
Rhodium-catalyzed sequential intermolecular hydroacylation and deconjugative isomerization toward diversified diketones†
Guirong
You
ab,
Zhi-Xin
Chang
b,
Jizhong
Yan
*a,
Chengcai
Xia
 b,
Fu-Rong
Li
b and
Hong-Shuang
Li
*b
b,
Fu-Rong
Li
b and
Hong-Shuang
Li
*b
aCollege of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: yjz@zjut.edu.cn
bInstitute of Pharmacology, School of Pharmaceutical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, Taian 271016, China. E-mail: hsli@sdfmu.edu.cn
First published on 3rd November 2020
Abstract
A rhodium-catalyzed tandem intermolecular hydroacylation of terminal alkyne-substituted secondary alcohols with chelating aldehydes and deconjugative isomerization of the resulting α,β-unsaturated ketones for the preparation of synthetically valuable 1,4-diketones is developed. Of note, the completely atom-economical protocol can be extended to the alkynyl alcohols with appropriate carbon chain length, from which 1,5-diketone and 1,6-diketone were obtained involving the short-range deconjugative isomerization. Given its robustness, broad substrate scope, and excellent regioselectivity, this approach provides a promising platform for the synthesis of diverse diketones.
Introduction
Transition–metal-catalyzed hydroacylation opens monumental opportunities for expeditious and atom-economical preparation of a myriad of α,β-unsaturated ketones,1 which are of high value in pharmaceuticals, agrochemicals, and fine chemicals. Especially appealing are those intermolecular hydroacylation reactions of alkyne-containing components with non-chelating and oxygen-, sulfur-, and nitrogen-chelating aldehydes involving metal catalysis (Scheme 1a).2 It is therefore not surprising that the catalytic hydroacylation of alkynes with chelating aldehydes is one of the most promising reaction classes that can lead to more complicated molecules through further decoration with heteroatom substituents in a tandem manner.3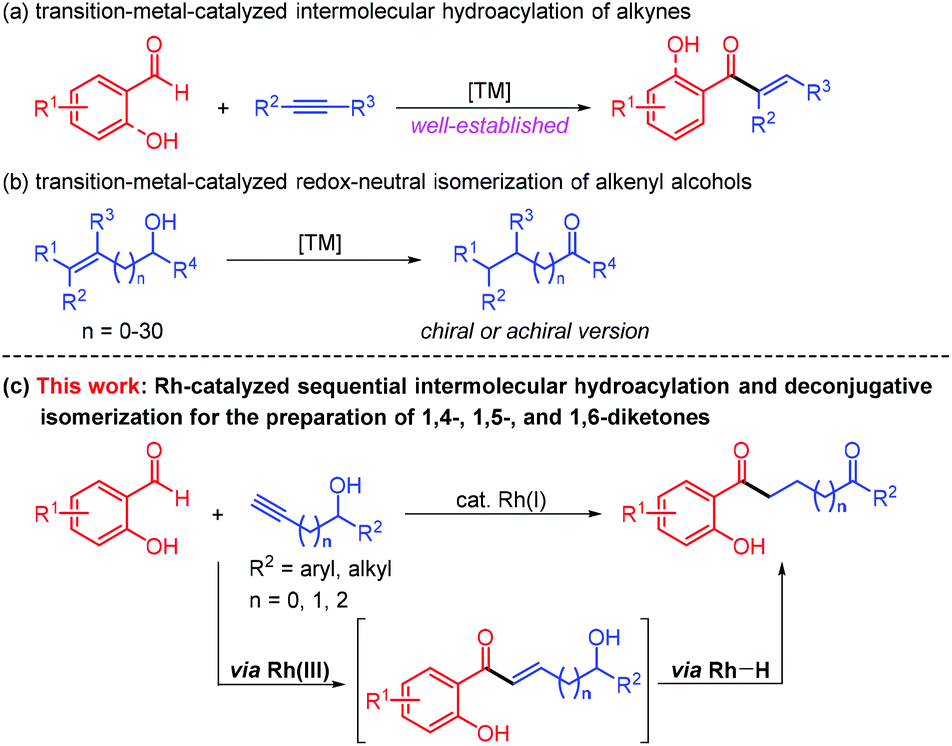 | ||
| Scheme 1 Background and synopsis of this work. | ||
The redox-neutral isomerization of alkenyl alcohols has provided a robust and practical alternative that enables the synthesis of versatile carbonyl compounds, demonstrating perfect atom- and step-economy and excellent functional group tolerance (Scheme 1b). In this respect, the catalytic isomerization of allylic alcohols to ketones relying on various transition–metal complexes is among the most commonly explored processes.4 A few illustrations of transition–metal-catalyzed short- and long-range isomerization of linear and branched alkenyl alcohols have also been successfully implemented through a chain-walking process consisting of successive migratory insertion/β-H elimination.5 For example, Yang, Zhao and co-workers very recently achieved a rhodium-catalyzed remote isomerization of alkene-tethered aliphatic and aromatic secondary alcohols into ketones under mild reaction conditions.6 Meanwhile, metal-catalyzed enantioselective redox isomerization reactions of allylic, homoallylic, and bishomoallylic secondary alcohols were described by Zhao and others.7 Although these protocols broadened the scope of the redox-neutral isomerization of alkenyl alcohols significantly, the deconjugative isomerization of α,β-unsaturated carbonyl compounds with the intramolecular alcohol moieties remains severely limited, and only an impressive palladium-catalyzed isomerization process with olefin migration achieved over thirty carbon atoms was disclosed by the Mazet group.8
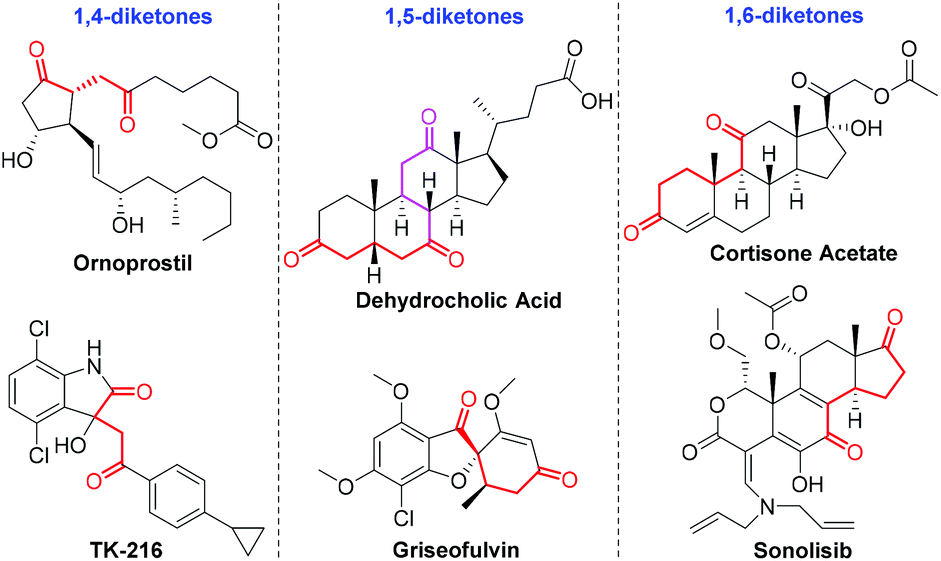
Diketones constitute a significant component in pharmaceutically active compounds and FDA-approved drugs (Fig. 1).9 Consequently, conscientious efforts from the organic chemistry community have been dedicated to the highly efficient and divergent synthesis of the diketones utilizing transition–metal-catalyzed hydroacylation. In 2019, Zhang and co-workers developed a rhodium-catalyzed intermolecular hydroacylation of alkynyl cyclobutanols with salicylaldehydes followed by semipinacol rearrangement to access 2-(2-oxoethyl)cyclopentanones in an atom-economical manner.10 A rhodium-catalyzed hydroacylation of alkenyl cyclobutanols with simple aldehydes for preparing 1,5-diketones via a ring-opening/transfer hydrogenation/hydroacylation cascade was also reported by the same group.11 It should be mentioned that the two examples were achieved by the unleashing of cyclobutyl ring strain. Despite these remarkable advances, the transformation involving the alkynyl alcohol components for the preparation of a diverse range of diketones by virtue of the transition–metal-catalyzed deconjugative isomerization has thus far not been reported to the best of our knowledge. In line with our group's continuing interest in developing rhodium(I)-catalyzed carbon–carbon bond forming reactions,12 we questioned whether the rhodium(I)-catalyzed intermolecular hydroacylation of terminal alkyne-substituted secondary alcohols with chelating aldehydes followed by the deconjugative isomerization of the resulting α,β-unsaturated ketones (Scheme 1c), which likely involves Rh(III) and Rh–H species, could be achieved via a one-pot reaction sequence. If successful, such a protocol for the intermolecular C–C bond formation would offer an opportunity for the discovery of the unusual deconjugative isomerization, and would provide a new platform for accessing diverse diketones.
 | ||
| Fig. 1 Selected pharmaceutically active molecules containing diketone scaffold. | ||
Results and discussion
Our investigations began by examining the reaction of salicylaldehyde (1a, 1.0 equiv.) with 1-phenylprop-2-yn-1-ol (2a, 1.5 equiv.) in the presence of the [Rh(COD)Cl]2/(p-Me-C6H4)3P/K2CO3 catalytic system under nitrogen atmosphere in anhydrous toluene (0.2 M) at 110 °C for 24 h. Encouragingly, the desired 1,4-diketone 3aa could be isolated in 33% yield (Table 1, entry 1). However, only a trace amount of the product was detected while using [Rh(C2H4)2Cl]2 as the catalyst (entry 2). Further screening of other Rh(I) catalysts demonstrated that [Rh(COD)OH]2 provided the optimal result (entries 3–6). On the other hand, using PPh3 as a ligand slightly decreased yield for 3aa was obtained (entry 7). In sharp contrast, no expected 1,4-diketone could be detected when tricyclohexylphosphane was introduced in the coupling reaction (entry 8), which might be attributed to the incapability of the formation of a stable coordinatively saturated rhodium complex. The employment of Xantphos, dppb, dppf, DPEPhos, and (±)-BINAP as ligands also provided the desired diketone, albeit with relatively lower yields (entries 9–13). We speculated that the bulkier phosphine ligands were not beneficial in promoting the model reaction. It was found that prolonging the reaction time from 24 to 36 h led to no improvement of the yield (entry 14). Elevating the temperature to 120 °C diminished the product yield to 43%, while lowering the temperature to 100 °C gave a catalytic performance comparable to that achieved at 110 °C (entries 15 and 16). Furthermore, performing the coupling reaction at a lower concentration (0.1 M) resulted in a similar yield of 3aa (entry 17). Switching the ratio of 1a to 2a suffered from an inferior chemical yield (entry 18). Finally, the 1,4-diketone yield was improved to 71% when the ratio of the aldehyde to the propargyl alcohol was set at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (entry 19).
:2 (entry 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Rh | Ligand | Temp. | Time | Yieldb (%) |
| a Unless specified otherwise, all reactions were performed with 1a (0.2 mmol, 1.0 equiv.) and 2a (1.5 equiv.) in anhydrous toluene (1.0 mL) under N2 atmosphere. b Yield of isolated product. c 2.0 mL of toluene (0.1 M) was used. d The reaction was performed using 1.5 equiv. of 1a and 1.0 equiv. of 2a (0.2 mmol). e 2.0 equiv. of 2a (0.4 mmol) was used. | |||||
| 1 | [Rh(COD)Cl]2 | (p-Me-C6H4)3P | 110 °C | 24 h | 33 |
| 2 | [Rh(C2H4)2Cl]2 | (p-Me-C6H4)3P | 110 °C | 24 h | Trace |
| 3 | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 110 °C | 24 h | 62 |
| 4 | Rh(PPh3)3Cl | (p-Me-C6H4)3P | 110 °C | 24 h | 26 |
| 5 | Rh(COD)2SbF6 | (p-Me-C6H4)3P | 110 °C | 24 h | 43 |
| 6 | Rh(COD)2OTf | (p-Me-C6H4)3P | 110 °C | 24 h | 35 |
| 7 | [Rh(COD)OH]2 | PPh3 | 110 °C | 24 h | 59 |
| 8 | [Rh(COD)OH]2 | PCy3 | 110 °C | 24 h | Trace |
| 9 | [Rh(COD)OH]2 | Xantphos | 110 °C | 24 h | 42 |
| 10 | [Rh(COD)OH]2 | dppb | 110 °C | 24 h | 44 |
| 11 | [Rh(COD)OH]2 | dppf | 110 °C | 24 h | 35 |
| 12 | [Rh(COD)OH]2 | DPEPhos | 110 °C | 24 h | 45 |
| 13 | [Rh(COD)OH]2 | (±)-BINAP | 110 °C | 24 h | 39 |
| 14 | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 110 °C | 36 h | 60 |
| 15 | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 120 °C | 24 h | 43 |
| 16 | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 100 °C | 24 h | 66 |
| 17c | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 110 °C | 24 h | 61 |
| 18d | [Rh(COD)OH]2 | (p-Me-C6H4)3P | 110 °C | 24 h | 47 |
| 19 | [Rh(COD)OH] 2 | (p-Me-C 6 H 4 ) 3 P | 110 °C | 24 h | 71 |
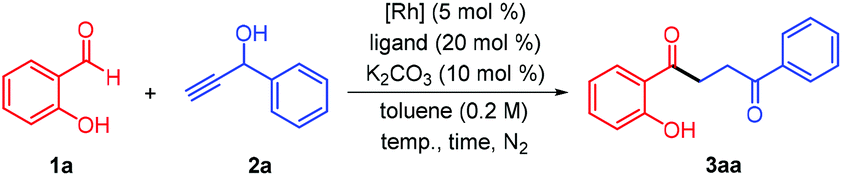
It would be of great significance in this novel rhodium-catalyzed methodology advancement to demonstrate the compatibility of diverse functional groups on the substrates, especially the alkynyl alcohols (Table 2). Accordingly, reactions of salicylaldehyde 1a with the alkynols bearing electron-donating groups at the para (3ab), ortho (3ac), and meta (3ad) positions on the benzene ring displayed good efficiencies under the standard reaction conditions, affording isolated 68%–79% yields of the 1,4-diketones. Meanwhile, electron-neutral 2-naphthyl-substituted alkynol was readily transformed into the desired product 3ae in 79% yield. Extraordinary functional group compatibility for the one-pot synthesis of the 1,4-diketones was observed, with various pharmaceutically relevant groups, including fluoro and chloro (3af), bromo (3ag), trifluoromethyl (3ah), and cyano (3ai), all being well-tolerated. In contrast, the substrate bearing a meta-nitro substituent resulted in only 24% yield of the 1,4-diketone (3aj). Although 2-pyridyl scaffold on the substrate (2k) could not be incorporated into the target product presumably due to the influence of the intramolecular hydrogen bond, 3-thienyl-substituted alkynyl alcohol successfully participated in this cascade reaction to furnish 3al in 68% yield. A discrepant substituent effect was observed in the alkynol reaction partner, since the reactivity of alkyl-substituted secondary alcohols (3am and 3an) was much lower than that of aryl-substituted ones, which potentially form the π–π conjugation between the carbonyl group and the aromatic ring. However, this protocol was not suitable for the transformation of 1-cyclohexylprop-2-yn-1-ol, from which an unidentified side-product was isolated (not shown). The influence of introducing a terminal substituent into the ethynyl moiety on the reaction was also investigated, but we found that the coupling reactions of the methyl- and phenyl-bearing materials (2o and 2p) failed. It is especially noteworthy that when homopropargyl alcohol was subjected to the standard catalytic conditions, the tandem reaction proceeded smoothly to yield the 1,5-diketone (3aq) in synthetically satisfactory yield, highlighting the broad utility of this strategy for the construction of high value-added chemicals. Although 1-phenylpent-4-yn-1-ol engaged in the coupling reaction sluggishly, the 1,6-diketone (3ar) was still afforded in 49% yield after 48 h.
| a Unless specified otherwise, all reactions were performed with salicylaldehyde 1a (0.2 mmol, 1.0 equiv.), alkynyl alcohol 2 (2.0 equiv.), [Rh(COD)OH]2 (5 mol%), (p-Me-C6H4)3P (20 mol%), and K2CO3 (10 mol%) under N2 atmosphere in 1.0 mL of anhydrous toluene at 110 °C for 24 h. b Isolated yield. c The reaction was performed on a 1 mmol scale using 2.5 mol% of [Rh(COD)OH]2 (see the ESI†). d The reaction was performed in 1.5 mL of toluene for 48 h. |
|---|

|
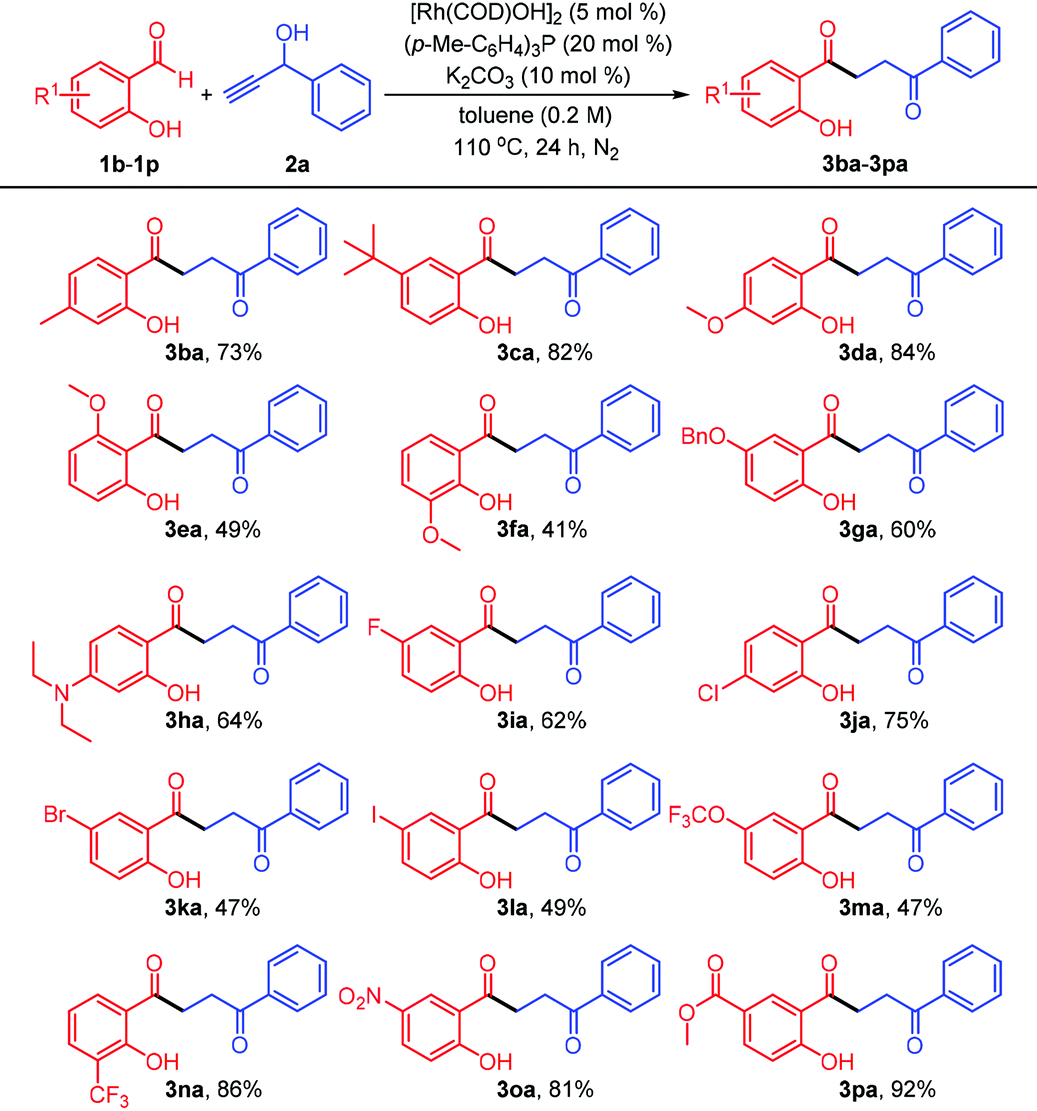
The scope of salicylaldehyde reactant was subsequently explored under the standard catalytic conditions (Table 3). Salicylaldehyde derivatives encompassing various electron-donating substituents, such as alkyl (3ba and 3ca), alkoxy (3da–3ga), and diethylamino (3ha), all gave the corresponding 1,4-diketone products, although the yields of 3ea and 3fa were remarkably lower than those from the reactions of meta-substituted components, presumably due to unfavorable steric hindrance of ortho-methoxy group. Unfortunately, the reaction with 2,4-dihydroxybenzaldehyde under rhodium(I) catalysis did not efficiently afford the expected 1,4-diketone (not shown). To our delight, the aldehydes bearing four kinds of halogen atoms were all compatible with this rhodium-catalyzed system. For instance, the fluoro- and chloro-substituted salicylaldehydes reacted smoothly with the propargyl alcohol 2a, providing the acceptable yields of 62% and 75%, respectively. The 5-bromo- and 5-iodosalicylaldehydes were also successfully coupled with the alkynol, leaving the bromo and iodo groups untouched (3ka and 3la) for further elaboration of more complex and value-added molecules. Notably, the substrates with electron-deficient groups did not influence the outcome of the transformation. Although exposure of the aldehyde containing a trifluoromethoxy group to the system delivered the 1,4-diketone (3ma) with a low yield, the components possessing trifluoromethyl (3na), nitro (3oa), and carbomethoxy (3pa) groups were converted into the desired compounds in good to excellent yields (up to 92%), demonstrating the robustness of this Rh(I)-catalyzed methodology.
| a Unless specified otherwise, all reactions were performed with salicylaldehyde 1 (0.2 mmol, 1.0 equiv.), 1-phenylprop-2-yn-1-ol 2a (2.0 equiv.), [Rh(COD)OH]2 (5 mol%), (p-Me-C6H4)3P (20 mol%), and K2CO3 (10 mol%) under N2 atmosphere in 1.0 mL of anhydrous toluene at 110 °C for 24 h. b Isolated yield. |
|---|
|
To provide further evidence for the mechanism of the rhodium-catalyzed intermolecular transformation, we performed a set of control experiments in combination with isotopic labeling studies (Scheme 2). In line with our previous observations,12a,c benzaldehyde (1q) was not amenable for this transformation (Scheme 2a). Subsequently, the progress of the catalytic reactions was monitored by 1H NMR spectroscopy. Although 1-phenylprop-2-yn-1-ol (2a) could be converted into α,β-unsaturated ketone (4) under the developed reaction conditions,13 the product was not detected in this rhodium-catalyzed system (Scheme 2b). More importantly, when the coupling of salicylaldehyde 1a with 2a was quenched after 5 min, the hydroacylation product 3aa′ was detected in 74% NMR yield, which could be further isomerized to the target 1,4-diketone 3aa in another reaction quenched after 30 min (Scheme 2c). These results unambiguously imply that the intermolecular hydroacylation proceeds readily prior to the deconjugative isomerization, consistent with the reported literature.10 Subjection of 5-tert-butylsalicylaldehyde-d (d-1c) instead of salicylaldehyde-d to the reaction conditions afforded 74% yield of d-3ca in which 34% and 32% deuteration were found at the α-C and β-C positions of the benzoyl group, respectively (Scheme 2d). Finally, the reaction of 1a with the deuterated alkynol d-2s was performed, and deuterium incorporation occurred at all sites on the alkyl chain of the 1,5-diketone (Scheme 2e). These results indicate that the reaction undergoes a chain-walking deconjugative isomerization process.6
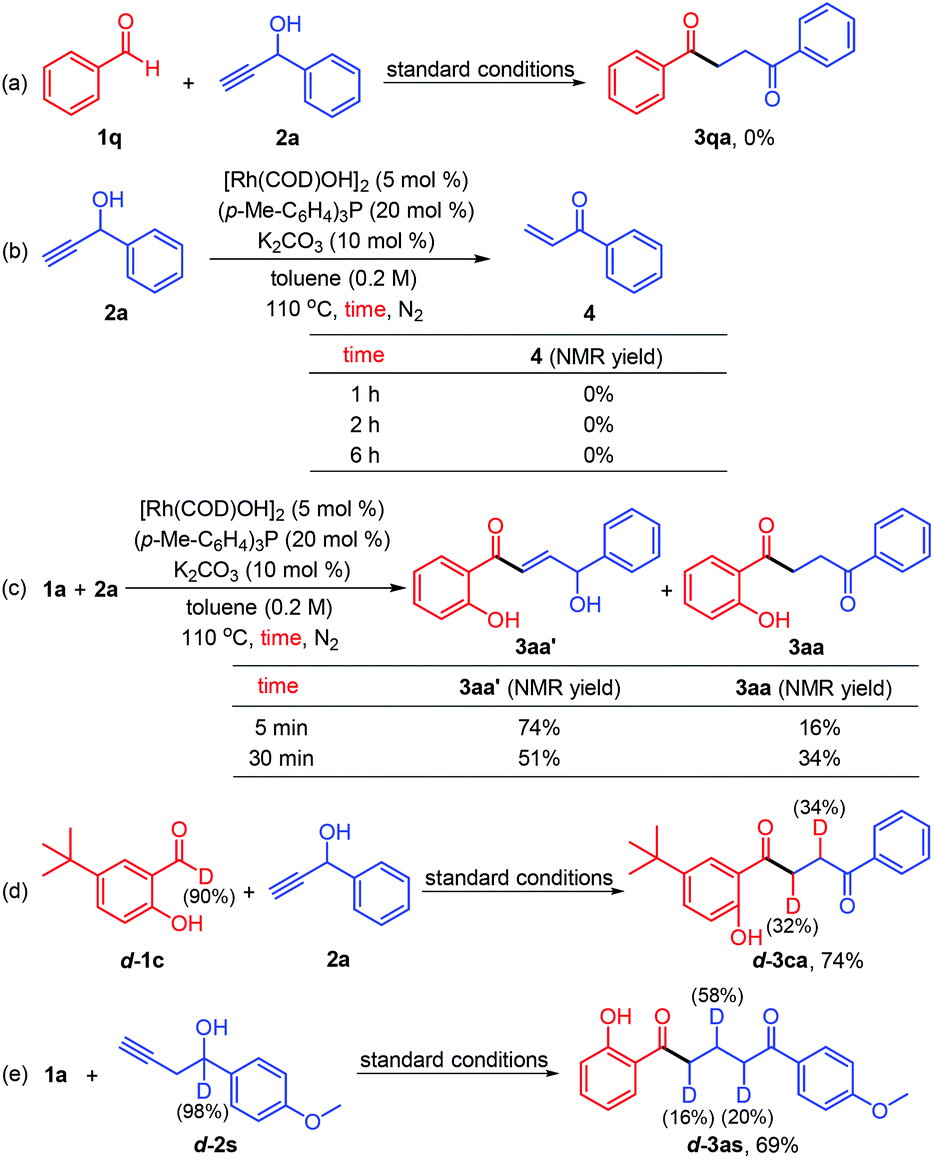 | ||
| Scheme 2 Mechanistic investigation. | ||
On the basis of an array of mechanistic evidence presented above, a plausible working hypothesis for this rhodium-catalyzed synthesis of diketones is described in Scheme 3. Initially, a typical intermolecular hydroacylation of the alkynyl alcohol 2q with salicylaldehyde 1a occurs rapidly to deliver α,β-unsaturated ketone IIIvia the acylrhodium(III) species.3 Following the reaction of the intermediate with the rhodium catalyst in the presence of K2CO3 to generate alkyloxyrhodium(I) intermediate IV,6,12b,14β-hydride elimination liberates the active Rh–H species, which can insert both in the α- and β-positions of the unsaturated ketone to afford Va and Vb, respectively. This process is also consistent with the palladium-catalyzed deconjugative isomerization, demonstrating the ambiphilic nature of the in situ generated metal hydride species.8 Note that Vb could be transformed into Va through a second β-H elimination/migratory insertion.6 Subsequently, β-H elimination of Va gives the allylic alcohol VI and reproduces the Rh–H species. Finally, the rhodium-catalyzed isomerization of the allylic alcohol leads to the formation of the target 1,5-diketone 3aq.4,6,7
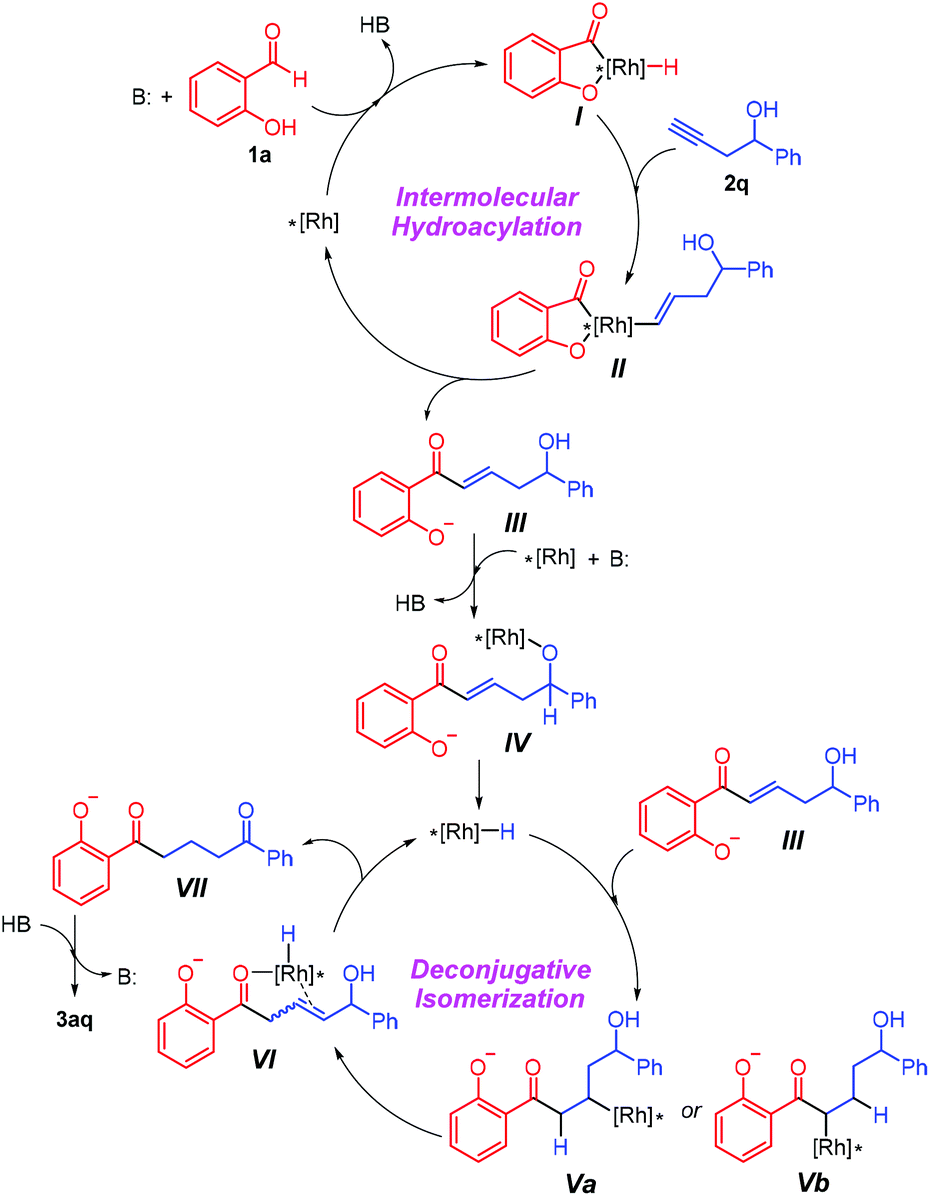 | ||
| Scheme 3 Plausible mechanistic proposal. | ||
Conclusions
In conclusion, a rhodium(I)-catalyzed highly efficient and completely atom-economical synthesis of 1,4-, 1,5-, and 1,6-diketones from readily accessible salicylaldehydes and terminal alkyne-substituted secondary alcohols is reported. The target products are usually obtained in good to excellent yields. Preliminary mechanistic studies reveal that the conversion might proceed in two partial reactions: the intermolecular hydroacylation via the Rh(III) species, followed by the deconjugative isomerization of the resulting α,β-unsaturated ketones via the Rh–H species. This one-pot protocol features good substitution pattern compatibility, broad substrate scope, and excellent regioselectivity, and we believe that it will turn out to be productive for further synthetic transformations providing heterocycle-containing molecules with potential pharmacological properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2019MB003), the Key Research and Development Program of Shandong Province (2018GGX109010), the High-Level Project Cultivation Program of TSMC (2018GCC16), and Academic Promotion Programme of Shandong First Medical University (2019LJ003).Notes and references
- For selected reviews, see: (a) A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker Jr. and L. M. Stanley, Recent Advances in Transition Metal-Catalysed Hydroacylation of Alkenes and Alkynes, Org. Chem. Front., 2016, 3, 639–644 RSC; (b) R. Guo and G. Zhang, Recent Advances in Intermolecular Hydroacylation of Alkenes with Aldehydes through Rhodium Catalysis, Synlett, 2018, 1801–1806 CAS; (c) Y. J. Park, J.-W. Park and C.-H. Jun, Metal-Organic Cooperative Catalysis in C-H and C-C Bond Activation and Its Concurrent Recovery, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS; (d) M. C. Willis, Transition Metal Catalyzed Alkene and Alkyne Hydroacylation, Chem. Rev., 2010, 110, 725–748 CrossRef CAS.
- For selected examples on transition–metal-catalyzed alkyne hydroacylation, see: (a) C. González-Rodríguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller and M. C. Willis, Rhodium-Catalyzed Branched-Selective Alkyne Hydroacylation: A Ligand-Controlled Regioselectivity Switch, Angew. Chem., Int. Ed., 2011, 50, 5134–5138 CrossRef; (b) C. González-Rodríguez, S. R. Parsons, A. L. Thompson and M. C. Willis, Rhodium-Catalysed Intermolecular Alkyne Hydroacylation: The Enantioselective Synthesis of α- and β-Substituted Ketones by Kinetic Resolution, Chem. – Eur. J., 2010, 16, 10950–10954 CrossRef; (c) K. Kokubo, K. Matsumasa, M. Miura and M. Nomura, Rhodium-Catalyzed Coupling Reaction of Salicyl Aldehydes with Alkynes via Cleavage of the Aldehyde C-H Bond, J. Org. Chem., 1997, 62, 4564–4565 CrossRef CAS; (d) V. M. Williams, J. C. Leung, R. L. Patman and M. J. Krische, Hydroacylation of 2-Butyne from the Alcohol or Aldehyde Oxidation Level via Ruthenium Catalyzed C-C Bond Forming Transfer Hydrogenation, Tetrahedron, 2009, 65, 5024–5029 CrossRef CAS; (e) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, R. L. Woodward, A. S. Weller and M. C. Willis, A Second-Generation Catalyst for Intermolecular Hydroacylation of Alkenes and Alkynes Using β-S-Substituted Aldehydes: The Role of a Hemilabile P-O-P Ligand, Angew. Chem., Int. Ed., 2006, 45, 7618–7622 CrossRef CAS; (f) M. C. Willis, H. E. Randell-Sly, R. L. Woodward, S. J. McNally and G. S. Currie, Rhodium-Catalyzed Intermolecular Chelation Controlled Alkene and Alkyne Hydroacylation: Synthetic Scope of β-S-Substituted Aldehyde Substrates, J. Org. Chem., 2006, 71, 5291–5297 CrossRef CAS; (g) G. L. Moxham, H. E. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Intermolecular Alkene and Alkyne Hydroacylation with β-S-Substituted Aldehydes: Mechanistic Insight into the Role of a Hemilabile P-O-P Ligand, Chem. – Eur. J., 2008, 14, 8383–8397 CrossRef CAS; (h) S. Hatanaka, Y. Obora and Y. Ishii, Iridium-Catalyzed Coupling Reaction of Primary Alcohols with 2-Alkynes Leading to Hydroacylation Products, Chem. – Eur. J., 2010, 16, 1883–1888 CrossRef CAS; (i) C.-H. Jun, H. Lee, J.-B. Hong and B.-I. Kwon, Efficient and Selective Hydroacylation of 1-Alkynes with Aldehydes by a Chelation-Assisted Catalytic System, Angew. Chem., Int. Ed., 2002, 41, 2146–2147 CrossRef CAS; (j) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, Intermolecular Hydroacylation: High Activity Rhodium Catalysts Containing Small-Bite-Angle Diphosphine Ligands, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS; (k) J. F. Hooper, S. Seo, F. R. Truscott, J. D. Neuhaus and M. C. Willis, α-Amino Aldehydes as Readily Available Chiral Aldehydes for Rh-Catalyzed Alkyne Hydroacylation, J. Am. Chem. Soc., 2016, 138, 1630–1634 CrossRef CAS; (l) J. Barwick-Silk, S. Hardy, M. C. Willis and A. S. Weller, Rh(DPEPhos)-Catalyzed Alkyne Hydroacylation Using β-Carbonyl-Substituted Aldehydes: Mechanistic Insight Leads to Low Catalyst Loadings that Enables Selective Catalysis on Gram-Scale, J. Am. Chem. Soc., 2018, 140, 7347–7357 CrossRef CAS; (m) T. J. Coxon, M. Fernández, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, Exploiting Carbonyl Groups to Control Intermolecular Rhodium-Catalyzed Alkene and Alkyne Hydroacylation, J. Am. Chem. Soc., 2017, 139, 10142–10149 CrossRef CAS; (n) S. R. Parsons, J. F. Hooper and M. C. Willis, O-Substituted Alkyl Aldehydes for Rhodium-Catalyzed Intermolecular Alkyne Hydroacylation: The Utility of Methylthiomethyl Ethers, Org. Lett., 2011, 13, 998–1000 CrossRef CAS.
- (a) P. Lenden, D. A. Entwistle and M. C. Willis, An Alkyne Hydroacylation Route to Highly Substituted Furans, Angew. Chem., Int. Ed., 2011, 50, 10657–10660 CrossRef CAS; (b) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, 2-Aminobenzaldehydes as Versatile Substrates for Rhodium-Catalyzed Alkyne Hydroacylation: Application to Dihydroquinolone Synthesis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS; (c) X.-W. Du and L. M. Stanley, Tandem Alkyne Hydroacylation and Oxo-Michael Addition: Diastereoselective Synthesis of 2,3-Disubstituted Chroman-4-ones and Fluorinated Derivatives, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS; (d) J. Yang and N. Yoshikai, Cobalt-Catalyzed Annulation of Salicylaldehydes and Alkynes to Form Chromones and 4-Chromanones, Angew. Chem., Int. Ed., 2016, 55, 2870–2874 CrossRef CAS.
- For selected reviews, see: (a) R. Uma, C. Crévisy and R. Grée, Transposition of Allylic Alcohols into Carbonyl Compounds Mediated by Transition Metal Complexes, Chem. Rev., 2003, 103, 27–52 CrossRef CAS; (b) P. Lorenzo-Luis, A. Romerosa and M. Serrano-Ruiz, Catalytic Isomerization of Allylic Alcohols in Water, ACS Catal., 2012, 2, 1079–1086 CrossRef CAS; (c) R. C. van der Drift, E. Bouwman and E. Drent, Homogeneously Catalyzed Isomerization of Allylic Alcohols to Carbonyl Compounds, J. Organomet. Chem., 2002, 650, 1–24 CrossRef CAS; (d) N. Ahlsten, A. Bartoszewicz and B. Martín-Matute, Allylic Alcohols as Synthetic Enolate Equivalents: Isomerisation and Tandem Reactions Catalysed by Transition Metal Complexes, Dalton Trans., 2012, 41, 1660–1670 RSC.
- (a) D. B. Grotjahn, C. R. Larsen, J. L. Gustafson, R. Nair and A. Sharma, Extensive Isomerization of Alkenes Using a Bifunctional Catalyst: An Alkene Zipper, J. Am. Chem. Soc., 2007, 129, 9592–9593 CrossRef CAS; (b) E. Larionov, L. Lin, L. Guénée and C. Mazet, Scope and Mechanism in Palladium-Catalyzed Isomerizations of Highly Substituted Allylic, Homoallylic, and Alkenyl Alcohols, J. Am. Chem. Soc., 2014, 136, 16882–16894 CrossRef CAS; (c) R. Damico and T. Logan, Isomerization of Unsaturated Alcohols with Iron Pentacarbonyl. Preparation of Ketones and Aldehydes, J. Org. Chem., 1967, 32, 2356–2358 CrossRef CAS.
- W. Dong, H. Yang, W. Yang and W. Zhao, Rhodium-Catalyzed Remote Isomerization of Alkenyl Alcohols to Ketones, Org. Lett., 2020, 22, 1265–1269 CrossRef CAS.
- (a) T.-L. Liu, T. W. Ng and Y. Zhao, Rhodium-Catalyzed Enantioselective Isomerization of Secondary Allylic Alcohols, J. Am. Chem. Soc., 2017, 139, 3643–3646 CrossRef CAS; (b) R.-Z. Huang, K. K. Lau, Z. Li, T.-L. Liu and Y. Zhao, Rhodium-Catalyzed Enantioconvergent Isomerization of Homoallylic and Bishomoallylic Secondary Alcohols, J. Am. Chem. Soc., 2018, 140, 14647–14654 CrossRef CAS and references cited therein.
- L. Lin, C. Romano and C. Mazet, Palladium-Catalyzed Long-Range Deconjugative Isomerization of Highly Substituted α,β-Unsaturated Carbonyl Compounds, J. Am. Chem. Soc., 2016, 138, 10344–10350 CrossRef CAS.
- (a) S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Recent Developments in the Synthesis of Prostaglandins and Analogues, Chem. Res., 2007, 107, 3286–3337 CAS; (b) S. Riva, R. Bovara, P. Pasta and G. Carrea, Preparative-Scale Regio- and Stereospecific Oxidoreduction of Cholic Acid and Dehydrocholic Acid Catalyzed by Hydroxysteroid Dehydrogenases, J. Org. Chem., 1986, 51, 2902–2906 CrossRef CAS; (c) J. M. Grandner, R. A. Cacho, Y. Tang and K. N. Houk, Mechanism of the P450-Catalyzed Oxidative Cyclization in the Biosynthesis of Griseofulvin, ACS Catal., 2016, 6, 4506–4511 CrossRef CAS; (d) S. Murahashi, T. Saito, H. Hanaoka, Y. Murakami, T. Naota, H. Kumobayashi and S. Akutagawa, Ruthenium-Catalyzed Oxidative Transformation of Alkenes to α-Ketols with Peracetic Acid. Simple Synthesis of Cortisone Acetate, J. Org. Chem., 1993, 58, 2929–2930 CrossRef CAS; (e) A. E. Garces and M. J. Stocks, Class 1 PI3 K Clinical Candidates and Recent Inhibitor Design Strategies: A Medicinal Chemistry Perspective, J. Med. Chem., 2019, 62, 4815–4850 CrossRef CAS.
- R. Guo, X. Mo and G. Zhang, Synthesis of 2(2-Oxo-2-phenylethyl)cyclopentanone by Rhodium-Catalyzed Tandem Alkynyl Cyclobutanols Hydroacylation and Semipinacol Rearrangement, Org. Lett., 2019, 21, 1263–1267 CrossRef CAS.
- R. Guo and G. Zhang, Expedient Synthesis of 1,5-Diketones by Rhodium-Catalyzed Hydroacylation Enabled by C–C Bond Cleavage, J. Am. Chem. Soc., 2017, 139, 12891–12894 CrossRef CAS.
- (a) H.-S. Li, S.-C. Lu, Z.-X. Chang, L. Hao, F.-R. Li and C. Xia, Rhodium-Catalyzed Ring-Opening Hydroacylation of Alkylidenecyclopropanes with Chelating Aldehydes for the Synthesis of γ,δ-Unsaturated Ketones, Org. Lett., 2020, 22, 5145–5150 CrossRef CAS; (b) H.-S. Li, G. Guo, R.-Z. Zhang and F. Li, Rhodium-Catalyzed Synthesis of α,β-Unsaturated Ketones through Sequential C–C Coupling and Redox Isomerization, Org. Lett., 2018, 20, 5040–5043 CrossRef CAS; (c) H.-S. Li, Y. Xiong and G. Zhang, Rhodium-Catalyzed Annulations of 1,3-Dienes and Salicylaldehydes/2-Hydroxybenzyl Alcohols Promoted by 2-Ethylacrolein, Adv. Synth. Catal., 2018, 360, 4246–4251 CrossRef CAS; (d) S.-C. Lu, Z.-X. Chang, Y.-L. Xiao and H.-S. Li, Regio- and Stereoselective Synthesis of 2-Hydroxymethyl-1,3-enynes by Rhodium-Catalyzed Decarboxylative C–C Coupling, Adv. Synth. Catal., 2019, 361, 4831–4836 CrossRef CAS.
- (a) J. Kaufmann, E. Jäckel and E. Haak, Ruthenium-Catalyzed Cascade Annulation of Indole with Propargyl Alcohols, Angew. Chem., Int. Ed., 2018, 57, 5908–5911 CrossRef CAS; (b) P. D. Hammen, A. C. Braisted and D. L. Northrup, Synthesis of vinyl and β-phthalimido ketones, Synth. Commun., 1991, 21, 2157–2163 CrossRef CAS; (c) R. F. Sweis, M. P. Schramm and S. A. Kozmin, Silver-Catalyzed [2 + 2] Cycloadditions of Siloxy Alkynes, J. Am. Chem. Soc., 2004, 126, 7442–7443 CrossRef CAS; (d) N. Thies, C. G. Hrib and E. Haak, Ruthenium-Catalyzed Functionalization of Pyrroles and Indoles with Propargyl Alcohols, Chem. – Eur. J., 2012, 18, 6302–6308 CrossRef CAS; (e) N. G. Kundu, S. K. Dasgupta, L. N. Chaudhuri, J. S. Mahanty, C. J. Spears and A. H. Shahinian, Synthesis and Biological Activities of E-5-(2-Acylvinyl)uracils, Eur. J. Med. Chem., 1993, 28, 473–479 CrossRef CAS; (f) D. A. Lomeli-Rosales, J. A. Delgado, M. Diaz de los Bernardos, S. Perez-Rodriguez, A. Gual, C. Claver and C. Godard, A General One-Pot Methodology for the Preparation of Mono- and Bimetallic Nanoparticles Supported on Carbon Nanotubes: Application in the Semi-hydrogenation of Alkynes and Acetylene, Chem. – Eur. J., 2019, 25, 8321–8331 CrossRef CAS.
- (a) H. Yamabe, A. Mizuno, H. Kusama and N. Iwasawa, Rh(I)-Catalyzed Cyclization of 1-Arylprop-2-yn-1-ol Derivatives Utilizing Rhodium 1,4-Migration, J. Am. Chem. Soc., 2005, 127, 3248–3249 CrossRef CAS; (b) R. Shintani, K. Okamoto and T. Hayashi, Rhodium-Catalyzed Isomerization of α-Arylpropargyl Alcohols to Indanones: Involvement of an Unexpected Reaction Cascade, J. Am. Chem. Soc., 2005, 127, 2872–2873 CrossRef CAS; (c) S. Omura, T. Fukuyama, J. Horiguchi, Y. Murakami and I. Ryu, Ruthenium Hydride-Catalyzed Addition of Aldehydes to Dienes Leading to β,γ-Unsaturated Ketones, J. Am. Chem. Soc., 2008, 130, 14094–14095 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qo01174f |
| This journal is © the Partner Organisations 2021 |