
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic role of metals supported on SBA-16 in hydrodeoxygenation of chemical compounds derived from biomass processing†
Paulina Szczyglewska a,
Agnieszka Feliczak-Guzika,
Mietek Jaroniecb and
Izabela Nowak*a
a,
Agnieszka Feliczak-Guzika,
Mietek Jaroniecb and
Izabela Nowak*a
aAdam Mickiewicz University, Poznań, Faculty of Chemistry, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: nowakiza@amu.edu.pl
bKent State University, Department of Chemistry and Biochemistry, Kent, Ohio 44242, USA. E-mail: jaroniec@kent.edu
First published on 3rd March 2021
Abstract
Hydrodeoxygenation (HDO) carried out at high temperatures and high hydrogen pressures is one of the alternative methods of upgrading pyrolytic oils from biomass, leading to high quality biofuels. To save energy, it is important to carry out catalytic processes under the mildest possible experimental conditions. The aim of our research was the synthesis of ordered mesoporous SBA-16 type silica materials modified with transition metal atoms (Ir, Ru, Pd, Pt), their physicochemical characterization and use as catalysts in hydrodeoxygenation of model chemicals (guaiacol, syringol, creosol). The HDO process was carried out under mild experimental conditions at temperatures in the range from 90 to 130 °C and hydrogen pressures in the range from 25 to 60 bar. The catalytic tests revealed differences in the catalytic properties of the samples studied. The catalytic systems used assured highly efficient transformations of the examined molecules as well as high selectivity towards chemical compounds with lower O/C ratio and higher H/C ratio as compared to those in the initial substrates. High activity of the catalysts containing precious metals in the experimental conditions applied suggests their potential to improve bio-oil production for biofuels.
Introduction
Development of the biofuel sector is a realistic alternative to the gradually depleting sources of fossil fuels. Increasing number of developed and developing countries consider biofuels to be of the key importance for reduction of the dependence on oil, greenhouse gas emissions and achievement of rural development goals.1,2 Currently, the production of biofuels is limited to the 1st generation biofuels, based on the well-established technologies, whose actual exploitation has been improved. The advantages of the 1st generation biofuels include, among others, the possibility of using their blends with oil-derived fuels in the existing internal combustion engines. In spite of numerous advantages of the fuels in question, they arouse high scepticism among scientists because they are obtained from edible plants and thus compete with the food market.3,4 Therefore, it is expected that the 2nd generation biofuels will become the dominant fuel products. Such fuels are produced from plant biomass (i.e., agricultural residues, organic waste or intentionally cultivated energy plantations) and use the majority of cheap and abundant non-food materials available from plants.3,5 Taking into account high bioavailability, low costs, low emission of harmful chemical compounds and inedible character, the lignocellulosic biomass has a high potential to be used for production of liquid fuels of the 2nd generation.6,7 According to literature data, scientists worldwide deal with this subject, which has resulted in a considerable number of reviews and research works. In order to obtain bioenergy from biomass of quality similar to that obtained from fossil fuel combustion, it is necessary to transform it into chemical compounds characterized by low molecular mass, hydrophobic nature and anaerobic structure.8 Bioenergy can be produced from lignocellulose biomass in various ways: by physical, thermochemical or biochemical methods.9 Special attention has been paid to pyrolysis, which is one of the technologies used for direct biomass liquefaction. In this process, as a result of high temperature action without access to air, macromolecular chemical compounds decompose to the chemical compounds of lower molecular weight from which solid, liquid or gas fuels can be obtained.10 However, the liquid pyrolytic oil produced is acidic (significant content of carboxylic acids), viscous and chemically unstable due to high content of oxygen functional groups (total oxygen content 35–50% by weight) and water (15–30% by weight).11,12 Moreover, pyrolytic oils derived from biomass do not mix with conventional hydrocarbon fuels and are too reactive to be fractionated by distillation. Thus, to make the pyrolysis process a viable technological platform for the production of hydrocarbon fuels from biomass the effective methods for improvement of pyrolytic oil through catalytic oxygen removal should first be developed.13 One of the most effective methods used to improve oxygen compounds is catalytic hydrodeoxygenation (HDO), which increases the energy value of bio-oil by transforming potential energy carriers into simple hydrocarbons.14,15 This is due to simultaneous deoxidation and hydrogenation of organic compounds in the presence of hydrogen gas supplied to the system under increased pressure and temperature.16,17 The key issue in the whole process is selection of an appropriate catalytic system, capable of reducing the O/C ratio, while increasing the H/C ratio.18 The catalysts used so far are of heterogeneous nature, consisting of a solid support and active substance dispersed on it.19,20 Precious metals are known for their ability to increase the efficiency of many advanced catalytic systems.21,22 The solid supports with incorporated species of these metals catalyse the hydrogenation reaction during hydrodeoxygenation, which also results in the removal of oxygen from the bio-oil. It is commonly known that precious metals are expensive, which results in costly production of the improved fuels. However, it should be noted that they are recyclable, and in addition, they are selective towards such chemicals as cyclohexane, methoxycyclohexane or aliphatic hydrocarbons. Three metals, i.e., ruthenium, palladium, platinum, have already been thoroughly tested in many catalytic reactions. Therefore, they are considered the most suitable metal catalysts to optimally remove oxygen and thus improve the efficiency of hydrocarbons production.23 The use of expensive noble metal catalysts in very small quantities is common in crude oil refining in such processes as hydrocracking, catalytic reforming or isomerization as well as in the production of specialized chemicals. Excellent catalytic efficiency of precious metals and available technologies of recycling of the used catalysts justify their application in various industrial catalytic processes despite higher cost in comparison to non-precious metals.24 Table 1 presents the literature data for hydrodeoxygenation reactions of selected model chemical compounds catalysed by various systems containing precious metals, such as: Ir, Ru, Pd, Pt.| Catalyst | Operating conditions | Model compound | Main reaction product | Selectivity [%] | Conversion [%] | Ref. | |
|---|---|---|---|---|---|---|---|
| T [°C] | p [bar] | ||||||
| Pd/Nb2O5 | 200 | 1 | Phenol | Cyclohexanone | 81 | 6 | 31 |
| 400 | Benzene | 100 | 10 | ||||
| Pd/C | 100 | 30 | Vanillin | p-Cresol | 95 | 100 | 24 |
| Pd/TiO2 | 220 | 30 | Guaiacol | Methoxycyclohexanol | 78 | 80 | 32 |
| Pd/C | 200 | 50 | Phenol | Cyclohexanol | 98 | 100 | 33 |
| Pd/SiO2 | 200 | 50 | Phenol | Cyclohexanol | 62 | 87 | 33 |
| Pd/SBA-16 | 90 | 25 | Phenol | 2,4-Dimethylhexane | 51 | 100 | 34 |
| Ru/SBA-15 | 300 | 45 | Phenol | Cyclohexanone | 57 | 4 | 35 |
| Ru/TiO2 | 61 | 4 | |||||
| Ru/MCM-41 | 38 | 16 | |||||
| Ru/H-Beta | 140 | 40 | Diphenyl ether | Cyclohexane | 67 | 55 | 36 |
| Ru/H-Beta | 100 | Cyclohexanol | 18 | 48 | |||
| Ru/C | 100 | 30 | Vanillin | Vanillin alcohol | 90 | 100 | 24 |
| Ru/SBA-16 | 130 | 60 | Phenol | 2,4-Dimethylhexane | 94 | 100 | 34 |
| Ru/MCM-41 | 130 | 60 | Anisole | 1-Methoxycyclohexene | 51 | 70 | 37 |
| Pt/C | 100 | 30 | Vanillin | Vanillin alcohol | 70 | 100 | 24 |
| Pt/TiO2 | 285 | 40 | Guaiacol | Cyclohexane | 44 | 70 | 38 |
| Pt/Al-MCM-48 | 250 | 40 | Guaiacol | Cyclohexane | 38 | 64 | 39 |
| Pt/H-Beta | 400 | 1 | Anisole | Benzene | 51 | 100 | 40 |
| Pt/SBA-16 | 90 | 25 | Phenol | 2,4-Dimethylhexane | 77 | 100 | 34 |
| Ir/SiO2 | 300 | 6 | Stearic acid | 1-Heptadecene | 26 | 5 | 41 |
| Ir/ZSM-5 | 200 | 30 | Phenol | Cyclohexane | 96 | 32 | 26 |
| Ir/SiO2 | 50 | 60 | Hydroxymethyl-furfural | 2,5-Bis(hydroxymethyl)furan | 100 | 60 | 42 |
Palladium is a valuable metal used repeatedly in the hydrodeoxygenation of pyrolytic oil.23 Very often it is a component of bifunctional and bimetallic catalysts. As can be seen in Table 1, the palladium catalysts are particularly highly active in hydrogenation of multiple bonds between carbon atoms. It has also been demonstrated that these catalysts are much less active in hydrogenation reactions of aromatic hydrocarbons and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, which is particularly true for aliphatic aldehydes and ketones.20 In turn, platinum is well known as a highly active metal in hydrogenation reactions at much lower temperatures than, for example, sulphide catalysts.25 Among many precious metals, Pt-based catalytic systems have been most frequently studied, whereas the catalytic activity of systems based on e.g., Ir have been rarely described.26 One of the reasons of high popularity of Pt catalysts is their good stability even under very demanding reaction conditions.27 Analysis of the data presented in Table 1 shows that the presence of platinum in a catalytic system often permits removal of oxygen atoms from chemical compounds subjected to the hydrodeoxygenation process. Iridium also has catalytic activity in HDO reactions of bio-oil. Nevertheless, its activity is not as good as that of Pd, Pt or Ru and therefore, as already mentioned, the studies of Ir catalysts have been noticeably rarer.28 As mentioned above, palladium and platinum are widely used components of heterogeneous catalysts; the application of ruthenium as another element from the group of precious metals is definitely less frequent. The application of ruthenium in hydrogenation reactions or Fischer–Tropsch synthesis is best known.29 Nevertheless, Ru-based catalysts are increasingly used in the production of biodiesel, which can also be deduced from the data summarized in Table 1. Due to the higher affinity of Ru to organic compounds containing oxygen atoms than other precious metals, the selectivity of Ru-based catalysts to such compounds is relatively high.28 The popularity of precious metals used especially in catalytic processes results from the fact that the d orbitals of these metals are not completely filled, while the support surface easily adsorbs reactive compounds and therefore, these metals exhibits high catalytic activity towards hydrogenation at low temperatures.28 Many precious metals are deposited on reducible oxides, such as CeO2, TiO2, ZrO2 and on non-reducible media, such as zeolites, silica.26 The solid supports with well-developed specific surface area and porosity are particularly widely used because such porous networks allow for high dispersion of metallic catalysts and additionally stabilize the whole catalyst.30
O bonds, which is particularly true for aliphatic aldehydes and ketones.20 In turn, platinum is well known as a highly active metal in hydrogenation reactions at much lower temperatures than, for example, sulphide catalysts.25 Among many precious metals, Pt-based catalytic systems have been most frequently studied, whereas the catalytic activity of systems based on e.g., Ir have been rarely described.26 One of the reasons of high popularity of Pt catalysts is their good stability even under very demanding reaction conditions.27 Analysis of the data presented in Table 1 shows that the presence of platinum in a catalytic system often permits removal of oxygen atoms from chemical compounds subjected to the hydrodeoxygenation process. Iridium also has catalytic activity in HDO reactions of bio-oil. Nevertheless, its activity is not as good as that of Pd, Pt or Ru and therefore, as already mentioned, the studies of Ir catalysts have been noticeably rarer.28 As mentioned above, palladium and platinum are widely used components of heterogeneous catalysts; the application of ruthenium as another element from the group of precious metals is definitely less frequent. The application of ruthenium in hydrogenation reactions or Fischer–Tropsch synthesis is best known.29 Nevertheless, Ru-based catalysts are increasingly used in the production of biodiesel, which can also be deduced from the data summarized in Table 1. Due to the higher affinity of Ru to organic compounds containing oxygen atoms than other precious metals, the selectivity of Ru-based catalysts to such compounds is relatively high.28 The popularity of precious metals used especially in catalytic processes results from the fact that the d orbitals of these metals are not completely filled, while the support surface easily adsorbs reactive compounds and therefore, these metals exhibits high catalytic activity towards hydrogenation at low temperatures.28 Many precious metals are deposited on reducible oxides, such as CeO2, TiO2, ZrO2 and on non-reducible media, such as zeolites, silica.26 The solid supports with well-developed specific surface area and porosity are particularly widely used because such porous networks allow for high dispersion of metallic catalysts and additionally stabilize the whole catalyst.30
In this work we examined the catalytic activity of the systems containing precious metals (Ir, Ru, Pd, Pt) dispersed in mesoporous SBA-16 support towards hydrodeoxygenation of guaiacol, syringol, creosol – key chemical compounds present in pyrolytic oil. The aim of this work was to synthesize the above-mentioned metallic catalysts and to study their catalytic performance in the hydrodeoxygenation reaction under mild experimental conditions (temperature 90–130 °C, hydrogen pressure 25–60 bar). The catalysts were comprehensively characterized by X-ray diffraction (XRD), low temperature nitrogen adsorption/desorption, transmission and scanning electron microscopy (TEM, SEM), scanning electron microscopy coupled with a mass detector (SEM/EDX). Inductively coupled plasma optical emission spectrometry (ICP-OES) and X-ray photoelectron spectroscopy (XPS) were also used. Special emphasis was placed on the comparison of the effectiveness of the catalysts with different active phases.
Materials and methods
Chemicals
All the listed chemicals were of analytical purity and were used as purchased without further purification. The chemicals used in the carrier synthesis were as follows: tetraethyl orthosilicate denoted as TEOS (Aldrich), copolymer of ethylene oxide and propylene oxide known as Pluronic F127 (Sigma) and hydrochloric acid (POCH). The chemicals used to modify the carriers were: hexachloroiridic acid (Alfa Aesar), ruthenium(III) chloride (Aldrich), palladium(II) chloride (Alfa Aesar), hexachloroplatinic acid (Alfa Aesar), ethanol (POCH). The chemicals used for reactivity tests were: syringol (Aldrich), guaiacol (SAFC), creosol (Sigma-Aldrich), decaline (Honeywell). The gases used were: Ar (Purity N5.0, Linde Gas Poland), H2 (Purity N5.0, Linde Gas Poland).Synthesis of SBA-16 material
SBA-16 was synthesized using Pluronic F127 (EO106PO70EO106) block copolymer as a structure directing agent and TEOS as a silicon source. In typical synthesis 4.8 g of Pluronic F127 were dissolved in 46.6 g of deionized water and 192.0 g of HCl (2 M). The prepared mixture was stirred at room temperature using a magnetic stirrer until complete dissolution of polymer (about 1 h). Then 20.2 g of TEOS were added in small portions and the whole mixture was stirred for 0.5 h without temperature change. After drying in a laboratory dryer (48 h, 90 °C), the solids were drained and then washed several times with deionised water. After drying at room temperature overnight, the products were calcined for 6 h at 550 °C. The resulting sample is denoted as SBA-16/K.Modification of SBA-16 material
The synthesized silica materials were subjected to modification by wetting impregnation to obtain samples with a metal content of 3% wt. All metal precursors, i.e., iridium (H2IrCl6·H2O), ruthenium (RuCl3·H2O), palladium (PdCl2) and platinum (H2PtCl6·6H2O) were dissolved in appropriate amount of ethyl alcohol and the solutions were left in ultrasonic cleaner for 1 h. Then the ethanol solution of the appropriate metal precursor was poured into the weighed portion of the SBA-16 support. The mixtures prepared in this way were tightly wrapped with polyolefin-paraffin foil Parafilm® and left for 24 h. After that time, the beaker with impregnated support was placed under the extractor (room temperature) until complete evaporation of ethanol. Then the obtained catalyst was dried for 1 h at 30 °C, 1 h at 40 °C, 18 h at 60 °C. The catalysts obtained in this way were reduced in the tube furnace in hydrogen atmosphere. The first stage of this process was to pass argon (50 cm3 min−1, 0.5 h) at room temperature through the catalyst to remove air from the system. Then the stream of inert gas was switched to hydrogen (50 cm3 min−1, 0.5 h) and the furnace heating was switched on. The catalysts were reduced for 3 h at temperature: 400 °C (iridium catalyst), 250 °C (ruthenium catalyst), 350 °C (palladium catalyst), 250 °C (platinum catalyst). The optimal temperature of metal precursor reduction was selected based on H2-TPR tests.Characterization of catalysts
X-ray diffraction data were collected at low angles to obtain information about structural properties of the synthesized materials, i.e., the degree of structural ordering. The XRD method was also used to determine the distance between parallel crystal lattice planes (d100) and the distances between mesopore centres (a0). The elementary cell parameters were determined from the formula: a0 = d√2. Low-angle X-ray scattering measurements were performed using a Bruker AXS D8 Advance having Johannson monochromator with CuKα radiation, which generated the wavelength of λ = 0.154 nm. All measurements were made with a step 0.02° in the range 2θ = 0.6–8°. Nitrogen adsorption/desorption isotherms and pore size distributions (PSD) were determined at −196 °C with a Quantachrome Nova 1200e analyser. All samples were degassed for 24 hours at 90 °C before the actual measurements. The specific surface area of the samples studied was determined using the BET method and the PSD functions were obtained using the BJH algorithm. Microscopic images of the materials were taken using a transmission electron microscope JEOL JEM-1200 EX II at 80 kV. This type of microscopy was used to determine the degree of ordering of mesopores and their distribution. The TEM image, after setting the scale and threshold for the whole image, were analyzed using “Analyze Particles” option to generate a table with data of the particle areas and diameters. These data were exported into Origin program to construct the histogram plots. The samples were also analysed using a Zeiss scanning electron microscope type EVO at 17 kV. The imaging data were used to evaluate the surface texture and morphology of all synthesized catalysts. Scanning electron microscopy with X-ray energy dispersion option was used to assess the structure of ordered silica modified with transition metal atoms. For this purpose, the LEO 1430 VP microscope (SEM measurement) and Quantax 200 with XFlash 4010 detector by Bruker AXS (EDX) were used. The content of transition metal was determined using inductively coupled plasma optical emission spectrometry with ICP-OES VISTA-MPX spectrometer by Varian. Prior to measurement the samples were mineralized in aqua regia in a microwave oven. Spectroscopic measurements of X-ray excited photoelectrons were performed using the SPECS UHV analysis system with AlKα radiation (E = 1.487 keV). All samples were neutralized using a low energy electron gun. The measurements permitted determination of the total content of metals in the analysed samples and their oxidation degree.Hydrodeoxygenation reaction
A high-pressure reactor (CAT 24 HEL) placed on a magnetic stirrer was used to carry out the hydrodeoxygenation reaction of the model substrates. The reactor was equipped with a thermocouple to control the process temperature and a manometer to determine the initial reaction pressure. A catalyst (0.05 g) and guaiacol (1 g) or creosol (1 g) were placed in a dedicated glass reaction vessel with a magnetic stirrer inside. As syringol was in solid state it was necessary to use a solvent, so a catalyst (0.005 g), syringol (0.05 g) and decaline (1 g) were placed in the vessel. The reaction mixtures prepared in this way were placed in a high-pressure reactor, which was then closed and rinsed three times with argon to remove air from inside the reactor. In the next step, the reactor was flushed three times with hydrogen and finally filled with hydrogen to 25 bar, 40 bar or 60 bar pressure. The reactions were carried out at different temperatures, i.e., 90 °C, 110 °C or 130 °C for 4 h and the reaction time was measured from the moment of achieving the desired temperature (stirring at 700 rpm). After 4 h the reactor was cooled, the accumulated gas was released and the reaction mixtures were centrifuged (Micro Star 17, VWR) to separate the liquid reaction products from the solid catalyst. After the centrifugation process, the reaction mixtures were transferred to 1.5 ml vials and subjected to chromatographic analysis.Chromatographic analysis
The reaction mixtures were analysed using two chromatographs: a VARIAN 3900 GC gas chromatograph equipped with automatic sample dispenser, flame ionization detector, 25 meter capillary column CPWAX57CB with internal diameter of 0.32 mm and 1.2 μm film and a gas chromatograph coupled to a VARIAN 4000 GC mass spectrometer equipped with an automatic sample dispenser, mass sensor and a similar capillary column (CPWAX57CB). The substrates and reaction products were identified in two ways by using: the retention times of commercially available standards and the NIST chemicals library data.Study of catalyst reuse
The procedure of regeneration of the SBA-16/3%Pd catalyst, previously used in the HDO process of guaiacol, was carried out for 4 hours at 130 °C under 60 bar hydrogen pressure. The catalyst was initially filtered through a high graded filter (to separate the catalyst from the liquid reaction products). Then the catalyst, which remained on the filter, was washed with ethanol, and finally dried in a laboratory dryer. The regenerated catalyst was reused in the guaiacol hydrodeoxygenation process under identical conditions. The same catalyst was used four times because the amount of the regenerated catalyst was sufficient to perform four tests only. The amount of guaiacol used in the reaction was adjusted in proportion to the amount of the remaining catalyst.Results and discussion
X-ray diffraction studies
Diffractograms of SBA-16 materials modified with transition metal species and SBA-16/K material are shown in Fig. S1 (see ESI†). The XRD profiles are very similar and characteristic for mesoporous SBA-16 structure (Im3m symmetry) with regularly arranged pores. Most often diffractograms of SBA-16 show the presence of three reflections with indices (110), (200) and (210).43–45 The diffractograms of the materials synthesized by us show a strong peak at a low angle 2θ = 0.82–0.88°. Its position corresponds to the peak observed for multi-walled particles and indexed by the plane (110). Only in the material modified with iridium, the intensity of the main reflection was lower than for the other materials, which indicates that this sample is poorly ordered. A weak intensity of higher-order reflections (which for SBA-16/Ir silica are completely undetectable) may be a consequence of the high ratio of the wall thickness to pore size or the roughness of the walls due to the large number of micropores in the structure of the samples studied.46In addition to the main reflection position, Table 2 summarises the parameters of the elementary cell. The values of the 2θ angle of all silicas corresponding to the first order reflection are similar, which implies similar distances between the parallel lattice planes (10.01–10.77 nm) and similar distances between the centres of mesopores (14.16–15.23 nm).
| Catalyst | 2θ [°] | d100 [nm] | a0 [nm] |
|---|---|---|---|
| SBA-16/K | 0.88 | 10.01 | 14.16 |
| SBA-16/3%Ir | 0.88 | 10.01 | 14.16 |
| SBA-16/3%Ru | 0.82 | 10.77 | 15.23 |
| SBA-16/3%Pd | 0.86 | 10.25 | 14.50 |
| SBA-16/3%Pt | 0.86 | 10.25 | 14.50 |
Low-temperature nitrogen sorption studies
The nitrogen adsorption/desorption isotherms for ordered mesoporous SBA-16 type silica materials modified with transition metal atoms and unmodified SBA-16 material are presented in Fig. 1. The shape of all isotherms is type IVa according to IUPAC classification,47 typical for SBA-16 samples. The observed sharp step at the relative pressure range p/p0 = 0.4–0.7 corresponds to capillary condensation occurring in uniform mesopores.43 The shape of the hysteresis loop indicates that the silicates tested contain pores with narrow openings and wide interiors (type H2 according to IUPAC classification).47,48 It can be also concluded that the modification of silica with metal atoms has no effect on the deterioration of the structure, which is indicated by the similarity of the isotherms of the unmodified and all modified materials.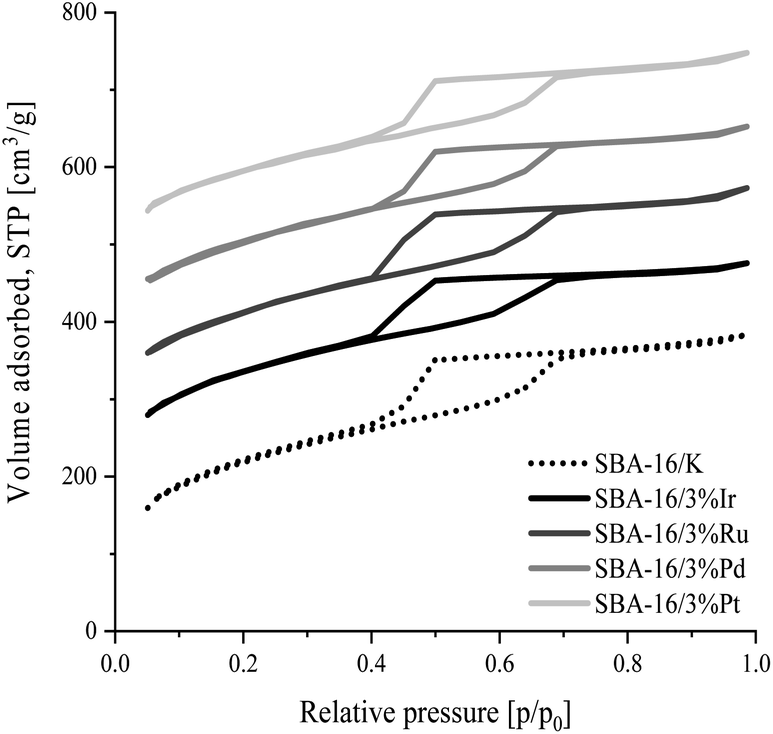 | ||
| Fig. 1 N2 adsorption/desorption isotherms measured for the SBA-16 materials modified with precious metal atoms and unmodified SBA-16 material. Each isotherm curve has been shifted successively by 100 cm3 g−1 from the previous one. | ||
The specific surface area, pore size and pore volume values estimated from the adsorption isotherm data for all synthesized samples are collected in Table 3. All samples have a large specific surface area in the range 705–849 m2 g−1. The total pore volume of the synthesized silicas is in the range from 0.54 to 0.59 cm3 g−1. In the materials tested the mesopore diameters corresponding to the maximum pore distribution functions are similar and are about 6.3 nm for all the samples except SBA-16/3%Ir, for which this dimension is about 5.5 nm. Also, all PSD curves in Fig. 2 show that the most of mesopores feature diameters in the range of 5–8 nm.
| Catalyst | Surface area [m2 g−1] | Pore size [nm] | Total pore volume [cm3 g−1] |
|---|---|---|---|
| SBA-16/K | 796 | 6.33 | 0.59 |
| SBA-16/3%Ir | 849 | 5.46 | 0.58 |
| SBA-16/3%Ru | 769 | 6.34 | 0.58 |
| SBA-16/3%Pd | 743 | 6.36 | 0.55 |
| SBA-16/3%Pt | 705 | 6.32 | 0.54 |
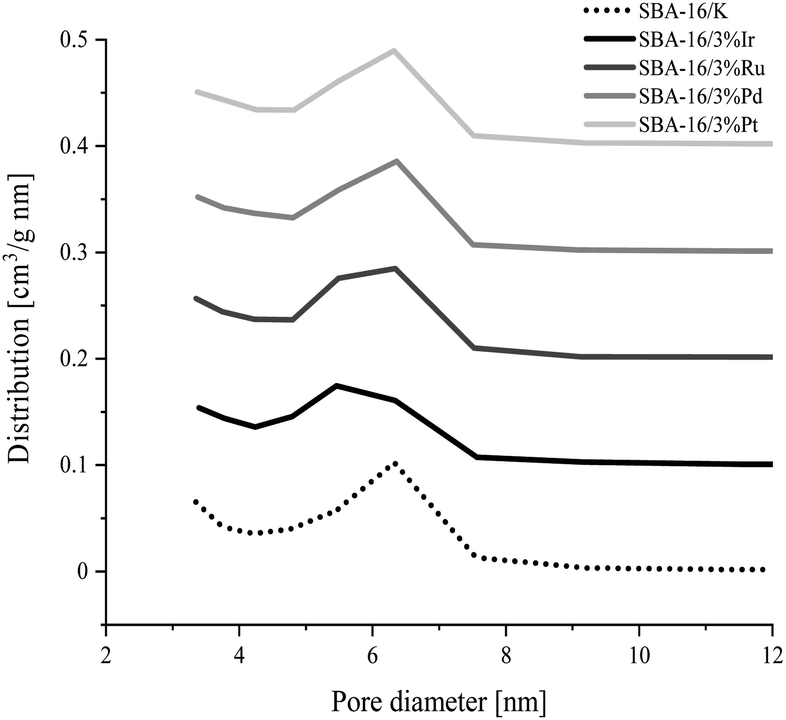 | ||
| Fig. 2 Pore size distribution (obtained by BJH method using the adsorption branch data) for the SBA-16 materials modified with transition metal atoms and unmodified SBA-16 material. Each PSD curve has been shifted successively by a fixed value of 0.1 cm3 g−1 nm−1 from the previous one. | ||
Transmission electron microscopy studies
Images from transmission electron microscope show that the synthesized materials are characterized by high structural ordering (Fig. S2, see ESI†). The TEM images are in agreement with the XRD analysis showing diffractograms of the silicas with characteristic reflections for ordered mesostructures. Additionally, dark spots visible on the images of the samples with metal species confirm the presence of precious metals introduced by impregnation method. The metal nanoparticles have approximately spherical shapes and exhibit relatively narrow and monomodal size distribution histograms as shown in ESI (Fig. S2†). These histograms were obtained by analyzing images with a few hundreds of individual nanoparticles. The presented distribution histograms of metallic nanoparticles were used to compute the mean particle size for each catalyst: Ir (3.52 ± 1.64 nm) ≅ Ru (3.82 ± 1.45 nm) ≈ Pd (4.63 ± 1.55 nm) ≤ Pt (8.02 ± 2.14 nm). Only histogram for platinum nanoparticles exhibits a broad distribution of the nanoparticle sizes, however low standard deviation can suggest that these nanoparticles are well dispersed in the SBA-16 silica.Scanning electron microscopy imaging
The observations made with the scanning electron microscope show, as expected, spherical nanoparticles (Fig. S3, see ESI†). All SEM images show a very similar morphology of mesostructured materials. Moreover, the materials studied are composed of particles of very similar sizes.Study of metal distribution by scanning electron microscopy coupled with X-ray energy dispersion
Table S1 (see ESI†) and Fig. 3 present the results of scanning electron microscopy with X-ray energy dispersion. The image obtained for the sample of mesoporous silica modified with iridium (Fig. 3a) clearly confirms the presence of metal on the surface. The results of this analysis also indicate an even distribution of the introduced metal species. The average iridium content in the sample is 5.34% – this value is higher than the assumed value (3%). No larger metal clusters are observed on the mesoporous matrix surface. Fig. 3b shows an image of the mesoporous silica impregnated with ruthenium. This image confirms that the process of impregnation of silica has been successful. The formation of ruthenium aggregates is observed on the surface of SBA-16 (point 2 marked on the image), which is reflected by the average percentage of ruthenium (about 12.2%) present on the surface of the examined fragment of the sample. This value is four times higher than the assumed one. However, it should be noted that the values obtained by the EDX method based on the SEM images represent local measurements and do not always reflect the situation for the entire sample. However, larger metal concentrations on the surface may have a catalytic effect. The subsequent photographs marked on Fig. 3c and d reflect structures of the silica modified with palladium and platinum, respectively. Both palladium and platinum modified silica samples show even distribution of crystallites. The average palladium content in the examined sample is 2.80%, while the platinum content is 4.30%. These results are not far from the assumed values. Moreover, similarly as for the samples containing iridium, no larger clusters of both metals are visible. | ||
| Fig. 3 Elemental maps showing the distribution of metal, silicon and oxygen on the SBA-16 support obtained with the use of EDX detector: (a) SBA-16/3%Ir, (b) SBA-16/3%Ru, (c) SBA-16/3%Pd, (d) SBA-16/3%Pt. | ||
Study of chemical composition by inductively coupled plasma optical emission spectrometry
Table 4 presents a comparison of the assumed percentage of the introduced metal with its actual content in the sample. The results show that the wetting impregnation method used for modification of silicates was successful and allowed for introduction of metal species in the quantities similar to those assumed.| Catalyst | Assumed metal content [%] | Measured metal content [%] |
|---|---|---|
| SBA-16/3%Ir | 3 | 2.81 |
| SBA-16/3%Ru | 3 | 2.33 |
| SBA-16/3%Pd | 3 | 2.72 |
| SBA-16/3%Pt | 3 | 2.72 |
X-ray photoelectron spectroscopy
XPS analysis was used to determine the presence and stoichiometric ratios of elements in subsurface layers of catalysts. By comparing these results with the results of volumetric analysis by ICP-OES (Table 4), one can comment on the metal incorporation into the SBA-16 mesostructure. If the percentage of metal in the surface layer is higher than the average value for the entire sample volume, then we can conclude that the pore penetration of metal clusters is negligible. The high specific surface area of the catalysts, resulting from its porosity, does not play a significant role then, and such a catalyst will work in a similar way as a bulk material (metal nanopowder). However, if the content of a given metal in the surface layer is the same or less than the average value for the entire sample volume, then we can conclude that the metal penetrated the pores effectively. Assuming a good dispersion of metal in the volume of samples, the porosity of such material will have a significant impact on the catalytic function. The latter case was observed if one compares data in Tables 4 and 5.| Catalyst | Metal content [wt%] | Binding energy (reference for metal) [eV] | Metal oxidation state | |
|---|---|---|---|---|
| Assumed | Measured | |||
| SBA-16/3%Ir | 3 | 0.48 | 59.8 (60.9 Ir4f7/2) | 0 |
| SBA-16/3%Ru | 3 | 0.13 | 278.3 (280.2 Ru3d5/2) | 0 |
| SBA-16/3%Pd | 3 | 0.18 | 334.9 (335.0 Pd3d5/2) | 0 |
| SBA-16/3%Pt | 3 | <0.10 | 70.4 (71.0 Pt4f7/2) | 0 |
Table 5 presents the results of XPS analysis of silica samples modified with transition metal ions. The percentage content of metals in the surface layer and their oxidation state was determined. For the catalysts modified with Ru, Pd, Pt, the minimum metal content in the surface layer was shown, which proves the effective penetration of metals into pores of the SBA-16 silica. Only in the case of the sample containing Ir, the measured metal content was higher than in the other samples and amounted to nearly 0.5%. Analysis of the data collected in this table, clearly shows that all metal species have been reduced to zero oxidation state, which confirms that the applied reduction temperature for the respective metal precursors has been well chosen. The analysis of the binding energy (BE) shows that all core levels of metals are not shifted to higher BE in comparison to the reference data. Thus, no metal oxides are present in the catalysts.
Results for the HDO reactions studied
Guaiacol
The products formed during hydrodeoxygenation of guaiacol on the SBA-16 catalysts containing precious metals are anisole, methoxyclohex-1-ene, methoxycyclohexane, 1,2-cyclohexanediol and 2-methylcyclohexanol (Scheme 1).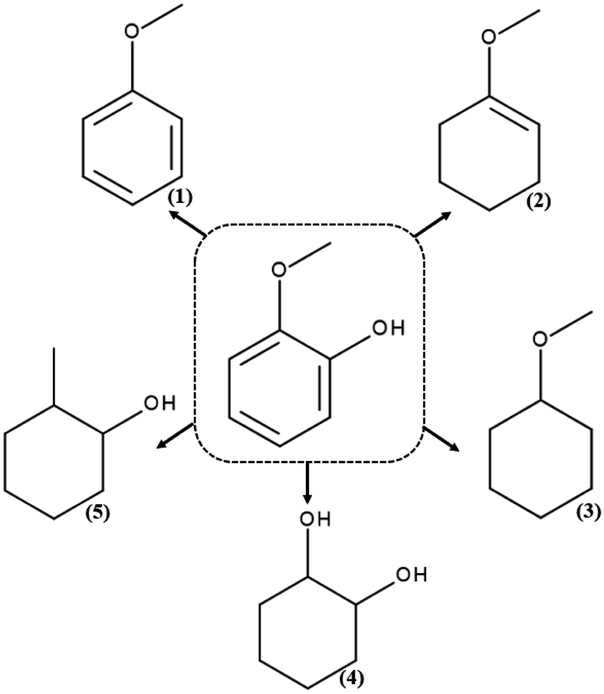 | ||
| Scheme 1 Products of the hydrodeoxygenation reaction of guaiacol: (1) anisole, (2) methoxycyclohex-1-ene, (3) methoxycyclohexane, (4) 1,2-cyclohexanediol, and (5) 2-methylcyclohexanol. | ||
The graph presented in Fig. 4a shows that the SBA-16 silica modified with iridium has no catalytic activity in the HDO guaiacol process. The catalyst did not show any improvement in the activity even under changed reaction conditions. In all cases, the conversion of guaiacol was less than 1% and the highest selectivity was obtained to methoxyclohex-1-ene and 1,2-cyclohexanediol (Table S2, see ESI†). Because of such low conversion, the results are not discussed as they are subject to a very high measurement error.
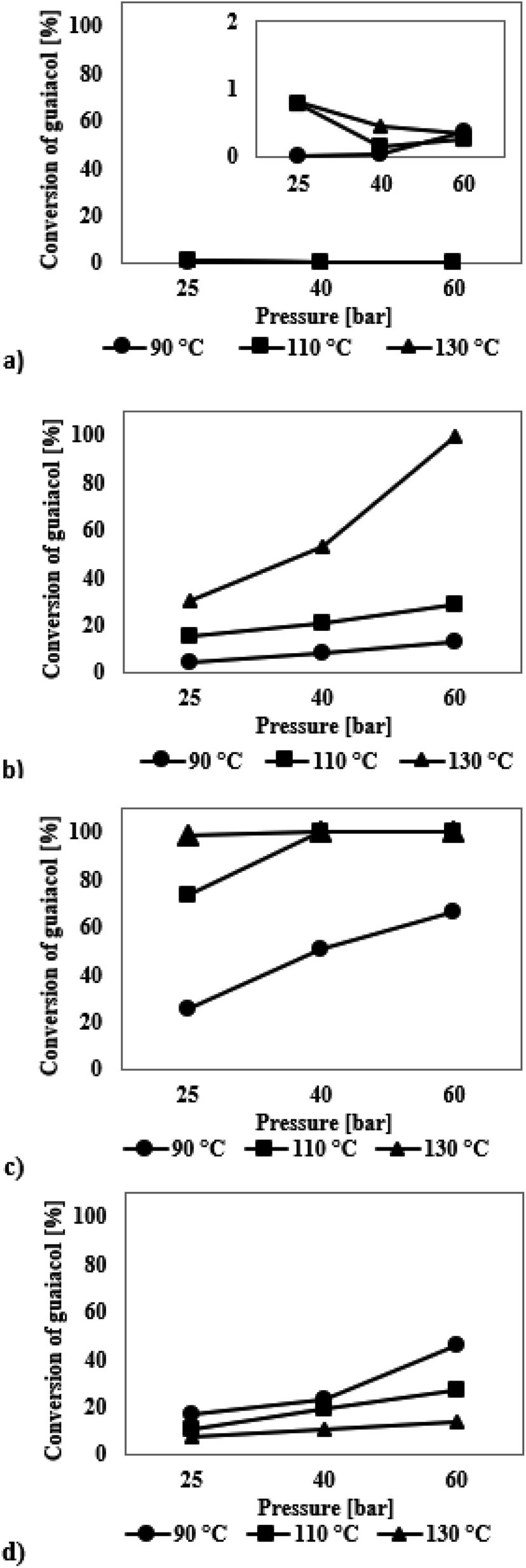 | ||
| Fig. 4 Guaiacol conversion in the HDO process at temperatures in the range of 90–130 °C and hydrogen pressure of 25–60 bar in the presence of catalysts modified with precious metals: (a) SBA-16/3%Ir, (b) SBA-16/3%Ru, (c) SBA-16/3%Pd, (d) SBA-16/3%Pt. | ||
Fig. 4b presents the results obtained for a series of HDO reactions for guaiacol on the ruthenium modified SBA-16. The catalytic tests performed show that the reaction leads to the formation of three products. The GC and GC-MS analysis shows the presence of methoxycyclohex-1-ene (formed in the highest amounts), 1,2-cyclohexanediol and 2-methylcyclohexanol (Table S2†). For processes carried out at 90 °C and 110 °C, the conversion of guaiacol did not exceed 30%. In turn, when the reaction temperature was 130 °C, 100% conversion of guaiacol took place. At 90 °C, the formation of methoxycyclohex-1-ene prevails, whereas the selectivity towards 2-methylcyclohexanol and 1,2-cyclohexanediol increase with increasing temperature.
The results presented in Fig. 4c show that the palladium catalyst deposited on SBA-16 exhibits a high activity in the HDO process of guaiacol. With increasing conversion of guaiacol, the selectivity to methoxyclohex-1-ene decreases and that to 1,2-cyclohexanediol increases (Table S2†). At 100% conversion of the model chemical, the selectivity to methoxyclohex-1-ene is 49–72% and to 1,2-cyclohexanediol 22–40%. Analysis of the data indicates that the values of hydrogen temperature and pressure had a significant influence on the process. No 2-methylcyclohexanol was found in the post-reaction mixture when palladium modified catalysts were used.
Analysis of the data presented in Fig. 4d shows that the platinum catalyst deposited on SBA-16 exhibits a moderate activity in the HDO process of guaiacol. In each case, the highest selectivity is observed towards methoxycyclohex-1-ene (Table S2†). Higher conversion of guaiacol is observed for the reactions carried out at 90 °C, at which the conversion of guaiacol increases with increasing hydrogen pressure. The maximum observed conversion was 46%.
The reported data for the HDO process of guaiacol show the highest selectivity to cyclohexane or cyclohexanol, however the temperature and pressure values used were much higher than those applied by us. Deutsch et al.49 in their HDO reaction of guaiacol on copper chromite (225–275 °C, hydrogen pressure 50 bar) reported methoxycyclohexane as one of the products. Its presence in the post-reaction mixture was explained by the probable occurrence of the alkylation process, which involved the addition of methyl group to the hydroxyl group with generation of 1,2-dimethoxybenzene. Then, anisole was formed due to demethoxylation, which was subsequently hydrogenated to methoxycyclohexane. In our study the highest selectivity is observed for methoxyclohex-1-ene – a chemical compound with a double bond. Such a reasoning may explain the presence of this compound in the post-reaction mixtures.
Syringol
GC and GC-MS chromatography of the products of hydrodeoxygenation reactions for syringol reveal the presence of four products: anisole, 1,2,3-trimethoxycyclohexane, 1,2-cyclohexanediol and cyclohexanol, whose structures are presented in Scheme 2.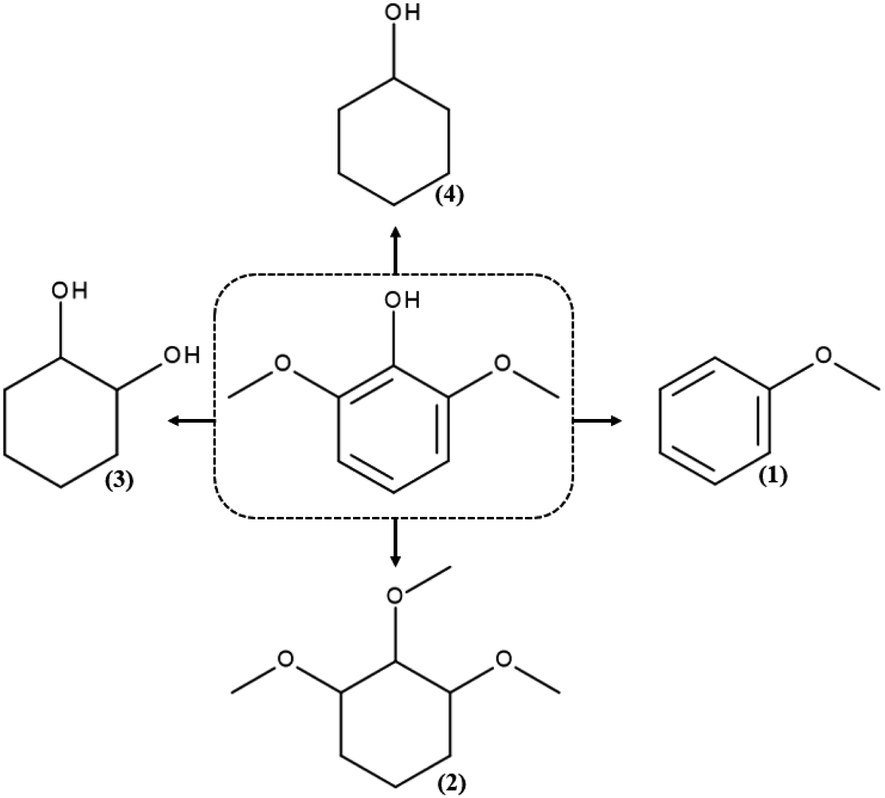 | ||
| Scheme 2 The products of the hydrodeoxygenation reaction for syringol: (1) anisole, (2) 1,2,3-trimethoxycyclohexane, (3) 1,2-cyclohexanediol, and (4) cyclohexanol. | ||
The iridium catalysts turned out to be completely ineffective in the process, which was concluded from the results presented in Fig. 5a. Only for the reaction at 130 °C at 60 bar, an increase in the conversion to 5% was observed, which still is an unsatisfactory result. Among the reaction products formed, only two chemicals were found: cyclohexanol and 1,2,3-trimethoxycyclohexane (Table S3, see ESI†). However, since the selectivity to the reaction products was much flawed due to low conversions, their comparison would be doubtful.
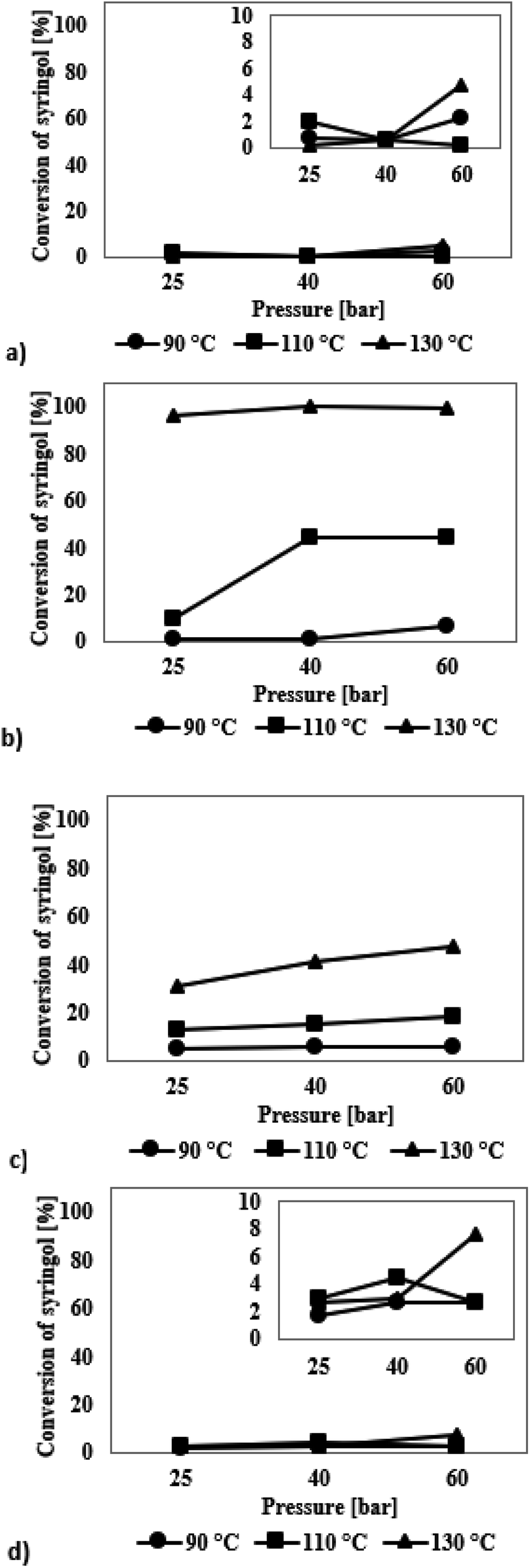 | ||
| Fig. 5 Syringol conversion in the HDO process at temperatures in the range of 90–130 °C and hydrogen pressure of 25–60 bar in the presence of catalysts modified with precious metals: (a) SBA-16/3%Ir, (b) SBA-16/3%Ru, (c) SBA-16/3%Pd, (d) SBA-16/3%Pt. | ||
The mesoporous silica material with ruthenium as the active phase shows the highest activity among all investigated catalysts in the process of syringol hydrodeoxygenation, which is illustrated in Fig. 5b. These catalysts were characterized by noticeably higher activity already at 110 °C, while at 130 °C almost 100% conversion of syringol was achieved. The product formed in the highest amounts in all the reactions is 1,2,3-trimethoxycyclohexane (Table S3†). Note that with increasing conversion of syringol, the amounts of anisole formed are higher at the expense of decreasing amounts of 1,2,3-trimethoxycyclohexane. Although anisole features an unsaturated bond system, it is a more desirable reaction product than 1,2,3-trimethoxycyclohexane because of a lower content of oxygen atoms in the structure. Analysis of the results shows that the amount of chemical compounds belonging to the group of alcohols increases with increasing pressure and temperature. Thus, the increase in temperature and pressure, apart from higher syringol conversion, also causes a total decrease in the O/C ratio of the post-reaction mixture, which is the desired effect.
The catalysts with palladium as the active phase exhibit a slightly worse activity than ruthenium catalysts in the HDO reaction of syringol as shown by the results in Fig. 5c. The highest syringol conversion of 48% is achieved for the process carried out at 130 °C and 60 bar. It was found that the selectivity of reaction products did not depend strictly on the applied reaction conditions; no significant differences in the selectivity are observed. The distribution of the products is like that for the reactions catalysed by the ruthenium-containing systems (Table S3†).
When a platinum catalyst is used, higher selectivity to anisole is achieved than that in the reactions catalysed by the systems containing Ir, Ru or Pd (Table S3†). Nevertheless, this catalyst is not very active in the HDO process of syringol, which is indicated by low degree of transformation reaching the maximum conversion of syringol of 5% only (Fig. 5d).
Under the applied reaction conditions, it was impossible to obtain a fully hydrodeoxygenated chemical compound – all the resulting molecules had oxygen atoms in their structure. The molecule of 1,2,3-trimethoxycyclohexane has the same number of oxygen atoms in its structure as that of the initial compound, but it has saturated bonds. In turn, 1,2-cyclohexanediol has one oxygen atom less than syringol and has a higher ratio of H/C atoms. The remaining compounds, i.e., anisole and cyclohexanol, have as much as 2 oxygen atoms less in the structure, which indicates a significant hydrodeoxygenation. To sum up, the total oxygen content because of the reactions carried out decreases while the ratio of H/C atoms increases. The chemical compound formed in the greatest amounts in the processes carried out in this study is 1,2,3-trimethoxycyclohexane is probably formed as a result of the methyl group attachment to the oxygen atom of the hydroxyl group and then the process of hydrogenation of the aromatic ring. In the available literature on the HDO reaction of syringol, we could not find a report on the reaction, in which 1,2,3-trimethoxycyclohexane is formed. Only Shu et al.50 reported for hydrodeoxygenation of syringol catalysed by Ni/Al2O3 (200 °C, 20 bar) formation of 1,2,3-trimethoxybenzene – an aromatic compound, as one of the products. The presence of this molecule in the post-reaction mixture was explained by the occurrence of side reactions, which were inevitable because of low activation energy of the main process.
Creosol
For the HDO reaction of creosol, five chemicals were identified: 1,2-cyclohexanediol, 1,2-dimethoxycyclohexane, 4-methylcyclohexanol, methoxycyclohexane and anisole (Scheme 3).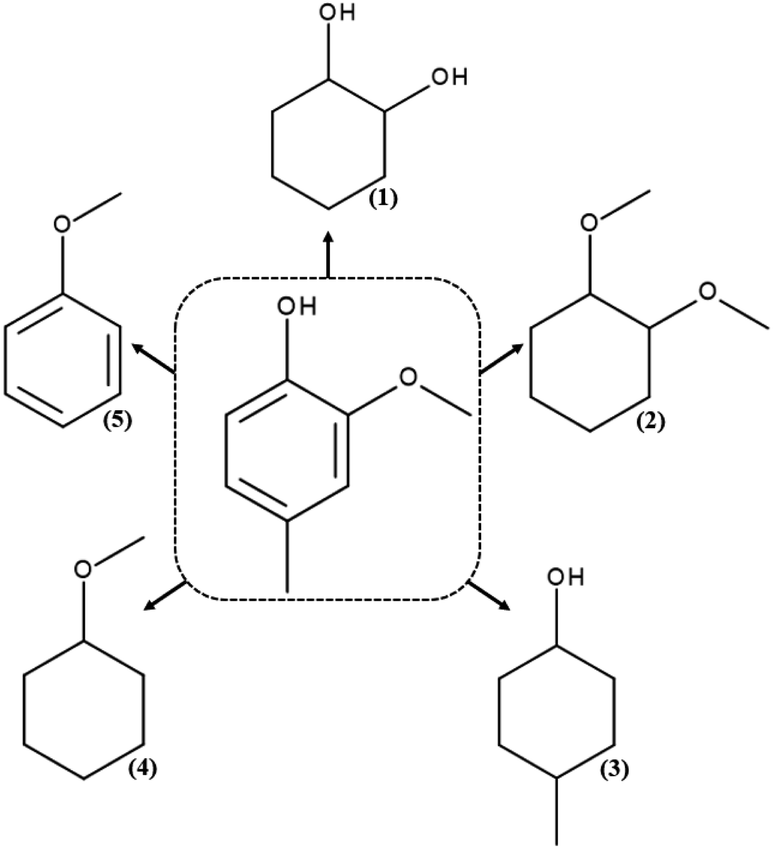 | ||
| Scheme 3 Products of hydrodeoxygenation reaction of creosol: (1) 1,2-cyclohexanediol, (2) 1,2-dimethoxycyclohexane, (3) 4-methylcyclohexanol, (4) methoxycyclohexane and (5) anisole. | ||
The graphs presented in Fig. 6a show the results of the HDO reaction of creosol in the presence of mesoporous SBA-16 silica modified with Ir. The iridium catalyst did not show any improvement in the activity under changing reaction conditions. The conversion of creosol ranged from 1 to 5%. In the presence of all catalytic systems studied three reaction products were identified: 4-methylcyclohexanol, 1,2-cyclohexanediol and anisole (Table S4, see ESI†). No formation of 1,2-dimethoxycyclohexane was observed. Due to the low degree of transformation of the model chemical compound, the selectivity values are charged with high error and are not discussed.
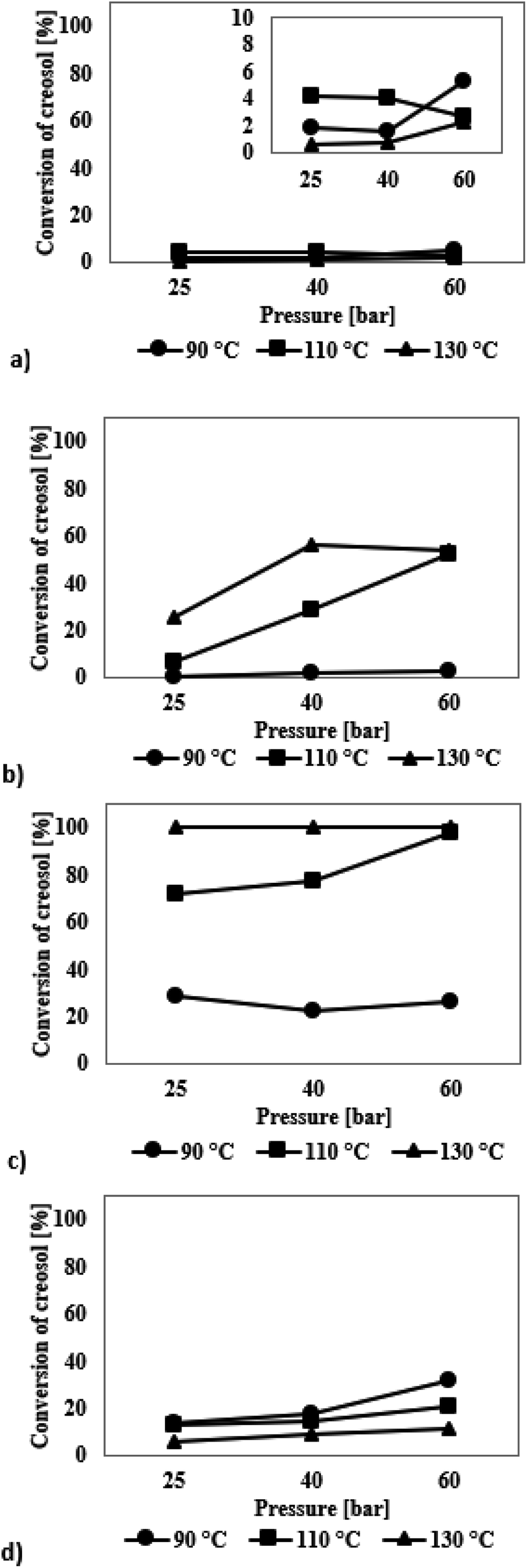 | ||
| Fig. 6 Creosol conversion in the HDO process at temperatures in the range of 90–130 °C and hydrogen pressure of 25–60 bar in the presence of catalysts modified with precious metals: (a) SBA-16/3%Ir, (b) SBA-16/3%Ru, (c) SBA-16/3%Pd, (d) SBA-16/3%Pt. | ||
The results of the HDO reaction of creosol catalysed by ruthenium-containing SBA-16 are shown in Fig. 6b. The catalyst used was very inactive during the processes carried out at 90 °C – the conversion of creosol did not exceed 3%. The activity of ruthenium-modified catalyst was recorded only at higher temperatures, where the maximum 56% conversion of creosol was achieved. In all the experiments the highest selectivity was obtained for 4-methylcyclohexanol and then for 1,2-cyclohexanediol (Table S4†). Note that with increasing conversion of creosol, the selectivity of catalysts towards 1,2-dimethoxycyclohexane decreases. For the process carried out at 130 °C, this compound was not formed in the amount greater than 2%.
Based on the results for the HDO reaction of creosol, catalysed by the systems containing palladium atoms (Fig. 6c), several conclusions can be drawn. Firstly, the conversion of creosol increases with increasing temperature and hydrogen pressure. Thanks to the catalyst used, the creosol molecule can be completely transformed in the process carried out at 110 °C and 60 bar hydrogen pressure and for the process carried out at 130 °C. Secondly, with decreasing conversion of creosol, the amounts of 1,2-dimethoxycyclohexane and 4-methylcyclohexanol increase (Table S4†). On the other hand, as the conversion of creosol increases, the selectivity to 4-methylcyclohexanol increases and that of 1,2-dimethoxycyclohexane decreases in favour of higher amounts of 1,2-cyclohexanediol. Higher values of pressure and temperature favour demethylation processes.
Results for the hydrodeoxygenation reaction of creosol on platinum catalysts are presented in Fig. 6d. As can be seen on this figure, platinum catalyst shows medium activity in the process. The conversion of creosol increases with increasing temperature and increasing hydrogen pressure. It reaches the value of 31% and is the highest among all conducted processes. The reaction product for which the highest selectivity is obtained is 4-methylcyclohexanol (Table S4†). Its amount increases with higher conversion degree. Note that the amount of 1,2-dimethoxycyclohexane decreases with increasing conversion of creosol. The main product of the HDO reaction of creosol in all conducted experiments is 4-methylcyclohexanol. The latter compound is probably formed as a result of the creosol demethoxylation and then hydrogenation of the aromatic ring. There are very few literature reports on HDO of creosol. Nevertheless, Deutsch et al.49 carried out the HDO process of creosol using copper chromite as a catalyst at 225–275 °C and 50 bar hydrogen pressure and also they obtained 4-methylcyclohexanol as the main product of the reaction. In turn, Jongerius et al.51 reported for the HDO reaction of creosol on CoMo/Al2O3 (300 °C and 50 bar) the formation of p-cresol, i.e., the equivalent of 4-methylcyclohexanol with aromatic character as the main product of the reaction.
Study of catalyst reuse
In conclusion, after the analysis of the data presented in Fig. 7, the SBA-16/3%Pd catalyst used four times in the HDO reaction of guaiacol shows similar activity. The conversion of guaiacol was 100% each time. It should be emphasized that the regeneration process causes a significant loss of the catalysts related to the loading and unloading of filters. Therefore, we were unable to carry out any more processes due to the large catalyst loss. Nevertheless, the catalyst destruction is compensated by its high catalytic performance, which makes the entire process profitable. Moreover, if the catalyst is used in the HDO process in larger amounts, its regeneration would be easier to carry out.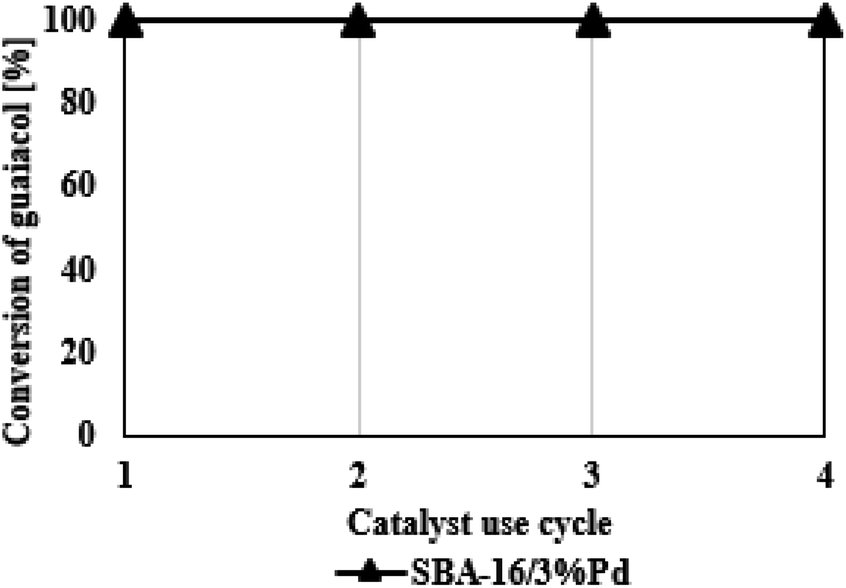 | ||
| Fig. 7 Reaction cycle using regenerated SBA-16/3%Pd catalyst. | ||
Reaction mechanism
In view of the complexity of the HDO process and understanding of the complete reaction network, an analysis of the calculated empirical data would be beneficial. However, the atomistic processes occurring at the catalyst surface are unclear because computational studies dealing with aromatic oxygenates are not as numerous as those dealing with aliphatic oxygenates.52 The adsorption energy (calculated by the standard Gaussian 16 program with Hartree–Fock method) on the palladium surface was in the following order: syringol < creosole < guiacol, the same as the catalytic activity. More detailed study of a “real” catalyst, which will contain defect sites is however necessary.Conclusions
The results of the experiments carried out in this study, including synthesis, physicochemical characterization and catalytic tests of ordered mesoporous silica materials with introduced precious metals on the surface allow for the following conclusions.(i) The synthesized materials are characterized by good structural and textural parameters, which is confirmed by XRD, TEM, SEM, SEM/EDX, low-temperature sorption N2, ICP-OES and XPS studies.
(ii) Among the HDO reactions of guaiacol studied in the presence of SBA-16 silica impregnated with precious metals, the highest selectivity was obtained towards methoxycyclohex-1-ene (molecule with higher degree of hydrogenation and deoxygenation than guaiacol).
The formation of 1,2-cyclohexanediol – a chemical compound with higher H/C ratio than that in guaiacol – was also observed in significant amounts. The influence of the type of active phase deposited on SBA-16 support on the reaction efficiency is reflected by the following order: Pd > Ru > Pt > Ir.
(iii) The model molecule, which is syringol, in the HDO process catalysed by SBA-16 type silica modified with precious metals is transformed to 1,2,3-trimethoxycyclohexane – a chemical compound with higher H/C ratio than that in the substrate. The formation of anisole in larger amounts is also observed, especially in the presence of the catalysts containing Ru and Pt. The influence of the active phase type deposited on SBA-16 on the reaction efficiency is reflected by the following order: Ru > Pd > Pt > Ir.
(iv) In the HDO reaction of creosol catalysed by SBA-16 type silica modified with precious metals the highest selectivity was observed for 4-methylcyclohexanol. Moreover, in the presence of silica modified with Pd ions a significant amount of 1,2-dimethoxycyclohexane was obtained, whereas in the presence of silica containing Ru species, the dominant product was 1,2-cyclohexanediol. The influence of the type of active phase deposited on SBA-16 on the reaction efficiency is reflected by the following order: Pd > Ru > Pt > Ir.
(v) The catalytic system, which showed the worst performance in all reactions is the silica modified with iridium. The reason for this is probably its less developed porous structure than those of the remaining catalysts, which is reflected by the XRD results. Diffractograms of the remaining catalytic systems show smaller crystallites and better metal dispersion, which translates directly into better catalytic performance. The XPS analysis also proved that iridium species are not impregnated effectively as in the case of the remaining metals (a large number of iridium atoms are identified in the surface layer, which indicates their weaker penetration into the pores). In the HDO reaction of guaiacol and creosol, the activity of the catalysts studied is as follows: Pd > Ru > Pt > Ir, whereas in the HDO reaction of syringol the ruthenium catalysts are the best (Ru > Pd > Pt > Ir). It is likely that the increase in the ruthenium activity is due to the fact that the catalyst consists of silica modified with this metal agglomerates (as shown in SEM/EDX images). Most likely, the ruthenium catalyst used for the HDO reaction of syringol showed much higher content of ruthenium than the metal content in other catalytic systems tested for the HDO reactions of guaiacol and syringol.
(vi) Higher temperature and higher hydrogen pressure improve the conversion and facilitate the formation of products with lower total oxygen content and higher hydrogen content. At higher temperatures, the mobility of metal species increases, and they have greater chance to overcome the energy barrier, resulting in the faster reaction rate. In turn, pressure has a significant impact on both the conversion of model chemicals and the distribution of the main products. Higher degrees of conversion of model molecules at higher pressures are natural. More hydrogen gas is present in the reaction environment, which means that more hydrogen molecules are bound to the active phase and react with the substrate.
The ordered mesoporous silica materials with appropriate structural parameters have a wide range of potential applications. With this in mind, we are inclined to assume that these materials can be used in the near future in catalytic production of biofuels, and thus they are beneficial for the environmental protection.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (project no: DEC 2013/10/M/ST5/00652).References
- F. Saladini, N. Patrizi, F. M. Poudyal, N. Marchettini and S. Bastianoni, Renewable Sustainable Energy Rev., 2016, 66, 221–227 CrossRef.
- M. V. Rodionova, R. S. Poudyal, I. Tiwari, R. A. Voloshin, S. K. Zharmukhamedov, H. G. Nam, B. K. Zayadan, B. D. Bruce, H. J. M. Hou and S. I. Allakhverdiev, Int. J. Hydrogen Energy, 2017, 42, 8450–8461 CrossRef CAS.
- T. Damartzis and A. Zabaniotou, Renewable Sustainable Energy Rev., 2011, 15, 366–378 CrossRef CAS.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS.
- S. Kaur, R. Sharma and S. Bansal, International Journal of Innovative Research in Science & Engineering, 2017, 3, 477–489 Search PubMed.
- F. Cheng and C. E. Brewer, Renewable Sustainable Energy Rev., 2017, 72, 673–722 CrossRef CAS.
- T. Demura and Z.-H. Ye, Curr. Opin. Plant Biol., 2010, 13, 299–304 CrossRef PubMed.
- Y. Wang, T. He, K. Liu, J. Wu and Y. Fang, Bioresour. Technol., 2012, 108, 280–284 CrossRef CAS PubMed.
- A. Sharma, V. Pareek and D. Zhang, Renewable Sustainable Energy Rev., 2015, 50, 1081–1096 CrossRef CAS.
- P. Roy and G. Dias, Renewable Sustainable Energy Rev., 2017, 77, 59–69 CrossRef CAS.
- T. M. H. Dabros, M. Z. Stummann, M. Høj, P. A. Jensen, J. D. Grunwaldt, J. Gabrielsen, P. M. Mortensen and A. D. Jensen, Prog. Energy Combust. Sci., 2018, 68, 268–309 CrossRef.
- M. Sharifzadeh, C. J. Richard, K. Liu, K. Hellgardt, D. Chadwick and N. Shah, Biomass Bioenergy, 2015, 76, 108–117 CrossRef CAS.
- A. J. Foster, P. T. M. Do and R. F. Lobo, Top. Catal., 2012, 55, 118–128 CrossRef CAS.
- E. F. Iliopoulou, E. V. Antonakou, S. A. Karakoulia, I. A. Vasalos, A. A. Lappas and K. S. Triantafyllidis, Chem. Eng. J., 2007, 134, 51–57 CrossRef CAS.
- M. Mohammad, H. T. Kandaramath, Z. Yaakob, S. Y. Chandra and K. Sopian, Renewable Sustainable Energy Rev., 2013, 22, 121–132 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- P. Dębek, A. Feliczak-Guzik and I. Nowak, Przem. Chem., 2016, 95/11, 2259–2263 Search PubMed.
- Y.-C. Lin, C.-L. Li, H.-P. Wan, H.-T. Lee and C.-F. Liu, Energy Fuels, 2011, 25, 890–896 CrossRef CAS.
- L. Faba, E. Dia and S. Ordónez, Renewable Sustainable Energy Rev., 2015, 51, 273–287 CrossRef CAS.
- D. Procházkowá, P. Zámostný, M. Bejblová, L. Červený and J. Čejka, Appl. Catal., A, 2007, 332, 56–64 CrossRef.
- F. Cheng and C. E. Brewer, Renewable Sustainable Energy Rev., 2017, 72, 673–722 CrossRef CAS.
- Z. He and X. Wang, Catal. Sustainable Energy, 2012, 1, 28–52 Search PubMed.
- A. S. Ouedraogo and P. R. Bhoi, J. Cleaner Prod., 2020, 253, 1–19 CrossRef.
- J. L. Santos, M. Alda-Onggar, V. Fedorov, M. Peurla, K. Ekräne, P. Mäki-Arvela, M. Á. Centeno and D. Y. Murzin, Appl. Catal., A, 2018, 561, 137–149 CrossRef CAS.
- J. Horáček, G. Št’ávová, V. Kelbichová and D. Kubička, Catal. Today, 2013, 204, 38–45 CrossRef.
- B. Pawelec, C. V. Loricera, C. Geantet, N. Mota, J. L. G. Fierro and R. M. Navarro, Mol. Catal., 2020, 482, 1–9 Search PubMed.
- M. S. Zanuttini, M. Gross, G. Marchetti and C. Querini, Appl. Catal., A, 2019, 587, 1–12 CrossRef.
- S. Chen, G. Zhou and C. Miao, Renewable Sustainable Energy Rev., 2019, 101, 568–589 CrossRef CAS.
- P. Kluson and L. Cerveny, Appl. Catal., A, 1995, 128, 13–31 CrossRef CAS.
- N. Arun, R. V. Sharma and A. K. Dalai, Renewable Sustainable Energy Rev., 2015, 48, 240–255 CrossRef CAS.
- A. M. Barrios, C. A. Teles, P. M. de Souza, R. C. Rabelo-Neto, G. Jacobs, B. H. Davis, L. E. P. Borges and F. B. Noronha, Catal. Today, 2018, 302, 115–124 CrossRef CAS.
- M. Lu, H. Du, B. Wei, J. Zhu, M. Li, Y. Shan and C. Song, Energy Fuels, 2017, 31, 10858–10865 CrossRef CAS.
- C. Zhao, J. He, A. A. Lemonidou, X. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS.
- A. Feliczak-Guzik, P. Szczyglewska and I. Nowak, Catal. Today, 2019, 325, 61–67 CrossRef CAS.
- C. Newman, X. Zhou, B. Goundie, I. T. Ghampson, R. A. Pollock, Z. Ross, M. C. Wheeler, R. W. Meulenberg, R. N. Austin and B. G. Frederick, Appl. Catal., A, 2014, 477, 64–74 CrossRef CAS.
- G. Yao, G. Wu, W. Dai, N. Guan and L. Li, Fuel, 2015, 150, 175–183 CrossRef CAS.
- P. Szczyglewska, A. Feliczak-Guzik and I. Nowak, Microporous Mesoporous Mater., 2020, 293, 1–6 CrossRef.
- Z. He, M. Hu and X. Wang, Catal. Today, 2018, 302, 136–145 CrossRef CAS.
- E. H. Lee, R.-S. Park, H. Kim, S. H. Park, S.-C. Jung, J.-K. Jeon, S. C. Kim and Y.-K. Park, J. Ind. Eng. Chem., 2016, 37, 18–21 CrossRef CAS.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS.
- M. Snare, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- R. J. Chimentāo, H. Oliva, J. Belmar, K. Morales, P. Mäki-Arvela, J. Wärna, D. Y. Murzin, J. L. G. Fierro, J. Llorca and D. Ruiz, Appl. Catal., B, 2019, 241, 270–283 CrossRef.
- A. Feliczak-Guzik, B. Jadach, H. Piotrowska, M. Murias, J. Lulek and I. Nowak, Microporous Mesoporous Mater., 2016, 220, 231–238 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- T.-W. Kim, R. Ryoo, M. Kruk, K. P. Gierszal, M. Jaroniec, S. Kamiya and O. Terasaki, J. Phys. Chem. B, 2004, 108, 11480–11489 CrossRef CAS.
- W. J. J. Stevens, K. Lebeau, M. Mertens, G. V. Tendeloo, P. Cool and E. F. Vansant, J. Phys. Chem. B, 2006, 110, 9183–9187 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- V. Meynen, P. Cool and E. F. Vansant, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS.
- K. L. Deutsch and B. H. Shanks, Appl. Catal., A, 2012, 447–448, 144–150 CrossRef CAS.
- R. Shu, Y. Xu, L. Ma, Q. Zhang, P. Chen and T. Wang, Catal. Commun., 2017, 91, 1–5 CrossRef CAS.
- A. I. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS.
- N. Guo, S. Caratzoulas, D. J. Doren, S. I. Sandler and D. G. Vlachos, Energy Environ. Sci., 2012, 5, 6703–6716 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06696f |
| This journal is © The Royal Society of Chemistry 2021 |