
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTable-top combined scanning X-ray small angle scattering and transmission microscopies of lipid vesicles dispersed in free-standing gel†
Francesco Scattarella a,
Emiliano Altamurab,
Paola Albaneseb,
Dritan Siliqia,
Massimo Ladisa‡
a,
Fabio Mavellib,
Cinzia Gianninia and
Davide Altamura*a
a,
Emiliano Altamurab,
Paola Albaneseb,
Dritan Siliqia,
Massimo Ladisa‡
a,
Fabio Mavellib,
Cinzia Gianninia and
Davide Altamura*a
aIstituto di Cristallografia – CNR, Via Amendola 122/O, 70126 Bari, Italy. E-mail: davide.altamura@ic.cnr.it
bChemistry Department University of Bari Aldo Moro, via Orabona 4, 70125 Bari, Italy
First published on 23rd December 2020
Abstract
A mm thick free-standing gel containing lipid vesicles made of 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) was studied by scanning Small Angle X-ray Scattering (SAXS) and X-ray Transmission (XT) microscopies. Raster scanning relatively large volumes, besides reducing the risk of radiation damage, allows signal integration, improving the signal-to-noise ratio (SNR), as well as high statistical significance of the dataset. The persistence of lipid vesicles in gel was demonstrated, while mapping their spatial distribution and concentration gradients. Information about lipid aggregation and packing, as well as about gel density gradients, was obtained. A posteriori confirmation of lipid presence in well-defined sample areas was obtained by studying the dried sample, featuring clear Bragg peaks from stacked bilayers. The comparison between wet and dry samples allowed it to be proved that lipids do not significantly migrate within the gel even upon drying, whereas bilayer curvature is lost by removing water, resulting in lipids packed in ordered lamellae. Suitable algorithms were successfully employed for enhancing transmission microscopy sensitivity to low absorbing objects, and allowing full SAXS intensity normalization as a general approach. In particular, data reduction includes normalization of the SAXS intensity against the local sample thickness derived from absorption contrast maps. The proposed study was demonstrated by a room-sized instrumentation, although equipped with a high brilliance X-ray micro-source, and is expected to be applicable to a wide variety of organic, inorganic, and multicomponent systems, including biomaterials. The employed routines for data reduction and microscopy, including Gaussian filter for contrast enhancement of low absorbing objects and a region growing segmentation algorithm to exclude no-sample regions, have been implemented and made freely available through the updated in-house developed software SUNBIM.
Introduction
Hydrogels1 are highly versatile formulations with widespread applications in several fields ranging from drug delivery systeims2,3 to tissue engineering,4,5 diagnostics6,7 and biotechnology.8 On the other hand, micro- and nano-artificial compartments and emulsions, based on low density contrast components, such as lipid structures in water solution, have great potential in several applications, in particular drug delivery,9 and have been profitably studied by Cryogenic Transmission Electron Microscopy (Cryo-TEM),10,11 Small Angle X-ray Scattering (SAXS), and more specifically Small Angle Neutron Scattering (SANS),12–14 being the small-wavelength scattering contrast particularly suited to study nanoscale objects. Although less than neutrons,13 hard X-rays feature sufficiently high penetration depth to study relatively thick samples with no need for specific sample preparation, non-negligible scattering contrast for light elements, but have on the other hand widespread availability both at synchrotron facilities and in traditional laboratories. Still, SAXS provides important information even in soft matter investigation, either by itself or in combination with SANS,10,13 although studies of complex systems with low scattering contrast such as lipid vesicles have been almost exclusively performed at synchrotron facilities (10,15–21 among others), except for a few examples,22 and to the best of our knowledge never as 2D microscopies on thick gel systems. On the other hand, laboratory equipment has been applied to Wide Angle X-ray Scattering studies of lipid vesicles, combined with synchrotron-SAXS/SANS data,10 or for SAXS studies mainly aimed at deriving the stacking periodicity of packed lipids from Bragg diffraction peaks11,23 (besides studies of high contrast nanoparticles, e.g. ref. 24). The scanning microscopy approach, possibly combining diffraction with other types of contrast, is specifically suited to investigate the spatial distribution of structural features over different length scales across extended samples,25–28 nowadays also available with advanced laboratory equipment,29–32,43 provided that proper software is available for data processing (e.g. ref. 33). In this work, combined scanning SAXS/XT (X-ray Transmission) microscopies were employed for the first time to the best of our knowledge to investigate the persistence of lipid vesicles of 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) obtained by extrusion techniques (≤100 nm) within a gel preparation, as well as their spatial distribution with lateral resolution of approximately 200 μm over a macroscopic sample region of order 10 mm2. The presence of vesicles was assessed, based on readapted traditional approaches involving the subtraction of the buffer contribution, as for bioSAXS data reduction.34 Moreover, since lipid distribution in such a thick gel can hardly be confirmed by alternative methods, the X-ray scattering contrast was again exploited in a self-consistent study to bypass the intrinsic ambiguity of SAXS profile fitting, by monitoring the clear Bragg diffraction signal resulting from stacked bilayers upon sample drying, providing the a posteriori confirmation of lipid presence in well-defined sample areas. Despite the low absorption of lipids, a XT microscopy with improved sensitivity, as well as lipid thickness evaluation based on absorption contrast, was achieved, encompassing in turn the case of (low) absorbing objects dispersed in a matrix, which gives further generality to this study. The scanning microscopy approach is shown to provide useful hints about vesicle interaction and aggregation (thoroughly studied in solution by Komorowski et al.15 for DOPC and other lipids), in semi-static configurations such as dispersions in gel, which can be in perspective studied as a function of different microenvironments, lipid composition and concentration. Despite the relatively low spatial resolution of the microscopies, diffraction contrast allowed to conclude that vesicles mainly concentrate towards the borders of the gel volume, following the expected gel density distribution, being with high probability in contact with each other even in the more diluted regions. The distribution of lipid concentration before and after sample drying indicated negligible vesicle diffusion through the gel network. Data reduction was carried out by specially developed routines aimed at comparing the gel embedding lipids with the corresponding bare gel used as a blank. Basically, from two datasets representing the transmitted and scattered X-ray intensity respectively, two more relevant microscopies were obtained, displaying the distributions of the relative thickness and of the vesicle concentration, respectively. The quantitative comparison between semi-solid samples having different arbitrary volumes was based on the normalization of the SAXS intensity against the local sample thickness. Avoiding supporting capillaries prevented significant sample handling in filling procedures possibly leading to vesicle alterations or damage. On the other hand, raster scanning relatively large volumes reduced the risk of radiation damage, which is to be considered in the perspective of using more and more brilliant laboratory X-ray sources, being nowadays available and under continuous development. The proposed platform is demonstrated for SAXS microscopy, based on room-sized instrumentation35 and is expected to be applicable to a wide variety of nanostructured systems. Moreover, it can be straightforwardly extended to Wide Angle X-ray Scattering (WAXS) data analysis and microscopy, to allow for atomic scale structural investigation as well. The routines have been implemented and made freely available through the in-house developed software SUNBIM.36Experimental
Vesicle preparation
Lipid vesicles were prepared following established procedure37 with some modifications. 100 μL of POPC 10 mM in chloroform were put in an 1.5 mL Eppendorf tube then dried under nitrogen flow and subsequently under vacuum for 3 hours to completely remove chloroform traces. 1 mL of K-phosphate buffer 20 mM pH 8 was added to hydrate the lipid film. Multilamellar vesicles were prepared by sonicating with an ultrasonic titan horn (3.5 mm diameter, Branson Sonifier 250, Danbury, CT) operating at 20 kHz. The instrument was set to 30 W in pulsed mode and duty cycle control 40% until the lipid film on the bottom of the tube disappeared. During ultrasound treatment, the Eppendorf tube containing the sample was kept in an ice/water bath to prevent overheating phenomena. The resulting multilamellar vesicle dispersion was then transferred into an extruder device (Avanti Mini Extruder produced by Avanti Polar Lipids, Inc., Alabama, USA) which allowed the extrusion of the multilamellar vesicles through standard 19 mm polycarbonate filters with track-etched uniform cylindrical 100 nm pores. The vesicles were passed through two (stacked) filters ten times, obtaining lipid compartments with a diameter ≤ 100 nm.Gel preparation
2 mL of K-phosphate buffer 20 mM pH 8 containing 40 mg of agar powder (Sigma-Aldrich # A1296) were heated under continuous stirring until complete dissolution of the solute obtaining a solution with 2% (w/v) of agar. GEL sample was prepared by diluting 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 previous solution (500 μL) with of fresh K-phosphate buffer 20 mM pH 8 (500 μL) obtaining a gel (after cooling) of agar 1%. POPC sample was prepared by diluting 1:1 previous solution (500 μL) with 500 μL of vesicles in K-phosphate buffer 20 mM pH 8 obtaining a vesicle-containing gel (after cooling) of agar 1%.
:1 previous solution (500 μL) with of fresh K-phosphate buffer 20 mM pH 8 (500 μL) obtaining a gel (after cooling) of agar 1%. POPC sample was prepared by diluting 1:1 previous solution (500 μL) with 500 μL of vesicles in K-phosphate buffer 20 mM pH 8 obtaining a vesicle-containing gel (after cooling) of agar 1%.
Scanning SAXS experiment
The samples consist of free-standing gel containing or not POPC vesicles, referred to as POPC and GEL sample, respectively. Samples were placed in ultralene sachets perpendicular to the primary X-ray beam. Scanning SAXS and XT microscopies were collected from a relatively large portion of the sample (about 10 mm2) with a 0.2 mm step and a 0.2 mm (diameter) beam footprint. A Rigaku three pinholes SAXS camera, coupled to a Fr-E+ superbright microsource through a focusing Confocal Max Flux (CMF 15-105) optics,35 was employed. The transmitted X-ray intensity was measured at each point of the raster scan by a pin diode in the beamstop. Due to beamstop, beam shape, and sample-to-detector distance (SDD ∼ 2.2 m) the actually usable Q-range was 0.15 ÷ 1.93 nm−1, where Q = (4π/λ)sinθ is the scattering vector modulus, θ is half the scattering angle, λ = 1.54 Å is the X-ray wavelength. The SAXS frames collected from each point on the sample were arranged in a 2D microscopy by using the in-house developed software SUNBIM.36 After performing angular calibration, dark current subtraction, and absorption correction (point-by-point normalization of the SAXS intensity to the transmission coefficient T) for all frames, the microscopy returned the spatial (2D) distribution of the scatterers, integrated along the sample thickness. A further normalization to the relative sample thickness (i.e. to the illuminated volume), allowed to map the relative abundance of the scatterers. Further microscopies were obtained by remapping SAXS intensity in specific Q-ranges corresponding to precise length scales, across the investigated sample area. Both SAXS and T maps were corrected for possible fluctuations of the primary beam intensity. Microscopies were collected while keeping the sample either at room pressure (wet samples), by means of kapton windows, or at about 5 × 10−2 mbar vacuum pressure (dried samples). In the case of the POPC sample, the same sample was studied in both conditions consecutively, without being removed. In the case of the bare gel, two different replicas were used for measurements at room and vacuum pressure, respectively.
X-ray diffraction (XRD)
XRD measurements were carried out on POPC bi-layers (control sample) deposited as chloroform solution on Silicon wafers and let dry at room pressure, by using a Bruker D8 Discover (CuKα X-ray source, λ = 1.54 Å) equipped with a sealed X-ray tube, a Göbel mirror, a scintillation point detector and an Eulerian cradle. The periodicity derived from the XRD experiment served as a reference for the Bragg peak appearing in the SAXS measurements.Results and discussion
Scanning SAXS/XT microscopies
The scanning transmission (T) and SAXS microscopies collected at room pressure and in vacuum from the bare gel and the gel embedding vesicles (referred to as GEL and POPC samples, respectively) are reported in Fig. 1 and 2, along with the derived maps of the relative sample thickness (n) and the normalized SAXS intensity (ISAXS/T/n), respectively. The normalization of the SAXS microscopy to n, required a masking procedure based on the Region Growing algorithm,41 to avoid singularities arising from the normalization to arbitrary low n values in the no-sample areas, as described in the ESI (Section E†). SAXS intensity is here integrated over the full accessible Q-range. Normalization to T(Isaxs/T) would yield the spatial distribution of the total amount of scatterers; the further normalization to n(Isaxs/T/n) thus yields the 2D spatial distribution of the relative scatterer concentration, as explained in the following.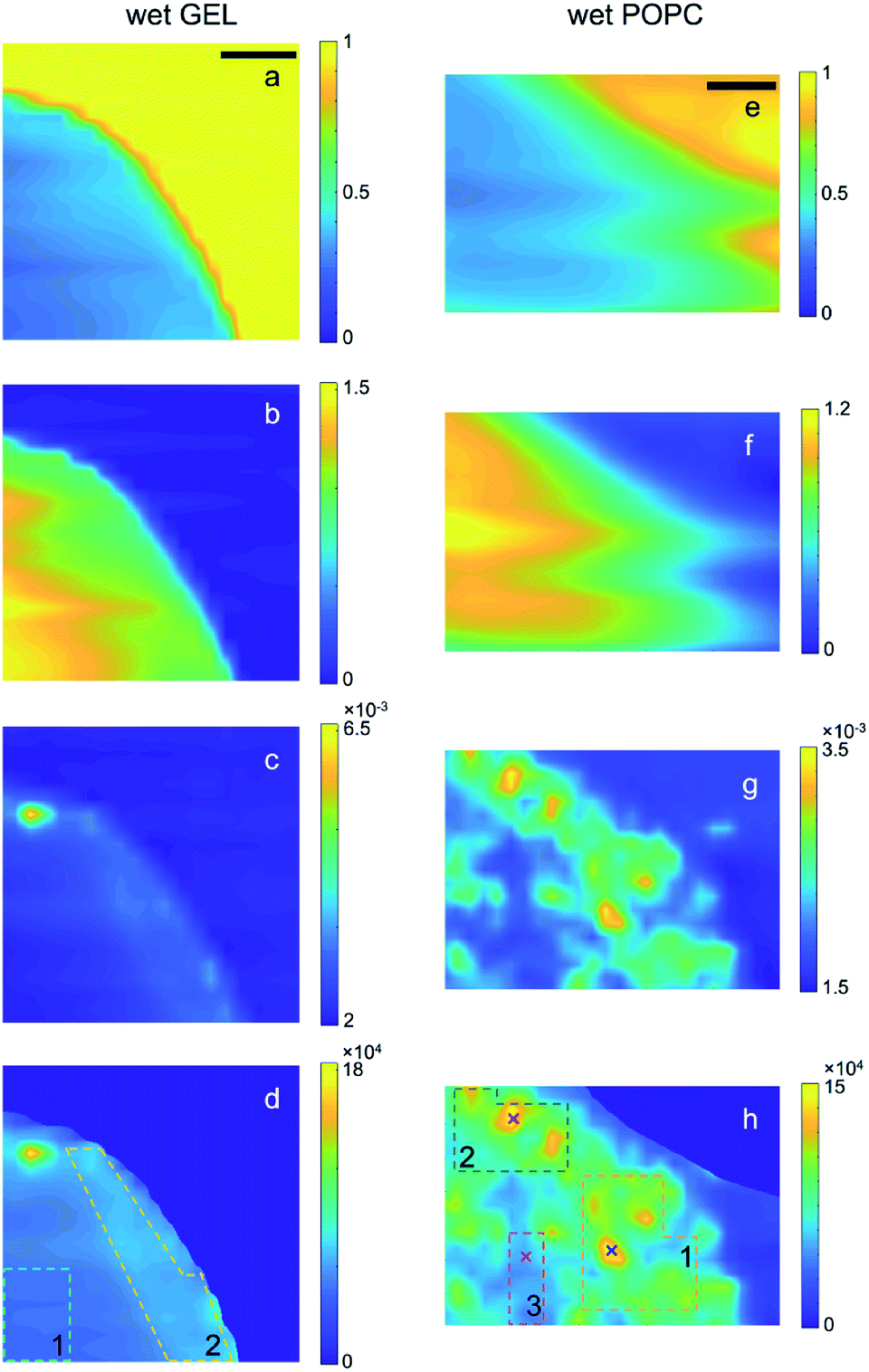 | ||
| Fig. 1 Transmission (a and e), relative thickness (b and f), raw SAXS (c and g) and normalized SAXS (d and h) intensity maps of the wet GEL and POPC samples, respectively. SAXS intensity of each pixel is integrated over the full accessible Q-range. In the case of raw SAXS maps (c and g), the intensity values in the colour bar are normalized to the mean value. In the case of the normalized SAXS maps (d and h), the absolute intensity values are reported and directly indicate the relative abundance of lipids. The dotted lines delimit the ROIs from which integrated SAXS profiles (in Fig. S1 and S2†) were extracted, whereas the (×) symbols indicate the points from which single SAXS profiles were extracted: the colour codes for GEL and POPC samples correspond to those in Fig. S1 and S2.† Scale bar: 1 mm. | ||
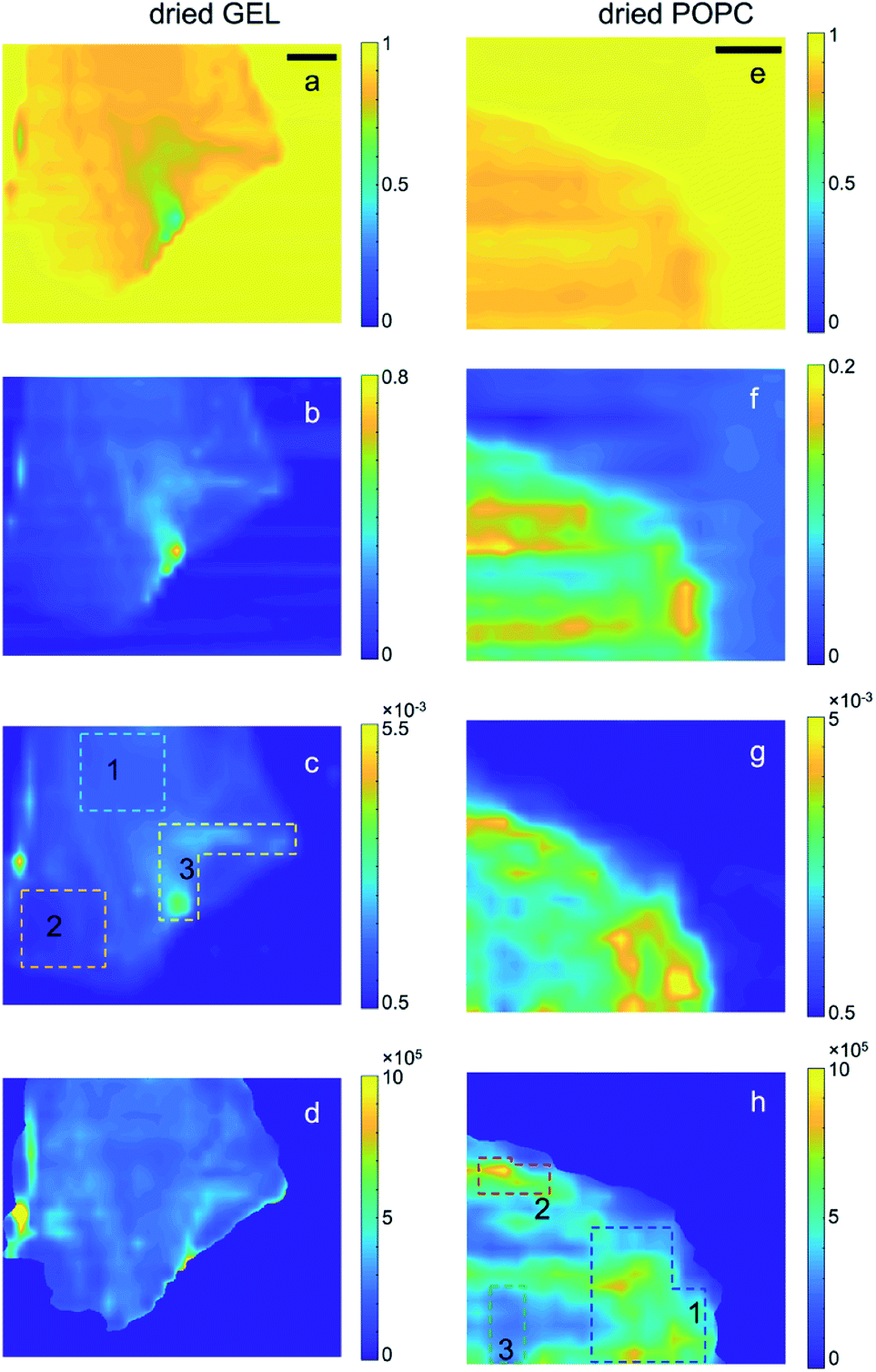 | ||
| Fig. 2 Transmission (a and e), relative thickness (b and f), raw SAXS (c and g) and normalized SAXS (d and h) intensity maps of the dried GEL and POPC samples, respectively; t (e) and n (f) maps were obtained after row by row compensation (see ESI: Section D†) of background irregularities and Gaussian filtering application. SAXS intensity in each pixel is integrated over the full accessible Q-range. In the case of raw SAXS maps (c and g), the intensity values in the colour bar are normalized to the mean value. In the case of the normalized SAXS maps (d and h), the absolute intensity values are reported and directly indicate the relative abundance of lipids. The dotted lines delimit the ROIs from which integrated SAXS profiles (in Fig. S7†) were extracted. The colour codes for gel and POPC samples correspond to those in Fig. S7a.† In the case of gel samples, ROIs were drawn on the as-collected SAXS microscopy, as features related to density inhomogeneities were levelled upon absorption/thickness correction. Scale bar: 1 mm. | ||
lnT) increases the relative SAXS contribution of the inner (thicker) regions of the samples compared to the outer (thinner) ones (panels c and d, g and h of Fig. 1 and 2), eventually returning the relative scattered intensity variation (per reference volume). From the point of view of the absorption contrast, both GEL and POPC samples show high homogeneity, with smooth gradients for T (Fig. 1a and e) and n (Fig. 1b and f), moving from the inner to the outer regions of the sample due to progressive thickness decrease. Such trends confirm in turn that the completely different SAXS features observed in Fig. 1g and h are due to the nanoscale structures of the additional POPC component rather than to microscale thickness/density inhomogeneities. Indeed, from the point of view of the scattering contrast, the GEL sample features by far a more homogeneous scattering distribution (Fig. 1c and d) than the POPC sample (Fig. 1g and h). There is only one high intensity spot in Fig. 1c and d, ascribed to a high density GEL region, probably containing agar aggregates, or an impurity (see discussion about Fig. S1†). Finally, the effect of thickness variations is highlighted in the comparison between the as collected SAXS (Fig. 1c and g) and normalized SAXS (Fig. 1d and h) intensity maps, the last featuring much smoother gradients (i.e. higher homogeneity of the X-ray intensity distribution per reference volume). Moreover, such intensity gradients are significantly different between GEL and POPC samples, staying below 20% across the gel matrix (Fig. 1d) (leaving out the aggregate), and being more than an order of magnitude larger in presence of lipids (Fig. 1h). The direct confirmation that Fig. 1g and h actually provide a visualization of the 2D distribution of POPC vesicles is given by the SAXS profile analysis (and following cross-check of the dried sample) from a morphological/structural point of view, as detailed in the following section (“SAXS analysis of wet samples”).The routines for such calculations have been implemented on purpose in the new release 3.0 of SUNBIM.36 Moreover, since the normalization allows to put the patterns averaged over any ROI on the same scale, it could be verified that the SAXS profiles from the outer and denser region of the GEL sample (ROI 2) better matched the intensity scale of the most populated regions (ROIs 1 and 2) of lipids in the POPC sample; similarly, the inner region (ROI 1) of the GEL sample matched the less populated region (ROI 3) of the POPC one. This could be expected based on surface segregation effects arising during gel formation in both samples. The SAXS profiles from GEL, to be used as the buffer profiles for given POPC ones, were then chosen accordingly (leading to the difference profiles in Fig. S1b and S2b†). In presence of isolated bright spots, single SAXS profiles from the relevant selected points were also extracted to check for the presence of aggregates and for their contribution to the areal averaged signal (see discussion about Fig. S1 and S2b†). The selected ROIs and points on the samples are indicated by dotted lines and symbols (×), respectively, in Fig. 1 and 2 (same colour code as in Fig. S1 and S2†). The comparison among areal averaged SAXS profiles from different ROIs of the POPC sample (Fig. S2b†) indicates similar morphological/structural features, with intensity scale differences related to lipid concentration. In particular, in the case of low POPC concentration regions, such as ROI 3 in Fig. 1h, the areal average provided an appreciable SAXS signal (magenta line in Fig. S2b†), where no appreciable scattering from lipids could be detected in single SAXS patterns. The density difference between the two GEL regions was exploited to have an insight in the gel structure: the SAXS difference profile between ROIs 2 and 1 of the GEL sample, could be fitted against a mass fractal model (Fig. S3†), with a 2.78 exponent. The careful choice of ROIs for the subtraction of gel scattering contribution allowed to have a morphological/structural insight into the lipids through the analysis of the SAXS difference profiles. The profile from ROI 2 in the POPC sample (subtracted by the correspondent ROI 2 GEL profile, green circles in Fig. S2b†), featuring the higher SNR, was fitted by using the program SasView39 either against a shape-independent model, so as to test data reliability and have an insight into the characteristic length scale of the scatterers, or against a shape-dependent model to figure out the possible lipid conformation and check vesicle subsistence within the gel. No assumptions were made about the size of vesicles, whereas a spherical shape was only assumed for the shape-dependent model. In both cases, no parameters were fixed in the fit. The two fits are reported in Fig. S4 and S5,† respectively. Based on the shape-independent model, assuming no polydispersity, a Rg value of 13 nm was obtained from the fitted power-law decay of the SAXS intensity as I(Q) ∼ Q−2, indicating disk-like objects with a larger dimension of Dmax = 35 nm. Due to the peculiar matrix embedding lipids, such outcome could be expected as the average contribution of deformed vesicles, with shape/size polydispersity. Based on the shape-dependent model, assuming a spherical shape and taking into account size polydispersity, either unilamellar or multilamellar core–shell vesicles were considered (Fig. S5†). A 22 ± 6 nm core radius (about 30% polydispersity) and a total radius of 23 ± 6 or 26 ± 7 nm were derived, respectively (about 50 nm diameter). Although none of the models perfectly fits the experimental data, the respective characteristic sizes are fully compatible and overlapping. Indeed, even in the multilamellar model, most of the vesicles (80%) are actually unilamellar. It is worth to note that, although the form factor related to the SAXS curves in Fig. S2b† does not allow unambiguous determination of 3D lipid structures due to the limited accessible Q-range, it is on the other hand strongly representative of single bilayer morphology, as well as of vesicles on the low side of size distribution. Since POPC bilayers are unlikely to float in solution, but are rather expected to self-assemble either in lamellas or in closed vesicles, the form factor returned by Fig. S2b, S4 and S5† indicating the subsistence of single bilayers leads to the reasonable conclusion that lipids kept their 3D morphology within the gel matrix, i.e. vesicles persist during gel formation, although with possible deviations from an exact spherical shape. That said, a main result is returned by the normalized SAXS difference profiles: the X-ray intensity plotted in Fig. S2b† is indeed proportional to the amount of lipids per volume unit (as is the intensity/colour scale of the quantitative microscopies in Fig. 1h and 2h), and therefore it directly indicates the relative abundance of lipids in the different ROIs. The difference between the average concentrations is thus simply given by the intensity difference between the average SAXS profiles relevant to ROIs 1,2,3 reported in Fig. S2b† (highlighted by colours in the legend), showing an increase of concentration by a factor of 3 from ROI 3 to ROI 2, and up to a factor of 5 from ROI 3 to ROI 1. Finally, in order to overcome the unavoidable ambiguity intrinsic in SAXS profile fitting, as discussed above, especially in the case of size/shape polydispersity of the scatterers, the a posteriori confirmation of lipid presence in well-defined sample areas was obtained by studying and comparing the same sample after drying (next paragraph), by monitoring the fingerprint provided by the clear Bragg diffraction signal resulting from tightly stacked bilayers.
Such conclusions are confirmed by extending the comparison between wet and dry samples to the whole microscopies. Notably, the spatial distribution of the contrast in the SAXS maps of Fig. 2h visibly follows the one observed in Fig. 1h, confirming that lipids were not significantly moving during gel drying. In particular, it can be recognized that the significantly different concentration of vesicles between ROI 3 (compared to the surrounding regions, as the bright ROIs 1 and 2) is largely preserved even upon gel drying, indicating low or no diffusion of lipids through the gel matrix.
Finally, by plotting and comparing the SAXS intensity maps in specific Q-ranges, it is in principle possible to visualize independently of the spatial distribution of the gel matrix, the POPC vesicles, as well as that of the docked bi-layers in the wet gel, respectively. Indeed, the Q-dependent SAXS maps allow in general to selectively visualize components in the specimen with different length scales, and their relative abundance, in a multimodal imaging approach42 (successfully applied to many previous studies of composite/hierarchical materials/tissues either with synchrotron25–28 or laboratory sources30). As shown in Fig. S2a,† three main Q-ranges can be identified, 0.15 ÷ 0.20 nm−1, 0.36 ÷ 0.90 nm−1, 0.92 ÷ 1.09 nm−1, corresponding to smaller or larger intensity difference between the POPC and GEL samples, the last two ranges being associated to morphological (form factor) and structural (Bragg-like diffraction) POPC scattering contribution, respectively. Similarly, in the dry samples three main Q-ranges were considered, 0.15 ÷ 0.45 nm−1, 0.45 ÷ 0.85 nm−1, 1.02 ÷ 1.20 nm−1, which, based on Fig. S7,† can be mainly related to lipid aggregates, gel matrix and stacked lipids, respectively. In Fig. 3 and 4, the SAXS microscopies of the POPC sample are re-mapped by separately considering only the SAXS intensity in each of the three Q-ranges (from left to right). Since POPC scattering barely emerges in the wet samples, only tiny differences are recognized among the three maps in Fig. 3 (bottom row). Nevertheless, an increasing relative contribution of the SAXS signal towards the high Q-ranges, is detected from the inner region of the sample (defined by the magenta dotted line). Based on Fig. S2a,† the high-Q-range contribution is expected to be mainly related to interacting bilayers; therefore, the observed trend as a function of Q provides the further conclusion that, compared to the surrounding brighter regions, the relative scattering contribution from the inner sample region is mainly due to interacting POPC vesicles (not forming big aggregates); to a smaller extent to lipid form factor; and lastly to cross-linked gel matrix. It is worth to note that the as-collected data (upper row in Fig. 3) cannot catch such fine quantitative details, yielding the opposite trend, due to absorption/thickness effects (as already recognized by comparing Fig. 1g and h). A further confirmation of this picture comes from the basically unchanged spatial distribution of lipids upon sample drying (Fig. 4), indicating that lipid diffusion is hindered by the gel matrix, and possibly by the consequent mutual interaction among neighbouring vesicles. The comparison between the two maps corresponding to the first Q-range (i.e. panels in the first column of Fig. 4), comprising most of the SAXS intensity, clearly evidences how the large SAXS intensity is produced by larger amount of lipids (which both absorb and scatter X-rays at lower Q values) and is therefore uniformed by the absorption/thickness correction (top and bottom left panels), except for a bright spot indicated by the white box, which is ascribed to high scattering aggregates in the specific Q-range. In the high-Q-range (SAXS maps in the right column), the intensity of such bright spot levels out to the surrounding region. On the other hand, further high intensity regions show up, as indicated by the black and magenta boxes, which are representative of bilayers without and with long range order, respectively.
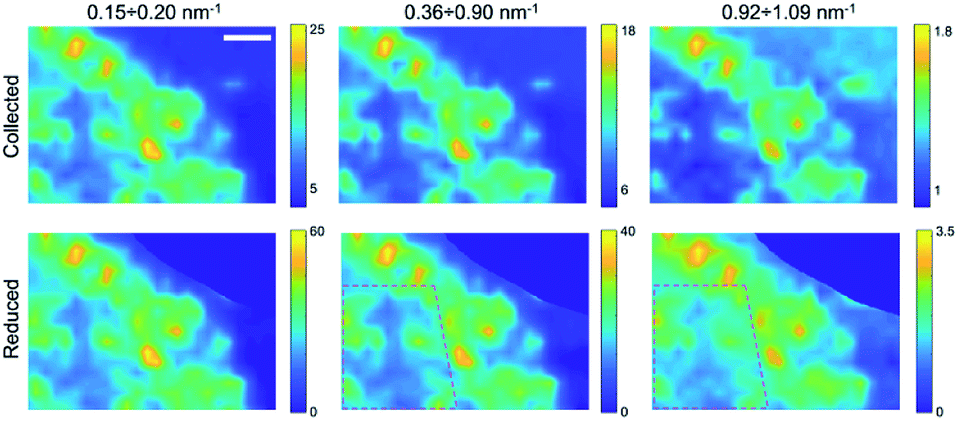 | ||
| Fig. 3 SAXS maps in different q-ranges for the wet POPC sample. From left to right (in both rows): 0.15 ÷ 0.20, 0.36 ÷ 0.90, 0.92 ÷ 1.09 nm−1. Upper row: as collected data; lower row: reduced data; in the area delimited by the dotted line, corresponding to low POPC concentration, the contrast is visibly changing as a function of the chosen Q-range, compared to the surrounding regions. Scale bar: 1 mm. | ||
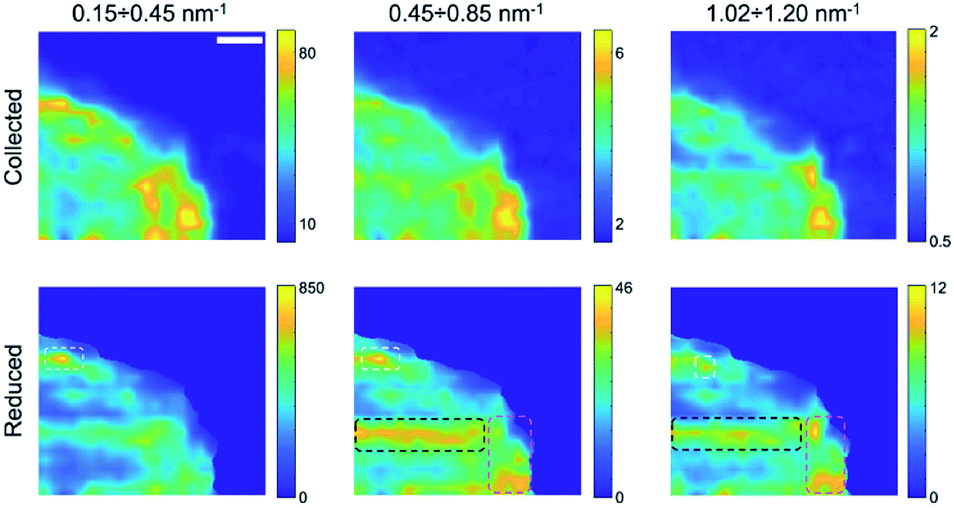 | ||
| Fig. 4 SAXS maps in different Q-ranges for the dried POPC sample. From left to right (in both rows): 0.15 ÷ 0.45, 0.45 ÷ 0.85, 1.02 ÷ 1.20 nm−1. Upper row: as collected data; lower row: reduced data; in the areas delimited by dotted lines, the contrast is visibly changing as a function of the chosen Q-range. Scale bar: 1 mm. | ||
For completeness, the Q-dependent SAXS microscopies of the dry GEL sample are reported in Fig. S9.† Again, only very tiny differences in the SAXS intensity are observed as a function of the Q-range, except for a big aggregate indicated by a black circle, producing high SAXS intensity only at low Q values. Such tiny differences confirm the much smaller weight of SAXS intensity gradients expected from the bare gel, underlying the signal collected from the POPC gelled sample.
The scanning SAXS microscopy approach is proposed as a platform allowing both the direct visualization of the spatial distribution of lipid nanostructures in semi-static configurations, such as dispersions in gel, and the study of features related to their interaction and aggregation, which can be in perspective performed as a function of different microenvironment and lipid composition and concentration. The potential of the Q-dependent sensitivity and possible automated selectivity strongly relies on the discrimination between small intensity variations, as well as between Q-related intensity variations over a sufficiently large Q-range, which are indeed the main limitations of laboratory grade instrumentation, compared to synchrotron facilities. By taking into account the normal overbooking of synchrotron facilities, the possibility of even slightly reducing such gaps would lead to significant improvement of in-house research, as well as of preliminary data quality preparatory to proposal submission for advanced synchrotron studies. To this aim, suitable routines have been here implemented and further ongoing studies are focused on the improvement of data processing, as well as on hardware upgrades. On the other hand, high contrast SANS methods will be hopefully exploited, as they would definitely provide peerless and complementary structural details of both vesicle morphology, as well as of the bilayer inner structure, although with the drawback of long time exposure and complicated timing due to limited availability.
Conclusions
Scanning SAXS microscopy is shown to provide useful hints about vesicle subsistence, interaction and aggregation in semi-static configurations, such as dispersions in gel, allowing both the direct visualization of the spatial distribution of vesicles, and the study of morphological parameters derived by fitting the relevant SAXS profiles, which can be in perspective performed as a function of different microenvironments, lipid composition and concentration. Lipids were found to mainly concentrate towards the borders of the gel volume, according to the gel density distribution, being most probably in contact with each other even in the more diluted regions. The comparison between wet and dry samples indicates that lipids do not significantly migrate within the gel even upon drying, whereas bilayer curvature is lost by removing water, resulting in lipids packed in ordered lamellae. The quantitative comparison between semi-solid samples having different arbitrary volumes was based on the normalization of the SAXS intensity against the local sample thickness derived from absorption contrast maps, resulting in a combined versatile approach: scanning SAXS/Transmission microscopy, providing maps of the spatial distribution of lipid nanostructures and sample thickness, respectively, finally combined in a 2D projection map of the distribution of scatterer concentration. The present study is demonstrated by room-sized instrumentation, and is proposed as a platform to study a wide variety of nanostructured systems. The routines to perform the complex analysis procedure here described, from data reduction to microscopy, have been implemented and made freely available through the updated in-house developed software SUNBIM. The potential of the Q-dependent sensitivity and possible automated selectivity strongly relies on the discrimination between small intensity variations and Q-related intensity variations over a sufficiently large Q-range: to this aim, ongoing studies are focused on further improvements of data processing, and hardware upgrades are envisaged.Authors contribution
F. S. developed the routines for data reduction and analysis, and implemented them in the software package SUNBIM in collaboration with D. S. and M. L.; E. A., P. A. and F. M. conceived and prepared samples, and discussed experimental strategy for X-ray characterization with C. G. and D. A.; D. S. and C. G. performed SAXS data analysis; D. A. collected X-ray data, conceived the methodology for data reduction and analysis and wrote the paper in collaboration with all authors. C. G. procured the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by funding from: MUR within the call PRIN2017, Project “A system approach for identifying connective tissue degeneration in diabetic analogues - SAPIENT”; PON “R&I”2014–2020 “Energie per l'Ambiente TARANTO – Tecnologie e processi per l'Abbattimento di inquinanti e la bonifica di siti contaminati con Recupero di mAterie prime e produzioNe di energia TOtally green” – Code: ARS01_00637 (CUP: B86C18000870005); and the two years (2020–2021) Bilateral agreement (CUP: B54I19005880001) between the Italian Consiglio Nazionale delle Ricerche (CNR) and the Royal Society (RS) of UK. This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView also contains code developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No 654000. Mr Rocco Lassandro is acknowledged for his technical support in the X-ray MicroImaging Laboratory. Mrs Brunella Maria Aresta is acknowledged for her administrative support to research.References
- J. Radhakrishnan, A. Subramanian, U. M. Krishnan and S. Sethuraman, Biomacromol, 2017, 18(1), 1–26 CrossRef CAS.
- H. R. Culver, J. R. Clegg and N. A. Peppas, Acc. Chem. Res., 2017, 50(2), 170–178 CrossRef CAS.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1(12), 16071 CrossRef CAS.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8(5), 607–626 CrossRef CAS.
- I. M. El-Sherbiny and M. H. Yacoub, Global Cardiology Science and Practice, 2013, 2013(3), 316–342 CrossRef.
- C. Diaferia, E. Gianolio and A. Accardo, J. Pept. Sci., 2019, 25(5), e3157, DOI:10.1002/psc.3157.
- E. Gallo, C. Diaferia, E. Di Gregorio, G. Morelli, E. Gianolio and A. Accardo, Pharmaceuticals, 2020, 13(2), 19 CrossRef CAS.
- S. Garg and A. Garg, Asian Journal of Biomaterial Research, 2016, 2(6), 163–170 Search PubMed.
- A. Heuschkel, S. Goebel and R. H. H. Neubert, J. Pharm. Sci., 2008, 97, 603 CrossRef.
- B. Eicher, D. Marquardt, F. A. Heberle, I. Letofsky-Papst, G. N. Rechberger, M.-S. Appavou, J. Katsaras and G. Pabst, Biophys. J., 2018, 114, 146–157 CrossRef CAS.
- H. Liu, L. Wang, Y. Hu, Z. Huang, Y. Sun, S. Dong and J. Hao, Chem. Commun., 2020, 56, 3484 RSC.
- V. Nele, M. N. Holme, U. Kauscher, M. R. Thomas, J. J. Doutch and M. M. Stevens, Langmuir, 2019, 35, 6064–6074 CrossRef CAS.
- J. M. Hollamby, Phys. Chem. Chem. Phys., 2013, 15, 10566 RSC.
- R. F. Tabor, J. Eastoe and I. Grillo, Soft Matter, 2009, 5, 2125–2129 RSC.
- K. Komorowski, A. Salditt, Y. Xu, H. Yavuz, M. Brennich, R. Jahn and T. Salditt, Biophys. J., 2018, 114, 1908–1920 CrossRef CAS.
- C. Seyrig, P. Le Griel, N. Cowieson, J. PErez and N. Baccile, J. Colloid Interface Sci., 2020, 580, 493–502 CrossRef CAS.
- M. R. Brzustowiczb and A. T. Brungera, J. Appl. Crystallogr., 2005, 38, 126–131 CrossRef.
- A. S. Poulos, M. Nania, P. Lapham, R. M. Miller, A. J. Smith, H. Tantawy, J. Caragay, J. Gummel, O. Ces, E. S. J. Robles and J. T. Cabral, Langmuir, 2016, 32, 5852–5861 CrossRef CAS.
- G. Ben Messaoud, P. Le Griel, D. Hermida-Merino and N. Baccile, Soft Matter, 2020, 16, 2540–2551 RSC.
- G. Pabst, M. Rappolt, H. Amenitsch and P. Laggner, Phys. Rev. E, 2000, 62, 4000–4009 CrossRef CAS.
- J. E. Nielsen, T. K. Lind, A. Lone, Y. Gerelli, P. R. Hansen, H. Jenssen, M. Cárdenas and R. Lund, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1355–1364 CrossRef CAS.
- G. Colafemmina, G. Palazzo, H. Mateos, S. Amin, A. Laure Fameau, U. Olsson and L. Gentile, Colloids Surf., A, 2020, 597, 124821 CrossRef CAS.
- G. Jacoby, et al., Sci. Rep., 2015, 5, 9481, DOI:10.1038/srep09481.
- M. Herbst, E. Hofmann and S. Förster, Langmuir, 2019, 35, 11702–11709 CrossRef CAS.
- C. Giannini, D. Siliqi, M. Ladisa, D. Altamura, A. Diaz, A. Beraudi, T. Sibillano, L. De Caro, S. Stea, F. Baruffaldi and O. Bunk, J. Appl. Crystallogr., 2014, 47, 1–8 CrossRef.
- C. Giannini, D. Siliqi, O. Bunk, A. Beraudi, M. Ladisa, D. Altamura, S. Stea and F. Baruffaldi, Sci. Rep., 2012, 2, 435 CrossRef CAS.
- I. Allegretta, R. Terzano, G. P. S. C. Giannini, M. Ladisa, V. Lutz-Bueno, A. Terzi, M. Ramella, L. Fusaro, D. Altamura, D. Siliqi, T. Sibillano, A. Diaz, F. Boccafoschi and O. Bunk, IUCrJ, 2019, 6, 267–276 CrossRef.
- R. Vanna, C. Morasso, B. Marcinn, F. Piccotti, E. Torti, D. Altamura, S. Albasini, M. Agozzino, L. Villani, L. Sorrentino, O. Bunk, F. Leporati, C. Giannini and F. Corsi, Cancer Res., 2020, 80(8), 1762–1772 CrossRef CAS.
- C. Giannini, M. Ladisa, D. Altamura, D. Siliqi, T. Sibillano and L. De Caro, Crystals, 2016, 6(87), 1–22 Search PubMed.
- D. Altamura, S. G. Pastore, M. G. Raucci, D. Siliqi, F. De Pascalis, M. Nacucchi, L. Ambrosio and C. Giannini, ACS Appl. Mater. Interfaces, 2016, 8, 8728–8736 CrossRef CAS.
- A. Procopio, E. Malucelli, A. Pacureanu, C. Cappadone, G. Farruggia, A. Sargenti, S. Castiglioni, D. Altamura, A. Sorrentino, C. Giannini, E. Pereiro, P. Cloetens, J. Maier and S. Iotti, ACS Cent. Sci., 2019, 5, 1449–1460 CrossRef CAS.
- J. M. Montes de Oca-Ávalosa, D. Altamura, R. J. Candal, F. Scattarella, D. Siliqi, C. Giannini and M. L. Herrera, Food Res. Int., 2018, 105, 129 CrossRef.
- D. Siliqi, L. De Caro, M. Ladisa, F. Scattarella, A. Mazzone, D. Altamura, T. Sibillano and C. Giannini, J. Appl. Crystallogr., 2016, 49, 1107–1114 CrossRef CAS.
- Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules, ed. D. I. Svergun, M. H. J. Koch, P. A. Timmins and P. Roland, May. Oxford University Press ( 2013) Search PubMed.
- D. Altamura, R. Lassandro, F. A. Vittoria, L. De Caro, D. Siliqi, M. Ladisa and C. Giannini, J. Appl. Crystallogr., 2012, 45, 869–873 CrossRef CAS.
- http://www.ba.ic.cnr.it/softwareic/sunbim/.
- M. J. Hope, M. B. Bally, G. Webb and P. R. Cullis, Biochim. Biophys. Acta, 1985 Jan 10, 812(1), 55–65, DOI:10.1016/0005-2736(85)90521-8.
- J. Als-Nielsen and D. McMorrow, Elements of Modern X-ray Physics, John Wiley and Sons, Ltd., New York, 2nd edn, 2011 Search PubMed.
- M. Doucet, et al., SasView Version 4.2.2, DOI:10.5281/zenodo.2652478.
- F. Scattarella, L. De Caro, D. Siliqi and E. Carlino, Crystals, 2017, 7, 186 CrossRef.
- W. K. Pratt, in Digital Image Processing, John Wiley & Sons, Inc., Los Altos, CA, USA, 4th edn, 2007 Search PubMed.
- O. Bunk, M. Bech, T. H. Jensen, R. Feidenhas’I, T. Binderup, A. Menzel and F. Pfeiffer, New J. Phys., 2009, 11, 123016 CrossRef.
- J. M. Montes-de-Oca-Ávalos, Food Packag. Shelf Life, 2020, 26, 100590 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Normalization of the SAXS intensity to sample absorption and thickness; SAXS 1D folding and averaging; SAXS profile analysis; Enhancement of T and n contrast in dry samples; Masking no-sample regions; SAXS & XRD of dry samples. See DOI: 10.1039/d0ra08581b |
| ‡ Present Addresses: Istituto per le Applicazioni del Calcolo “Mauro Picone”-CNR, Via Amendola 122/O, 70126 Bari, Italy. |
| This journal is © The Royal Society of Chemistry 2021 |