
Regioselective and stereoselective cleavages of P–S/C–S bonds by lithium and the formation of P-stereogenic functional phosphine derivatives†
Xiao-Ning
Wang‡
,
Zhan-Cai
Li‡
,
Yu
Zhang
,
Bing-Xia
Yan
,
Hong-Xing
Zheng
*,
Qiang
Li
 * and
Chang-Qiu
Zhao
*
* and
Chang-Qiu
Zhao
*
College of Chemistry and Chemical Engineering, Liaocheng University, No.1, Hunan Road, Liaocheng, Shandong 252059, China. E-mail: zhenghongxing@lcu.edu.cn; tiamochem@hotmail.com; literabc@hotmail.com
First published on 12th October 2020
Abstract
Various menthyl-containing phosphinothioates were prepared. Using these compounds, an unusual phosphorus-promoted cleavage of the C–S bond with lithium-naphthalene was examined. The C–S/P–S cleavages could be controlled by the amount of naphthalene and temperature applied. Also, the P–S cleavage was confirmed to retain the configuration about the P and this feature was used for achieving a stereospecific formation of the C–P bond.
Introduction
Because of their important biological activity, namely inhibition of acetylcholine esterase, phosphonothioates and related compounds are widely used as agricultural chemicals such as pesticides, herbicides and insecticides.1 These compounds are also well known as neurotoxins.2 Species containing P–S bonds, especially P-stereogenic species, are usually used as substrates to explore the metabolism and degradation of these substances in organisms or other natural settings.Besides their applications in physiology and biochemistry, P-stereogenic P–S species also have wide applications in chemistry. For example, they can be used as chemical shift solvating reagents in the analysis of chiral substances.3 Compounds containing P–S bonds (denoted here as simply “P–S species”) can be used as precursors for preparing P-stereogenic compounds via nucleophilic substitutions with alkoxide or alkyl anions as attacking reagents that, respectively, invert or retain the configuration on the phosphorus.4 The stereochemical integrity on phosphorus during the reactions of the P–S species enable the use of sulfur to protect the phosphine via formation of a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S or P–S bond.5 Because of the facile formation and cleavage of P–S bonds, the P-stereogenic P–S species have advantages for easy and versatile applications regarding their conversions to various chiral substances.6,7
S or P–S bond.5 Because of the facile formation and cleavage of P–S bonds, the P-stereogenic P–S species have advantages for easy and versatile applications regarding their conversions to various chiral substances.6,7
Traditionally, P-stereogenic P–S species are prepared via kinetic resolution.2a H–P(O) compounds such as H-phosphinates and secondary phosphine oxides could be converted to P–S compounds via sulfurization. For example, Mislow and co-workers obtained diastereomerically enriched phosphonothioates from a mixture of two stereoisomers of chiral H–P(O) species.8 The very first preparations of (RP)-t-BuPhP(S)OH involved the sulfurization of racemic H–P species and kinetic resolution of the products.3 Several years ago, we reported the preparations of phosphonothioates via the sulfurization of menthyl H-phenylphosphinate.9 The stereoselective conversions of P–H bonds to P–Cl bonds, followed by their reactions with thiols, also afforded optically pure phosphonothioates, as reported by Han.10
Most of the above preparations relied on the use of optically pure H–P(O) compounds, but the poor availability of these compounds restricted the feasibility to acquire P-stereogenic P–S species.11 Additionally, the application of P–S species usually involved their conversions to P–C bonds via nucleophilic reagents.12 The reverse conversion of the P–S bond to the P–H bond had rarely been studied.5a When the optically pure P–S species were obtained conveniently, the conversion of the P–S bond to the P–H bond was hoped to provide an effective route to the preparation of more versatile H–P species.
In the current work, we developed a facile preparation of P-stereogenic P–S species and achieved conversions of these species to functional secondary phosphine oxides. Menthyl(2-hydroxy biphenyl)phosphinothioic acid was obtained as a single stereoisomer, which was alkylated to form the corresponding ester. In contrast to the stability of the C–S bond when in the presence of a metallic reagent, a phosphorus-promoted cleavage of the C–S bond of phosphinothioate, occurring simultaneously with the cleavage of the C–P bond, was observed when the phosphinothioate was treated with lithium (Chart 1). The regioselective cleavage of the P–S bond afforded a secondary phosphine oxide, which was reacted with an electrophilic reagent to form various P-stereogenic functional phosphine derivatives.
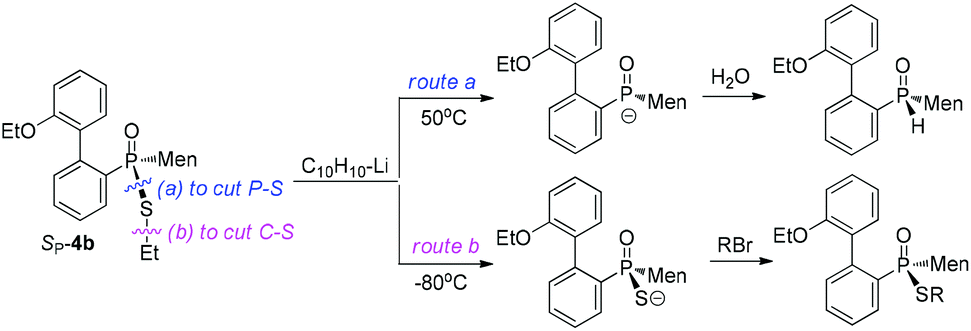 | ||
| Chart 1 Regioselective cleavages of P–S/C–S bonds of phosphonothioate. | ||
Results and discussion
The reaction of 6-chloro-6H-dibenzo[c,e][1,2]oxaphosphinine 1 (CDOP) with (L)-menthyl magnesium chloride,13 followed by treatment of the product of this reaction with sulfur, afforded 6-menthyl-6H-dibenzo[c,e][1,2] oxaphosphinine 6-sulfide 2/2′, as a mixture of two diastereomers. After subjecting this mixture to alkali hydrolysis, a mixture of RP-3′ and SP-3 in a 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65 ratio was obtained, according to the two peaks observed at 76.3 and 82.7 ppm in the 31P-NMR spectrum of the product of the hydrolysis.14SP-3, which produced a signal at 82.7 ppm, was obtained from the recrystallization of the mixture (Scheme 1) and its structure was confirmed using X-ray diffraction (Fig. 1).
:65 ratio was obtained, according to the two peaks observed at 76.3 and 82.7 ppm in the 31P-NMR spectrum of the product of the hydrolysis.14SP-3, which produced a signal at 82.7 ppm, was obtained from the recrystallization of the mixture (Scheme 1) and its structure was confirmed using X-ray diffraction (Fig. 1).
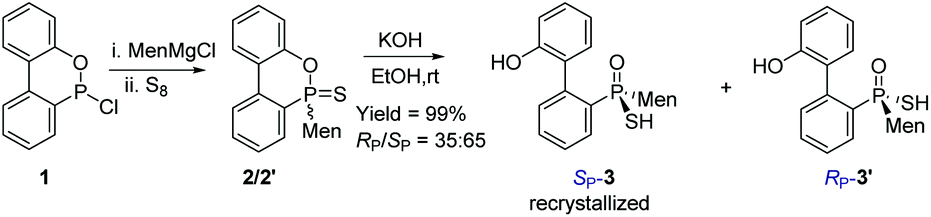 | ||
| Scheme 1 Preparation of optically pure SP-3. | ||
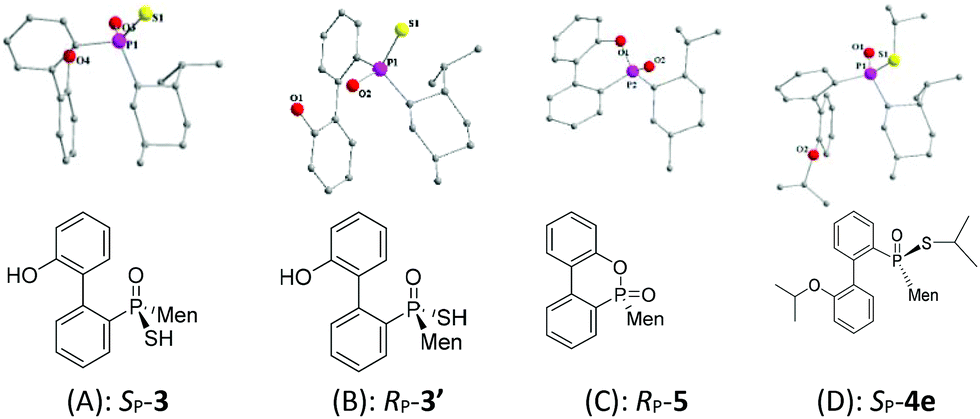 | ||
| Fig. 1 X-ray diffraction structures of (A) SP-3, (B) RP-3′, (C) RP- and (D) SP-4e. | ||
When SP-3 was stirred with ethyl bromide in acetonitrile in the presence of potassium hydroxide, SP-4b was afforded; here, the alkylation occurred on both sulfur and 2′-OH. In the 31P-NMR spectrum of this product, two single peaks were observed, at 65.84 and 60.42 ppm and were assigned to the two stereoisomers resulting from the axial chirality.13,14 The alkylation did not involve phosphorus.
An intramolecularly cyclized RP-5 was detected when the alkylation was carried out in the presence of potassium carbonate. In the presence of methyl iodide, RP-5 formed in near 100% yield, either with potassium hydroxide or potassium carbonate as the base (Scheme 2). It was proposed that sulfur was alkylated before 2′-hydroxyl was alkylated and the resulting alkylthio group was substituted with an oxygen anion to afford RP-5. In fact, heating the solution of SP-3 also afforded RP-5, in which the sulfur atom was similarly substituted as a leaving group. The structure of RP-5 was confirmed using X-ray diffraction (Fig. 1), which indicated that the substitution of the alkylthio or sulfur group occurred by way of a P-retained mechanism according to Berry pseudo-rotation (BPR) theory.15
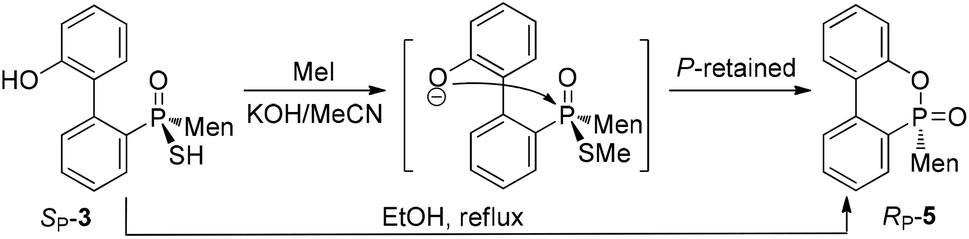 | ||
| Scheme 2 Cyclization of SP-3 to form RP-5. | ||
After the reaction of CDOP with menthyl magnesium chloride, the product was oxidized by exposing it to the air to afford 5/5′ as a mixture of two stereoisomers. Heating the mother liquid shown in Scheme 1 in refluxing ethanol also formed the mixture. After subjecting this mixture to recrystallization, SP-5′ was obtained and was then converted to RP-3′ by treating it with Lawson's reagent and subsequent hydrolysis (Scheme 3). The stereochemistry on phosphorus during the conversions was confirmed using X-ray diffraction (Fig. 1).
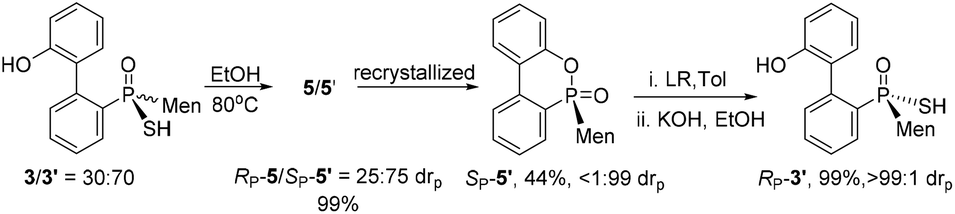 | ||
| Scheme 3 Preparation of optically pure RP-3′. | ||
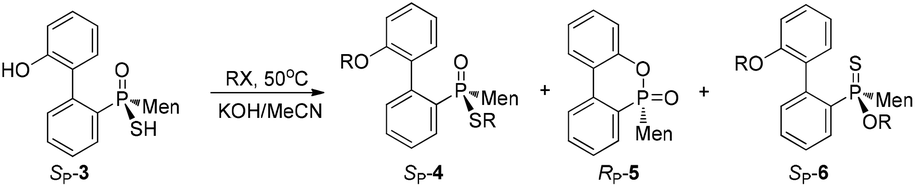
In addition to methyl iodide, aliphatic alkyl bromides were used for the O,S-alkylation of SP-3, affording SP-4 as the major product (entries 2–7, Table 1). During the process, another by-product, namely SP-6, was detected; here, the oxygen of phosphinothioic acid was alkylated. When benzyl bromide was made to react with SP-3, SP-6 was formed as the major product (entries 8 and 9).16 The benzyl-containing SP-4 was obtained from the reaction with benzyl chloride (entries 10–12, Table 1).
|
|
|||
|---|---|---|---|
| Entry | RX | S P-4, Yield % (drA)a | Other products, yielda (%) |
| a Typical procedure: Bromoethane (61.3 μL, 0.822 mmol) was added to a solution of SP-3 (80.0 mg, 0.205 mmol) and potassium hydroxide (14.9 mg, 0.225 mmol) in acetonitrile (1 mL) and the resulting mixture was stirred at 50 °C for 10 hours. The yields and drA values were estimated from the corresponding 31P{1H} NMR spectra of 4, which gave two 31P NMR signals that were assigned as two axial stereoisomers. The axial absolute configuration was not confirmed. b The reaction was carried out at rt in the presence of potassium carbonate. c The O-alkylated product was formed. | |||
| 1 | MeI | R P-5, 99b | |
| 2 | EtBr |
4b, 98 (80:20) |
|
| 3 | nBuBr |
4c, 68 (80:20) |
R P-5, 2; SP-6c, 15 |
| 4 | iBuBr |
4d, 72 (80:20) |
R P-5, 2; SP-6d, 4 |
| 5 | iPrBr |
4e, 95 (81:19) |
|
| 6 | AllylBr |
4f, 77 (82:18) |
S P-6f, 10 |
| 7 | PhCH2CH2Br |
4g, 67 (80:20) |
R P-5, 22 |
| 8 | PhCH2Br | S P-6h, 84c | |
| 9 | oMeC6H4CH2Br | S P-6i, 29c; RP-5, 40 | |
| 10 | pMeC6H4CH2Cl |
4j, 74 (81:19) |
S P-6j, 26 |
| 11 | mMeOC6H4CH2Cl |
4k, 43 (83:17) |
S P-6k, 45 |
| 12 | pClC6H4CH2Cl |
4l, 58 (76:24) |
S P-6l, 40 |
Cleavages of the P–S bond of SP-4b were attempted with Grignard reagents. When SP-4b was stirred with n-butyl magnesium bromide in THF at 50 °C, the reaction did not proceed. At 90 °C in toluene, the substitution of the ethylthio group with an n-butyl group afforded RP-7c in 34% yield. When methyl magnesium iodide was used, RP-7a was similarly obtained in a low yield of 35%. The retention of the configuration on phosphorus was confirmed by comparing this product to RP-7b obtained vide infra, which was consistent with the reported substitution of the alkylthio group on the phosphorus (Scheme 4).12,15
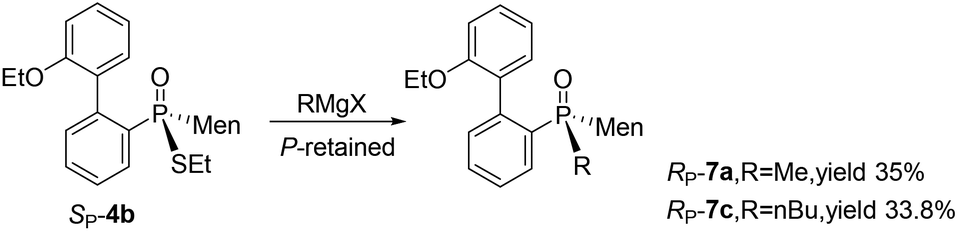 | ||
| Scheme 4 Cleavages of the P–S bond of SP-4b with Grignard reagents. | ||
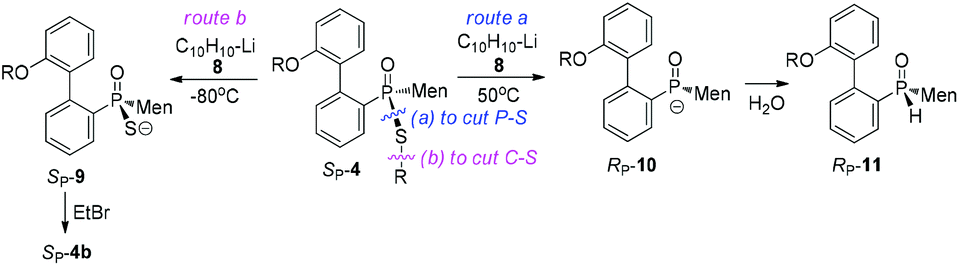
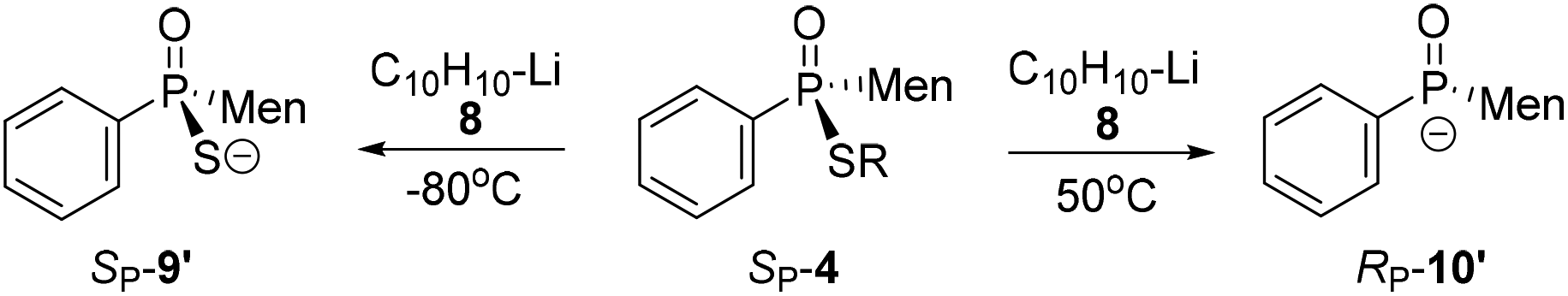
Another strategy explored to break the P–S or C–S bond of SP-4 involved using lithium or lithium-naphthalene 8. When SP-4b was treated with lithium, cleavage of the Et–S bond occurred and afforded 9, which after quenching yielded a peak at 97.97 ppm in its 31P NMR spectrum. When 9 was subjected to S-alkylation with ethyl bromide, it converted back to SP-4b, according to the observation of the peaks at 65.84/60.42 ppm. Meanwhile, the cleavage of the P–S bond formed 10, which was confirmed by the detection of secondary phosphine oxide RP-11 after quenching. RP-11 showed peaks at 36.63 and 33.87 ppm in its 31P NMR spectrum, a result similar to those for the reported compounds.12,13 Thus the P-stereochemistry-retained cleavage of the P–S bond was confirmed. In the absence of naphthalene, the reaction was sluggish and showed only a 57% conversion of SP-4b and the formations of 9 and 10 were detected in a ratio of 46:54 (entry 1 in Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | R of SP-4 | Naph. (equiv.) | Temp. | Conversion of SP-4% (SP-9/RP-10)a |
|
a Typical procedure: a solution of lithium-naphthalene 8 in THF (lithium:naphthalene = 1:1.5, 0.23 ml, 1 M, 0.23 mmol) was added to a solution of SP-4b (50 mg, 0.112 mmol) in THF (1 ml) at the indicated temperature. The yield and SP-9/RP-10 ratio were estimated from the peaks in the corresponding 31P{1H} NMR spectrum.
b Most of the SP-4m was consumed and two unidentified peaks at 52.17 and 49.34 ppm were observed.
|
||||
| 1 | 4b, Et | 0 | 0 °C-rt | 57 (46:54) |
| 2 | 4b, Et | 0.9 | Rt | 99 (58:42) |
| 3 | 4b, Et | 0.9 | 50 °C | 89 (56:44) |
| 4 | 4b, Et | 0.9 | −80 °C | 69 (99:1) |
| 5 | 4b, Et | 1.5 | Rt | 99 (6:94) |
| 6 | 4b, Et | 1.5 | 50 °C | 99 (1:99) |
| 7 | 4b, Et | 1.5 | −80 °C | 84 (99:1) |
| 8 | 4e, iPr | 0.9 | −80 °C | 60 (99:1) |
| 9 | 4j, pMeC6H4CH2 | 0.9 | −80 °C | 99 (99:1) |
| 10 | 4g, PhCH2CH2 | 0.9 | −80 °C | 99 (99:1) |
| 11 | 4m, Ph | 0.9 | Rt | 98 (1:99) |
| 12 | 4m, Ph | 1.5 | Rt | 93 (1:99) |
| 13 | 4m, Ph | 0.9 | −80 °C | 96b |
In the presence of 0.9 equivalent naphthalene, naphthalene-Li 8 reacted with SP-4b at room temperature, giving an excellent conversion, with the detection of 9/10 in a ratio of 50:50 (entry 2, Table 2). Carrying out the reaction at an elevated temperature did not change the ratio (entry 3, Table 2). At −80 °C, it was the C–S bond that was predominantly cleaved, as indicated by the observed 99:1 ratio of 9/10 and 69% conversion of SP-4b (entry 4, Table 2).
In the presence of excess naphthalene, the cleavage of the P–S bond predominantly occurred. When 1.5 equivalents of naphthalene-Li 8 reacted with SP-4b at room temperature, a 6:94 ratio of 9/10 resulted. The ratio was improved to 1:99 at 50 °C (entries 5 and 6, Table 2). However, when the reaction was carried out at −80 °C, 9 was still formed as the major product (entry 7, Table 2).
Similar to the observation for entry 7 in Table 2, the use of S-isopropyl-substituted SP-4e predominantly generated 9 at −80 °C (entry 8, Table 2). The cleavage of the C–S bond was also observed for S-phenylethyl- or S-benzyl-substituted SP-4g or SP-4j (entries 9 and 10, Table 2). The results indicated that the cleavage of the C–S bond predominantly occurred in the presence of insufficient naphthalene or at low temperature.
The facile cleavage of the C–S bond by lithium was unusual. As a comparison, 1,2-diethyldisulfide was treated with 8 under the same conditions as used in entry 2 of Table 2. The generated lithium ethylthiolate was captured with benzyl bromide to afford ethyl(phenyl)sulfide 12 (Scheme 5). The cleavage of the Et–S bond was not observed during the process.
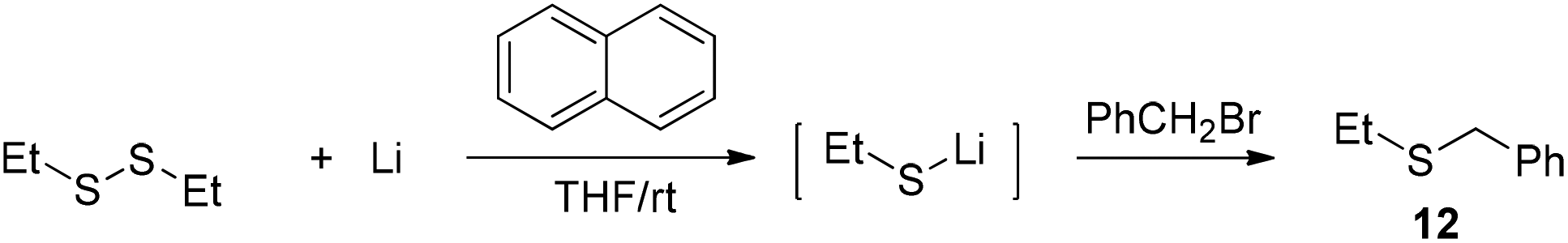 | ||
| Scheme 5 Cleavage of the S–S bond of 1,2-diethyldisulfane. | ||
S-Aromatic-substituted SP-4m, which was prepared from the reaction of 10 with diphenyldisulfide, exhibited a behavior different than the behaviors of SP-4b. Only the P–S bond of SP-4m was cut at room temperature, whether or not naphthalene was supplied in excess. When the reaction was carried out at −80 °C, neither the formation of 9 nor of 10 was obvious (entries 11–13, Table 2).17
The C–S cleavage was further confirmed via the reaction of various phenylphosphinothioates with 8. Similar cleavages of C–S/P–S bonds were observed. At −80 °C, 4n (S-methyl) and 4o (S-Et) predominantly afforded the C–S-cleaved product 9′. For S-phenyl-substituted 4p, 9′ was similarly not detected (Table 3, runs 1–3). When the reaction was carried out at 50 °C, the P–S cleavage took place to form 10′. For each of 4n and 4o, the cleavage of the C–S bond occurred simultaneously with the P–S cleavage, as indicated by the 9′/10′ ratio being about 50:50, which was slightly different than the results in Table 2. The reaction using 4p predominantly afforded 10′, although 9′ was also detected (runs 4–6).
|
|
||||
|---|---|---|---|---|
| Entry | R of Sp-4 | Naph. (equiv.) | Temp. | Conversion of Sp-4% (Sp-9′/Rp-10′) |
|
a Major byproduct was observed at 49 ppm on 31P NMR spectrum whose structure was not confirmed.
b
4o was used in 67:33 dr, and 9′ was detected as two stereoisomers in the corresponding ratio.
|
||||
| 1 | 4n, Me | 0.9 | −80 °C | 94 (99:1)a |
| 2 | 4o, Et | 0.9 | −80 °C | 94 (99:1)a,b |
| 3 | 4p, Ph | 0.9 | −80 °C | 86a |
| 4 | 4n, Me | 1.5 | 50 °C | 99 (42:58) |
| 5 | 4o, Et | 1.5 | 50 °C | 99 (47:53) |
| 6 | 4p, Ph | 1.5 | 50 °C | 98 (9:91)a |
The α-carbon of the S-isopropyl group in SP-4e was thought to be subjected to a level of spatial hindrance greater than that of S-ethyl in SP-4b. The attack of the α-carbon, or the sulfur, of 4e was supposed to be more hindered than that of 4b. However, SP-4b and SP-4e gave similar results when treated with 8 (entries 7 and 8), which indicated that the cleavage of the C–S bond did not occur via a direct nucleophilic attack on the α-carbon or sulfur.
Instead, the adjacent phosphorus was then proposed to be involved in the cleavage of C–S bond. The mechanism for this proposal is shown in Scheme 6. According to this proposal, in the presence of 8, the phosphorus of SP-4b was attacked by lithium, probably on its vacant d-orbital, forming intermediate 14. After the negative charge on the O substituent of the phosphorus was transferred to the S-alkyl substituent, via a possible cyclic transition state, the alkyl moiety dissociated from the sulfur to afford 15, which was then converted to 9. For 4j, a relatively stable benzyl anion was removed, also affording 9 (entry 9 of Table 2). A similar electron-transfer to the phenylthio group cannot occur, so that SP-4m did not afford 9 at either −80 °C or room temperature.
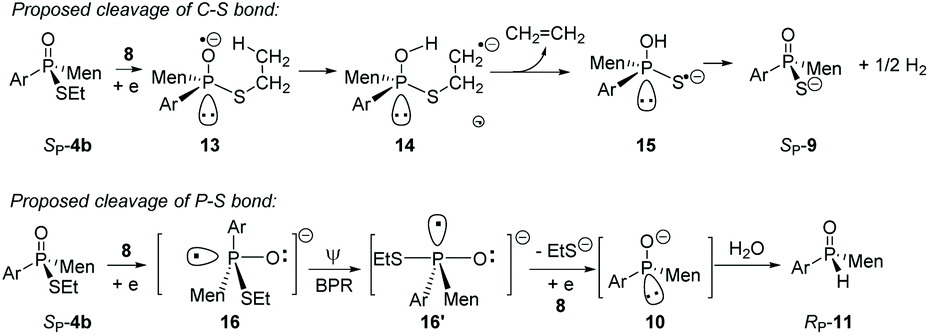 | ||
| Scheme 6 Proposed mechanism for the cleavages of P–S and C–S bonds of SP-4b. | ||
In the case of P–S cleavage, according to the proposed mechanism, 16 was converted to 16′via a Berry pseudo-rotation (BPR). Dissociation of the alkylthio substituent from the axial position of the bipyramidal structure of 16′ afforded phosphorus anion 10 in a P-stereochemistry-retained manner. As previously reported, the BPR could be stopped at a low temperature, which was consistent with the P–S cleavage having only occurred at a relatively high temperature.15 The configuration of the phosphorus anion was stabilized by the presence of the menthyl group, so that 10 did not epimerize and 11 formed with excellent stereoselectivity.18
Metallic lithium has been thought to be more active than naphthalene-lithium 8. However, the poor dispersion of metallic lithium led to a heterogeneous reaction and low conversion of SP-4b. When naphthalene was used in excess, the reduced activity of lithium also resulted in a more regioselective attack of the P–S/C–S bond.
Subsequent alkylations of SP-10 with various alkyl halides were performed and the results are summarized in Table 4. Aliphatic primary alkyl halides reacted smoothly with SP-10 to afford RP-7a to RP-7d. As seen in ESI†RP-7a and RP-7c obtained herein yielded the same spectral data as that obtained in Scheme 4. Secondary alkyl halides such as cyclohexyl bromide did not react with SP-10, but allyl chloride could be used for the alkylation and formed RP-7e. The reaction of SP-10 with variously substituted benzyl halides afforded 7f to 7k in good to excellent yields.
|
|
|||
|---|---|---|---|
| R | 7 yield %, (drA) | R | 7 yield %, (drA) |
| Me |
7a, 82, (69:31) |
oMeC6H4CH2 |
7g, 56%, (72:28) |
| Et |
7b, 85, (53:47) |
pMeC6H4CH2 |
7h, 96, (64:36) |
| nBu |
7c, 94, (51:49) |
p-tBuC6H4CH2 |
7i, 88, (70:30) |
| iBu |
7d, 95, (43:57) |
mMeOC6H4CH2 |
7j, 86, (63:37) |
| Allyl |
7e, 50, (84:16) |
oCIC6H4CH2 |
7k, 72, (74:26) |
| PhCH2 |
7f, 85, (65:35) |
pCIC6H4CH2 |
7l, 90, (64:36) |
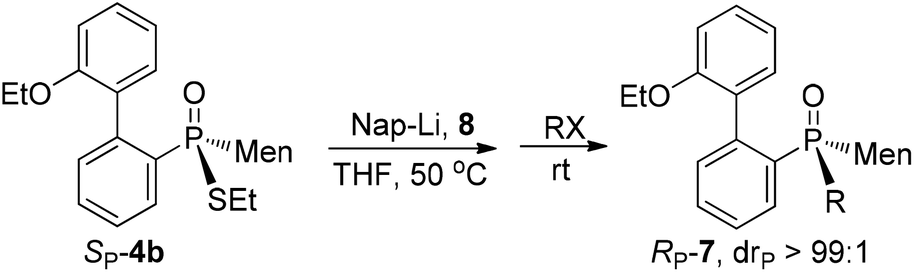
After cleavage of the P–S bond of SP-4b, the resulting SP-10 was reacted with formaldehyde to afford α-hydroxy phosphine oxide RP-17. The configuration about the phosphorus was thought to have been retained, based on the reported addition of secondary phosphine oxide to aldehyde (Scheme 7).19
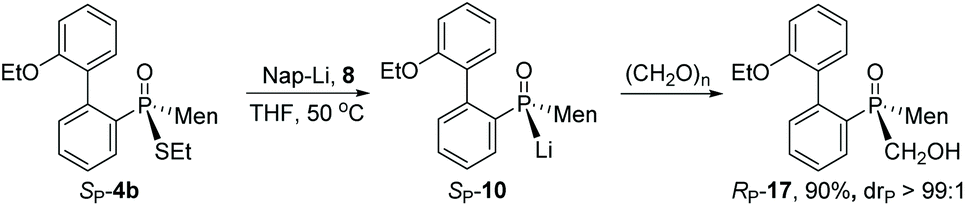 | ||
| Scheme 7 Reaction of 10 with formaldehyde. | ||
The stereochemistry during the above conversions was confirmed using X-ray diffraction. As seen in Fig. 1, the structures of both SP-3 and RP-3′ were confirmed and SP-3 and RP-3′ were converted, respectively, to RP-5 and SP-5′ upon being heated; and the structure of RP-5 was also confirmed. During the cyclization, the configuration about the phosphorus was retained. The S-alkylation of SP-3 did not involve the phosphorus and thus 4e displayed the same SP-structure as did SP-3. Upon comparing the X-ray structures of SP-4e and RP-7b,13 the replacement of the alkylthio group with an alkyl group, via either a Grignard reagent or lithium, was confirmed to have occurred in a P-stereochemistry-retained manner.
Conclusions
Menthyl (2-hydroxy biphenyl)phosphinothioic S-acid 3 was formed from the treatment of a P(III) specie using sulfur and alkali hydrolysis. After subjecting the product of the hydrolysis to recrystallization, the SP-stereoisomer was successfully obtained and could be converted to various optically pure O,S-alkylated SP-4 molecules. The regioselectivities of the cleavages of the P–S/C–S bonds of SP-4 were studied using lithium or naphthalene lithium and were found to depend on the relative amounts of naphthalene/lithium used and on the temperature applied. At low temperature or in the absence of naphthalene, the cleavage of the C–S bond occurred. In the presence of excess naphthalene or at a relatively high temperature, the P–S bond was cleaved. The unusual cleavage of C–S bond was thought to be promoted by the adjacent phosphorus. The cleavage of the P–S bond could be used for the formation of a C–P bond in a P-stereochemistry-retained manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Natural Science Foundation of China (grant no. 21802062) and the Natural Science Foundation of Shandong Province (grant no. ZR2016BM18 and ZR2018PB008).Notes and references
- (a) C. E. Berkman, D. A. Quinn and C. M. Thompson, Interaction of Acetylcholinesterase with the Enantiomers of Malaoxon and Isomalathion, Chem. Res. Toxicol., 1993, 6, 724–730 Search PubMed; (b) C. E. Berkman, S. Ryu, D. A. Quinn and C. M. Thompson, Kinetics of the Postinhibitory Reactions of Acetylcholinesterase Poisoned by chiral Isomalathion: a Surprising Nonreactivation Induced by the RP Stereoisomers, Chem. Res. Toxicol., 1993, 6, 28–32 Search PubMed; (c) C. M. Thompson, S. Ryu and C. E. Berkman, Consequence of phosphorus stereochemistry upon the postinhibitory reaction kinetics of acetylcholinesterase poisoned by phosphorothiolates, J. Am. Chem. Soc., 1992, 114, 10710–10715 CrossRef CAS; (d) Y. C. Yang, J. A. Baker and J. R. Ward, Decontamination of chemical warfare agents, Chem. Rev., 1992, 92, 1729–1743 CrossRef CAS; (e) J. M. Battershill, P. M. Edwards and M. K. Johnson, Toxicological assessment of isomeric pesticides: a strategy for chiral organophosphorus (OP) compounds for delayed polyneuropathy in a regulatory setting, Food Chem. Toxicol., 2004, 42, 1279–1285 CrossRef CAS.
- (a) L. Y. Kuo, D. C. Baker, A. K. Dortignacq and K. M. Dill, Phosphonothiotate Hydrolysis by Molybdocene Dichlorides: Importance of Metal Interaction with the Sulfur of the Thiolate Leaving Group, Organometallics, 2013, 32, 4759–4765 CrossRef CAS; (b) B. B. Dhar, D. R. Edwards and R. S. Brown, A Study of the Kinetics of La3+−Promoted Methanolysis of S-Aryl Methylphosphonothioates: Possible Methodology for Decontamination of EA 2192, the Toxic Byproduct of VX Hydrolysis, Inorg. Chem., 2011, 50, 3071–3077 CrossRef CAS; (c) L. Bromberg, N. Pomerantz, H. Schreuder-Gibson and T. A. Hatton, Degradation of Chemical Threats by Brominated Polymer Networks, Ind. Eng. Chem. Res., 2014, 53, 18761–18774 CrossRef CAS; (d) I. Onyido, K. Swierczek, J. Purcell and A. C. Hengge, A Concerted Mechanism for the Transfer of the Thiophosphinoyl Group from Aryl Dimethylphosphinothioate Esters to Oxyanionic Nucleophiles in Aqueous Solution, J. Am. Chem. Soc., 2005, 127, 7703–7711 CrossRef CAS.
- (a) R. K. Haynes, T. L. Au-Yeung, W. K. Chan, W. L. Lam, Z. Y. Li, L. Y. Yeung, A. S. C. Chan, P. Li, M. Koen, C. R. Mitchell and S. C. Vonwiller, Reaction of Metallated tert–Butyl(phenyl)phosphane Oxide with Electrophiles as a Route to Functionalized Tertiary Phosphane Oxides: Alkylation Reactions, Eur. J. Org. Chem., 2000, 3205–3216 CrossRef CAS; (b) W. Perlikowska, M. Gouygou, M. Mikolajczyk and J. C. Daran, Enantiomerically pure disulfides: key compounds in the kinetic resolution of chiral PIII-derivatives with stereogenic phosphorus, Tetrahedron: Asymmetry, 2004, 15, 3519–3530 CrossRef CAS; (c) A. Alexakis and A. Chauvin, TADDOL organophosphorus derivatising agents for the determination of the enantiomeric excess of chiral alcohols and carboxylic acids by 31P and 1H NMR spectroscopy, Tetrahedron: Asymmetry, 2000, 11, 4245–4248 CrossRef CAS; (d) A. Alexakis, J. C. Frutos, S. Mutti and P. Mangeney, Chiral Diamines for a New Protocol To Determine the Enantiomeric Composition of Alcohols, Thiols, and Amines by 31P, 1H, 13C, and 19F NMR, J. Org. Chem., 1994, 59, 3326–3334 CrossRef CAS; (e) S. Reymond, J. M. Brunel and G. Buono, New chiral organophosphorus derivatizing agent for the determination of enantiomeric composition of chloro- and bromohydrins by 31P NMR spectroscopy, Tetrahedron: Asymmetry, 2000, 11, 1273–1278 CrossRef CAS; (f) Z. Skrzypczynski and J. Michalski, Stereoselective Synthesis and Stereochemistry of Optically Active tert-Butylphenylphosphine Sulfide, J. Org. Chem., 1988, 53, 4549–4551 CrossRef CAS; (g) R. K. Haynes, R. N. Freeman, C. R. Mitchell and S. C. Vonwiller, Preparation of Enantiomerically Pure Tertiary Phosphine Oxides from, and Assay of Enantiomeric Purity with, (RP)- and (SP)-tert-Butylphenylphosphinothioic Acids, J. Org. Chem., 1994, 59, 2919–2921 CrossRef CAS.
- (a) W. B. Farnham, K. Mislow, N. Mandel and J. Donohue, Stereochemistry of Methanolysis of Menthyl S-Methyl Phenylphosphonothioate, J. Chem. Soc., Chem. Commun., 1972, 120–121 RSC; (b) T. Kawashima, S. Kojima and N. Inamoto, The Optically Active Phosphinodithioates. Synthesis and Conversion to the Optically Active Phosphine Sulfides, Chem. Lett., 1989, 18, 849–852 CrossRef; (c) K. E. Debruin, C. W. Tang, D. M. Johnson and R. L. Wilde, Kinetic facial selectivity in nucleophilic displacements at tetracoordinate phosphorus: kinetics and stereochemistry in the reaction of sodium ethoxide with O,S-dimethyl phenylphosphonothioate, J. Am. Chem. Soc., 1989, 111, 5871–5879 CrossRef CAS.
- (a) T. Miura, H. Yamada, S. Kikuchi and T. Imamoto, Synthesis and Reactions of Optically Active Secondary Dialkylphosphine-Boranes, J. Org. Chem., 2000, 65, 1877–1880 CrossRef CAS; (b) Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Synthesis of Unsymmetrical Tertiary Phosphine Oxides via Sequential Substitution Reaction of Phosphonic Acid Dithioesters with Grignard Reagents, Org. Lett., 2017, 19, 3899–3902 CrossRef CAS.
- (a) K. E. G. Zon and K. DeBruin, Naumann and K. Mislow, Stereospecific desulfurization of acyclic phosphine sulfides with hexachlorodisilane and the alkaline hydrolysis of monoalkoxy- and monoalkylthiophosphonium salts, J. Am. Chem. Soc., 1969, 91, 7023–7027 CrossRef; (b) E. J. Corey, Z. L. Chen and G. J. Tanoury, A new and highly enantioselective synthetic route to P-chiral phosphines and diphosphines, J. Am. Chem. Soc., 1993, 115, 11000–11001 CrossRef CAS; (c) W. Tang, W. Wang, Y. Chi and X. Zhang, A Bisphosphepine Ligand with Stereogenic Phosphorus Centers for the Practical Synthesis of β-Aryl-β-Amino Acids by Asymmetric Hydrogenation, Angew. Chem., Int. Ed., 2003, 42, 3509–3511 CrossRef CAS.
- J. Omelanczuk and M. Mikolajczyk, Unexpected Opposite Stereochemistries and Different Mechanisms of Nucleophilic Substitution Reactions of Homochiral tertButylphenylthiophosphinoyl Chloride and Bromide, J. Chem. Soc., Chem. Commun., 1994, 2223–2224 RSC.
- W. B. Farnham, R. K. Murray and K. Mislow, Retention stereochemistry in a Grignard displacement reaction at chiral phosphorus. Absolute configuration of O-menthyl S-methyl phenylphosphonothiolate, J. Am. Chem. Soc., 1971, 93, 3792–3793 CrossRef.
- W.-M. Wang, L.-J. Liu, L. Yao, F.-J. Meng, Y.-M. Sun, C.-Q. Zhao, Q. Xu and L.-B. Han, Stereospecific Preparations of P-Stereogenic Phosphonothioates and Phosphonoselenoates, J. Org. Chem., 2016, 81, 6843–6847 CrossRef CAS.
- G. Wang, R.-W. Shen, Q. Xu, M. Goto, Y.-F. Zhao and L.-B. Han, Stereospecific Coupling of H-Phosphinates and Secondary Phosphine Oxides with Amines and Alcohols: A General Method for the Preparation of Optically Active Organophosphorus Acid Derivatives, J. Org. Chem., 2010, 75, 3890–3892 CrossRef CAS.
- (a) Q. Xu, C.-Q. Zhao and L.-B. Han, Stereospecific Nucleophilic Substitution of Optically Pure H-Phosphinates: A General Way for the Preparation of Chiral P-Stereogenic Phosphine Oxides, J. Am. Chem. Soc., 2008, 130, 12648–12655 CrossRef CAS; (b) W.-M. Wang, L.-J. Liu, C.- Q. Zhao and L.-B. Han, Diastereoselective Hydrolysis of Asymmetric P–Cl Species and Synthesis of Optically Pure (RP)–(-)–Menthyl H–Phenylphosphinate, Eur. J. Org. Chem., 2015, 2342–2345 CrossRef CAS; (c) J.-J. Ye, B.-X. Yan, J.-P. Wang, J.-H. Wen, Y. Zhang, M.-R. Qiu, Q. Li and C.-Q. Zhao, The construction of three C–P bonds of P-stereogenic tertiary phosphines containing (L)-menthyl, Org. Chem. Front., 2020, 7, 2063–2068 RSC.
- L.-J. Liu, W.-M. Wang, L. Yao, F.-J. Meng, Y.-M. Sun, Q. Li, C.-Q. Zhao and L.-B. Han, Reinvestigation of the Substitutions Reaction of Stereogenic Phosphoryl Compounds: Stereochemistry, Mechanism, and Applications, J. Org. Chem., 2017, 82, 11990–12002 CrossRef CAS.
- Y. Zhang, S.-Z. Nie, J.-J. Ye, J.-P. Wang, M.-M. Zhou, Q. Li and C.-Q. Zhao, Functional Phosphine Derivatives Having Stationary and Flexible Chiralities: Their Preparation and Chirality Controlling, J. Org. Chem., 2019, 84, 8423–8439 CrossRef CAS.
- The two chiral centers on phosphorus and axis derived four stereoisomers. We believed the axial chirality was controlled by P-chirality via the H-bonding between PO and hydroxyl. Therefore only two signals of 3 were observed on 31P NMR spectrum. The similar case could be found in the ref. 13.
- (a) R. S. Berry, Correlation of Rates of Intramolecular Tunneling Processes, with Application to Some Group V Compounds, J. Chem. Phys., 1960, 32, 933–938 CrossRef CAS; (b) L. Y. Kuo and S. K. Glazier, Inorg. Chem., 2012, 51, 328–335 CrossRef CAS; (c) K. E. Debruin, C. l. W. Tang, D. M. Johnson and R. L. Wilde, Kinetic facial selectivity in nucleophilic displacements at tetracoordinate phosphorus: kinetics and stereochemistry in the reaction of sodium ethoxide with O,S-dimethyl phenylphosphonothioate, J. Am. Chem. Soc., 1989, 111, 5871–5879 CrossRef CAS; (d) J. Seckute, J. L. Menke, R. J. Emnett, E. V. Patterson and C. J. Cramer, Ab Initio Molecular Orbital and Density Functional Studies on the Solvolysis of Sarin and O,S-Dimethyl Methylphosphonothiolate, a VX-like Compound, J. Org. Chem., 2005, 70, 8649–8660 CrossRef CAS; (e) J. L. Menke and E. V. Patterson, Quantum mechanical calculations on the reation of ethoxide anion with O,S-dimethyl methylphosphonothiolate, J. Mol. Struct.: THEOCHEM, 2007, 811, 281–291 CrossRef CAS.
- The configuration on P of 6 was inferred based on SP-3 that was converted to 6 without variation of P-configuration. The alkylation product of P–OH probably was P(O)OR or P(S)OR, which was not confirmed.
- New signals at 52.17 and 49.34 ppm on 31P NMR spectrum were observed, which were not ascribed to 9 or 10.
- S.-Z. Nie, Z.-Y. Zhou, J.-P. Wang, H. Yan, J.-H. Wen, J.-J. Ye, Y.-Y. Cui and C.-Q. Zhao, Nonepimerizing Alkylation of H−P Species to Stereospecifically Generate P-Stereogenic Phosphine Oxides: A Shortcut to Bidentate Tertiary Phosphine Ligands, J. Org. Chem., 2017, 82, 9425–9434 CrossRef CAS.
-
R
P-15 was obtained as a mixture of axial stereoisomers, in a ratio of 53:47, as seen its 1H NMR spectrum. Similar reaction could be found in the reference. H. Zhang, Y.-M. Sun, L. Yao, S.-Y. Ji, C.-Q. Zhao and L.-B. Han, Stereogenic Phosphorus-Induced Diastereoselective Formation of Chiral Carbon during Nucleophilic Addition of Chiral H-P Species to Aldehydes or Ketones, Chem. – Asian J., 2014, 9, 1329–1333 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1975582, 1975583, 1975584 and 2010827. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo00891e |
| ‡ These two authors contributed equally to this work |
| This journal is © the Partner Organisations 2021 |