclization precursor 130. Using Snider's conditions, they successfully constructed the 6/6 trans-fused AB-ring skeleton via a Mn(III)-mediated radical cyclization. PDC oxidized the primary alcohol into carboxylic acid followed by esterification with diazomethane giving diester 132. The ketone group was diastereoselectively reduced by NaBH4 due to its less hindered Re-face, and the resulting secondary alcohol was trapped by AcCl affording acetate 133. They planned to use the Pd-catalyzed bislactonization strategy to construct the CD-rings. Thus, three steps of conventional manipulation were performed, namely ozonolysis, Grieco elimination and Tebbe olefination, from compound 133. The diacid 137 was obtained through saponification of 136 with sodium propanethiolate. Then the key tetracyclic podolactone 139 was successfully obtained through the aforementioned bislactonization strategy. Finally, desaturation of 139 gave 3β-CJ-14445 (29) by Grieco elimination. This was the proposed structure of wentilactone B (28) in a previous report; however, the natural sample's NMR spectra did not match those of the synthetic sample. The authors speculated that the OH group should be at C2 instead of the C3 position. In this synthesis, the authors also demonstrated a substrate-controlled asymmetric radical cyclization strategy to obtain the chiral compound 144. They installed (−)-8-phenylmenthol as a chiral auxiliary to induce the diastereoselectivity of the cyclization step. The acyclic precursor with a chiral auxiliary was treated with Mn(OAc)3 and Cu(OAc)2 to afford the cyclization products 144 and 145 with a ratio of 2:1. The synthesis took them about 20 steps to achieve 3β-CJ-14445 (29).
 |
| | Scheme 9 Synthetic efforts to A-ring functionalized congeners of tetranorditerpene dilactone by Barreo in 2002. | |
2.9 Collective asymmetric total synthesis of tetranorditerpene dilactones by Hanessian
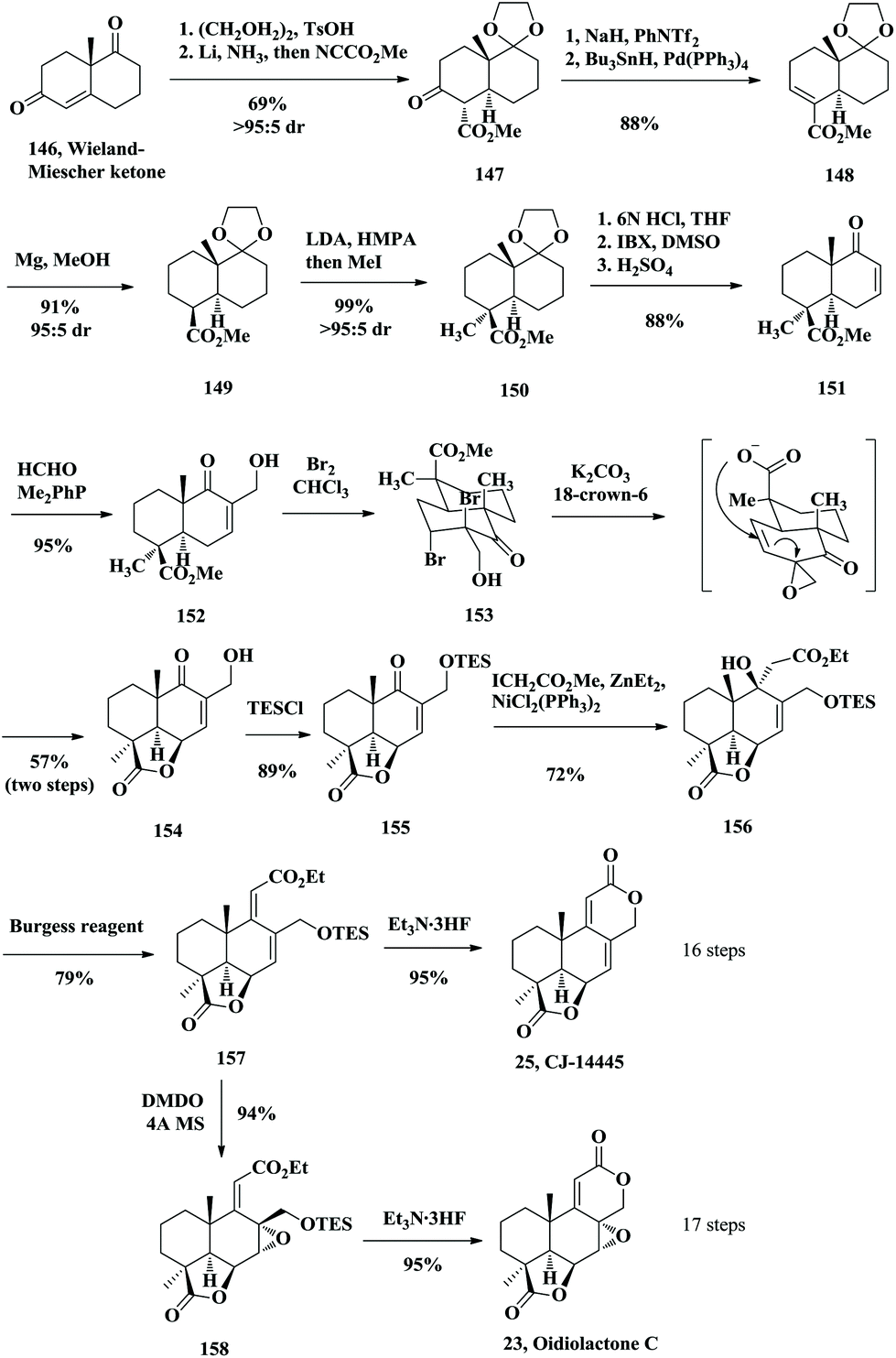
In 2009, the Hanessian group reported the asymmetric total synthesis of seven norditerpenoid dilactones. Their strategy involved three key steps including a Morita–Baylis–Hillman reaction, a γ-lactone through bromolactonization and an efficient catalytic Reformatsky-type reaction (Scheme 10).30 The process of asymmetric synthesis of norditerpenoid dilactones started from the optically pure Wieland–Miescher ketone 146. Protection of the unconjugated ketone and following 1,4-reduction with Li/NH3 and then SN2 reaction afforded 147. From 147, kinetically controlled enol triflation followed by Stille cross-coupling gave 148. Conjugated reduction of 148 with Mg produced 149 followed by the introduction of a methyl group at the α position of the ester group in an expected stereocontrolled manner giving rise to 150. Deprotection of the ketone and oxidation by IBX gave enone 151. Introduction of the hydroxymethyl branch at C8 through Morita–Baylis–Hillman reaction generated 152. Treatment of 152 with Br2 gave rise to the dibromide 153 followed by elimination under basic conditions and SN2′ cyclization to obtain the tricyclic γ-lactone core 154. To construct the C ring, they introduced the carbethoxy alkyl chain at C9 through an intermolecular catalytic Reformatsky-type reaction to afford a common intermediate 156 as a single isomer. Treatment of 156 with the Burgess reagent gave the unsaturated easter 157. Desilylation with Et3N·3HF followed by in situ lactonization provided the natural product CJ-14445 (25). Epoxidation of 157 with DMDO and lactonization upon treatment with Et3N·3HF generated oidiolactone C (23) in excellent yield (Scheme 10).
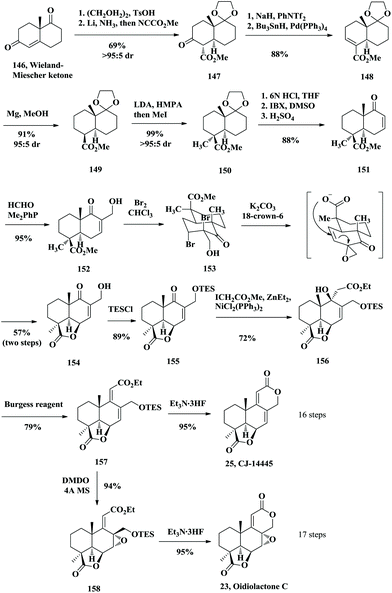 |
| | Scheme 10 Asymmetric total synthesis of CJ-14445 and oidiolactone C by Hanessian in 2009. | |
Mild TES deprotection of 158 with CSA gave a primary alcohol, which was oxidized with the Dess–Martin periodinane reagent producing the aldehyde 159 (Scheme 11). Then lactonization under the acidic conditions provided oidiolactone D (24). 159 was converted into oidiolactone A (21) by treatment with catalytic TsOH in MeOH. The common intermediate 157 was treated with CSA followed by oxidation with the Dess–Martin periodinane reagent to generate the aldehyde 160. Dehydration of the tertiary alcohol with the Burgess reagent and lactonization under acidic conditions subsequently provided LL-Z1271γ (26), a common intermediate that was converted to nagilactone F (5) and oidiolactone B (22).
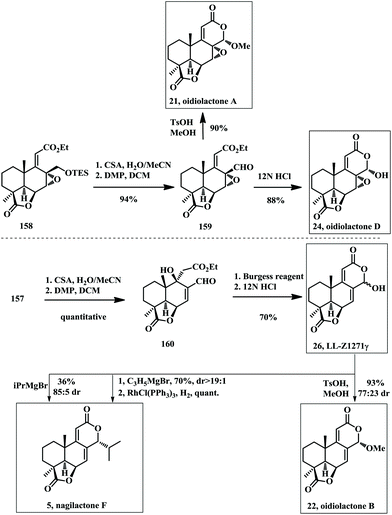 |
| | Scheme 11 Asymmetric total synthesis of LL-Z1277γ, oidiolactone A, B, and D and nagilactone F by Hanessian in 2009. | |
3. Conclusion
In conclusion, there are ten reported total syntheses methods towards norditerpenoid dilactone family members. Novel strategies and new reaction development en route to target-oriented synthesis have been demonstrated. However, there are very few synthetic studies focusing on the A-ring functionalized congeners, provided their biological importance towards lung carcinoma and life-threatening microbial infection. Another observation is that, chronologically speaking, the semi-synthesis or natural product-based strategy had been slowly replaced by the Wieland–Mischer ketone based retro-synthesis. But there is still no catalytic asymmetric total synthesis yet. In addition, the efficiency of constructing the tetracyclic core motif is still far from satisfactory, not to mention a unified strategy to access all the family members. We believe that novel strategies and reaction sequences need to be conceived and applied to solve the problems of norditerpene dilactone synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank NSFC (No. 81991522, U1706213, 81973232, and 82122063), Shandong Science Fund for Distinguished Young Scholars (ZR2020JQ32), and the Fundamental Research Funds for the Central Universities (202041003) for financial support. The project was partially funded by Taishan Youth Scholar (tsqn20161012 & tsqn201812013), Youth Innovation Plan of Shandong Province (2019KJM004) and Jinan Municipal Government fund (2019GXRC039).
References
- J. A. Hembree, C. Chang, J. L. McLaughlin, J. M. Cassady, D. J. Watts, E. Wenkert and S. F. Fonseca, J. Phytochem., 1979, 18, 1691 CrossRef CAS.
- T. Hosoe, K. Nozawa, T. C. Lumley, R. S. Currah, K. Fukushima, K. Takizawa, M. Miyaji and K.-I. Kawai, Chem. Pharm. Bull., 1999, 47, 1591 CrossRef CAS PubMed.
- M. Zhang, B. P. Ying and I. Kubo, J. Nat. Prod., 1992, 55, 1057 CrossRef CAS.
- P. Singh, G. B. Russell, Y. Hayashi, R. T. Gallagher and S. Fredercksen, Entomol. Exp. Appl., 1979, 25, 121 CrossRef CAS.
-
(a) M. N. Galbraith, D. H. S. Horn, J. M. Sasse and D. Adamson, J. Chem. Soc., 1970, 170 CAS;
(b)
Y. Hayashi and T. Sakan, Proc. 8th Int. Conf. Plant Growth Substances, 1974, 525;
(c)
A. Rahman, Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier Science B.V., 2013, Vol. 28, pp. 453–514 Search PubMed.
- T. Hosoe, K. Nozawa, T. C. Lumley, R. S. Currah, K. Fukushima, K. Takizawa, M. Miyaji and K. Kawai, Chem. Pharm. Bull., 1999, 47, 1591 CrossRef CAS PubMed.
- K. Ichikawa, T. Inagaki, H. Kachi-Tonai, Y. Kojima, T. Nakamura, H. Nishida, Y. Ueno, P. Binding, C. A. Gabel, V. Lucas, P. A. McNiff and N. Kojima, Biochem. Biophys. Res. Commun., 2001, 286, 697 CrossRef CAS PubMed.
- J. W. Dorner, R. J. Cole, J. S. P. Springer, R. H. Cox, H. Cutlers and D. T. Wicklow, Phytochemistry, 1980, 19, 1157 CrossRef CAS.
- H.-F. Sun, X.-M. Li, L. Meng, C.-M. Cui, S.-S. Gao, C.-S. Li, C.-G. Huang and B.-G. Wang, J. Nat. Prod., 2012, 75, 148 CrossRef CAS PubMed.
- C. Lv, Y. Hong, L. Miao, C. Li, G. Xu, S. Wei, B. Wang, C. Huang and B. Jiao, Cell Death Dis., 2013, 4, e952 CrossRef CAS PubMed.
- Z. Zhang, L. Miao, C. Lv, H. Sun, S. Wei, B. Wang, C. Huang and B. Jiao, Cell Death Dis., 2013, 4, e657 CrossRef CAS PubMed.
- G. A. Ellestad, R. H. Evans and M. P. Kunstmann, J. Am. Chem. Soc., 1970, 91, 2134 CrossRef PubMed.
- M. Adinolfi, L. ManEoni, G. Barone and G. Laonipr, Tetrahedron Lett., 1972, 8, 695 CrossRef.
- W. Cocker, B. E. Gross, S. R. Ruff, J. T. Edwards and T. F. Halley, J. Chem. Soc., 1953, 2540 RSC.
- S. C. Welch, C. P. Hagan, D. H. White, W. P. Fleming and J. W. Trotter, J. Am. Chem. Soc., 1977, 99, 549 CrossRef CAS PubMed.
-
(a) T. A. Spencer, T. D. Weaver, R. M. Villarica, R. J. Friary, J. Posler and M. A. Schwartz, J. Org. Chem., 1968, 33, 712 CrossRef CAS;
(b) T. A. Spencer, R. J. Friary, W. W. Schmiegal, J. F. Simeone and D. S. Watter, J. Org. Chem., 1968, 33, 719 CrossRef CAS.
- R. M. Coatesand and J. E. Shaw, J. Org. Chem., 1970, 35, 2597 CrossRef.
- Y. Hayashi, T. Matsumoto, M. Nishizawa, M. Togami, T. Hyono, N. Nishikawa and M. Uemura, J. Org. Chem., 1982, 47, 3428–3423 CrossRef CAS.
- J. T. A. Reuvers and A. de Groot, J. Org. Chem., 1986, 51, 4594 CrossRef CAS.
- J. T. A. Reuvers and A. de Groat, J. Org. Chem., 1984, 49, 1110 CrossRef CAS.
- S. D. Burke, S. M. S. Strickland and T. H. Powner, J. Org. Chem., 1983, 48, 454 CrossRef CAS.
- S. D. Burke, S. M. S. Strickland, H. M. Organ and L. A. Silks, Tetrahedron Lett., 1989, 30, 6303 CrossRef CAS.
- T. Nakata, S. Nagoa and T. Oishi, Tetrahedron Lett., 1985, 26, 6465 CrossRef CAS.
- S. D. Burke, M. E. Kort, S. M. S. Strickland, H. M. Organ and L. A. Silks, Tetrahedron Lett., 1994, 35, 1503 CrossRef CAS.
-
(a) J. Conception, C. Francisco, R. Hernandez, J. Salazar and E. Suárez, Tetrahedron Lett., 1984, 25, 1953 CrossRef;
(b) R. Hernández, J. J. Marrero, E. Suárez and A. Perales, Tetrahedron Lett., 1988, 29, 5979 CrossRef.
- A. F. Barrero, J. F. Sánchez, J. Elmerabet, D. Jiménez-Gonzélez, F. A. Macías and A. M. Simonet, Tetrahedron, 1999, 55, 7289 CrossRef CAS.
- A. F. Barrero, S. Arseniyadis, J. F. Q. Moral, M. M. Herrador, M. Valdivia and D. Jiménez, J. Org. Chem., 2002, 67, 2501 CrossRef CAS PubMed.
- T. Hosoe, K. Nozawa, T. C. Lumley, R. S. Currah, K. Fulushima, K. Takizawa, M. Miyaji and K. Kawai, Chem. Pharm. Bull., 1999, 47, 1591 CrossRef CAS PubMed.
- A. F. Barrero, M. M. Herrador, J. Q. Moral and M. V. Valdivia, Org. Lett., 2002, 4, 1379 CrossRef CAS PubMed.
- S. Hanessian, N. Boyer, G. J. Reddy and B. Deschênes-Simard, Org. Lett., 2009, 11, 4640 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab