DOI:
10.1039/D0RA08744K
(Paper)
RSC Adv., 2021,
11, 5629-5642
Formation pathways of polycyclic aromatic hydrocarbons (PAHs) in butane or butadiene flames†
Received
14th October 2020
, Accepted 17th January 2021
First published on 2nd February 2021
Abstract
The reaction pathways from phenyl radicals to phenanthrene (A3) and pyrene (A4) via C2H3 and C4H4 additions were investigated using the G3(MP2, CC) method. Rate constants of elementary reactions were calculated. The influence of additions, H-abstraction ways and reactive sites on the reaction rates were considered. These polycyclic aromatic hydrocarbon (PAH) formation pathways were used to improve the combustion chemistry model for C4 fuels, and the results from the improved model and the original model were compared with experimental data. H atoms are important for PAH formation owing to their influential roles in the production of aromatic radicals and stable aromatic structures. C2H3 and C4H4 addition reactions can occur at low temperature, and need less energy than C2H2 addition. The PAH formation pathways determined from G3 calculations, which were used to improve the model, were effective in promoting PAH formations in this model. Comparison of PAH formation in butane and butadiene flames showed both the C2H3 and C4H4 addition pathways included in this work can improve the formation of PAHs in butadiene and butane flames. C4H4 addition pathways in a butane flame were better for PAH formation than C2H3 addition.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs), which mainly come from the incomplete combustion of fuel, are extremely harmful to humans and the environment. So far, the most commonly-used addition for PAH growth is C2H2, and the most popular PAH formation mechanism is H-abstraction-C2H2-addition (HACA).1–5 However, the HACA mechanism underestimates the concentrations of PAHs and soot compared with the experimental results.6–9 This underestimation may be explained by two main reasons. Firstly, there are many non-acetylene organic compounds (e.g. CH3, C3H3, C3H4, C3H6, C4H2, C4H4, C4H6 and C5H8) that also contribute to PAH growth in various flame configurations.8–13 Secondly, many PAH formation reactions in the HACA mechanism are reversible.14 Hence, more additions in PAH formation should be considered to perfect the PAH formation mechanism.
Many non-acetylene additions have been proved experimentally to be important for PAHs formation. C4H4 addition can effectively lead to PAHs formation.15 C2H3 addition is critical for the formation of PAHs and soot.16 The hydrogen abstraction/vinyl radical addition (HAVA) mechanism may be the best complement to the HACA mechanism, and the phenyl addition/cyclization (PAC) mechanism can promote the growth of molecular mass and ring number of PAHs without the limit of reactive sites.10,17 Hence, the additions of C2H3, C4H4 and C6H5 contribute greatly to PAHs formation, and more research on PAHs formation from C2H3, C4H4 and C6H5 additions is needed.
Butane and butadiene, two light hydrocarbons, are both beneficial to the environment and can relieve energy shortage. In this research, PAHs formation routes via C2H3, C4H4 and C6H5 additions within all reaction rates of elementary reactions in the modified Arrhenius equation were studied and coupled to a chemical kinetic model for butadiene and butanol. The effects of additions, reaction types and reaction sites were considered during PAHs formation.
2. Methods
All molecular structures involved in the reactions under study were optimized by using the hybrid B3LYP functional with the 6-311++G(d,p) basis set.18–20 Vibrational frequency was calculated at the same level to identify the optimized structures as local minimum or first-point saddle points and to provide their zero-point vibrational energy (ZPE). Intrinsic reaction coordinate calculations were implemented to ensure that the transition states connect to relevant reactants and products correctly. The final single-point energies of all species were determined using the G3(MP2, CC) method,21,22 which is expected to generate relative energies of various species within the accuracy of 1–2 kcal mol−1 (ref. 23) and is extensively used to study PAHs growth mechanisms.21,24–28 The intermediate species and transition structures found in this study were all closed shell singlets or open shell doubles. Molecular properties of chemical species (CS) and transition states (TS) are shown in ESI.† The G3(MP2, CC) energies are calculated as follows:| | |
E[G3(MP2, CC)] = E[CCSD/6-311++(d,p)] + ΔE(MP2) +ΔE(SO) + ΔE(HLC) + E(ZPE)
| (1) |
where ΔE(MP2) = E[MP2/6-311G++(3df,2p)] − E[MP2/6-311G++(d,p)] is the basis set correction; ΔE(SO) is the spin–orbit correction, included for atomic species only. For molecules, higher level correction (HLC) is ΔE(HLC) = −Anb − B(na − nb) with A = 9.279 mHartrees and B = 4.471 mHartrees, and for atoms, is ΔE(HLC) = −Cnb − D(na − nb) with C = 9.345 mHartrees and D = 2.021 mHartrees, where na and nb are the numbers of a and b valence electrons, respectively. All calculations were performed on Gaussian 09.29
Based on the calculated potential energy surface (PES) and molecular characteristics, Rice–Ramsperger–Kassel–Marcus (RRKM) and transitional state theory (TST) were used to determine reaction rates by ChemRate program.30 Noticeably, none of the rate constants were arbitrarily changed or intuitively estimated to match the computed and the experimental results. The calculated PAHs formation pathways were used to update Hansen's mechanism31 which consists of 216 species connected via 1028 reactions. Then the updated mechanism was used to simulate the premixed 1,3-butadiene or butane flame and to discuss the roles of our pathways in PAHs formation under 1,3-butadiene or butane flame.
3. Results and discussion
3.1 PAHs formation pathways
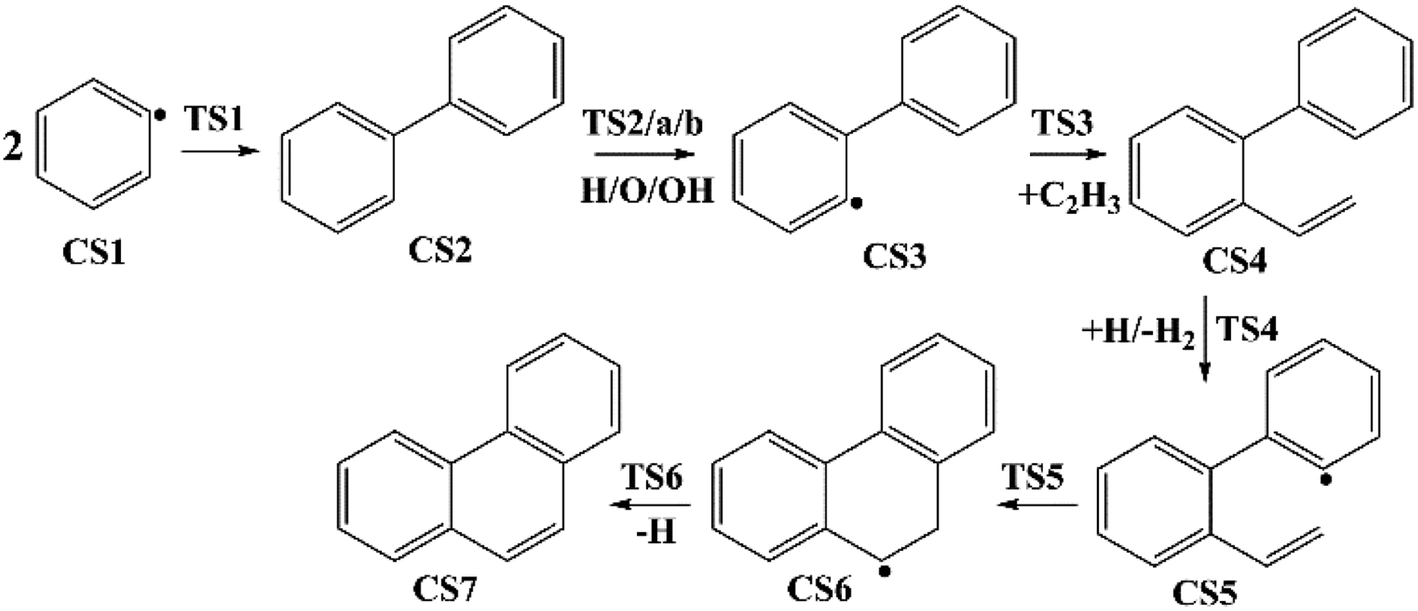
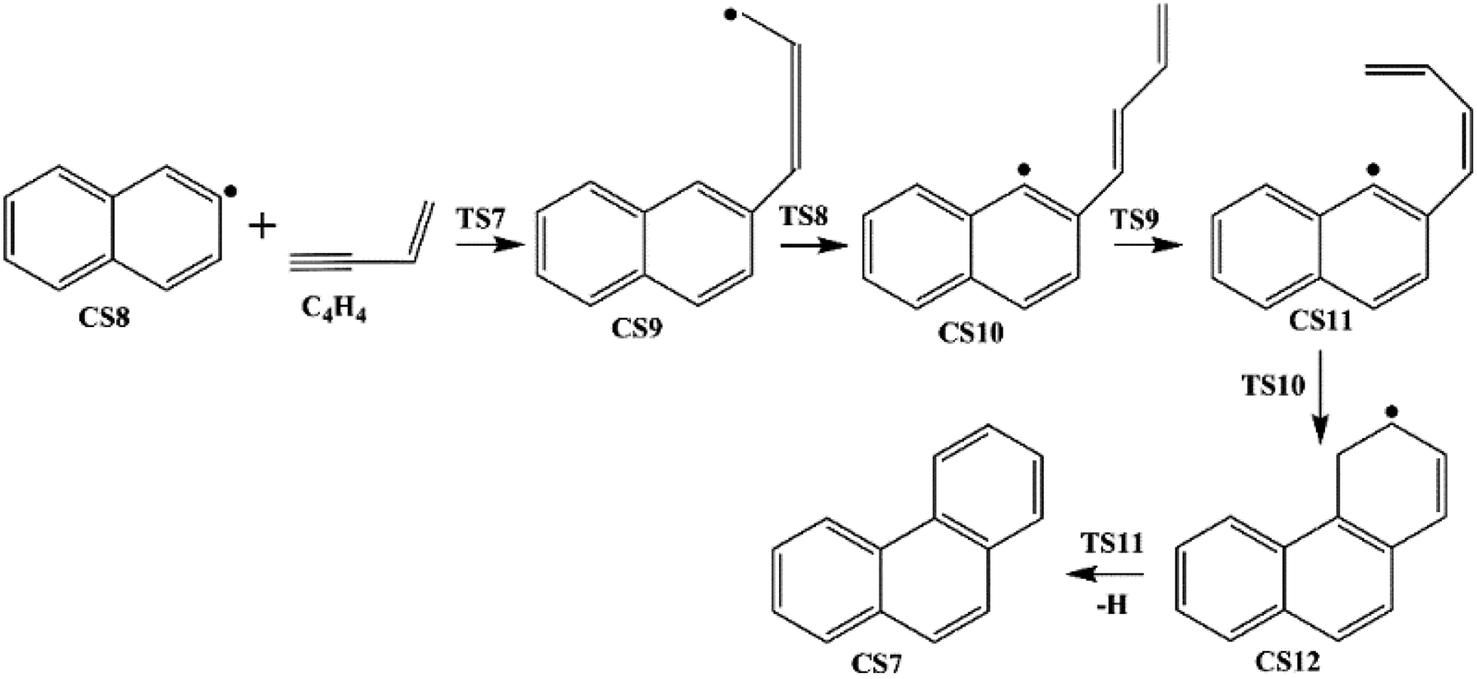
Four PAHs formation pathways were studied (Fig. 1–4): (i) formation of phenanthrene via once addition of phenyl radical (C6H5) and twice additions of C2H3 onto phenyl radical; (ii) formation of phenanthrene via once addition of C4H4 onto naphthyl radical; (iii) formation of pyrene via once addition of C2H3 onto phenanthryl radical; (iv) formation of pyrene via once addition of C4H4 onto 1-ethynylnaphthyl radical. Comparisons between H abstraction reactions of biphenyl via H atom, O atom or OH radical at same reactive site were studied. The effects of reactive sites to hydrogen abstraction, carbon addition, ring closure, and hydrogen atom loss reactions were investigated. The barrier heights and reaction energies for all steps are collected in Table S1.† Meanwhile, to understand the thermodynamics of the studied reactions better, the enthalpies and Gibbs free energies are illustrated in Tables S2 and S3.† The molecular geometries, vibrational frequencies, moments of inertia, and rotational constants of the chemical species (CS) and transition states (TS) involved in all routes are shown in Table S5.†
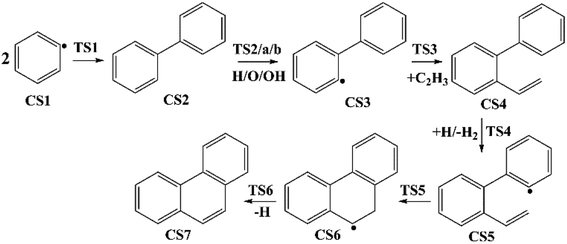 |
| | Fig. 1 Phenanthrene formation pathways from phenyl radical via C6H5 and C2H3 additions. | |
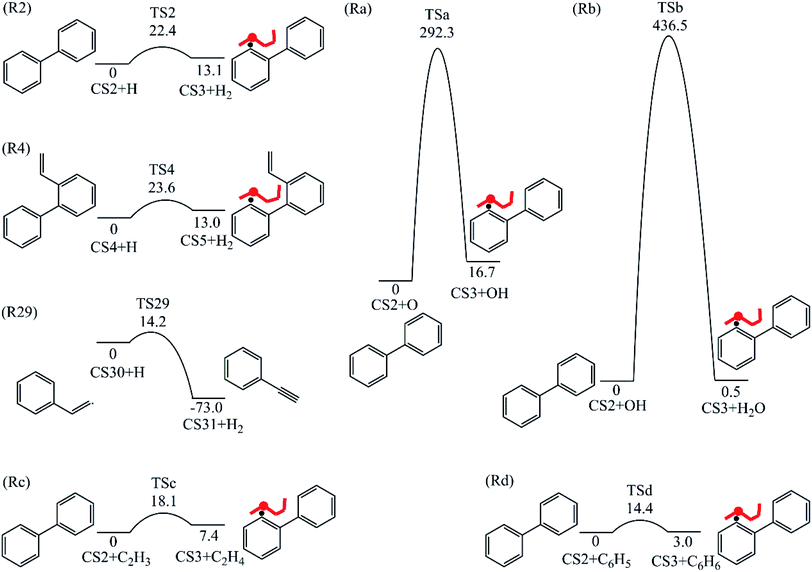
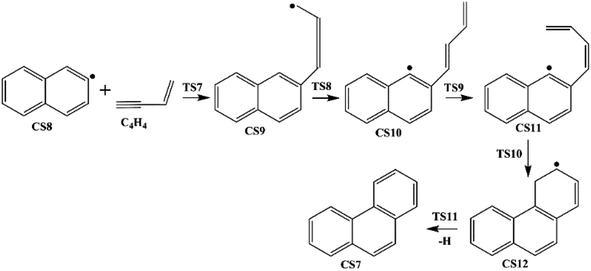 |
| | Fig. 2 Phenanthrene formation pathways from naphthyl radical via C4H4 addition. | |
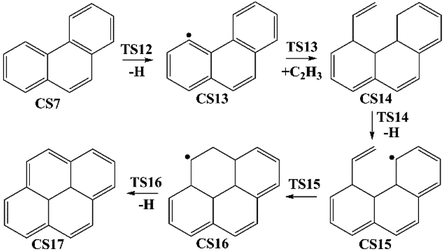 |
| | Fig. 3 Pyrene formation pathways from phenanthryl radical via C2H3 addition. | |
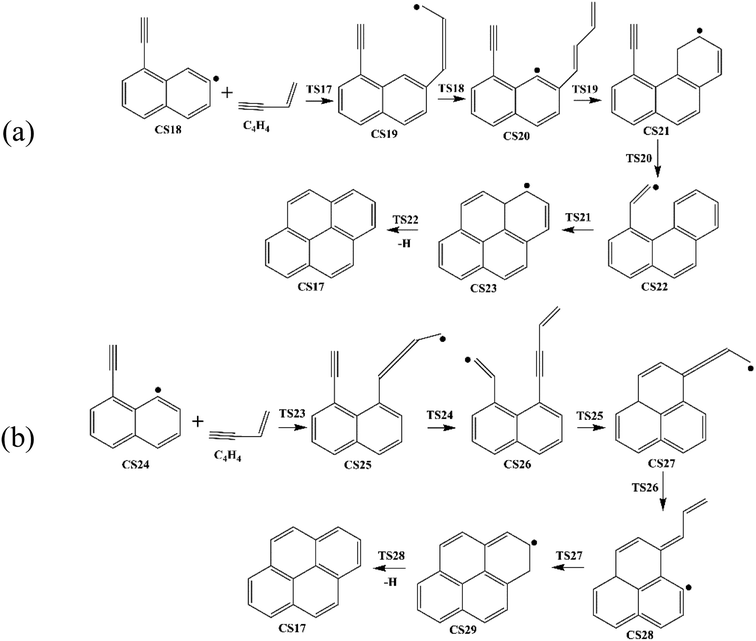 |
| | Fig. 4 Pyrene formation pathways via C4H4 addition onto different sites of 1-ethynylnaphthyl radical. | |
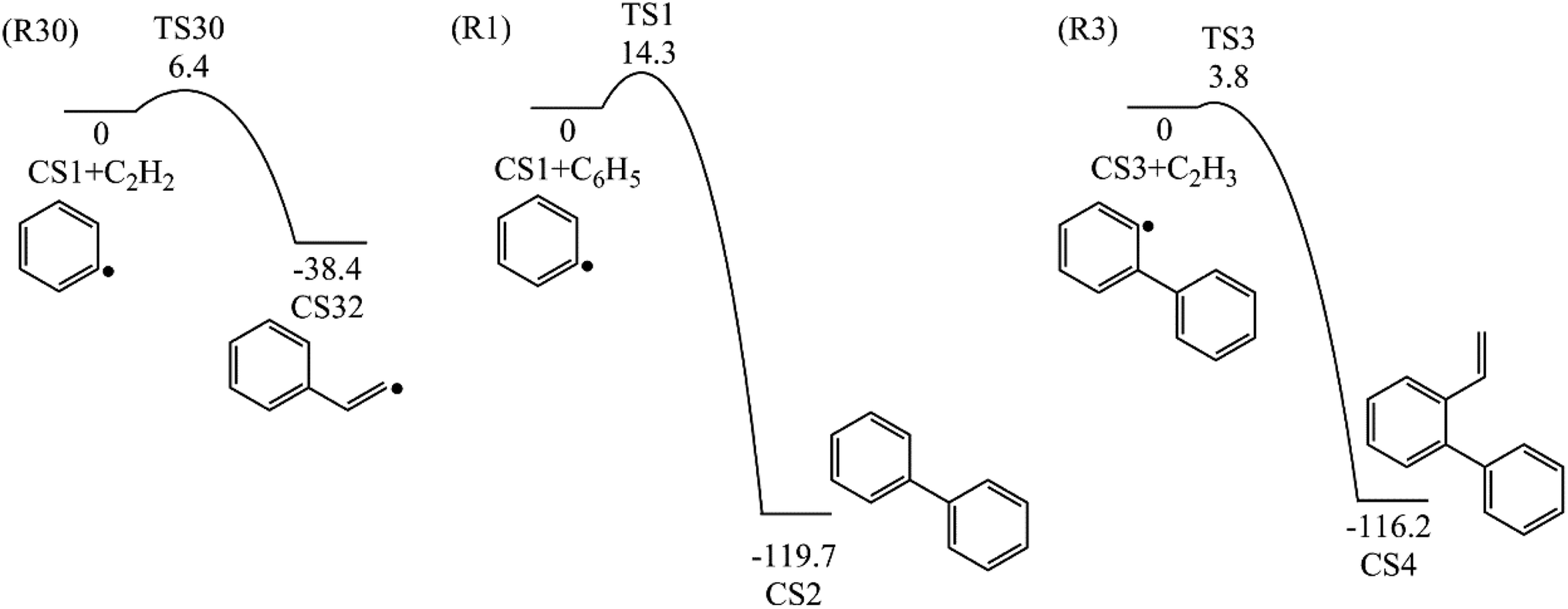
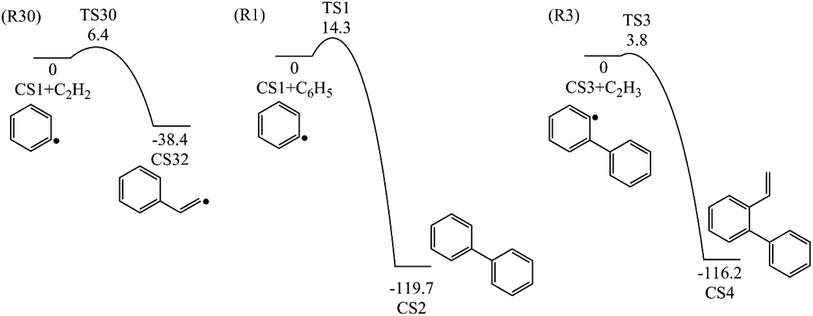
3.1.1 Phenanthrene formation pathway via C2H3 additions. As shown in Fig. 1, phenanthrene was formed through H-abstractions, C-additions and ring formation reactions, and the barrier heights and reaction energies were determined in Table 1. The initial association of two C6H5 additions results in the formation of CS2, biphenyl. This thermodynamically and kinetically favorable process is highly exothermic (119.7 kcal mol−1) with a barrier of 14.3 kcal mol−1, leading to a quick increase of ring number. In Fig. 6, C2H2, and especially C6H5 and C2H3 addition reactions are all exothermic. Besides, C2H3 addition reaction occurs more easily than C2H2 or C6H5 addition reaction.
Table 1 Barrier heights and reaction energies for steps involved in phenanthrene formation pathways from phenyl radical via C6H5 and C2H3 additions computed at the G3(MP2, CC) level
| No. |
Reaction |
Barriers/kcal mol−1 |
Reaction heats/kcal mol−1 |
| Carbon addition |
| R1 |
2CS1 = CS2 |
14.3 |
−119.7 |
| R3 |
CS3 + C2H3 = CS4 |
3.8 |
−116.2 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Hydrogen abstraction |
| Ra |
CS2 + O = CS3 + OH |
292.3 |
16.7 |
| Rb |
CS2 + OH = CS3 + H2O |
436.5 |
0.5 |
| Rc |
CS2 + C2H3 = CS3 + C2H4 |
18.1 |
7.4 |
| Rd |
CS2 + C6H5 = CS3 + C6H6 |
14.4 |
3.0 |
| R2 |
CS2 + H = CS3 + H2 |
22.4 |
13.1 |
| R4 |
CS4 + H = CS5 + H2 |
23.6 |
13.0 |
|
| Ring formation |
| R5 |
CS5 = CS6 |
7.3 |
−39.7 |
|
| Hydrogen loss and (or) disproportionation |
| R6 |
CS6 = CS7 + H |
27.9 |
16.5 |
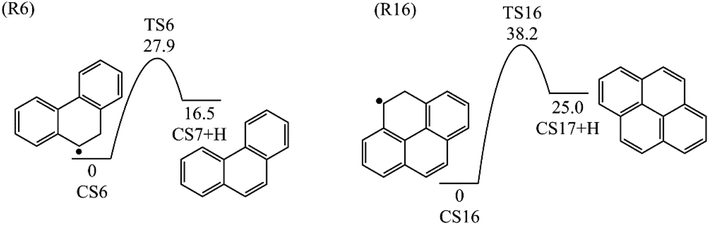
H-abstractions may produce singlet PAH intermediates such as CS31 and CS7, or radicals such as CS3 and CS5.32 Most of the former H-abstractions are exothermic, and most of the later H-abstractions are endothermic, providing precondition for the possible carbon additions or cyclization reactions. To explore the easier formation of larger PAHs molecules, we calculated and compared five radical site formation processes via H, O, OH, C2H3 and C6H5 respectively to provide a prime radical formation process. The barriers and reaction heats were shown in Table 1 and all H-abstraction reactions with assistance of H, O, OH, C2H3 and C6H5 in this pathway together with H-abstraction reaction R29 from literature32 were compared in Fig. 5. Comparison of R2, Ra, Rb, Rc and Rd showed the barriers of the H-abstraction reactions via O and OH radicals were much higher than those via H, C2H3 and C6H5 radicals. This means that, depending on the concentrations of H, C2H3 and C6H5 radicals in combustion environments, H, C2H3 and C6H5 radicals can much more easily initiate H-abstractions than O and OH radicals. However, the concentration H radicals in combustion environments is generally higher than those of C2H3 and C6H5 radicals.8–10 Thus, H radical is much easier to initiate H-abstractions than O, OH, C2H3 and C6H5 radicals. Little difference between H-abstractions via TS2 and TS4 was found in barriers or reaction heats, which is mainly because of the same reaction types and reactive sites between armchair and free edges.33 CS6 was formed via a cyclization reaction after H-abstraction, and formed CS7, phenanthrene, by emitting a H atom.
 |
| | Fig. 5 H-abstractions via the assistance of H, O, OH, C2H3 or C6H5 radicals. | |
 |
| | Fig. 6 C2H2, C6H5 and C2H3 addition reactions. | |
3.1.2 Phenanthrene formation pathway via C4H4 addition. This pathway describes the phenanthrene formation process initiated by C4H4 addition onto naphthyl radical. The energies of the intermediate species and the transition states are relative to the total energy of CS8 and C4H4 (Fig. 7). The addition of C4H4 leads to CS9 with a barrier of 3.4 kcal mol−1, which is lower than that of C2H2 addition, RI. After that, the reaction CS9 → CS10 is internal hydrogen abstraction via migration of a hydrogen atom from the aromatic ring to the second carbon of the C4 fragment, producing a butadiene chain with a barrier of 39.1 kcal mol−1. Then to prepare for the cyclization, CS10 undergoes cis–trans isomerization, forming CS11 with a barrier of 38.6 kcal mol−1. Due to its structural characteristics, the CS11 adduct can be cyclized, producing the phenanthrene precursor CS12, which yields phenanthrene after hydrogen atom elimination. The barrier of the cyclization step CS11 → CS12 is only 0.4 kcal mol−1, which can be ignored easily. Also, the reaction is highly exothermic by 65.1 kcal mol−1. The hydrogen atom elimination step CS12 → CS7 + H overcomes a barrier of 40.5 kcal mol−1, and is the rate determining step of this pathway.
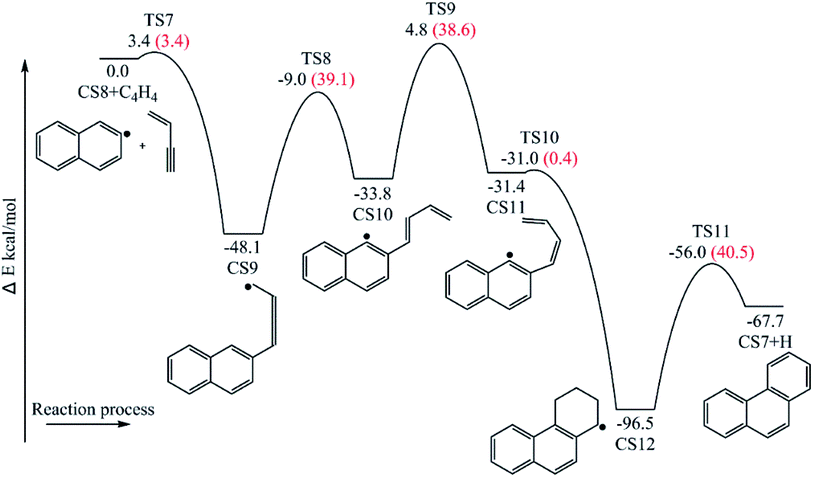 |
| | Fig. 7 Phenanthrene formation initiated by C4H4 addition onto naphthyl radical. | |
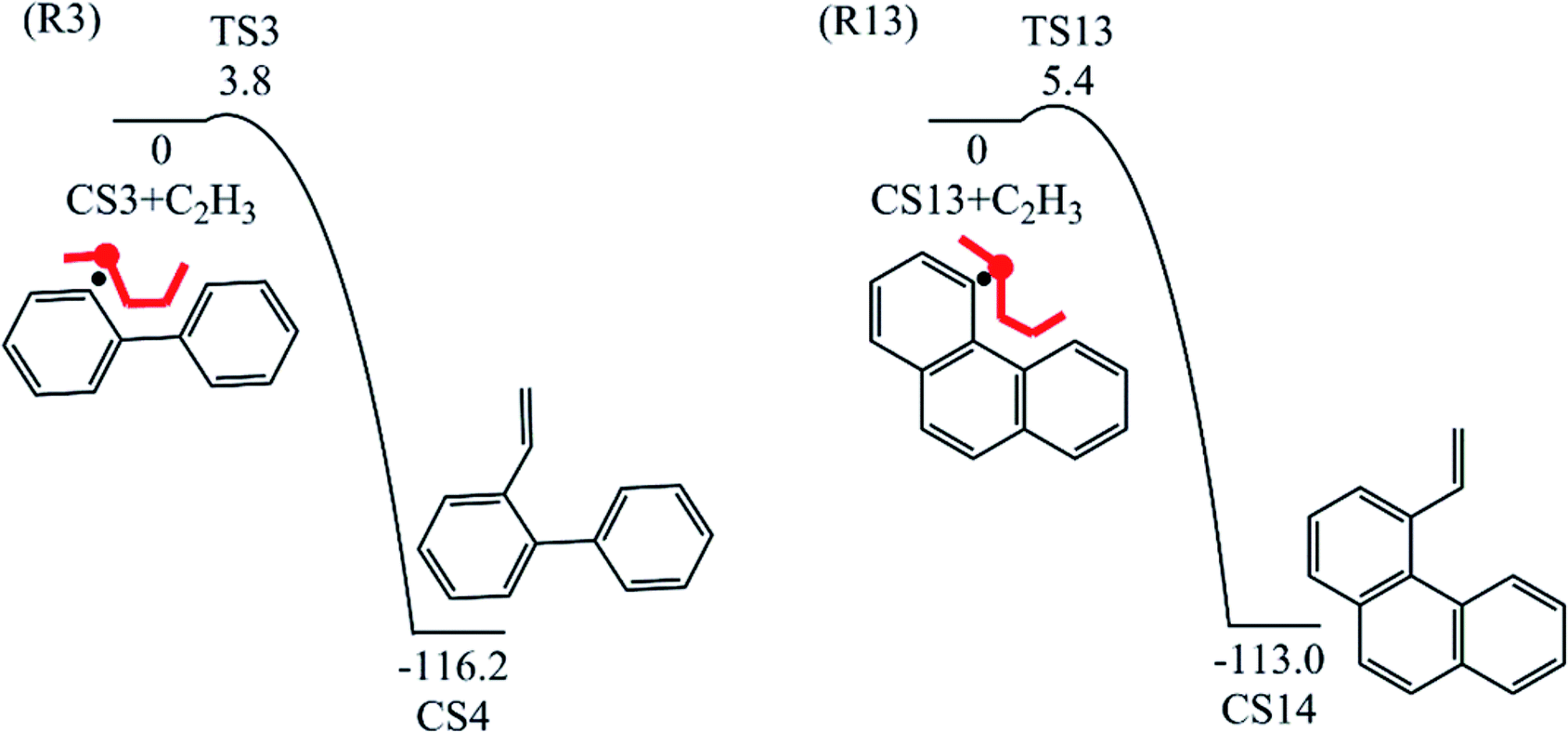
3.1.3 Pyrene formation pathway via C2H3 additions. The reaction process from phenanthrene to pyrene via C2H3 addition and the energy of this process were shown in Fig. 3 and Table 2. The C2H3 addition reaction is followed by activation of phenanthrene via H-abstraction, forming a radical structure, CS13. After that, the C2H3 addition onto CS13 is highly exothermic with a barrier of only 5.4 kcal mol−1. Then CS14 undergoes H-abstraction and cyclization, producing CS16, with barriers of 31.0 and 5.3 kcal mol−1, respectively. At last, CS16 emits a H atom to produce CS17, pyrene. This reaction is a rate-determining step of this pathway, overcoming a barrier of 38.2 kcal mol−1.
Table 2 Barrier heights and reaction energies for steps involved in pyrene formation pathway from phenanthrene via C2H3 addition computed at the G3(MP2, CC) level
| No. |
Reaction |
Barrier/kcal mol−1 |
Reaction heat/kcal mol−1 |
| R13 |
CS13 + C2H3 = CS14 |
5.4 |
−113.0 |
| R14 |
CS14 + H = CS15 + H2 |
31.0 |
11.9 |
| R15 |
CS15 = CS16 |
5.3 |
−53.2 |
| R16 |
CS16 = CS17 + H |
38.2 |
25.0 |
Fig. 8 compares two C2H3 additions onto CS3 and CS13 separately. Little difference is found in the barrier heights or reaction energy between these two addition steps. This may be because the reactive sites on CS3 and CS13 are similar between “armchair” and “free edge” as shown by red lines in Fig. 8.
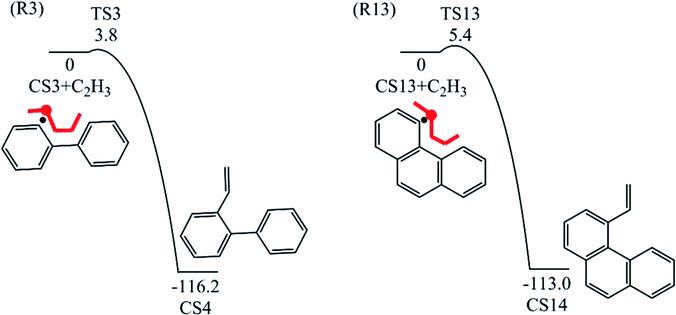 |
| | Fig. 8 C2H3 addition reactions onto CS3 and CS13. | |
Fig. 9 shows two hydrogen atom loss reactions that produce phenanthrene and pyrene, respectively. Compared with H-abstractions via the assistance of H atom, these two H atom loss reactions without the assistance of H atom need to overcome higher barriers and are both endothermic. Thus, the assistance of H atom is beneficial for H loss.
 |
| | Fig. 9 H atom loss reactions. | |
Furthermore, both reaction CS15 → CS16 and reaction CS5 → CS6 are ring formation processes that occur at the edge of vinyl group and the carbon of aromatic ring between “armchair” and “free edge” (Fig. 1, 3 and 10). These two reactions are both exothermic with low barriers.
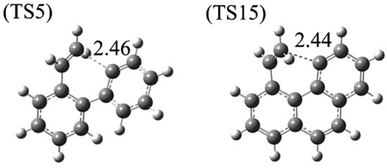 |
| | Fig. 10 Structures of transition states of R5 and R15. | |
3.1.4 Pyrene formation pathways via C4H4 addition onto CS18. Fig. 11 provides the energy diagram for pyrene formation pathway initiated from the C4H4 addition onto CS18. The energies of the intermediate species and the transition states are relative to the total energy of CS18 and C4H4. The C4H4 addition onto the radical site of CS18 to form CS19 requires a barrier of 0.4 kcal mol−1, which is 3.0 kcal mol−1 lower than the C4H4 addition step above, CS8 + C4H4 → CS9. This may be because of the existence of the acetylene group in CS18. After that, the reaction CS19 → CS20 is internal hydrogen abstraction via migration of a hydrogen atom, producing a butadiene chain with a barrier of 39.0 kcal mol−1. The energetics of this reaction is rather similar to that observed for another internal hydrogen migration step CS9 → CS10. The barrier heights range within 39.0–39.1 kcal mol−1, and reaction exothermicities are about 11.6–14.3 kcal mol−1. After that, cyclization occurs at the end of the butadiene group and the radical site of aromatic ring, CS20 → CS21, which requires an activation energy of 7.9 kcal mol−1, forming a six-membered ring. Later, another cyclization process takes place at the vinyl radical group and the newly-formed ring, forming the forth ring structure, CS23, with a barrier of 7.1 kcal mol−1. Finally, adduct CS23 eliminates an “extra” hydrogen atom to produce CS17, pyrene.
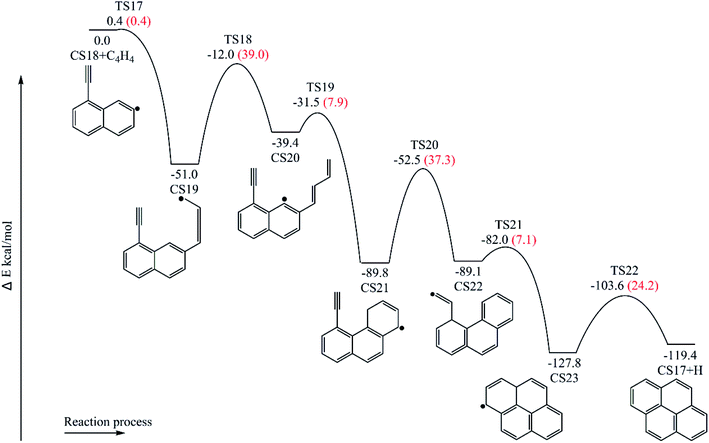 |
| | Fig. 11 Pyrene formation pathways via C4H4 addition onto CS18. | |
Fig. 12 provides the energy diagram for pyrene formation pathway initiated from the C4H4 addition onto CS24. The energies of the intermediate species and the transition states are relative to the total energy of CS18 and C4H4. The C4H4 addition onto the radical site of CS24 to produce CS25 requires a barrier of 4.2 kcal mol−1, which is higher than C4H4 addition steps via TS7 and TS17. This may be because the reactive site for C4H4 addition is near the shared carbons of two aromatic rings. The exothermicity of this addition reaction is 48.2 kcal mol−1, which differs by only 0.1 and 2.8 kcal mol−1 with the other two C4H4 addition steps via TS7 and TS17, respectively, but is significantly lower than the reaction energies for C6H5 and C2H3 addition reactions, R1, R3 and R13. Combined with Fig. 6 and 8, in the addition reactions, the same added species may contribute equally to similar reaction kinetics, just like C2H3 and C4H4. After the addition of C4H4, the internal hydrogen migration from C4 group to acetylene group adjusts the structure of CS25 to adduct CS26, which yields CS27 within three aromatic rings through cyclization via TS25. At this point, another internal hydrogen migration via TS26 has a large thermodynamic driving force for the subsequent formation of PAHs, CS29. At last, pyrene is formed through the elimination of an “extra” hydrogen atom from adduct CS29. In this pathway, the structure of TS25, which is the transition state for CS26 → CS27, was not found. Thus, the highest barrier of this pathway is 41.8 kcal mol−1 so far, and the rate-deciding step is the dehydrogenation, CS29 → CS17 + H.
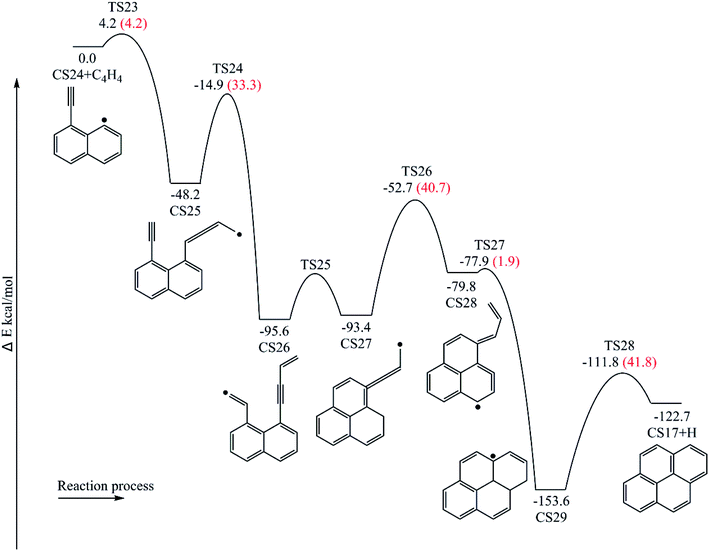 |
| | Fig. 12 Pyrene formation pathway via C4H4 addition onto CS24. | |
3.2 Elementary reaction rate constants
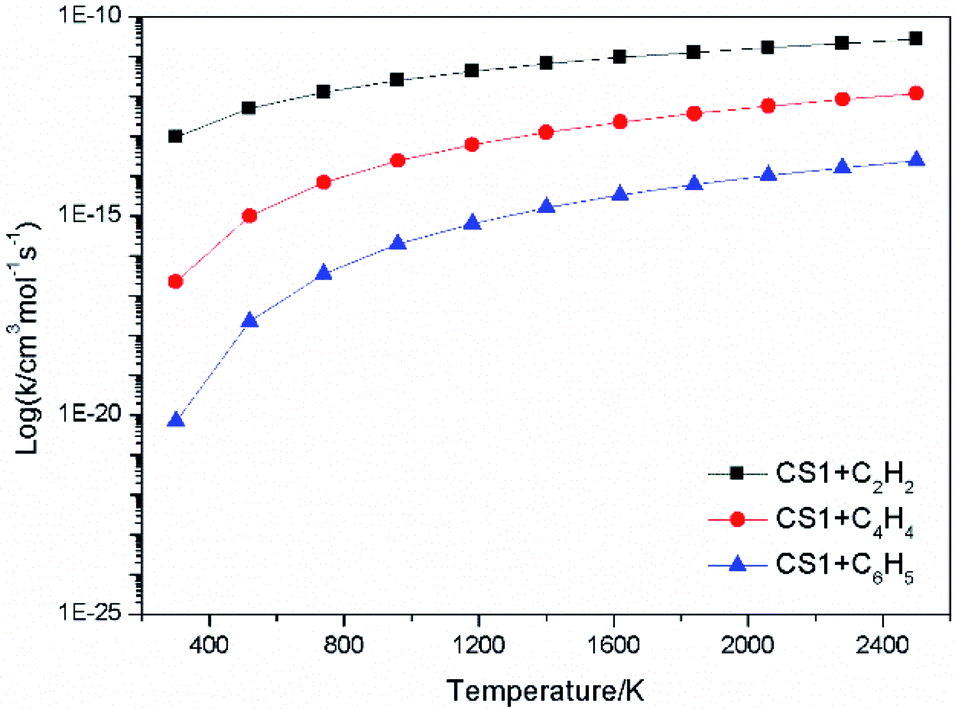
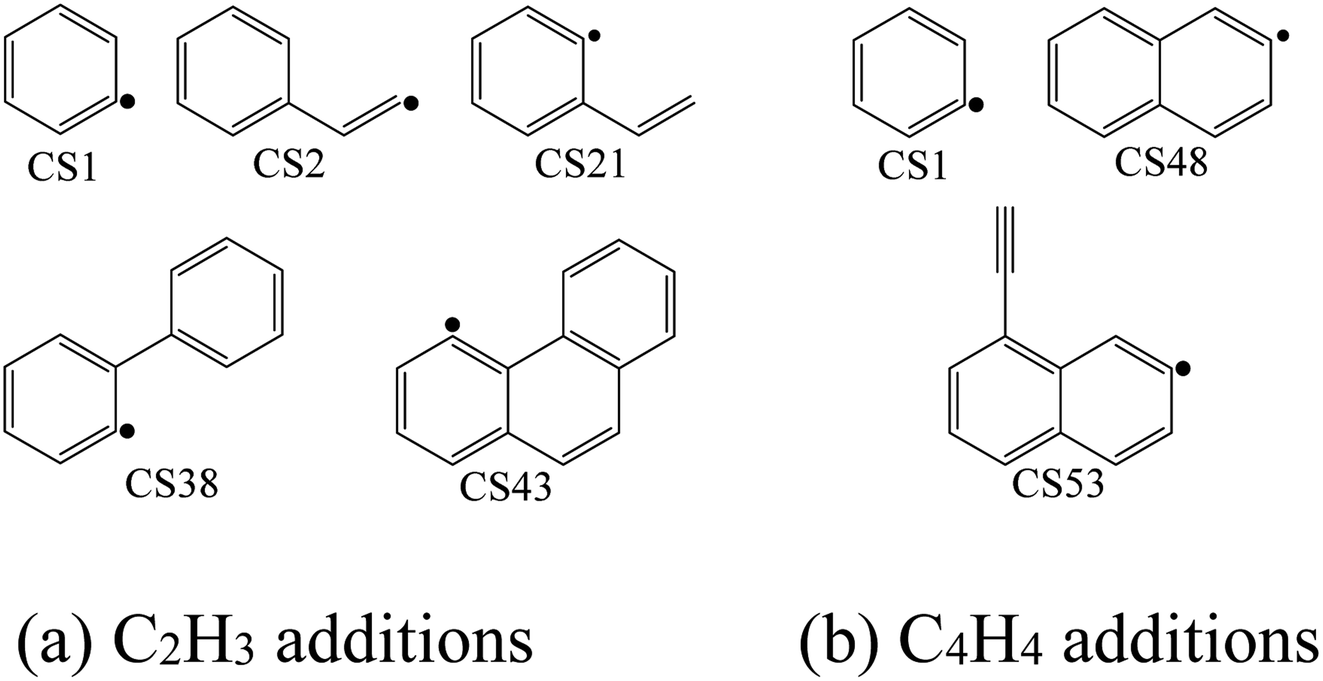
The high-pressure limit rate constants of the elementary reactions involved above are listed in Table 3 ranging from 300 to 2500 K. Fig. 13 and 14 demonstrate the rate constants for C2H2, C4H4 and C6H5 additions, and for C2H3 and C4H4 additions along temperature respectively. Fig. 15 shows the aromatic structures for C2H3 and C4H4 additions. Results imply that reactions R48 and R53 almost share the same rate constants at different temperatures, which is because the reactive sites of CS38 and CS43 are both armchair sites. Moreover, for C4H4 additions, the rate constant added onto CS1 is the fastest, followed by CS48, and that of CS53 is the last. All reactive sites on CS1, CS48 and CS53 are free edges with no adjacent groups, and the molecular mass of CS1 is the lightest, followed by CS48, and that of CS53 is the heaviest. Thus, we believe C4H4 can be more easily added onto aromatic structures with light mass.
Table 3 Elementary reaction rate constants in the modified Arrhenius equation form of ATnexp(−E/RT) with the units of cal, K, mol, cm and s, evaluated at 300–2500 K
| No. |
Reaction |
A |
n |
E |
| Phenanthrene formation pathway via C2H3 addition |
| R1 |
2CS1 → CS2 |
5.00 × 10−14 |
−0.3 |
8.2 |
| R2 |
CS2 + H → CS3 + H2 |
4.00 × 10−16 |
1.8 |
23 |
| R3 |
CS3 + C2H3 → CS4 |
5.00 × 10−11 |
−1.1 |
42.4 |
| R4 |
CS4 + H → CS5 + H2 |
4.00 × 10−16 |
1.8 |
23.8 |
| R5 |
CS5 → CS6 |
4.00 × 1012 |
0.4 |
10.3 |
| R6 |
CS6 → CS7 + H |
3.20 × 1010 |
1 |
42.3 |
|
| Phenanthrene formation pathway via C4H4 addition |
| R7 |
CS8 + C4H4 → CS9 |
3.40 × 1016 |
−9.6 |
89.9 |
| R8 |
CS9 → CS10 |
8.40 × 1062 |
−15.2 |
57.2 |
| R9 |
CS10 → CS11 |
3.90 × 1030 |
−5.9 |
28.9 |
| R10 |
CS11 → CS12 |
6.10 × 1012 |
0.1 |
1.5 |
| R11 |
CS12 → CS7 + H |
2.00 × 1011 |
0.8 |
29.5 |
|
| Pyrene formation pathway via C2H3 addition |
| R12 |
CS7 + H → CS13 + H2 |
— |
— |
— |
| R13 |
CS13 + C2H3 → CS14 |
5.90 × 1010 |
−1.4 |
44.6 |
| R14 |
CS14 + H → CS15 + H2 |
2.50 × 10−16 |
1.8 |
27.5 |
| R15 |
CS15 → CS16 |
2.50 × 1012 |
0.1 |
5.8 |
| R16 |
CS16 → CS17 + H |
5.00 × 1010 |
1.0 |
43.6 |
|
| Pyrene formation pathways via C4H4 addition |
| R17 |
CS18 + C4H4 → CS19 |
4.30 × 1039 |
−10.4 |
90.4 |
| R18 |
CS19 → CS20 |
4.00 × 1011 |
0.4 |
42.2 |
| R19 |
CS20 → CS21 |
5.80 × 1011 |
0.1 |
8 |
| R20 |
CS21 → CS22 |
8.70 × 1010 |
0.5 |
38.9 |
| R21 |
CS22 → CS23 |
1.60 × 1012 |
0.1 |
7.9 |
| R22 |
CS23 → CS17 + H |
1.80 × 1011 |
0.8 |
29.5 |
| R23 |
CS24 + C4H4 → CS25 |
8.60 × 1049 |
−13.9 |
94.7 |
| R24 |
CS25 → CS26 |
1.20 × 1032 |
0.5 |
36.6 |
| R25 |
CS26 → CS27 |
— |
— |
— |
| R26 |
CS27 → CS28 |
3.30 × 1032 |
0.4 |
44.1 |
| R27 |
CS28 → CS29 |
3.10 × 1032 |
0.6 |
2.3 |
| R28 |
CS29 → CS17 + H |
8.90 × 1030 |
0.6 |
47.5 |
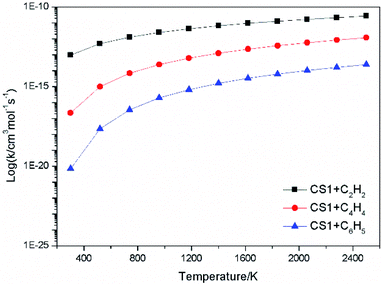 |
| | Fig. 13 Elementary reaction rate constants for C2H2, C4H4 and C6H5 additions. | |
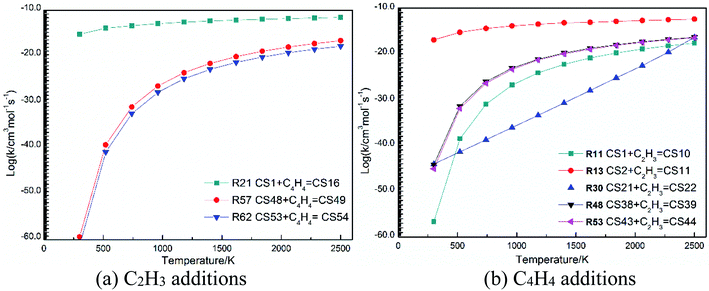 |
| | Fig. 14 Elementary reaction rate constants for C2H3 and C4H4 additions along temperature. | |
 |
| | Fig. 15 Aromatic structures for C2H3 and C4H4 additions. | |
3.3 Kinetic modeling
The mechanism investigated here was improved from Hansen's mechanism31 by the quantum chemical calculations reported before32 and in this work. The improvements of the new mechanism are shown in Fig. 16. All computations for laminar flames were performed with the code PREMIX from CHEMKIN II.34 Thermodynamic and transport data for the species involved in the mechanism were taken from Hansen's mechanism31 or evaluated by applying group additives rules.
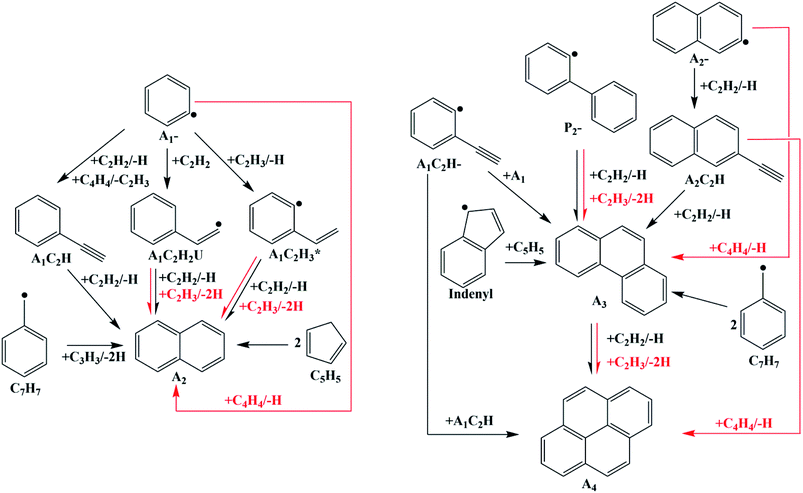 |
| | Fig. 16 PAHs formation process in the improved mechanism (black lines: from Hansen's mechanism; red lines: from quantum chemical calculations reported before32 and in this study). | |
The improved mechanism was verified in premixed butane and butadiene flames separately. Species concentrations determined from these flames were compared with the experimental results and the simulation results from Hansen's mechanism. Thereby, the influence of new PAHs formation routes on predicting PAHs formation was analyzed.
3.3.1 Species concentration in premixed 1,3-butadiene flame. With the improved chemical mechanism, we simulated the variation in concentrations of the main intermediates and PAHs in the premixed 1,3-butadiene flame. All modelling parameters were cited from experiments,35 and all results were shown in Fig. 17 compared with experiments35 and simulation of Hansen's mechanism.31
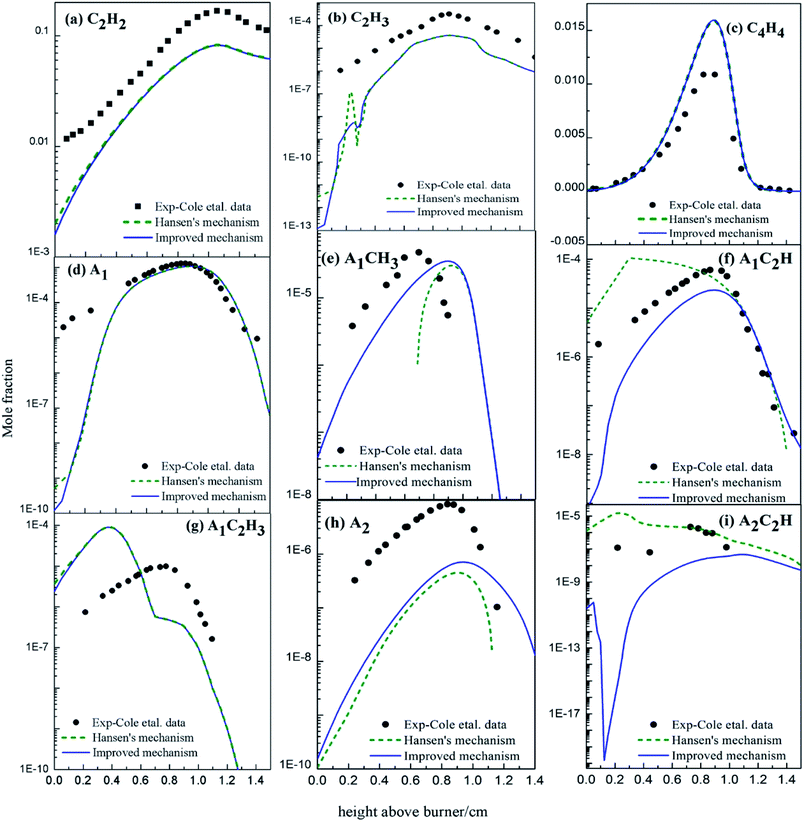 |
| | Fig. 17 Comparison of calculated and experimental data35 of mole fraction profiles for major products in butadiene flames (black dots: from experiments;35 dashed lines: from Hansen's mechanism;31 solid lines: from the improved mechanism). | |
The mole concentrations of C2H2, C2H3, C4H4, A1 and A1C2H3 decrease at low temperature (Fig. 17). Among them, the consumption of C2H3, C4H4, A1 and A1C2H3 may be because of the newly-added PAHs formation routes, and the C2H2 concentration decreases probably because of the transition of C2H2 into C2H3 and C4H4 for chemical equilibrium. Specifically, the added PAHs formation routes do contribute to PAHs formation, especially at low temperature.
The mole concentrations of A1CH3 and A2 increase, but those of A1C2H and A2C2H decrease compared with the results of Hansen's mechanism. A2 forms from HACA routes, conjugation of A1CH2 radical and C3H3 radical, or conjugation of two cyclopentadienyl radical in Hansen's mechanism (Fig. 16). A2 formation routes via C2H3 and C4H4 additions are newly-added in this study. Hence, the increase of the A2 mole concentration compared to the results of Hansen's mechanism is mainly due to the new added routes. As a consequence of chemical equilibrium, the mole concentration of A1CH3 also increases. Since A1C2H and A2C2H are both reactants in the new added PAHs formation routes, their mole concentrations decrease compared with the results of Hansen's mechanism.
3.3.2 Species concentration in premixed butane flame. The variations in the concentrations of main intermediates and PAHs in the premixed butane flame were simulated according to experiments,36 Hansen's mechanism31 and the improved mechanism in this study. All results were shown in Fig. 18.
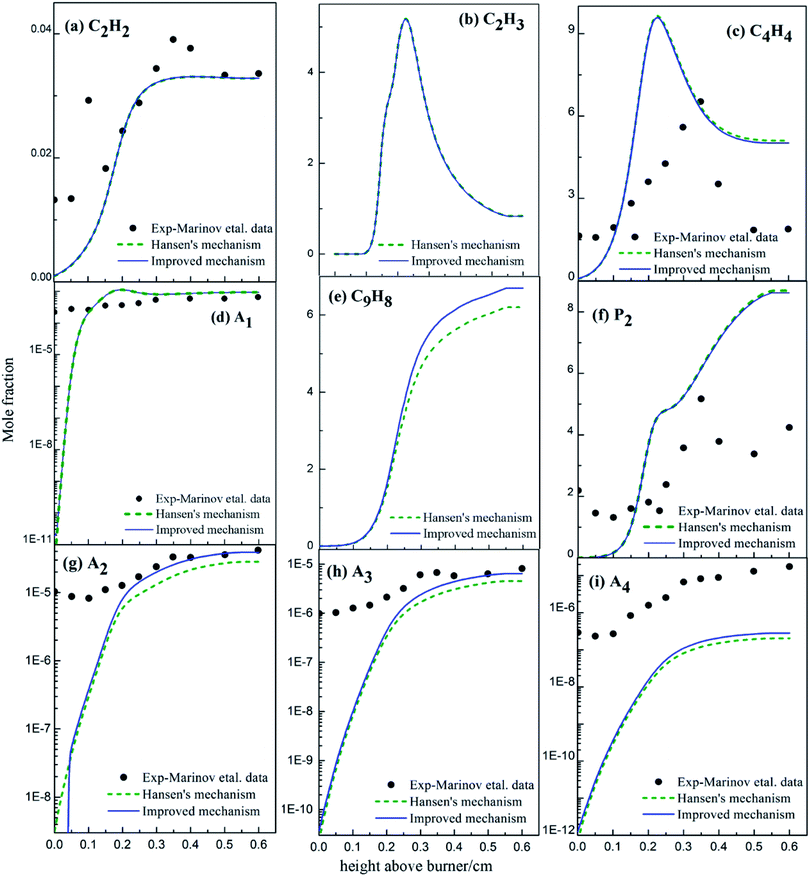 |
| | Fig. 18 Comparison of calculated and experimental data36 of mole fraction profiles for major products in butadiene flames (black dots: from experiments;36 dashed lines: from Hansen's mechanism;31 solid lines: from the improved mechanism). | |
The C2H3 mole concentration from the improved mechanism agrees well with the data from Hansen's mechanism (Fig. 18). Namely, the newly-added PAHs formation routes via C2H3 addition were not brought into full play in butane flame. Compared with the modelling results from Hansen's mechanism, the mole concentration of C4H4 from the improved mechanism decreases, and those of A2, A3 and A4 from the improved mechanism increase. However, at low temperature, the concentration of A2 decreases obviously compared with Hansen's results for the newly-added A3 formation routes from A2. Thus, the newly-added PAHs formation routes via C4H4 addition play key roles in butane flame.
The mole concentration of C9H8 from the improved mechanism increases compared with results from Hansen's mechanism, because the newly-added A3 formation routes improve the formation of A3, and a part of A3 forms indenyl and cyclopentadienyl due to the chemical equilibrium. Hence, the newly-added PAHs formation routes, especially the C4H4 addition routes, contribute in butane flame.
Comprehensive comparison about the added PAHs formation routes and the modelling results of 1,3-butadiene and butane flame shows that both C2H3 and C4H4 addition routes benefit PAHs formation in 1,3-butadiene flame, but C4H4 addition routes benefit PAHs formations more than C2H3 additions in butane flame. This is mainly because of the geometries of 1,3-butadiene and butane.
4. Conclusions
Detailed A3 and A4 formation routes via C2H3 and C4H4 addition reactions onto aromatic radicals were investigated with the G3(MP2, CC) method. The influences of reaction sites, reaction types and additions to PAHs formation rates were discussed. The PAHs formation routes gained in this study and reported before were used to improve an existing mechanism. This improved mechanism was verified and compared to experimental results and the modelling results of the original mechanism. The contributions of C2H3 and C4H4 addition routes to PAHs formation in both butadiene and butane flame were studied.
(1) Compared with C2H2 addition, C2H3 and C4H4 addition reactions occurred more easily at the radicals of aromatics; C2H3, C4H4 and C6H5 additions were more irreversible, and the formed PAH geometries were more irreversible. All these addition reactions easily occurred in flame, and were verified to produce PAHs at low temperature in both butadiene and butane flame.
(2) H atoms are important for PAHs formation. On one hand, almost all H-abstractions with the assistance of H atoms need barriers about 30 kcal mol−1, and disproportionations need to overcome high barriers and are highly endothermic. That is, H atoms decreased the energies for radical production, and made the reactions more irreversible. On the other hand, H loss with the assistance of O atom and OH radical needs to overcome much higher barriers than that with the assistance of H atom. In other words, H atoms more easily induce H loss than O atoms and OH radicals.
(3) The C2H3 and C4H4 addition routes are both beneficial to PAHs formation in 1,3-butadiene flame, and the C4H4 addition route benefits PAHs formation in butane flame. This is mainly because C4H4 can be effectively formed from continuous dehydrogenations of 1,3-butadiene and butane. C2H3 can be formed easily from the breakage of C–C in 1,3-butadiene structure, but difficultly from the corresponding reactions of butane molecules.
Conflicts of interest
There are no conflicts to declare.
References
- H. Wang and M. Frenklach, Calculations of Rate Coefficients for the Chemically Activated Reactions of Acetylene with Vinylic and Aromatic Radicals, J. Phys. Chem., 1994, 98, 11465–11489 CrossRef CAS.
- M. Frenklach and H. Wang, Detailed modeling of soot particle nucleation and growth, Symp., 1991, 23, 1559–1566 Search PubMed.
- M. Frenklach, D. W. Clary, W. C. Gardiner and S. E. Stein, Detailed kinetic modeling of soot formation in shock-tube pyrolysis of acetylene, Symp., 1985, 20, 887–901 Search PubMed.
- J. D. Bittner and J. B. Howard, Composition profiles and reaction mechanisms in a near-sooting premixed benzene/oxygen/argon flame, Symp., 1981, 18, 1105–1116 Search PubMed.
- J. Appel, H. Bockhorn and M. Frenklach, Kinetic modeling of soot formation with detailed chemistry and physics: laminar premixed flames of C2 hydrocarbons, Combust. Flame, 2000, 121, 122–136 CrossRef CAS.
- V. V. Kislov, A. I. Sadovnikov and A. M. Mebel, Formation mechanism of polycyclic aromatic hydrocarbons beyond the second aromatic ring, J. Phys. Chem. A, 2013, 117, 4794–4816 CrossRef CAS.
- A. Violi, A. D'Anna and A. D'Alessio, Modeling of particulate formation in combustion and pyrolysis, Chem. Eng. Sci., 1999, 54, 3433–3442 CrossRef CAS.
- Y. Li, L. Wei, Z. Tian, B. Yang, J. Wang, T. Zhang and F. Qi, A comprehensive experimental study of low-pressure premixed C3-oxygenated hydrocarbon flames with tunable synchrotron photoionization, Combust. Flame, 2008, 152, 336–359 CrossRef CAS.
- B. Yang, Y. Li, L. Wei, C. Huang, J. Wang, Z. Tian, R. Yang, L. Sheng, Y. Zhang and F. Qi, An experimental study of the premixed benzene/oxygen/argon flame with tunable synchrotron photoionization, Proc. Combust. Inst., 2007, 31, 555–563 CrossRef.
- B. Shukla and M. Koshi, A novel route for PAH growth in HACA based mechanisms, Combust. Flame, 2012, 159, 3589–3596 CrossRef CAS.
- J. Yang, L. Zhao, W. Yuan, F. Qi and Y. Li, Experimental and kinetic modeling investigation on laminar premixed benzene flames with various equivalence ratios, Proc. Combust. Inst., 2015, 35, 855–862 CrossRef CAS.
- W. Yuan, Y. Li, P. Dagaut, J. Yang and F. Qi, Experimental and kinetic modeling study of styrene combustion, Combust. Flame, 2015, 162, 1868–1883 CrossRef CAS.
- W. Yuan, Y. Li, P. Dagaut, J. Yang and F. Qi, Investigation on the pyrolysis and oxidation of toluene over a wide range conditions. I. Flow reactor pyrolysis and jet stirred reactor oxidation, Combust. Flame, 2015, 162, 3–21 CrossRef CAS.
- H. Wang, Formation of nascent soot and other condensed-phase materials in flames, Proc. Combust. Inst., 2011, 33, 41–67 CrossRef CAS.
- D. S. Parker, B. B. Dangi, R. I. Kaiser, A. Jamal, M. N. Ryazantsev, K. Morokuma, A. Korte and W. Sander, An experimental and theoretical study on the formation of 2-methylnaphthalene (C11H10/C11H3D7) in the reactions of the para-tolyl (C7H7) and para-tolyl-d7 (C7D7) with vinylacetylene (C4H4), J. Phys. Chem. A, 2014, 118, 2709–2718 CrossRef CAS.
- K. O. Johansson, M. P. Head-Gordon, P. E. Schrader, K. R. Wilson and H. A. Michelsen, Resonance-stabilized hydrocarbon-radical chain reactions may explain soot inception and growth, Science, 2018, 361, 997–1000 CrossRef CAS.
- B. Shukla and M. Koshi, A highly efficient growth mechanism of polycyclic aromatic hydrocarbons, Phys. Chem. Chem. Phys., 2010, 12, 2427–2437 RSC.
- A. D. Becke, Density-functional thermochemistry. I. The effect of the exchange-only gradient correction, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. I. Kaiser, B. B. Dangi, T. Yang, D. S. Parker and A. M. Mebel, Reaction dynamics of the 4-methylphenyl radical (p-tolyl) with 1,2-butadiene (1-methylallene): are methyl groups purely spectators?, J. Phys. Chem. A, 2014, 118, 6181–6190 CrossRef CAS.
- T. Yang, L. Muzangwa, D. S. Parker, R. I. Kaiser and A. M. Mebel, Formation of 2- and 1-methyl-1,4-dihydronaphthalene isomers via the crossed beam reactions of phenyl radicals (C6H5) with isoprene (CH2C(CH3)CHCH2) and 1,3-pentadiene (CH2CHCHCHCH3), Phys. Chem. Chem. Phys., 2015, 17, 530–540 RSC.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, A. G. Baboul and J. A. Pople, Gaussian-3 theory using coupled cluster energies, Chem. Phys. Lett., 1999, 314, 101–107 CrossRef CAS.
- C. W. Bauschlicher, A. Ricca and M. Rosi, Mechanisms for the growth of polycyclic aromatic hydrocarbon (PAH) cations, Chem. Phys. Lett., 2002, 355, 159–163 CrossRef CAS.
- P. Ghesquière, D. Talbi and A. Karton, The reaction of the benzene cation with acetylenes for the growth of PAHs in the interstellar medium, Chem. Phys. Lett., 2014, 595, 13–19 CrossRef.
- R. I. Kaiser, D. S. Parker, M. Goswami, F. Zhang, V. V. Kislov, A. M. Mebel, J. Aguilera-Iparraguirre and W. H. Green, Crossed beam reaction of phenyl and D5-phenyl radicals with propene and deuterated counterparts-competing atomic hydrogen and methyl loss pathways, Phys. Chem. Chem. Phys., 2012, 14, 720–729 RSC.
- D. S. Parker, F. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera and A. M. Mebel, On the formation of phenyldiacetylene (C6H5CCCCH) and D5-phenyldiacetylene (C6D5CCCCH) studied under single collision conditions, Phys. Chem. Chem. Phys., 2012, 14, 2997–3003 RSC.
- T. Yang, L. Muzangwa, R. I. Kaiser, A. Jamal and K. Morokuma, A combined crossed molecular beam and theoretical investigation of the reaction of the meta-tolyl radical with vinylacetylene-toward the formation of methylnaphthalenes, Phys. Chem. Chem. Phys., 2015, 17, 21564–21575 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- V. Mokrushin, V. Bedanov, W. Tsang, M. Zachariah and V. Knyazev, ChemRate 1.5.8, NIST, Gaithersburg, MD, 2006 Search PubMed.
- N. Hansen, M. Braun-Unkhoff, T. Kathrotia, A. Lucassen and B. Yang, Understanding the reaction pathways in premixed flames fueled by blends of 1,3-butadiene and n-butanol, Proc. Combust. Inst., 2015, 35, 771–778 CrossRef CAS.
- M. Wei, T. Zhang, S. Li, G. Guo and D. Zhang, Naphthalene formation pathways from phenyl radical via vinyl radical (C2H3) and vinylacetylene (C4H4): computational studies on reaction mechanisms and kinetics, Can. J. Chem., 2017, 95, 816–823 CrossRef CAS.
- Y. Wang and S. H. Chung, Soot formation in laminar counterflow flames, Prog. Energy Combust. Sci., 2019, 74, 152–238 CrossRef.
- R. J. Kee, F. M. Rupley and J. A. Miller, CHEMKIN-II: A FORTRAN Chemical Kinetics Package for the Analysis of Gas-phase Chemical Kinetics, Sandia, 1989 Search PubMed.
- J. A. Cole, J. D. Bittner, J. P. Longwell and J. B. Howard, Formation mechanisms of aromatic compounds in aliphatic flames, Combust. Flame, 1984, 56, 51–70 CrossRef CAS.
- N. M. Marinov, W. J. Pitz, C. K. Westbrook, A. M. Vincitore, M. J. Castaldi, S. M. Senkan and C. F. Melius, Aromatic and Polycyclic Aromatic Hydrocarbon Formation in a Laminar Premixed n-Butane Flame, Combust. Flame, 1998, 114, 192–213 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08744k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a