
Nitrogen and oxygen tailoring of a solid carbon active site for two-electron selectivity electrocatalysis†
Hongyang
Shao‡
a,
Quan
Zhuang‡
a,
Hongda
Gao
b,
Yin
Wang
*a,
Lei
Ji
a,
Xia
Wang
a,
Tingting
Zhang
a,
Limei
Duan
*a,
Jie
Bai
 b,
Zhiqiang
Niu
c and
Jinghai
Liu
*a
b,
Zhiqiang
Niu
c and
Jinghai
Liu
*a
aInner Mongolia Key Laboratory of Carbon Nanomaterials, Nano Innovation Institute (NII), College of Chemistry and Materials Science, Inner Mongolia University for Nationalities Tongliao 028000, People's Republic of China. E-mail: jhliu2008@sinano.ac.cn; wy19890703@126.com; duanlmxie@126.com
bChemical Engineering College, Inner Mongolia University of Technology, Huhhot 010051, People's Republic of China
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China
First published on 29th October 2020
Abstract
The synthesis of hydrogen peroxide (H2O2) via two-electron (2e−) oxygen reduction reaction (ORR) electrocatalysis has garnered extensive attention as an appealing green and safe production technology. However, the key progress is still strongly dependent on developing highly active, selective and stable electrocatalysts. Here, guided by first-principles calculations, we developed a rational design for nitrogen (N) and oxygen (O) tailoring of carbon materials by precisely controlling the pyridinic N (Pydn-N) structure and the oxygen doping. The second nearest C atoms of Pydn-N were identified to be the active site, and this is suppressed owing to its local chemical environment and increase in the oxygen dopant content. Experimentally, high-temperature hydrogenation (HTH) regulates the Pydn-N content and synchronous oxygen removal for N,O co-doped carbons (CNO-x). The increment of Pydn-N from 37.42% up to 39.73%, along with oxygen removal from 5.24 at% down to 3.80 at% after HTH, promotes the H2O2 selectivity and electron transfer number (n) to 82% and 2.36. The H2O2 production rate is stable at around 200 mmol gcat−1 h−1 and a faradaic efficiency (F.E.) of up to 80% was recorded during the initial 4 h. The long-term H2O2 production further highlights the significance of sustaining the Pydn-N structure, modulating the solid carbon active site.
1. Introduction
Hydrogen peroxide (H2O2), as a green, versatile and multifunctional chemical, has been extensively applied in pulp and paper bleaching, medicine and environmental protection.1–3 The global market demand for H2O2 is enormous, projected to reach 6000 kilotons in 2024.4 However, the traditional anthraquinone process for the synthesis of H2O2 is not a green process and generates substantial waste chemicals.5,6 Alternatively, H2O2 production through the oxygen reduction reaction (ORR) is highly desirable with the advantages of green precursors and renewable energy sources.7–9 However, the ORR involves two competitive processes in which the oxygen (O2) can be converted to H2O through a four-electron (4e−) pathway or a two-electron (2e−) one to form H2O2.10 For H2O2 production, owing to the difficult control in triggering the 2e− pathway and the subsequent suppression of the following 2e− step, electrocatalysts with a high activity and selectivity are a prerequisite for ORR for the 2e− process.11 Therefore, the rational design of a catalyst with a high selectivity, activity and stability is the key to the electrochemical synthesis of H2O2 through the ORR.To overcome the dilemmas for electrocatalysts in terms of compatible activity and selectivity during the 2e− ORR process, a substantial number of studies have focused on noble metals and their alloys (such as Pt,12–15 Pd,16,17 Pt–Hg18 and Pd–Hg19,20), which exhibit a high efficiency for H2O2 production both in rate and selectivity. However, their nature, in terms of the scarcity and inferior stability of noble metal-based catalysts, hinders their large-scale application.11,21 Cost-effective carbon materials with a nanoscale two-dimensional (2D) morphology and chemically functionalized surface have recently been regarded as promising catalysts for electrochemical synthesis of H2O2 through the ORR.10,22–24 Currently, heteroatom doping is an effective route to regulate the electronic structure of carbon nanomaterials and simultaneously provide catalytic active sites for the ORR, thus enabling this approach to tailor the catalytic activity and selectivity towards H2O2 electrosynthesis.25–28 In particular, nitrogen (N) doping plays an important role in enhancing the ORR activity and selectivity for H2O2 production.29–31 For the N-doped carbon catalyst, the N atoms with a higher electronegativity create a net positive charge on the adjacent carbon atoms to influence the adsorption properties toward the intermediate (OOH* for 2e− process).10,25 Specifically, the contents and bonding states of nitrogen are crucial to affecting the catalytic activity and selectivity. Fellinger et al. reported an N-doped mesoporous carbon material with a favoured 2e− ORR process, which was derived from the incremental nitrogen content and the pyrrolic nitrogen (Pyli-N) sites, which had a radical character. Moreover, pure pyridinic N (Pydn-N) doped graphene has been reported to be a good catalyst towards the synthesis of H2O2 with a high selectivity.32 It is preferential to accurately regulate the local chemical environment of the carbon atoms using N doping and to further elaborately study their correlations with the catalytic active site and selectivity. In addition to the N doping, the oxygen (O) is also considered to be an effective heteroatom for improving the selectivity of 2e− ORR.33–36 Cui and co-workers demonstrated the oxidized O-CNTs with the active site of the carbon atoms adjacent to several oxygen functional groups, showing an excellent activity and high selectivity toward H2O2 electrosynthesis.5
Notably, hydrogenation can modulate and redistribute the content and type of N atoms and partly remove the O atoms37 for doped carbon materials. Inspired by this research progress, a large challenge remains in the design and synthesis of N and O co-doped carbon materials by chemically tailoring the catalytic active site for green and safe H2O2 production and to explore the mechanism of the local N and O microstructure determined active site for selective 2e− ORR.
Herein, we report the density functional theory (DFT) calculation guided rational design of N and O tailored carbon materials by precisely controlling the Pydn-N structure and oxygen doping for H2O2 synthesis. The second nearest C atoms of Pydn-N are predicted to act as the active site for 2e− ORR. A series of N,O co-doped carbon materials (CNO-x) were synthesized, and following high-temperature hydrogenation (HTH) under Ar/H2 atmosphere, were used to prepare the CNO-x-H. It was sufficiently demonstrated that HTH can generally regulate the Pydn-N structure and content, and the synchronous oxygen removal. The increment of the Pydn-N structure along with the oxygen removal for CNO-x-H, obviously promotes the H2O2 selectivity and electron transfer number (n) during the ORR. We also observed that the CNO-glu with a higher Pydn-N ratio exhibited a better H2O2 selectivity in comparison with the CNO-cyc and CNO-cel, while the CNO-glu-H, CNO-cyc-H and CNO-cel-H showed no difference in the Pydn-N ratio and 2e− selectivity. The H2O2 production rate is stable during the initial 4 h. Long-term H2O2 production operation deteriorated the activity of CNO-x-H owing to breaking of the solid carbon active site caused by reduction of the Pydn-N structure.
2. Experimental
2.1 First-principles calculations
The DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP).38,39 Core electrons are described using the projector augmented wave (PAW) pseudopotentials.40 We utilized the BEEF-vdW exchange correlation functional, which uses the vdW-DF2 nonlocal correlation energy and potential to estimate the effect of van der Waals interactions and this has been shown to accurately describe the chemisorption and physisorption properties on graphene.41 The plane-wave kinetic energy cutoff of 400 eV with a Gaussian smearing width of 0.1 eV, and Monkhorst–Pack k-point grids of 3 × 3 × 1 were adopted to ensure the convergence of the total-energy calculations. In addition, the convergence criterion of energy and forces were set to be 10−5 eV and 0.01 eV Å−1, respectively. The constructed model structures were built by doping different types of N and O atoms into the 3 × 4 supercell of the orthogonal unit cell of single-layer graphite. The optimized lattice parameters for the model systems are 21.3 × 16.3 Å. All the single layer structures were separated by 20 Å of vacuum space perpendicular to the slab surface. Further details on the DFT calculations are provided in the ESI.†2.2 Synthesis of CNO-x (x = glu, cyc and cel)
The CNO-x (x = glu, cyc and cel) were prepared according to the method described in our previous study.42 1 g of glucose and 10 g of urea were mixed and ground using a mortar. Then, the mixture was placed in a tightly sealed crucible and calcined in a muffle furnace. The calcination temperature was first set at 550 °C for 1 h, then it was reduced down to 200 °C for 0.5 h. Subsequently, the temperature was raised to 900 °C and maintained for 1 h. The finally obtained black powder was named as CNO-glu. CNO-cyc and CNO-cel were prepared using the same method with cyclodextrin and cellulose being used separately to replace glucose.2.3 Synthesis of CNO-x-H (x = glu, cyc and cel)
All these CNO-x-H materials were obtained by hydrogenation treatment, in which CNO-glu, CNO-cyc and CNO-cel were placed in a tube furnace at 500 °C for 1 h under an Ar/H2 atmosphere, respectively.2.4 Characterizations
The morphology was observed using scanning electron microscopy (SEM, Hitachi S-4800, 15 kV) and transmission electron microscopy (TEM, JEOL JEM F200). The X-ray diffractometry (XRD) patterns were obtained using a SmartLab (Rigaku 9 kW). The Raman spectra analysis were recorded on a Renishaw Invia Raman spectrometer. X-ray photoelectron spectroscopy (XPS) spectra were conducted with an ESCALAB 250 X-ray photoelectron spectrometer using Al Kα radiation as an excitation source with a pass energy of 30 eV.2.5 Electrochemical characterization
The electrochemical performances were measured using a multichannel electrochemical workstation (Bio-Logic VMP-3) with a three-electrode system. The catalyst ink was prepared by dispersing 2 mg of the as-prepared catalyst in 1 ml ethanol with 30 μl of 5 wt% Nafion. A rotating ring-disk electrode (RRDE, 5 mm) was used as a working electrode (10 μL of catalyst ink was dropped onto the disk electrode). A platinum wire electrode and a KCl saturated Ag/AgCl electrode (or a Hg/HgO electrode) served as the counter electrode and the reference one, respectively. The activity and selectivity were investigated using linear sweep voltammetry (LSV) measurements in an O2-saturated 0.1 M KOH electrolyte. During the testing process, a potential of 1.5 V versus reversible hydrogen electrode (RHE) was applied on the ring electrode. The working electrode worked under a speed of 1600 rpm. The electron transfer number (n) and the H2O2 selectivity were calculated using the following formulas: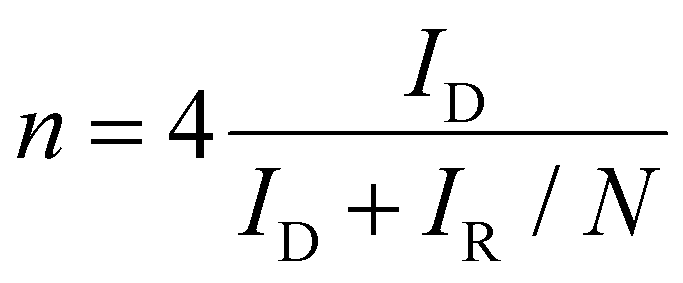 | (1) |
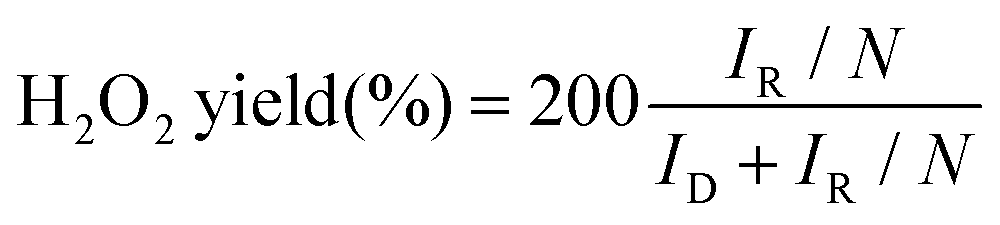 | (2) |
In which, IR is the ring current, ID is the disk current and N is the collection efficiency of the ring electrode (N = 0.38).
The Nernst equation for converting the reference electrode (Ag/AgCl or Hg/HgO) potential into the reversible hydrogen electrode (RHE) potential was formulated according to the following formulas:
| E(RHE) = E(Ag/AgCl) + 0.0591 pH + 0.1976 | (3) |
| E(RHE) = E(Hg/HgO) + 0.0591 pH + 0.098 | (4) |
Electrocatalytical H2O2 production was performed in an H-cell with a Nafion 117 membrane as a separator. A carbon fibre paper (area 1 × 1 cm2) loaded with 1 mg of CNO-glu-H as the working electrode and a KCl saturated Ag/AgCl electrode (or Hg/HgO) as the reference electrode was placed in the cathode compartment. A platinum sheet electrode as the counter electrode was placed in the anode compartment. Both the cathode and anode compartment were filled with 35 mL 0.1 M KOH solution. The chronoamperometry curves applied at 0.5 V versus RHE were recorded in an O2-saturated electrolyte.
2.6 H2O2 concentration measurements
The cerium sulfate (Ce(SO4)2) titration method was employed to measure the H2O2 concentration based on the mechanism that the yellow transparent Ce4+ can be reduced to colourless Ce3+ by H2O2. Thus, the content of H2O2 can be calculated based on the consumption of Ce4+ determined using a ultraviolet-visible (UV-Vis) spectrometer (UV2310II, Techcomp). 404 mg of Ce(SO4)2 was dissolved in 500 mL of 0.5 M H2SO4 to prepare 10 mM of Ce(SO4)2 solution. For the calibration curve, a series of H2O2 solutions with known concentration were respectively added to Ce(SO4)2 solution and measured using the UV-Vis spectrometer (Fig. S8†). 1 mL of the electrolyte after the chronoamperometry measurement was added to the Ce(SO4)2 solution to calibrate the concentration of H2O2.3. Results and discussion
DFT calculations were employed to explore the catalytic activity and active site for the two-electron (2e−) ORR to H2O2 on different types of N and O atoms doped carbon model structures. The model configurations are shown in Fig. 1A, in which all models contain one Pydn-N and Pyli-N on the zigzag and armchair edges, respectively. In addition, one O atom has been introduced at the edge or skeletal location of the graphene-like framework, except for the model labelled “Edge_C” without O doping. To understand the catalytic activities of all model structures, the binding energies of the reaction intermediates to the active sites (the carbon atoms denoted by dashed magenta circles in Fig. 1A) were calculated using DFT. The 2e− ORR comprises two elementary steps:| O2 + * + (H+ + e−) → OOH* | (5) |
| OOH* + (H+ + e−) → H2O2 + * | (6) |
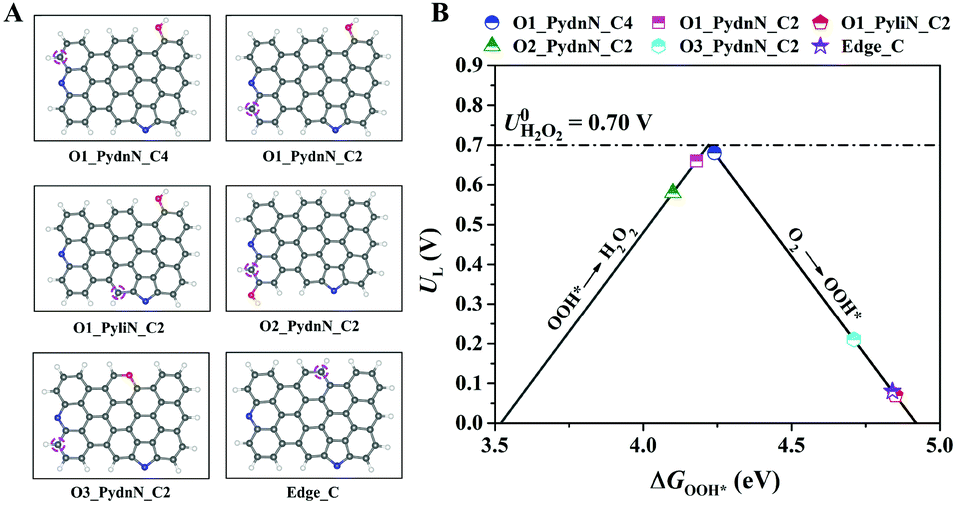 | ||
| Fig. 1 Theoretical prediction for the 2e− ORR activity for different model systems. (A) Schematic diagram of the topological models for the N and O tailoring of carbons examined in this study. Colour code: C, grey; N, blue; O, red; H, white. The carbon atoms denoted by dashed magenta circles are the active sites under investigation. (B) Calculated catalytic activity for 2e− ORR to H2O2 displayed with the limiting potential plotted as a function of ΔGOOH*. The equilibrium potential for the two-electron ORR is shown as the dashed black line. | ||
Here, * denotes the active sites. Eqn (5) represents the direct proton/electron transfer process to the adsorbed molecular O2 to produce OOH*. Furthermore, eqn (6) describes the production of H2O2via the second proton/electron transfer. Obviously, the pathway involves only OOH* as the reaction intermediate. Fig. 1B shows the volcano plot for the two-electron ORR activity with the descriptor of ΔGOOH*. The calculated limiting potential (UL) can be defined as the highest potential at which all the reaction steps are downhill in free energy. In addition, the theoretical overpotential is defined as the difference between the limiting potential and the equilibrium potential (U = 0.7 V vs. RHE).5 For the two-electron ORR, the overpotential is either due to the protonation of oxygen (eqn (5)) or the reduction of OOH* to form H2O2 (eqn (6)). The positioning of the structures at the left side of the volcano plot present strong binding toward OOH*, hence, eqn (6) is their rate-determining step. However, the ones located at the right side weakly bind OOH*, thus, eqn (5) is the rate-determining one. According to the calculations results, the second nearest carbon atom of Pyli-N (see model O1_PyliN_C2) and the carbon atom far from the N atoms (see model Edge_C) exhibit no significant contribution to the 2e− ORR. In comparison, the second nearest carbon atoms of Pydn-N (see model O1_PydnN_C4 and O1_PydnN_C2) are highly active for the 2e− ORR with overpotentials of 0.04 and 0.02 V, respectively. Moreover, when the location of the doped O atom is close to Pydn-N (see model O2_PydnN_C2), the overpotential increases to 0.12 V, and the overpotential further rises to 0.63 V when an O atom occurs in the skeletal location. Therefore, the second nearest C atoms of Pydn-N are predicted to be the active site for the 2e− ORR, in which the catalytic activity would also be affected by the configuration and location of the doped O atom. Furthermore, the 2e− ORR activity of the model structures with various O doping amounts have also been investigated. Models with no O, or only one O atom doped at an edge location, are predicted to give a superior 2e− ORR performance (Fig. S1A–D†). As for the models with more O atoms (Fig. S1E–H†), the 2e− ORR pathway is impractical owing to the uphill free energy in the first elementary step in eqn (5). Consequently, a rational design strategy for N and O tailored carbon materials for 2e− ORR to H2O2 production will promote the Pydn-N structure and related content, and reduce oxygen doping to precisely control its chemical location.
Based on the DFT calculations, we focused on designing and synthesizing a series of N,O co-doped carbon materials and tailoring the catalytic active site of C atoms by regulating the content of Pydn-N and oxygen atoms. It has been reported that various types of N structures would be affected by high-temperature treatment and the oxygen atoms would be removed under the H2 reducing atmosphere.28,37 Hence, the hydrogenation temperature is critical in modulating the C atom active site for the 2e− ORR performance improvement. The CNO-x (x = glu, cyc and cel) was synthesized using a one-step self-supporting solid-state pyrolysis (OSSP) technique, in which temperature terrace calcination was used to construct graphene-like oxygenated carbon nitride (OCN) materials.42 As shown in Scheme 1, at the first stage at 550 °C, the graphitic carbon nitride (g-C3N4) was produced by polycondensation of urea to serve as the 2D layered template. Simultaneously, amorphous carbon pitch with oxygen-containing functional groups was produced from glucose (cyclodextrin and cellulose). At the second stage at 900 °C, the g-C3N4 with amino groups reacted with the carbonyl groups of carbon pitch through the Maillard reaction43 to chemically graft both components. Moreover, the evolved gases from the second stage expanded into the interlayers and finally generated the CNO-x nanosheets. Subsequently, the CNO-x was further calcinated by HTH37 under an Ar/H2 atmosphere to remove the O atoms to prepare the CNO-x-H.
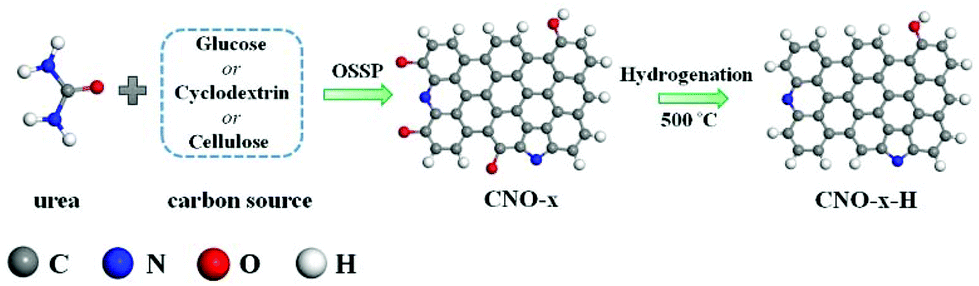 | ||
| Scheme 1 Schematic illustration of CNO-x-H prepared by OSSP followed by HTH. | ||
The morphology and microstructure were first investigated using SEM and TEM. According to Fig. 2A and B, both CNO-glu and CNO-glu-H present a two-dimensional (2D) nanosheet morphology with abundant wrinkles. In addition, the CNO-glu-H exhibits a much more wrinkled defect structure than that of CNO-glu, which was further confirmed by the TEM images (Fig. 2C and D). The elemental mapping analysis shows that the C, N and O elements were uniformly distributed among the 2D nanosheet, but the O content decreased for CNO-glu-H after the hydrogenation treatment. Then, XRD was carried out to identify the phase structure. As shown in Fig. 2E, the diffraction peaks centred at 26.2° and 42.2° are indexed to the (002) and (100) plane of graphite (JCPDS no. 75-1621), which demonstrates that no change in the graphitic phase and condensation state occurs after the HTH process. Following this, the Raman spectrum was measured to analyse the defect microstructure features. As shown in Fig. 2F, the Raman spectra between 1000 to 2000 cm−1 are deconvoluted into four bands, including impurity (I), in-plane defect (D), interstitial defects (D′′) and graphitic structure (G).44,45 The intensity ratio of D and G (ID/IG), an indicator for estimating the degree of disorder in carbon materials, in CNO-glu-H decreases slightly from 1.28 to 1.03, indicating the suppression of the in-plane defect by taking away the O atoms after HTH treatment.
 | ||
| Fig. 2 SEM images of (A) CNO-glu and (B) CNO-glu-H. TEM and elemental mapping images of (C) CNO-glu and (D) CNO-glu-H. (E) XRD patterns of CNO-glu and CNO-glu-H. (F) Raman spectra of CNO-glu and CNO-glu-H. | ||
Moreover, XPS analysis was performed to detect the elemental constituents and bonding state of C, N and O. As shown in Table 1, around 13.23 at% of nitrogen and 5.24 at% of oxygen were measured in CNO-glu. After HTH treatment, the N content in CNO-glu-H increases slightly to 13.55 at%, while the O decreases to 3.80 at%. High resolution XPS spectroscopy further demonstrates the existence of multiple types of C and N bonding states. The deconvolution C 1s spectrum shown in Fig. 3A and B reveal four peaks: carbon in graphite (C–C) at 284.7 eV, carbon bound to nitrogen (C–N) at 285.7 eV, carbon singly bound to oxygen (C–O) at 286.6 eV, and carbon bound to two N/O atoms (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/N–CN) at 288 eV.46 The N 1s spectrum can be deconvoluted into four bands (Fig. 3C and D): Pydn-N at 398.4 eV; Pyli-N at 399.9 eV; graphitic N (Grap-N) at 401 eV; and oxidized N at 402.2 eV.47,48 The ratios of the different C and N types are summarized in Tables 1 and S1.† For CNO-glu, the content of C–C, C–N, C–O and O–CO/N–CN is approximately 62.16%, 16.05%, 15.00% and 6.79%, respectively. For CNO-glu-H, the C–N concentration increases to 21.17% while the C–O concentration decreases to 10.41%, in accordance with the variation of the elemental constituent ratio. Notably, the content of Pydn-N in CNO-glu-H increases from 37.42% to 39.73% after HTH treatment. A similar phenomenon for elemental constituents and bonding states was also observed for CNO-glu-H. These results sufficiently demonstrate that the HTH can experimentally regulate the Pydn-N structure and content, and is synchronous to the removal of doping oxygen.
O/N–CN) at 288 eV.46 The N 1s spectrum can be deconvoluted into four bands (Fig. 3C and D): Pydn-N at 398.4 eV; Pyli-N at 399.9 eV; graphitic N (Grap-N) at 401 eV; and oxidized N at 402.2 eV.47,48 The ratios of the different C and N types are summarized in Tables 1 and S1.† For CNO-glu, the content of C–C, C–N, C–O and O–CO/N–CN is approximately 62.16%, 16.05%, 15.00% and 6.79%, respectively. For CNO-glu-H, the C–N concentration increases to 21.17% while the C–O concentration decreases to 10.41%, in accordance with the variation of the elemental constituent ratio. Notably, the content of Pydn-N in CNO-glu-H increases from 37.42% to 39.73% after HTH treatment. A similar phenomenon for elemental constituents and bonding states was also observed for CNO-glu-H. These results sufficiently demonstrate that the HTH can experimentally regulate the Pydn-N structure and content, and is synchronous to the removal of doping oxygen.
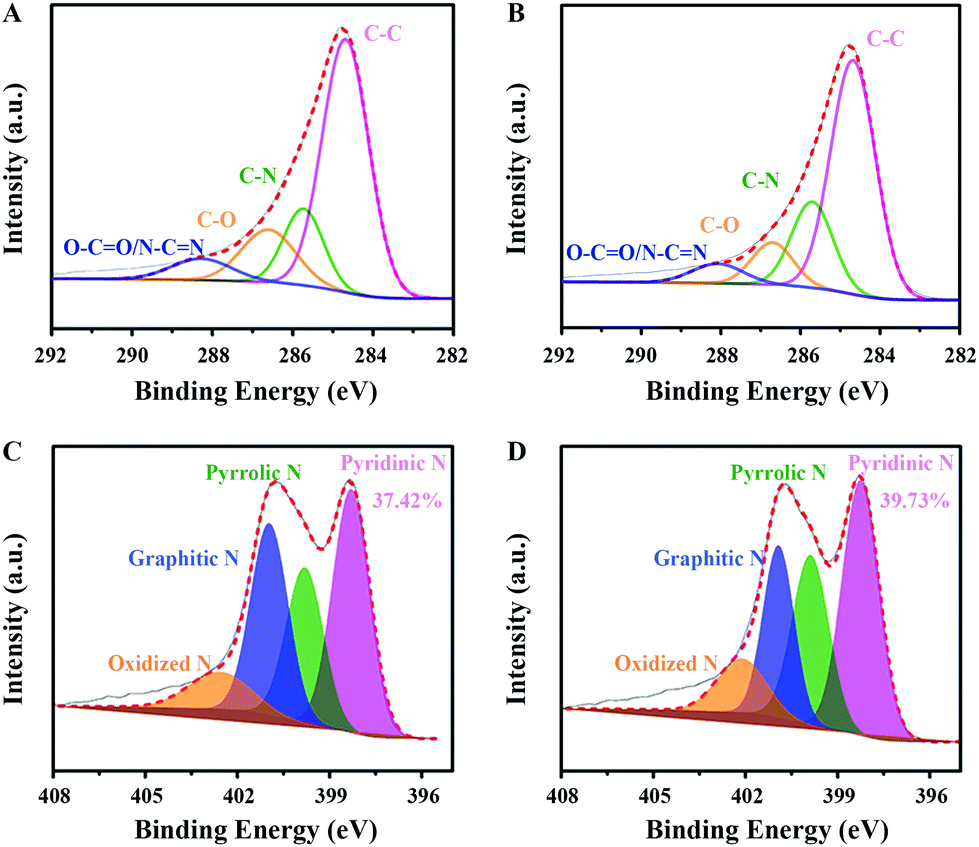 | ||
| Fig. 3 Pydn-N structure and content determined by XPS. (A) and (B) High-resolution C 1s spectrum of CNO-glu and CNO-glu-H. (C) and (D) High-resolution N 1s spectrum of CNO-glu and CNO-glu-H. | ||
| Sample | Composition (at%) | N 1s bonding types (at%) | |||||
|---|---|---|---|---|---|---|---|
| C | N | O | Pyridinic N | Pyrrolic N | Graphitic N | Oxided N | |
| CNO-glu | 81.53 | 13.23 | 5.24 | 37.42 | 24.54 | 31.45 | 6.59 |
| CNO-glu-H | 82.65 | 13.55 | 3.8 | 39.73 | 26.50 | 22.58 | 11.19 |
| CNO-cyc | 81.54 | 12.37 | 6.08 | 36.72 | 25.25 | 29.48 | 8.55 |
| CNO-cyc-H | 83.12 | 12.86 | 4.03 | 39.47 | 25.76 | 25.19 | 9.6 |
| CNO-cel | 86.05 | 8.25 | 5.7 | 35.96 | 26.67 | 28.47 | 8.88 |
| CNO-cel-H | 84.83 | 10.81 | 4.36 | 40.43 | 23.04 | 28.88 | 7.66 |
The 2e− ORR activity and selectivity were investigated using a RRDE. The H2O2 oxidation currents (orange lines) were measured on a platinum ring electrode held at 1.5 V versus RHE, along with the oxygen reduction currents (cyan lines) measured on a disk electrode loaded the CNO catalysts. As shown in Fig. 4A, the CNO-glu exhibits catalytic activity for 2e− ORR and an onset potential at 0.73 V versus RHE (defined as the potential37 at a current density of 0.5 mA cm−2). The electron transfer number and H2O2 selectivity were calculated using eqn (1) and (2), respectively. The ORR takes place at the disk electrode, and the H2O2 produced at the disk electrode is radially transferred to the concentric platinum ring electrode by the forced convection caused by the rotating motion of the electrode. Subsequently, H2O2 is reoxidized back to O2 at the platinum ring electrode. We can calculate the electron transfer number and H2O2 selectivity according to the disk current (ID) and ring current (IR), which indicates the fraction of O2 used for H2O2. The CNO-glu displays a selectivity of 68% for 2e− ORR with a transfer electron number of 2.64 at 0.5 V versus RHE (Fig. 4B). For the CNO-glu-H, as shown in Fig. 4C, a similar onset potential of 0.73 V versus RHE was performed, which indicates similar active sites present in both catalysts. Interestingly, the H2O2 selectivity and electron transfer number changed to 82% and 2.36 (Fig. 4D), which is better than some of the reported 2e− ORR carbon catalysts (Table S2†). The catalytic performance of CNO-glu-H was also measured using a Hg/HgO reference electrode (Fig. S2†) to eliminate the effects of the reference in alkine solution. The CNO-glu-H shows a similar 2e− ORR performances for the Hg/HgO reference electrode in the 0.1 M KOH solution. As predicted, the 2e− ORR catalytic activity of CNO-glu is obviously improved after HTH treatment. This experimental demonstration directly denotes the key roles of tailoring the Pydn-N structure and oxygen dopants in promoting the solid carbon active sites for selective H2O2 production, which are in good accordance with the predictions from the DFT calculations.
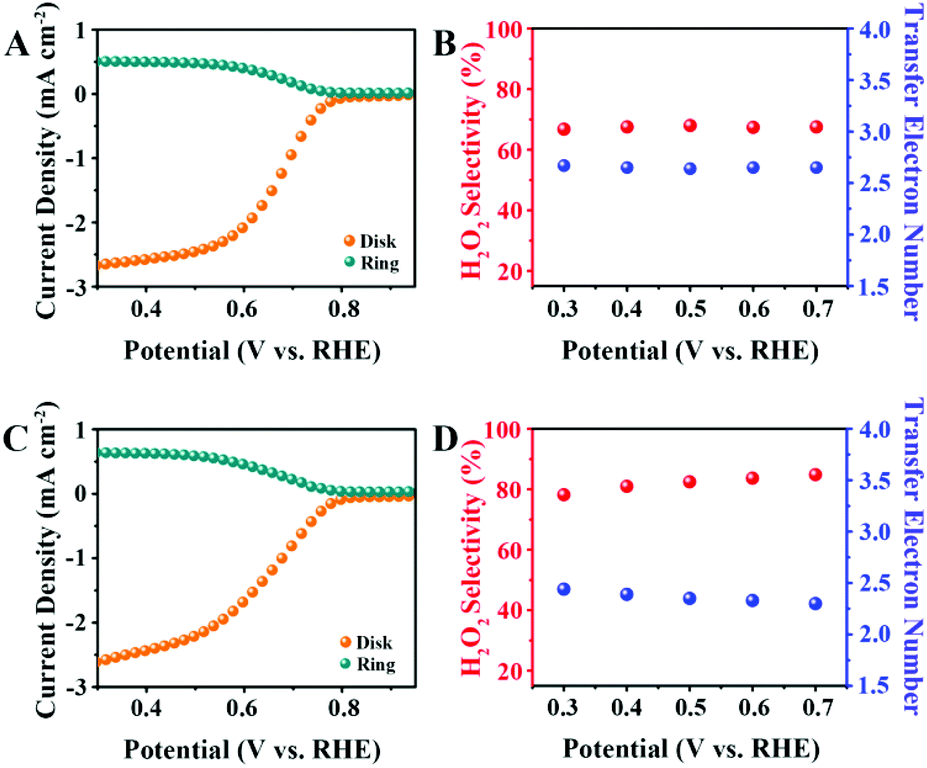 | ||
| Fig. 4 N and O tailoring of the solid carbon active site for 2e− ORR selectivity. (A) Polarization curves (orange line) and simultaneous H2O2 detection currents at the ring electrode (cyan line) for CNO-glu in O2-saturated 0.1 M KOH solution at 1600 rpm. (B) H2O2 selectivity and transfer electron number for CNO-glu. (C) Polarization curves (orange line) and simultaneous H2O2 detection currents at the ring electrode (cyan line) for CNO-glu-H in O2-saturated 0.1 M KOH solution at 1600 rpm. (D) H2O2 selectivity and transfer electron number for CNO-glu-H. | ||
To demonstrate the general principle of precisely tailoring the Pydn-N structure and content, and strictly controlling the oxygen dopants using the HTH approach for N,O co-doped carbon to improve the 2e− ORR, we further examined two other CNO-cyc and CNO-cel samples. Similar to CNO-glu-H, the CNO-cyc-H and CNO-cel-H show the same trends in the structural, morphological and constituent evolution. A more wrinkled surface (Fig. S3†) without phase change (Fig. S4†) was observed after HTH treatment. For Raman spectra (Fig. S5†), the CNO-cyc-H and CNO-cel-H also display a reduced ID/IG of 1.19 and 1.18 in comparison to that of CNO-cyc (1.21) and CNO-cel (1.20), indicating a similar suppression of the in-plane defects. In detail, the XPS survey results show that the O content from 6.08 at% for CNO-cyc and 5.7 at% for the CNO-cel decreases down to 4.03 at% for CNO-cyc-H and 4.36 at% for the CNO-cel-H. The N content, from 12.37 at% for CNO-cyc and 8.25 at% for CNO-cel, increases to 12.86 at% for CNO-cyc-H and 10.81 at% for CNO-cel-H (Fig. S6†). The Pydn-N ratio rises up to 39.47% for CNO-cyc-H and 40.43% for CNO-cel-H, relative to that of 36.72% for CNO-cyc and 35.96% for CNO-cel (Fig. 5C–F and Table 1). The C 1s spectrum exhibits four similar deconvoluted peaks attributed to the C–C, C–N, C–O, and O–CO/N–CN bonds for the CNO-cyc-H and CNO-cel-H, but presents significantly more C–N bonds than CNO-cyc and CNO-cel owing to the removal of O atoms after HTH treatment (Fig. S7†).
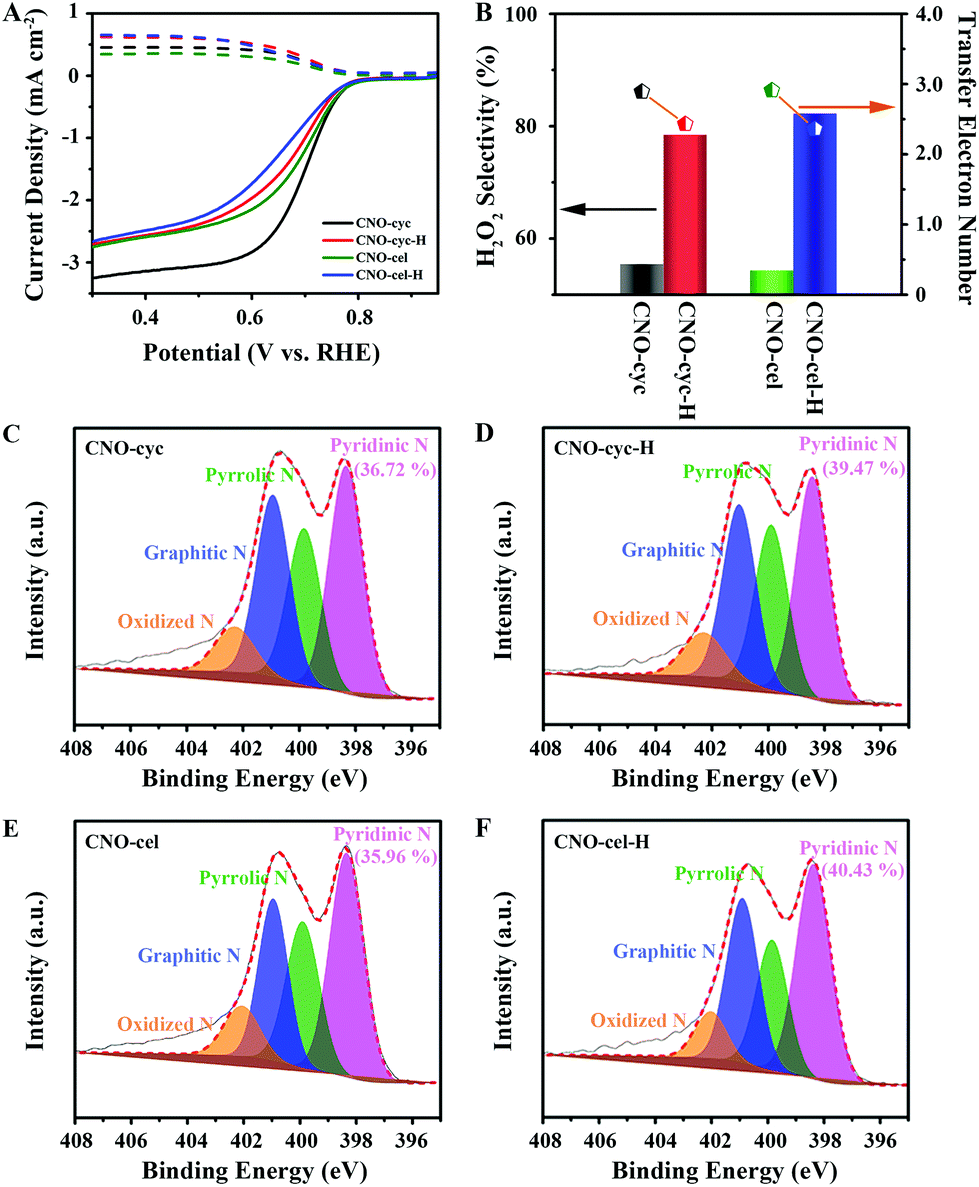 | ||
| Fig. 5 General principle of Pydn-N structure modulating carbon active site. (A) Polarization curves (solid lines) and simultaneous H2O2 detection currents at the ring electrode (dashed lines) in O2-saturated 0.1 M KOH solution at 1600 rpm for CNO-cyc, CNO-cel, CNO-cyc-H and CNO-cel-H. (B) H2O2 selectivity and transfer electron number for CNO-cyc, CNO-cel, CNO-cyc-H and CNO-cel-H. High-resolution N 1s peaks of (C) CNO-cyc, (D) CNO-cel, (E) CNO-cyc-H and (F) CNO-cel-H. | ||
In addition, according to the RRDE results shown in Fig. 5A, these four catalysts (CNO-cyc and CNO-cyc-H, CNO-cel and CNO-cel-H) display a similar onset potential, polarization current and 2e− ORR activity. Similar to the CNO-glu-H series catalyst, the CNO-cyc-H and CNO-cel-H exhibits a higher H2O2 selectivity (column charts) and 2e− ORR pathway (point segment charts) (see Fig. 5B). The H2O2 selectivity increases from 55.4% for CNO-cyc and 54.3% for CNO-cel to 78.5% for CNO-cyc-H and 82.3% for CNO-cel-H, respectively. Simultaneously, the electron transfer number for CNO-cyc and CNO-cel decreases down to 2.43 for CNO-cyc-H and 2.36 for CNO-cel-H from 2.89 and 2.91, respectively. This is consistent with the observations for CNO-glu and CNO-glu-H. Moreover, the effects of the structure and content of Pydn-N and the oxygen dopants on the solid carbon active sites for selective 2e− ORR are consequently confirmed. Interestingly, the CNO-glu has a higher Pydn-N ratio with a better H2O2 selectivity among the initial three catalysts (CNO-glu, CNO-cyc and CNO-cel), while the CNO-glu-H, CNO-cyc-H and CNO-cel-H show no difference in the Pydn-N ratio and 2e− selectivity. This analysis experimentally confirms our DFT prediction that the second nearest C atom of Pydn-N is a highly active site for 2e− ORR and the reducing O content is further beneficial to the 2e− ORR process.
Finally, as critical criteria for practical applications, the H2O2 production rate and long-term stability were also examined. The H2O2 production rate on CNO-glu-H was measured using an H-cell with chronoamperometry operation for hours. As shown in Fig. 6A, the H2O2 production rate is stable around 200 mmol gcat−1 h−1 (the calibration curve of H2O2 is shown in Fig. S8†) and the faradaic efficiency (F.E.) is maintained at up to 80% during the initial 4 h. However, an obvious decrease occurs down to 126 mmol gcat−1 h−1 at the 5th hour, indicating that the catalyst has an inferior catalytic stability above a 4 h long-term extension. The i–t curves show the disk current loss of CNO-glu-H is 26% after 5 h and 31% after 6 h (Fig. 6B). In addition, SEM and XPS analysis were employed to investigate the morphology and composition information during the stability test. The SEM image in Fig. 6C reveals that the CNO-glu-H retains a two-dimensional (2D) morphology without obvious change. However, the N 1s spectrum of CNO-glu-H displays an evident decrease of Pydn-N from 39.73% to 30.88% after the 5 h stability test (Fig. 6D), which further demonstrates the remarkable tailoring function of Pydn-N in determining the solid carbon active site for 2e− ORR. After the long-term chronoamperometry operation, the Pydn-N structure was partly destroyed leading to the reduction of the Pydn-N tailored solid C active sites for the 2e− ORR process. Hence, it is still a big challenge for these carbon electrocatalysts to sustain the Pydn-N structure modulating active site, namely the second nearest C atom of Pydn-N, under long-term H2O2 production operation conditions. A novel strategy for dynamic active site regeneration still needs to be explored in the future.
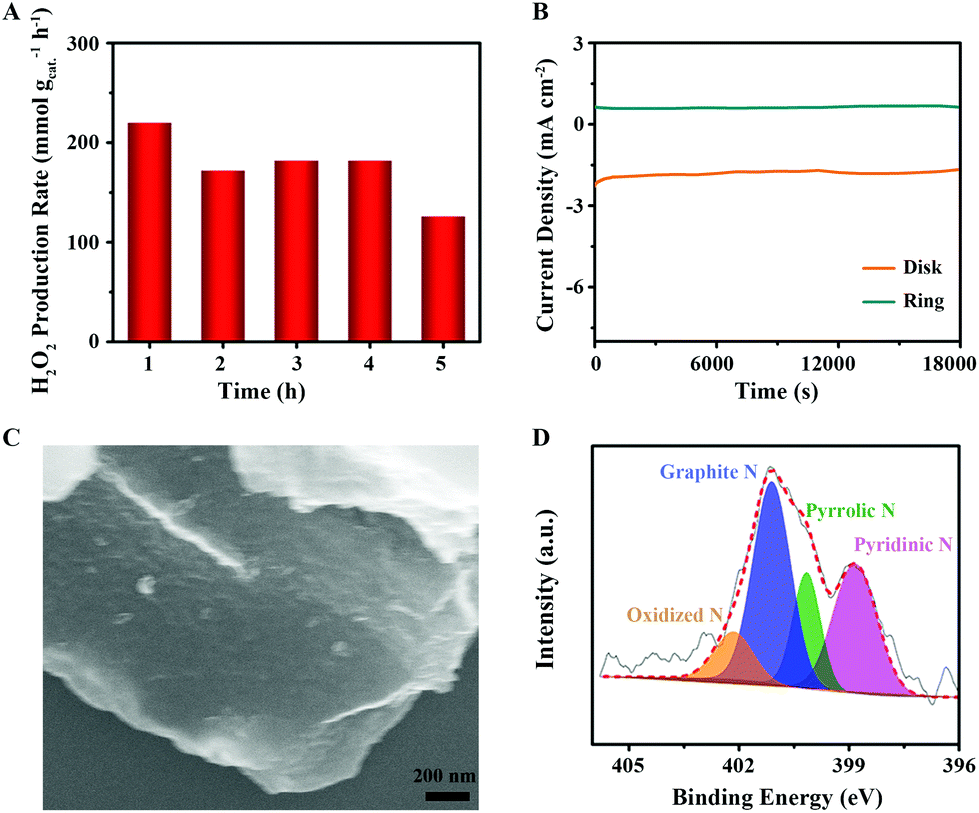 | ||
| Fig. 6 H2O2 production and long-term stability. (A) H2O2 production rate on CNO-glu-H at various electrolysis extensions. (B) Current–time (i–t) chronoamperometric responses of CNO-glu-H in O2-saturated 0.1 M KOH solution at 1600 rpm. (C) SEM image of CNO-glu-H after 5 h chronoamperometry test. (D) High-resolution N 1s spectrum of CNO-glu-H after 5 h H2O2 production test. | ||
4. Conclusions
In this work, we have theoretically and experimentally demonstrated the modulation of Pydn-N and O dopants on a solid carbon active site for 2e− ORR selectivity. The second nearest C atoms of Pydn-N are the predicted active sites, and would be suppressed upon an increase in the doped oxygen or reducing the Pydn-N structure. A novel strategy for dynamic active site regeneration still needs to be explored to sustain the Pydn-N structure modulated active site under long-term H2O2 production operation conditions. This work provides a prospective and practical avenue to combine theoretical predications with experimental demonstrations to develop advanced carbons with active sites for 2e− ORR electrocatalysis for green and safe H2O2 synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21961024, 21961025 and 11904187), the Inner Mongolia Natural Science Foundation (No. 2018JQ05, 2019BS02007), the Inner Mongolia Autonomous Region Science & Technology Planning Project for Applied Technology Research and Development (No. 2019GG261), Incentive Funding from Nano Innovation Institute (NII) of Inner Mongolia University for Nationalities (IMUN), the Inner Mongolia Autonomous Region Incentive Funding Guided Project for Science & Technology Innovation (2016), the Inner Mongolia Autonomous Region Funding Project for Science & Technology Achievement Transformation (No. CGZH2018156), the Doctoral Scientific Research Foundation of Inner Mongolia University for Nationalities (No. BS480, BS437) and the Scientific Research Program of Inner Mongolia University for Nationalities (No. NMDYB19040).Notes and references
- C. A. Martinez-Huitle and S. Ferro, Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- I. Yamanaka, T. Onizawa, S. Takenaka and K. Otsuka, Direct and continuous production of hydrogen peroxide with 93% selectivity using a fuel-cell system, Angew. Chem., Int. Ed., 2003, 42, 3653–3655 CrossRef CAS.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts, Nat. Catal., 2018, 1, 282–290 CrossRef.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- W. Zhou, X. Meng, J. Gao and A. N. Alshawabkeh, Hydrogen peroxide generation from O2 electroreduction for environmental remediation: A state-of-the-art review, Chemosphere, 2019, 225, 588–607 CrossRef CAS.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Toward the Decentralized Electrochemical Production of H2O2: A Focus on the Catalysis, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- E. Pizzutilo, S. J. Freakley, S. Cherevko, S. Venkatesan, G. J. Hutchings, C. H. Liebscher, G. Dehm and K. J. J. Mayrhofer, Gold–Palladium Bimetallic Catalyst Stability: Consequences for Hydrogen Peroxide Selectivity, ACS Catal., 2017, 7, 5699–5705 CrossRef CAS.
- Y. Lei, S. Lei and L. Piao, Study on the preparation of H2O2 by photocatalytic reduction of oxygen, Prog. Chem., 2020 DOI:10.7536/PC200463.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Selective Electrochemical H2O2 Production through Two-Electron Oxygen Electrochemistry, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, Defective Carbon-Based Materials for the Electrochemical Synthesis of Hydrogen Peroxide, ACS Sustainable Chem. Eng., 2017, 6, 311–317 CrossRef.
- C. M. Sanchez-Sanchez and A. J. Bard, Hydrogen peroxide production in the oxygen reduction reaction at different electrocatalysts as quantified by scanning electrochemical microscopy, Anal. Chem., 2009, 81, 8094–8100 CrossRef CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, High-Concentration Single Atomic Pt Sites on Hollow CuSx for Selective O2 Reduction to H2O2 in Acid Solution, Chem, 2019, 5, 2099–2110 CAS.
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, Support Effects in Single-Atom Platinum Catalysts for Electrochemical Oxygen Reduction, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Single-Atom Catalyst of Platinum Supported on Titanium Nitride for Selective Electrochemical Reactions, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS.
- D. C. Ford, A. U. Nilekar, Y. Xu and M. Mavrikakis, Partial and complete reduction of O2 by hydrogen on transition metal surfaces, Surf. Sci., 2010, 604, 1565–1575 CrossRef CAS.
- J. S. Jirkovsky, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H2O2 production, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. Stephens and J. Rossmeisl, Enabling direct H2O2 production through rational electrocatalyst design, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Trends in the electrochemical synthesis of H2O2: enhancing activity and selectivity by electrocatalytic site engineering, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS.
- D. Deiana, A. Verdaguer-Casadevall, P. Malacrida, I. E. L. Stephens, I. Chorkendorff, J. B. Wagner and T. W. Hansen, Determination of Core-Shell Structures in Pd-Hg Nanoparticles by STEM-EDX, ChemCatChem, 2015, 7, 3748–3752 CrossRef CAS.
- X. Xiao, T. Wang, J. Bai, F. Li, T. Ma and Y. Chen, Enhancing the Selectivity of H2O2 Electrogeneration by Steric Hindrance Effect, ACS Appl. Mater. Interfaces, 2018, 10, 42534–42541 CrossRef CAS.
- T. P. Fellinger, F. Hasche, P. Strasser and M. Antonietti, Mesoporous nitrogen-doped carbon for the electrocatalytic synthesis of hydrogen peroxide, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS.
- Y. Sun, S. Li, Z. P. Jovanov, D. Bernsmeier, H. Wang, B. Paul, X. Wang, S. Kuhl and P. Strasser, Structure, Activity, and Faradaic Efficiency of Nitrogen-Doped Porous Carbon Catalysts for Direct Electrochemical Hydrogen Peroxide Production, ChemSusChem, 2018, 11, 3388–3395 CrossRef CAS.
- J. Park, Y. Nabae, T. Hayakawa and M. a. Kakimoto, Highly Selective Two-Electron Oxygen Reduction Catalyzed by Mesoporous Nitrogen-Doped Carbon, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells, ACS Nano, 2010, 4, 1321 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts, Science, 2016, 351, 361 CrossRef CAS.
- K. Zhao, Y. Su, X. Quan, Y. Liu, S. Chen and H. Yu, Enhanced H2O2 production by selective electrochemical reduction of O2 on fluorine-doped hierarchically porous carbon, J. Catal., 2018, 357, 118–126 CrossRef.
- Y. H. Lee, F. Li, K. H. Chang, C. C. Hu and T. Ohsaka, Novel synthesis of N-doped porous carbons from collagen for electrocatalytic production of H2O2, Appl. Catal., B, 2012, 126, 208–214 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. R. Cuenya and P. Strasser, Efficient Electrochemical Hydrogen Peroxide Production from Molecular Oxygen on Nitrogen-Doped Mesoporous Carbon Catalysts, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, N-Doped Graphitized Carbon Nanohorns as a Forefront Electrocatalyst in Highly Selective O2 Reduction to H2O2, Chem, 2018, 4, 106–123 CAS.
- L. Han, Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, In-Plane Carbon Lattice-Defect Regulating Electrochemical Oxygen Reduction to Hydrogen Peroxide Production over Nitrogen-Doped Graphene, ACS Catal., 2019, 9, 1283–1288 CrossRef CAS.
- B. Zheng, X. L. Cai, Y. Zhou and X. H. Xia, Pure Pyridinic Nitrogen-Doped Single-Layer Graphene Catalyzes Two-Electron Transfer Process of Oxygen Reduction Reaction, ChemElectroChem, 2016, 3, 2036–2042 CrossRef CAS.
- Y. J. Sa, J. H. Kim and S. H. Joo, Active Edge-Site-Rich Carbon Nanocatalysts with Enhanced Electron Transfer for Efficient Electrochemical Hydrogen Peroxide Production, Angew. Chem., Int. Ed., 2019, 58, 1100–1105 CrossRef CAS.
- G. F. Han, F. Li, W. Zou, M. Karamad, J. P. Jeon, S. W. Kim, S. J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J. B. Baek, Building and identifying highly active oxygenated groups in carbon materials for oxygen reduction to H2O2, Nat. Commun., 2020, 11, 2209 CrossRef CAS.
- K. H. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B. J. Su, J. M. Chen, D. S. Su, W. Qi and S. Guo, Highly Selective Hydrogen Peroxide Electrosynthesis on Carbon: In Situ Interface Engineering with Surfactants, Chem, 2020, 6, 1443–1458 CAS.
- X. Lu, D. Wang, K. H. Wu, X. Guo and W. Qi, Oxygen reduction to hydrogen peroxide on oxidized nanocarbon: Identification and quantification of active sites, J. Colloid Interface Sci., 2020, 573, 376–383 CrossRef CAS.
- S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund, D. Sokaras, T. R. Kim, Y. Liu, X. Yan, E. Nilsson, R. Sinclair, J. K. Norskov, T. F. Jaramillo and Z. Bao, Designing Boron Nitride Islands in Carbon Materials for Efficient Electrochemical Synthesis of Hydrogen Peroxide, J. Am. Chem. Soc., 2018, 140, 7851–7859 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. Wellendorff, K. Lundgaard, A. Møgelhøj, V. Petzold, D. Landis, J. Nørskov, T. Bligaard and K. Jacobsen, Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou, Z. Liu, W. Chen, L. Liu, S. Feng and Y. Zhang, A Graphene-like Oxygenated Carbon Nitride Material for Improved Cycle-Life Lithium/Sulfur Batteries, Nano Lett., 2015, 15, 5137–5142 CrossRef CAS.
- J. E. Hodge, Dehydrated Foods, Chemistry of Browning Reactions in Model Systems, J. Agric. Food Chem., 1953, 1, 625–651 CrossRef.
- H. Shao, X. Zhang, H. Huang, K. Zhang, M. Wang, C. Zhang, Y. Yang, M. Wen and W. Zheng, Magnetron Sputtering Deposition Cu@Onion-like N-C as High-Performance Electrocatalysts for Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2017, 9, 41945–41954 CrossRef CAS.
- R. Zhou and S. Z. Qiao, An Fe/N co-doped graphitic carbon bulb for high-performance oxygen reduction reaction, Chem. Commun., 2015, 51, 7516–7519 RSC.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis, ACS Nano, 2011, 5, 4350 CrossRef CAS.
- C. Zhang, W. Zhang, S. Yu, D. Wang, W. Zhang, W. Zheng, M. Wen, H. Tian, K. Huang, S. Feng and J. J. Bentzen, Cover Picture: Unlocking the Electrocatalytic Activity of Chemically Inert Amorphous Carbon-Nitrogen for Oxygen Reduction: Discerning and Refactoring Chaotic Bonds (ChemElectroChem 6/2017), ChemElectroChem, 2017, 4, 1266–1266 CrossRef CAS.
- C. Hu, Y. Zhou, R. Ma, Q. Liu and J. Wang, Reactive template synthesis of nitrogen-doped graphene-like carbon nanosheets derived from hydroxypropyl methylcellulose and dicyandiamide as efficient oxygen reduction electrocatalysts, J. Power Sources, 2017, 345, 120–130 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qi01089h |
| ‡ These two authors contributed equally. |
| This journal is © the Partner Organisations 2021 |