
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorus modification of cobalt–iron nanoparticles embedded in a nitrogen-doped carbon network for oxygen reduction reaction†
Rui Zhang,
Zheng Wang,
Lin Zhu,
Weixin Lv * and
Wei Wang*
* and
Wei Wang*
School of Chemistry and Chemical Engineering, Yancheng Institute of Technology, Yancheng 224051, China. E-mail: lvweixin@ycit.edu.cn; wangw@ycit.edu.cn
First published on 3rd March 2021
Abstract
For the electrochemical reduction of oxygen the development of heteroatom-doped carbon-based transition metal catalysts has become a recognized strategy to replace traditional noble metal catalysts. In this work a catalyst consisting of CoFe nanoparticles encapsulated in N-doped carbon-based materials (NC) supported by carbon nanotubes (CNTs), i.e. Fe3Co1@NC/CNTs, was modified via treatment with a phosphate salt to synthesize a P-Fe3Co1@NC/CNTs catalyst. The P-Fe3Co1@NC/CNTs exhibits with 5.29 mA cm−2 an enhanced current density which is comparable to a Pt/C catalyst. In addition, a stability and methanol resistance better than the Pt/C catalyst were observed which is ascribed to the carbon encapsulation and the synergies between the two transition metals. Finally, the reaction mechanism of P-doping was studied and discussed. These results provide possible directions for carbon-based catalysts and doping with heteroatoms for the improvement of catalytic activity. Moreover, the zinc–air battery assembled with P-Fe3Co1@NC/CNTs as the air-cathode exhibited a high-power density of 73 mW cm−2, which is comparable to that of Pt/C (71 mW cm−2) and a specific capacity of 763 mA h g−1. The prepared catalyst could potentially serve to take the place of precious metal catalysts in rechargeable Zn–air batteries.
1. Introduction
Fuel cells (FCs) are widely regarded as one of the most promising energy conversion devices to mitigate energy and environmental crises. Especially, a Zn–air battery is the most prominent energy storage device among the different metal–air batteries because of its higher energy and power density, economic viability, safety, and environmental friendliness.1 The electrochemical performance of Zn–air batteries depends greatly on the kinetics of the oxygen reduction reaction (ORR) at the cathode.2,3 This reaction is a kinetically sluggish multi-electron transfer process; hence, highly active catalysts are required for fast progress of ORR.4 At present, ORR catalysts are still dependent on precious metal catalysts like Pt, but their high cost and poor stability seriously limit the large-scale application of fuel cells.5–7 Therefore, it is of great importance to develop low cost ORR catalysts with high activity and stability.N-Doped carbon-based materials can enhance the surface activity of carbon materials and have therefore been extensively studied as high performance ORR catalysts.8 The doping with nitrogen favors the adsorption of oxygen which facilitates the ORR by lowering the electrochemical potential.9,10 As the closest congener of nitrogen, phosphorus has also been found as an appropriate dopant for tuning the surface reactivity of graphene.11 Similarly, the introduction of phosphorus induces structural defects into the carbon matrix and increases the electron delocalization due to the good electron donating properties of phosphorus, which results in materials with enriched active sites and improved conductivity and charge transfer.12,13 Therefore, adding an appropriate amount of phosphorus into the material can improve its electrochemical activity. Jing et al. prepared P-doped CNTs encapsulated nickel hybrids via a one-step chemical vapor deposition followed by P-doping treatment with NaH2PO4 as the phosphorus source. The obtained catalyst showed excellent hydrogen evolution reaction (HER) performance.14 Recently, some researchers have investigated the doping of carbon materials with both phosphorus and nitrogen atoms to obtain a more active catalyst.15–17 Hu et al. prepared FeP/N, P-doped carbon nanosheets, via a one-pot pyrolysis process by using 1,2-ethylenediphosphonic acid as the phosphorus source and melamine as the nitrogen source. The obtained catalyst showed excellent ORR performance.18
In recent years, the combination of transition metal-based materials (Fe,19,20 Co,21,22 Ni,23,24 etc.) and a carbon matrix (e.g. carbon nanotubes, graphene) was proposed as a prospective strategy not only to improve the stability of metal-based materials but also to endow the hybrids with a favorable electrocatalytic activity for ORR.25 However, the exposed metal nanoparticles are still vulnerable to the influence of external factors resulting in agglomeration similar to Pt/C catalyst and consequently in a reduction of their electrochemical activity.9 Based on this observation, it is important to confine the nanoparticles into a limited space (encapsulate metal nanoparticles in an N-doped carbon layer structure), to maintain their excellent electrochemical activities.26,27 Deng et al. synthesized a FeCo alloy catalyst encapsulated in pod-like carbon nanotubes and found through theoretical calculations that the carbon wall thickness of carbon-encapsulated metal catalysts significantly affects the dissociative adsorption of O2 on the outermost surface of CNTs. Reducing the carbon wall thickness around the metal would promote the ORR activity significantly and introducing nitrogen atoms into the carbon lattice could further enhance the dissociative adsorption of O2.28 Hu et al. successfully synthesized FeCo nanorods embedded in N-doped CNTs and conducted electrochemical tests indicating a high stability and electrocatalytic activity for oxygen evolution reaction (OER) and ORR. The authors ascribed their observation to the presence of a higher number of metal–NCNT heterointerfaces resulting in more catalytically active sites for ORR and OER.29 However, the mechanism by which ORR occurs when such nitrogenous carbon layers bind to transition metals and the nature of the specific active sites are still controversial. While some authors believe that the transition metal particles are not part of the active site but can promote the formation of the catalytically active site,30–33 others reported that transition metals are indeed part of ORR active sites.34,35 In the light of these different points of view, further research and exploration of ORR catalysts needs to be conducted.
In this paper, we synthesized ORR catalysts consisting of transition metal particles encapsulated into N-doped carbon-based materials (NC) and used a simple procedure to dope the surface of the catalysts with phosphorus. The resulting enhanced ORR performance and the catalytic kinetics of the P-doped catalysts were investigated.
2. Experimental section
2.1 Synthesis of functional CNTs
CNTs were dispersed in 200 mL mixed solution containing 10% HNO3 and 30% H2O2 with a volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and stirred at 60 °C for 5 h, then washed with deionized water, finally dried at 80 °C for 24 h to synthesize functional CNTs.
:1 and stirred at 60 °C for 5 h, then washed with deionized water, finally dried at 80 °C for 24 h to synthesize functional CNTs.
As a result of the above reactions, CNTs were modified by a certain amount of carboxyl and hydroxyl groups, which acted as the active centers to promote the next reaction with iron(II) phthalocyanine (FePc), cobalt(II) phthalocyanine (CoPc) and melamine (C3H6N6, >99%).
2.2 Synthesis of P-Fe3Co1@NC/CNTs, Fe3Co1@NC/CNTs, Fe3Co1@NC and P-Fe3Co1@NC catalysts
100 mg functional CNTs, 150 mg FePc (Aladdin, >97%) and 50 mg CoPc (Aladdin, >95%) were dispersed in an ultrasonic for 1 h in 75 mL N,N-dimethylformamide (DMF) (Aladdin, 99%). The suspension was then stirred for 6 h. After adding melamine (6 g) the suspension was stirred for 1 h.After filtration the obtained solid was rinsed three times with DMF and dried in vacuo at 80 °C for 24 h. The material was calcined at 420 °C for 2 h and at 750 °C for 1 h in a N2 atmosphere. Finally, the material was pickled with 0.5 M H2SO4 at 80 °C for 8 h to remove any unreacted transition metal in the material to obtain the catalyst Fe3Co1@NC/CNTs. The P-doped catalyst P-Fe3Co1@NC/CNTs was obtained by heating Fe3Co1@NC/CNTs with sodium dihydrogen phosphate powder in a N2 atmosphere at 300 °C for 30 min.
For further study and comparison, Fe3Co1@NC and P-Fe3Co1@NC were synthesized using the same method mentioned above without the addition of functional CNTs.
2.3 Characterization
The crystal structures of the samples were characterized via X-ray diffraction (XRD, Bluker Rigaku D/MAX 2200 diffractometer with Cu Kα). The morphologies and composition of the samples were investigated via transmission electron microscopy (TEM, JEM2100F). X-ray photoelectron spectroscopy (XPS) measurements were carried out using an ESCALAB 250Xi electron spectrometer.2.4 Electrochemical test
All electrochemical measurements were made using a CHI 760E electrochemistry workstation (CH Instrument, Shanghai, China) in a standard three-electrode system. A platinum electrode was used as the counter electrode, and a Hg/HgO (1 M KOH) was used as the reference electrode. All the potentials were initially measured against Hg/HgO electrode and then converted to reversible hydrogen electrode (RHE) using the relation: ERHE = EHg/HgO + 0.098 + 0.059 × pH. 0.1 M KOH solution was used as the electrolyte (170 mL). Before each experiment, O2 gas (99.99%) was aerated into the electrolyte for at least 40 min to reach oxygen saturation. The working electrode was prepared by coating 20 μL of well dispersed catalyst ink on the surface of the pre-cleaned rotating disk electrode (RDE, 5 mm in diameter) and dried under ambient conditions. The catalyst ink was prepared with 4 mg of as-prepared catalyst sample, 985 μL ethanol and 15 μL of Nafion solution (DuPont, D-520, 5 wt%) after sonication for 1 h. For comparison, 20 wt% Pt/C (Johnson Matthey) ink was formed like other catalyst inks with a similar procedure. The linear sweep voltammetry (LSV) curves were evaluated at a sweep rate of 5 mV s−1 at different rotation rates from 400 to 2025 rpm. Long-time and methanol crossover measurements were conducted in O2-saturated solution under a rotating speed of 1600 rpm. For the anti-methanol tests, the current was recorded when 1 M methanol was added into the electrolyte at 400 s.The apparent electron transfer number (n) for a typical ORR is calculated from the Koutecky–Levich (K–L) equations:
| 1/J = 1/JK + 1/JL = 1/JK + 1/Bω1/2 | (1) |
| B = 0.62nFC0D02/3ν−1/6 | (2) |
485 C mol−1); C0 is the volume concentration of O2 (1.2 × 10−3 mol L−1); D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1); ν is the kinematic viscosity of electrolyte (0.01 cm2 s−1).
For the rotating ring disk electrode (RRDE) tests, the ring potential is set at 1.3 V constantly. The transferred electron number (n) and the yield of HO2− during the ORR process on the base of RRDE can be calculated using the following equations:
| n = 4ID/(ID + IR/N) | (3) |
| HO2− (%) = 200IR/N/(ID + IR/N) | (4) |
2.5 Zn–air battery measurement
Home-assembled Zn–air battery was fabricated by the as-prepared catalysts or 20% Pt/C as cathode and a polished zinc foil (2 cm2) as anode with 6 M KOH as electrolyte. Discharge polarization curves were measured by linear sweep voltammetry from 1.5 to 0.4 V at a scan rate of 5 mV s−1. Discharge current and the corresponding power density were normalized to the effective surface area of air-cathode electrode.3. Results and discussions
A comparison of the X-ray diffraction (XRD) patterns of Fe3Co1@NC/CNTs and P-Fe3Co1@NC/CNTs (Fig. 1a) does not reveal any change after doping with phosphorus. A peak centered at 26.2° corresponds to the (002) plane of the carbon atom structure of CNTs. A relatively large peak observed at 44.7° can be assigned to the (111) and (002) plane of the Fe and Co nanoparticles and the (110) plane of the metal compounds Co7Fe3 and CoFe. The occurrence of this peak indicates the presence of alloys and elemental metals in Fe3Co1@NC/CNTs, which is still present in the spectrum of P-Fe3Co1@NC/CNTs indicating that the doping with phosphorus does not change the existing form of transition metals in the material. It is notable that a smaller peak appears at 43.7°, which can be attributed to the (111) plane of the transition metal nitrides Co5.47N and Fe3N. Since some studies have shown that CoxN and FexN are also active sites for ORR, the presence of transition metal nitride also provides an explanation for the catalytic ORR activity of the synthesized Fe3Co1@NC/CNTs and P-Fe3Co1@NC/CNTs materials.36,37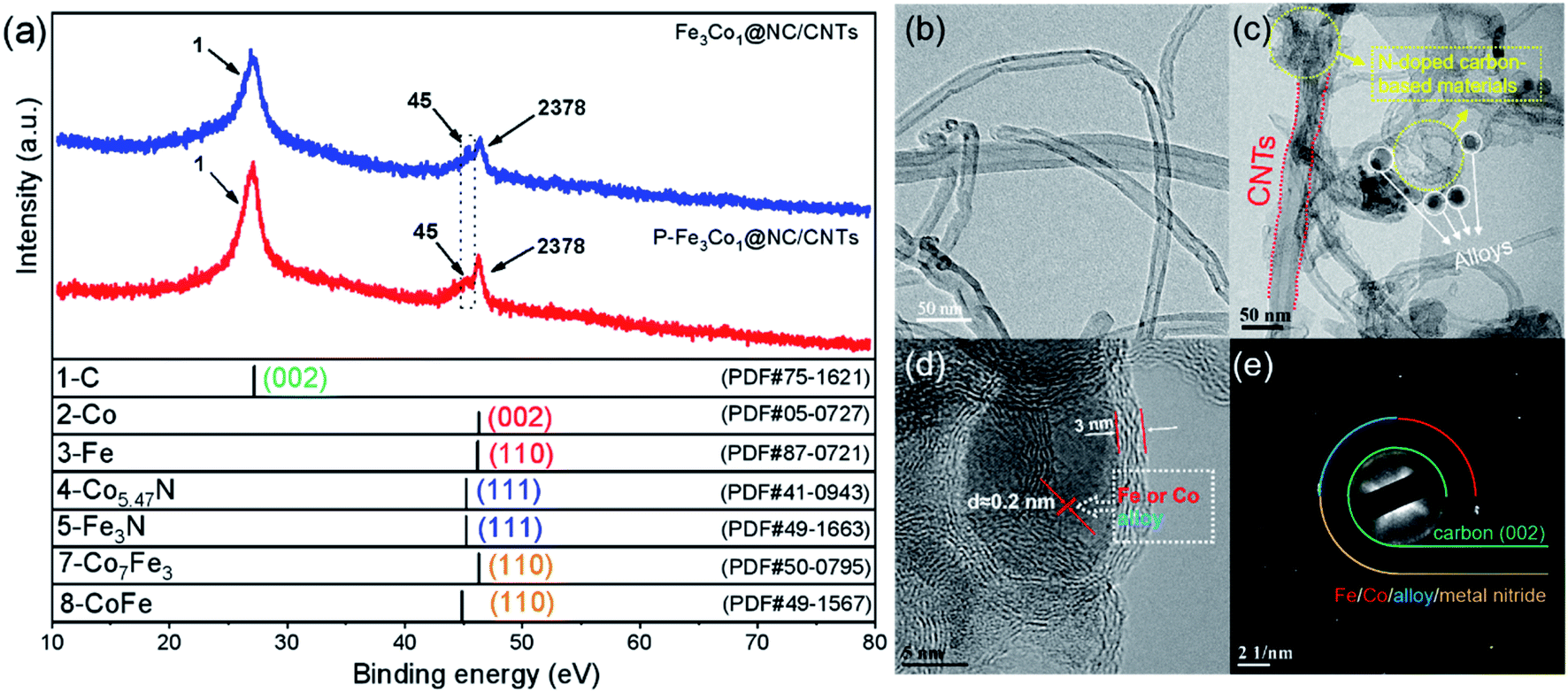 | ||
| Fig. 1 (a) XRD patterns of Fe3Co1@NC/CNTs and P-Fe3Co1@NC/CNTs. (b) TEM images of CNTs. (c and d) TEM images of P-Fe3Co1@NC/CNTs. (e) SAED of P-Fe3Co1@NC/CNTs. | ||
The TEM image of CNTs in Fig. 2a shows the smooth surface and typical hollow structure of this material. The TEM images of P-Fe3Co1@NC/CNTs in Fig. 2b and c depict that black particles are encapsulated in the bamboo-like nitrogenous carbon-based material with a thin tube wall (3 nm). This interpenetrated conductivity network structure is favorable for electron transport within the CNTs.26 It is worth noting that the ԁ-spacing of these black particles is about 2 nm, which corresponds to the ԁ-spacing of the metallic elements Fe and Co and their alloys Co7Fe3 and CoFe as calculated from the XRD pattern shown in Fig. 1a. The results of the selected area electron diffraction (SAED) pattern of P-Fe3Co1@NC/CNTs presented in Fig. 1d are consistent with the XRD results and confirm the (002) crystal plane belonging to C, and the respective planes to the metallic elements Fe, Co and their alloys, Co7Fe3 and CoFe, as well as the nitrides of two transition metals, Fe3N and Co5.47N.
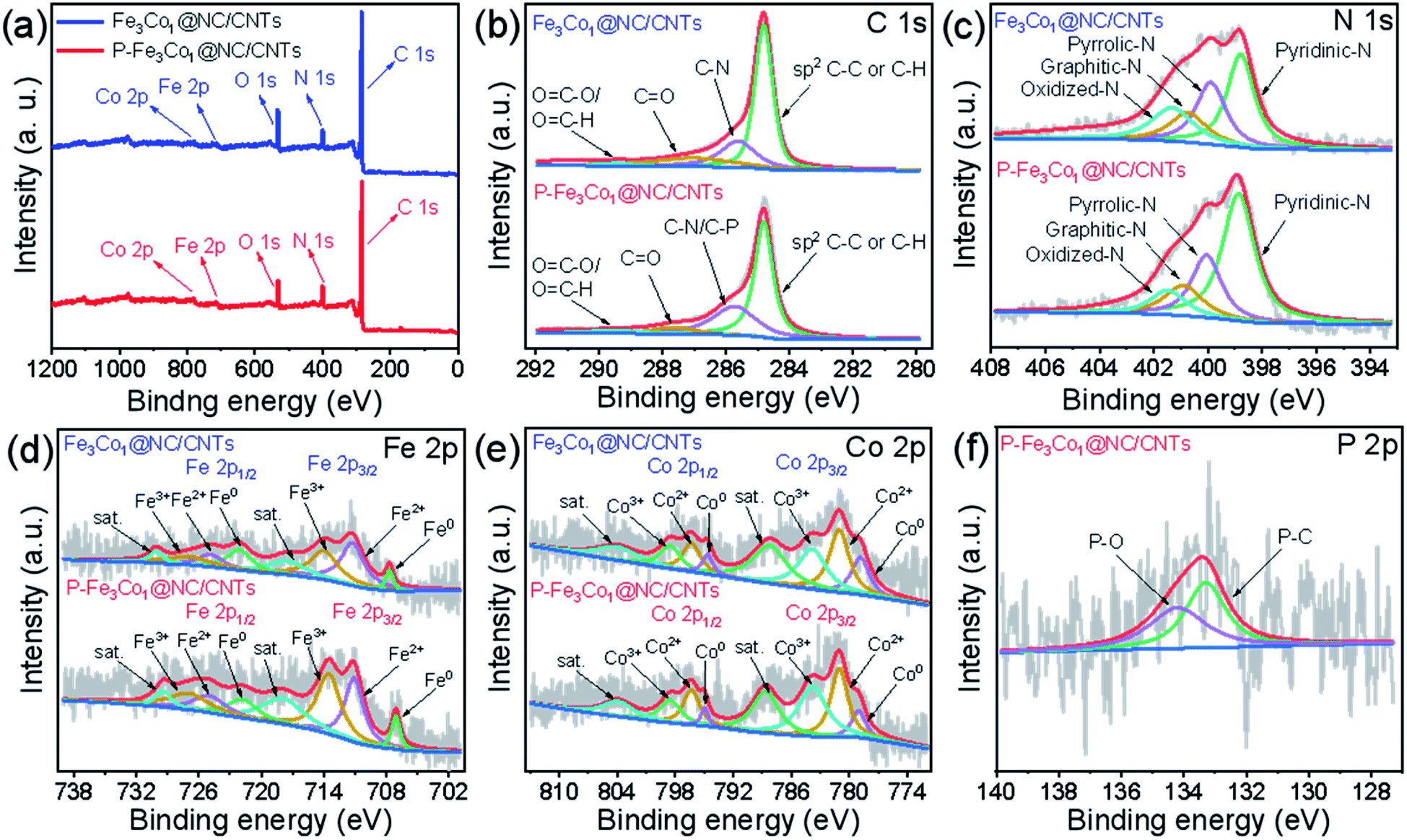 | ||
| Fig. 2 (a) XPS survey spectra of P-Fe3Co1@NC/CNTs and Fe3Co1@NC/CNTs. High-resolution XPS spectra of C 1s (b), N 1s (c), Fe 2p (d), Co 2p (e) and P 2p (f) for P-Fe3Co1@NC/CNTs. | ||
It was preliminarily proved by TEM imaging that the used preparation method could successfully encapsulate transition metal particles in the NC which compound with CNTs. The morphology NC is not homogeneous, showing bamboo-like tubular, lamellar or spherical shape. Such NC have been shown to exhibit a higher activity compared to common CNTs.38–40 In addition, the thinner wall can ease penetration of electrons from the metal cores to the carbon surface. These features could be responsible for the observed superior ORR performance.
The element composition and chemical environment of P-Fe3Co1@NC/CNTs and Fe3Co1@NC/CNTs have been studied by X-ray photoelectron spectroscopy (XPS). The spectra presented in Fig. 2a indicate that P-Fe3Co1@NC/CNTs and Fe3Co1@NC/CNTs contain C, O, N, Fe, and Co elements. The quantitative analysis resulted in a nitrogen content in P-Fe3Co1@NC/CNTs and Fe3Co1@NC/CNTs of 10.28 at% and 8.87 at%, respectively. The higher nitrogen content in P-Fe3Co1@NC/CNTs could result in a higher ORR performance of this material.
As illustrated in Fig. 2b, the C 1s spectra of P-Fe3Co1@NC/CNTs and Fe3Co1@NC/CNTs can be deconvoluted with four components corresponding to the graphitized C, C–N (C–P), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO/N–CO atoms or functional groups.9 In the two materials, graphitized C can be assigned to the majority of peaks with total peak areas of 54.6% and 53.8%, respectively, in line with the bonding type of CNTs. It is worth noting that the XPS spectra of P-Fe3Co1@NC/CNTs indicate C–N/C–P bonds at 285.73 eV due to the doping with phosphorus.41 The increase in the peak area proves that phosphorus was successfully incorporated in the C structure to form C–P bonds.
O, and O–CO/N–CO atoms or functional groups.9 In the two materials, graphitized C can be assigned to the majority of peaks with total peak areas of 54.6% and 53.8%, respectively, in line with the bonding type of CNTs. It is worth noting that the XPS spectra of P-Fe3Co1@NC/CNTs indicate C–N/C–P bonds at 285.73 eV due to the doping with phosphorus.41 The increase in the peak area proves that phosphorus was successfully incorporated in the C structure to form C–P bonds.
The deconvolution of the N 1s spectrum (Fig. 2c) results in patterns corresponding to four kinds of nitrogen atoms: pyridinic N (398.8 eV), pyrrolic N (400.1 eV), graphitic N (400.9 eV) and oxidized N (401.5 eV).42,43 The content of pyridinic N is the highest and increases from 37.0% of the total amount of nitrogen to 51.5% after doping with phosphorus. As the widely accepted electroactive site, the electron-accepting pyridinic N species can impart a relatively high positive charge density on the neighboring sp2-bonded C atoms, facilitate the adsorption of reactants (e.g., OH−, O2), and promote the electron transfer between the catalyst surface and reaction intermediates, thus efficiently catalyzing ORR with favorable reaction kinetics.8 It is generally believed that for ORR the catalytic activity decreases in the order pyridinic N > pyrrolic N > graphitic N > oxidized N. Therefore, the higher pyridinic N content in P-Fe3Co1@NC/CNTs can be responsible for its improved ORR activity.
The peaks in the Fe 2p XPS-spectrum (Fig. 2d) centred at 707.1, 711.0, and 713.5 eV can be attributed to the Fe0, Fe2+, and Fe3+ 2p3/2 states, respectively, whereas peaks centred at 721.8, 724.8, and 727.0 eV correspond to the Fe0, Fe2+, and Fe3+ 2p1/2 states, respectively. Satellite peaks denoted as “sat.” are observed at 717.9 and 729.4 eV.44,45 Similarly, in the Co 2p XPS-spectrum (Fig. 2e), the peaks at 779.0, 781.0, and 783.7 eV refer to the Co0, Co2+, and Co3+ 2p3/2 states and the peaks at 794.8, 796.3, and 798.5 eV to the Co0, Co2+, and Co3+ 2p1/2 states, respectively. Two satellite peaks are also present at 788.6 and 803.9 eV.42,46 Finally, the P 2p XPS-spectrum (Fig. 2f) indicates that P–O (134.2 eV) and P–C (133.3 eV) are the two forms of doped phosphorus in P-Fe3Co1@NC/CNTs.41
The XPS analysis reveals that the two transition metals contained in the two materials before and after phosphorus doping were present both as zero-valent metal elements and in other oxidation states, which proves that this method could successfully insert the metal elements and their alloys into NC. Furthermore, the observation of the P–C bond peak proves the specific existence of this form of phosphorus in the material.
To evaluate the electrocatalytic ORR activities of Fe3Co1@NC/CNTs and P-Fe3Co1@NC/CNTs with different metal ratios, linear sweep voltammetry (LSV) was conducted in O2-saturated 0.1 M KOH solution. Referring to the spectra shown in Fig. 3a and c for the undoped and P-doped materials, respectively, in the catalysis of ORR the onset potential and half-wave potential decreases at a rotation rate of 1600 rpm from Eonset = 0.877 V and E1/2 = 0.814 V for Fe3Co1@NC/CNTs to Eonset = 0.860 V and E1/2 = 0.802 V for P-Fe3Co1@NC/CNTs. However, it is worth noting that P-Fe3Co1@NC/CNTs shows with JL = 5.29 mA cm−2 a higher limiting current density at 1600 rpm than Fe3Co1@NC/CNTs (JL = 4.91 mA cm−2) due to the doping with phosphorus. To further study the catalytic mechanism of the two catalysts in ORR before and after phosphorus doping and the influence of phosphorus doping on the reaction path of P-Fe3Co1@NC/CNTs, K–L curves were fitted for the two catalysts to calculate the apparent transfer electron number in ORR.
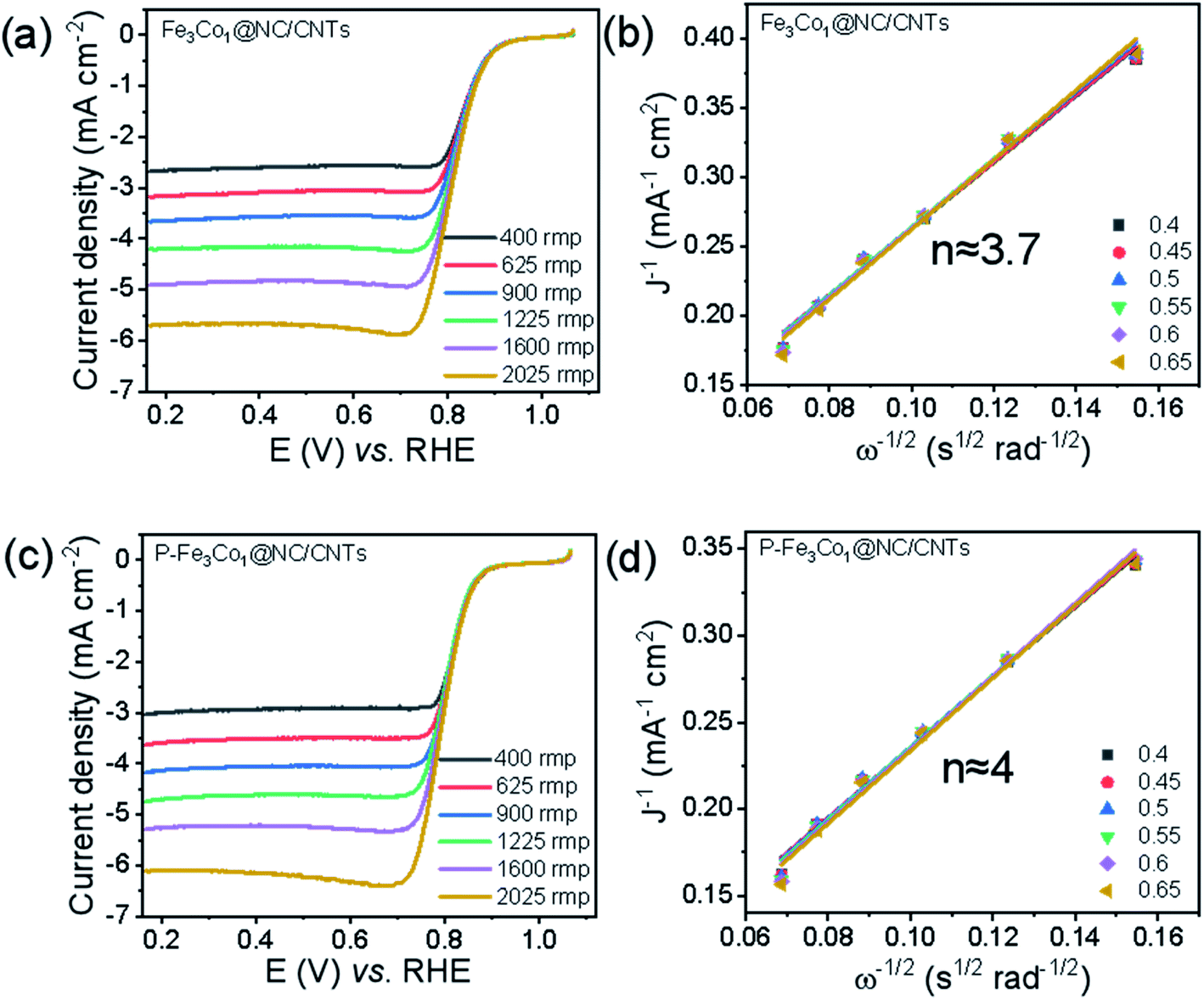 | ||
| Fig. 3 LSV curves at various rotation rates for Fe3Co1@NC/CNTs (a), P-Fe3Co1@NC/CNTs (c). The insets in figure (b) and (d) refer to the corresponding K–L plots. | ||
The K–L curves of Fe3Co1@NC/CNTs (Fig. 3b) and P-Fe3Co1@NC/CNTs (Fig. 3d) can be approximated with a straight line with a good value for the goodness of fit. The comparison shows that the apparent electron transfer number of Fe3Co1@NC/CNTs is 3.7, which indicates that the reaction proceeds by a path dominated by a four-electron transfer step but mixed with two and four electron steps in the ORR. However, P-Fe3Co1@NC/CNTs after phosphorus doping exhibits a higher apparent transfer electron number (n ≈ 4) demonstrating that phosphorus doping improved the four-electron step selectivity of the catalyst during ORR and thereby further inducing the four-electron reaction path.
When comparing the LSV curves of the different materials (Fig. 4a) the P-doped P-Fe3Co1@NC/CNTs exhibits a higher limit current density than Fe3Co1@NC/CNTs, CNTs and the traditional commercial 20% Pt/C.
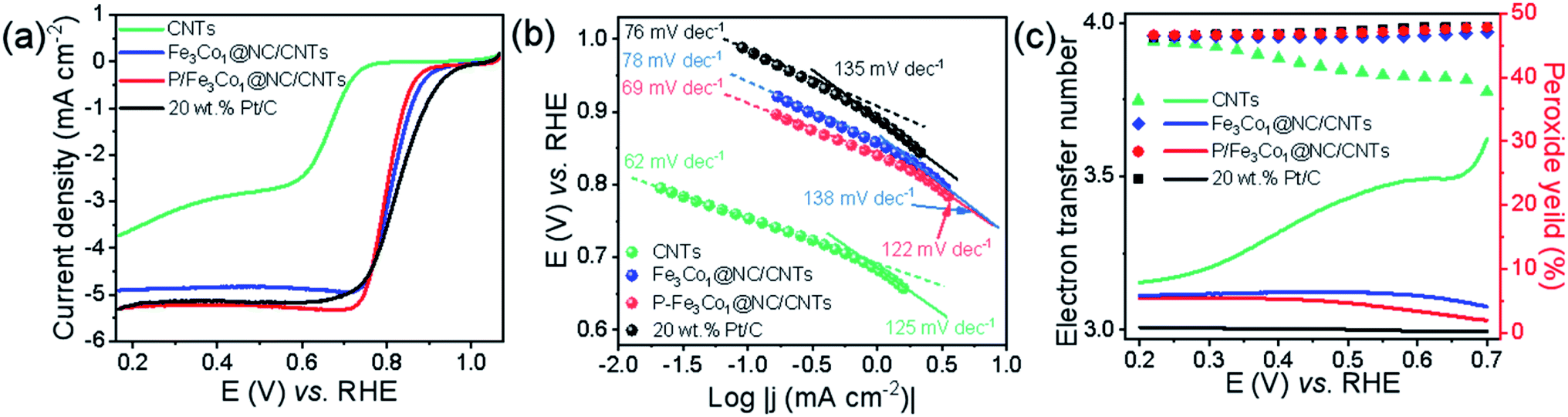 | ||
| Fig. 4 (a) LSV curves of different component catalysts at the rotation rate of 1600 rpm. (b) Corresponding Tafel plots. (c) HO2− yields (top panel) and electron transfer numbers (bottom panel) of RRDE measurements. | ||
More information on the oxygen adsorption mechanism and oxygen reduction kinetics of the catalysts can be deducted from the diffusion-corrected Tafel curve (Fig. 4b). In the low current density region, several catalysts, i.e., P-Fe3Co1@NC/CNTs (69 mV dec−1) and CNTs (62 mV dec−1), exhibit lower Tafel slopes than 20% Pt/C (76 mV dec−1) and Fe3Co1@NC/CNTs (78 mV dec−1) in ORR. But in the high current density region, P-Fe3Co1@NC/CNTs (122 mV dec−1) has the lowest Tafel slope among the catalysts above. A lower value of Tafel slope implies faster kinetics indicating that the catalyst can quickly reach a higher catalytic current density under a lower applied potential. It can be concluded that P doping of Fe3Co1@NC/CNTs increases the limit current density and simultaneously improves the ORR dynamics. This result is in good agreement with the results of LSV curves presented in Fig. 4a.
In order to more accurately study the specific reaction paths and reaction mechanisms of different catalysts during ORR, rotating ring-disk electrode (RRDE) experiments were conducted. One can conclude from the results depicted in Fig. 4c, that the reaction path selectivity of the materials with encapsulated transition metal particles were significantly improved under ORR conditions as evidenced by a higher number of transferred electrons and a lower peroxide yield compared to CNTs. A higher transfer electron number increases energy efficiency, and a lower peroxide formation can effectively prevent the catalyst from being eroded.
It is worth noting that the introduction of phosphorus voids the electroneutrality leading to high positive spin density and atomic charge density for carbon atoms, which renders those carbon atoms adjacent to P as the catalytic sites for ORR. Consequently, P-Fe3Co1@NC/CNTs exhibits a number of transferred electrons very close to 4 similar to 20% Pt/C and a low hydrogen peroxide yield (<5%) indicating that P-Fe3Co1@NC/CNTs shows a good selectivity for the four-electron reaction pathway.
The stability and methanol tolerance of the catalyst are also important indices for its electrochemical performance and its commercialization potential.47 As shown in Fig. 5a, after 25000 s of the ORR, P-Fe3Co1@NC/CNTs still maintained a highly effective oxygen reduction activity with an activity loss of only 5.2%. However, the electrocatalytic activity of Pt/C catalyst decreased ∼30% after 25000 s of electrocatalytic reaction, which was mainly due to the aggregation of Pt nanoparticles and the dissociation of Pt nanoparticles from the carbon supports. The much slower decay in the current density of P-Fe3Co1@NC/CNTs is a consequence of the synergistic contribution arising from the Fe–Co nanoparticles and their coating materials of NC, which well avoids the structure variation of the P-Fe3Co1@NC/CNTs and suppresses the agglomeration/dissolution of the Fe3Co1 nanoparticles during the ORR.
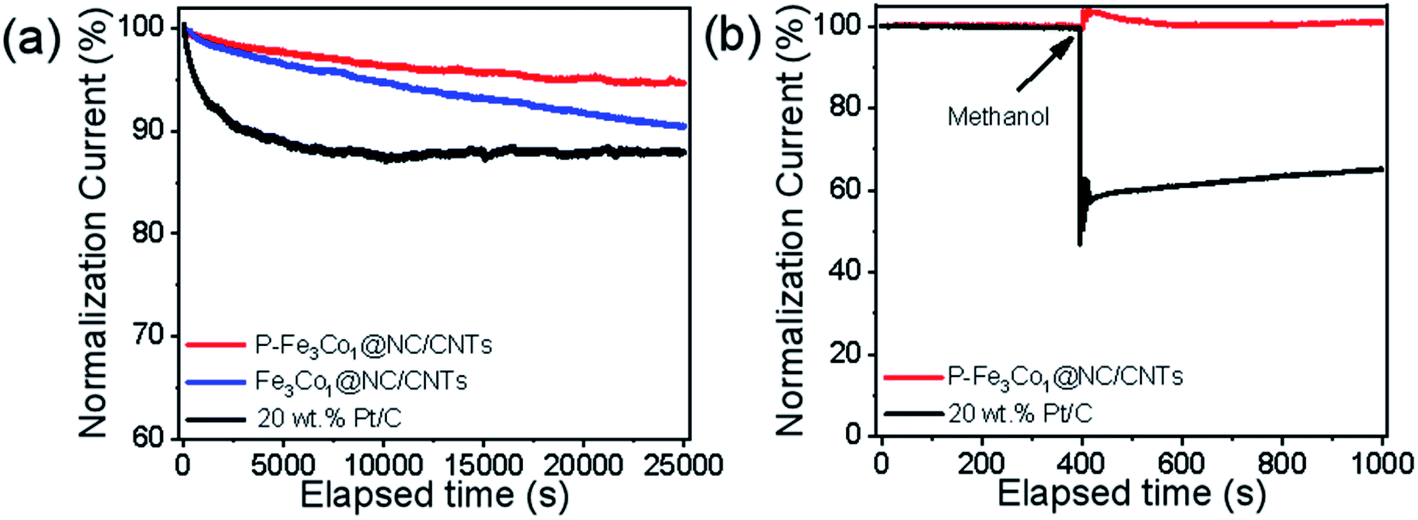 | ||
| Fig. 5 (a) Durability evaluation at 0.6 V in an O2-saturated 0.1 M KOH solution. (b) Chronoamperometric response of ORR for P-Fe3Co1@NC/CNTs and Pt/C by introducing 1 M methanol solution after 400 s. | ||
The chronoamperometric response of ORR to 1 M methanol solution (Fig. 5b) indicates a dramatic decrease in the activity of the commercial Pt/C immediately after the introduction of methanol, which oxidizes and blocks active sites on the catalyst. However, the effect of methanol on P-Fe3Co1@NC/CNTs was negligible which demonstrates the higher durability of this catalyst to methanol in ORR. These results indicate the advantage of metal particles being wrapped by carbon materials and the synergistic effects of both transition metals and of phosphorus and carbon. The above series of electrochemical tests demonstrates the superior performance of P-Fe3Co1@NC/CNTs which is inexpensive and simple to synthesize thereby showing a great potential for its use in the development of fuel cells.
To elucidate the location of phosphorus doping and the mechanistic role of phosphorus in promoting the ORR of such materials, CNTs and Fe3Co1@NC were doped with phosphorus by the same method and their ORR performance tested. The obtained P-CNTs exhibits a higher initial potential and half-wave potential (Fig. 6a), and its limit current density (JL) increased from 3.81 to 4.21 mA cm−2 indicating that the ORR performance of CNTs can be improved by P-doping. The LSV curves shown in Fig. 6b do not reveal any obvious difference in the ORR performance of Fe3Co1@NC before and after doping with phosphorus. Therefore, we speculate that the reason for the excellent ORR activity of P-Fe3Co1@NC/CNTs is mainly caused by the increased activity of CNTs after P-doping. To further prove this hypothesis, the ORR performance of the Fe3Co1@NC composite catalysts with different supports, CNTs and P-doped CNTs, were compared. Referring to Fig. 6c, Fe3Co1@NC/P-CNTs did not show any obvious activity improvement compared to Fe3Co1@NC/CNTs, while both their ORR performance was lower than that of P-Fe3Co1@NC/CNTs. This result indicates that P-doping of Fe3Co1@NC/CNTs composites have obvious advantages compared to treating the CNTs alone with phosphorus.
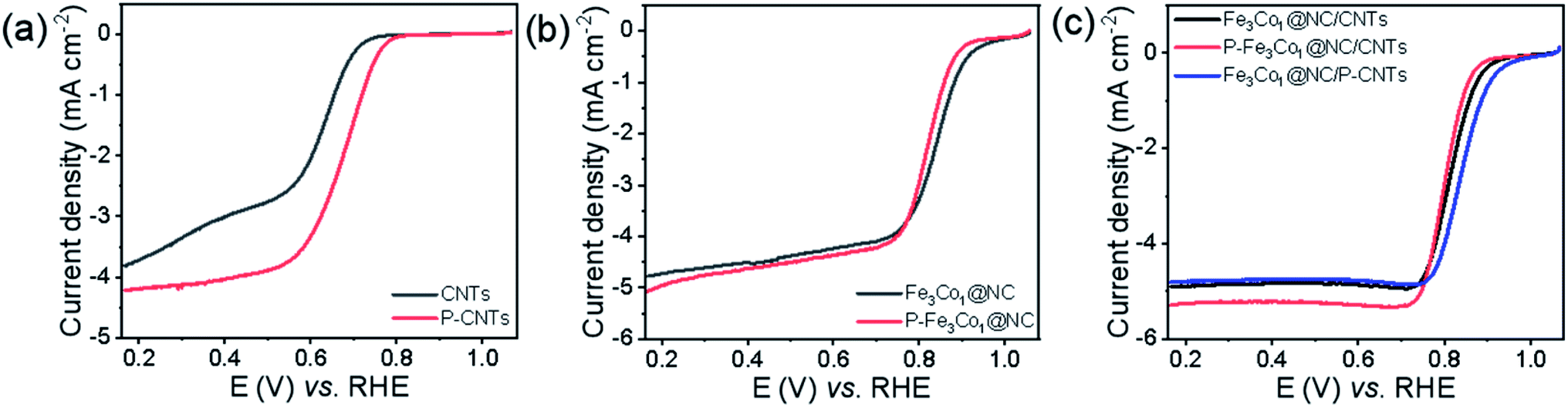 | ||
| Fig. 6 LSV curves in the O2-saturated 0.1 M KOH solution at a rotation rate of 1600 rpm. | ||
During the P-doping process, NaH2PO4 produced phosphine gas (PH3) during heating at 300 °C, which reacted with the material. The possible reaction process is illustrated in Fig. 7. PH3 molecules can be embedded into the skeleton of CNTs at the vacancy of defective carbon layers through the depicted reactions, which is consistent with the XPS results of phosphorus being mainly bound to the material by a P–C bond. For the Fe3Co1@NC catalyst the ORR performance did not improve significantly after P-doping. For both P-Fe3Co1@NC and P-CNTs the phosphorus content were about 0.8 at% as determined by XPS. While these results indicate that phosphorus can be doped into Fe3Co1@NC and CNTs, as a new hypothesis it can be proposed that the defective Fe3Co1@NC and CNTs can react with one PH3 molecule when two defect sites are adjacent to each other. In this case, Fe3Co1@NC and CNTs can be joined together by P atoms and attain favorable synergies.
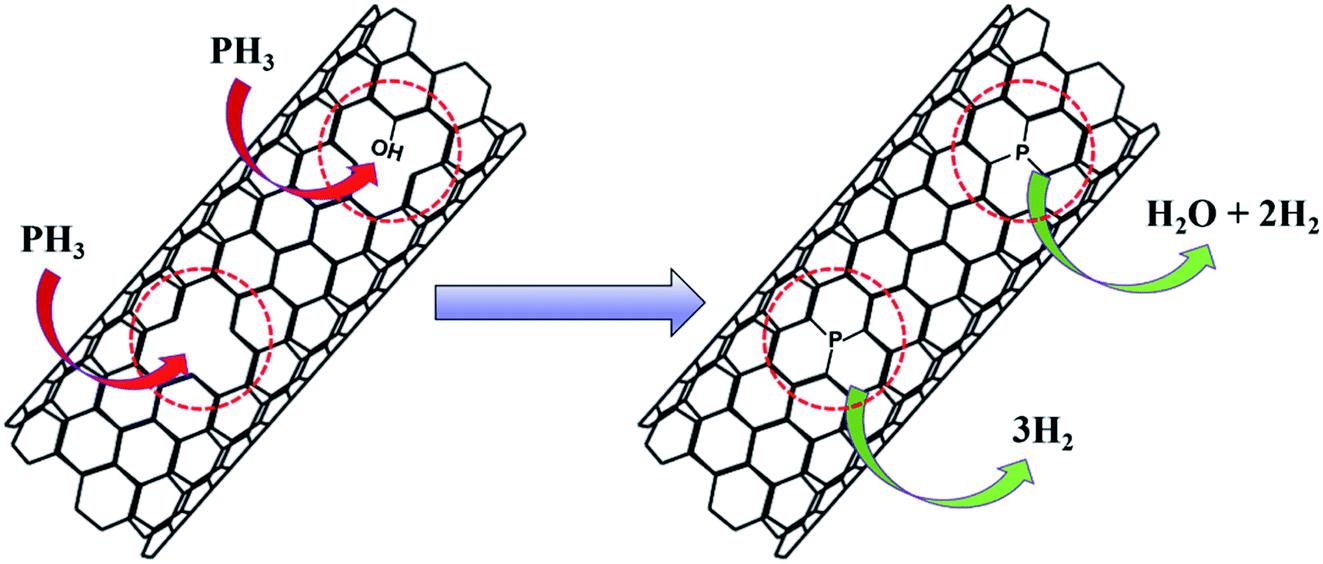 | ||
| Fig. 7 Schematic diagram of P element doping mechanism. | ||
Additionally, Zn–air battery performances were tested with P-Fe3Co1@NC/CNTs acting as the catalyst of the air cathode in comparison with the coupled noble metal Pt/C catalyst. Fig. 8a shows that the battery containing the P-Fe3Co1@NC/CNTs affords an open-circuit voltage of 1.466 V. Multiple Zn–air batteries connected in series can thus be fabricated and integrated into circuits to meet specific energy/power needs for various applications. One proof-of-concept illustration is to power a light emitting diode (LED, 2 V) with excellent operation stability (Fig. 8b).
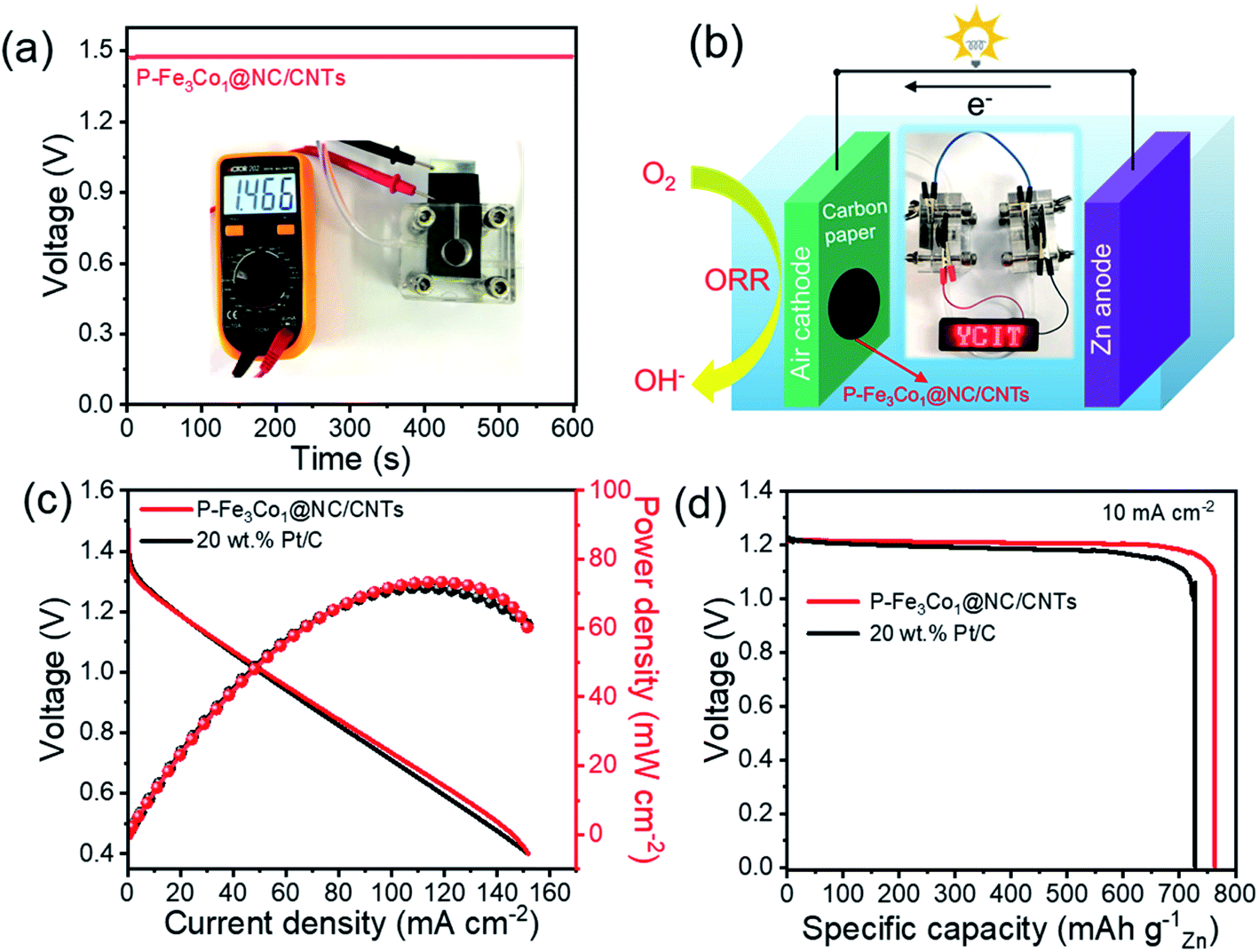 | ||
| Fig. 8 (a) Open circuit plots with inset showing the photograph of home-made Zn–air battery with an open circuit voltage of 1.47 V. (b) Schematic representation of a rechargeable Zn–air battery. (c) Polarization curves and corresponding power density plots of the Zn–air batteries with P-Fe3Co1@NC/CNTs and Pt/C catalysts. (d) Discharge curves of the primary Zn–air battery with P-Fe3Co1@NC/CNTs catalyst until complete consumption of Zn at 10 mA cm−2 current densities. Specific capacity was normalized to the mass of consumed Zn. | ||
Fig. 8c reproduces the power density curves for Zn–air batteries with P-Fe3Co1@NC/CNTs and Pt/C catalysts. The maximum power density of the Zn–air battery using P-Fe3Co1@NC/CNTs catalyst was calculated to be 73 mW cm−2, which is comparable to that of Pt/C catalyst (71 mW cm−2). The galvanostatic discharge curves at 10 mA cm−2 are presented in Fig. 8d. The battery with the P-Fe3Co1@NC/CNTs catalyst and that with the Pt/C catalyst showed voltage plateaus both around 1.20 V, with specific capacity of 763 and 732 mA h g−1, respectively, when normalized to the weight of consumed zinc electrode.
4. Conclusions
In summary, using simple and inexpensive methods a series of transition metal catalysts encapsulated in bamboo-like nitrogenous carbon-based material was prepared and the surface of the materials successfully doped with phosphorus. Based on the applied analytical methods and comparisons, it is proved that phosphorus atoms are incorporated at the defect locations of CNTs by means of P–C bonds. The superior electrocatalytic activity of P-Fe3Co1@NC/CNTs can be attributed to (i) the synergistic interaction of the two metals in P-Fe3Co1@NC/CNTs promoting their ORR activity; (ii) the higher pyridinic-N content; (iii) a small amount of metal nitride in P-Fe3Co1@NC/CNTs providing more ORR active sites; (iv) the presence of CNTs facilitating the formation of the interpenetrated conductivity network structure which improves the electrical conductivity of the P-Fe3Co1@NC/CNTs to promote a fast electron transport and rapid reaction kinetics during the catalytic process and which increases the dispersion of the material, consequently preventing P-Fe3Co1@NC/CNTs deactivation by suppressing agglomeration in the long-term cycle test; (v) the encapsulation of transition metal alloy particles into highly active carbon-based material effectively prevented the erosion and diminished the influence of external solution on the alloy particles thereby improving the stability of P-Fe3Co1@NC/CNTs; in addition, the thinner wall facilitates the transfer of electrons from the environment to the alloy particles through its surface; (vi) P-doping improved the surface structure of carbon-based material, thus increasing the number of surface active sites of P-Fe3Co1@NC/CNTs and promoting its ORR activity. Moreover, the Zn–air battery assembled with P-Fe3Co1@NC/CNTs catalyst exhibits a high open circuit voltage and excellent long-term cycling performance. These highly efficient ORR catalysts with encapsulated alloy particles in a confined space and with the incorporation of heteroatoms, offer a new concept for future clean energy catalyst and new opportunities for the further development of fuel cell.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21906142 and 21876144).References
- O. Z. Sharaf and M. F. Orhan, Renewable Sustainable Energy Rev., 2014, 32, 810–853 CrossRef CAS.
- S. Yasuda, Y. Uchibori, M. Wakeshima, Y. Hinatsu, H. Ogawa, M. Yano and H. Asaoka, RSC Adv., 2018, 8, 37600–37605 RSC.
- S. H. Tai and B. K. Chang, RSC Adv., 2019, 9, 6035–6041 RSC.
- N. Yamada, D. Kowalski, A. Koyama, C. Zhu, Y. Aoki and H. Habazaki, RSC Adv., 2019, 9, 3726–3733 RSC.
- S. Y. Shan, J. Luo, J. F. Wu, N. Kang, W. Zhao, H. Cronk, Y. G. Zhao, P. Joseph, V. Petkov and C. J. Zhong, RSC Adv., 2014, 4, 42654–42669 RSC.
- T. Meng, A. Nsabimana, Z. Y. Liu, H. X. Jia, S. Y. An, H. Wang and Y. F. Zhang, J. Colloid Interface Sci., 2020, 579, 12–20 CrossRef CAS PubMed.
- A. Zadick, L. Dubau, N. Sergent, G. Berthomé and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS.
- C. Y. Su, H. Cheng, W. Li, Z. Q. Liu, N. Li, Z. Hou, F. Q. Bai, H. X. Zhang and T. Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- S. S. Li, W. H. Chen, H. Z. Pan, Y. W. Cao, Z. Q. Jiang, X. N. Tian, X. G. Hao, T. Maiyalagan and Z. J. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 8530–8541 CrossRef CAS.
- B. K. Barman and K. K. Nanda, ACS Sustainable Chem. Eng., 2018, 6, 12736–12745 CrossRef CAS.
- N. Mohammadi-Rad, J. J. Sardroodi and M. D. Esrafili, J. Mol. Graphics Modell., 2020, 100, 107647 CrossRef CAS PubMed.
- T. Y. Ma, J. R. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- W. Lei, Y. P. Deng, G. R. Li, Z. P. Cano, X. L. Wang, D. Luo, Y. S. Liu, D. Wang and Z. W. Chen, ACS Catal., 2018, 8, 2464–2472 CrossRef CAS.
- S. Y. Jing, D. R. Wang, S. B. Yin, J. J. Lu, P. K. Shen and P. Tsiakaras, Electrochim. Acta, 2019, 298, 142–149 CrossRef CAS.
- Z. T. Li, W. N. Zhao, C. Z. Yin, L. Q. Wei, W. T. Wu, Z. P. Hu and M. B. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 44519–44528 CrossRef CAS PubMed.
- H. L. Jiang, Y. H. Zhu, Q. Feng, Y. H. Su, X. L. Yang and C. Z. Li, Chem.–Eur. J., 2014, 20, 3106–3112 CrossRef CAS PubMed.
- J. Q. Chi, W. K. Gao, J. H. Lin, B. Dong, K. L. Yan, J. F. Qin, B. Liu, Y. M. Chai and C. G. Liu, ChemSusChem, 2018, 11, 743–752 CrossRef CAS PubMed.
- K. Hu, Z. H. Xiao, Y. Cheng, D. F. Yan, R. Chen, J. Huo and S. Y. Wang, Electrochim. Acta, 2017, 254, 280–286 CrossRef CAS.
- Y. Wang, W. Chen, Y. Chen, B. Wei, L. H. Chen, L. S. Peng, R. Xiang, J. Li, Z. C. Wang and Z. D. Wei, J. Mater. Chem. A, 2018, 6, 8405–8412 RSC.
- Y. C. Wang, Y. J. Lai, L. Song, Z. Y. Zhou, J. G. Liu, Q. Wang, X. D. Yang, C. Chen, W. Shi, Y. P. Zheng, M. Rauf and S. G. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed.
- C.-L. Zhang, B.-R. Lu, F.-H. Cao, Z.-Y. Wu, W. Zhang, H.-P. Cong and S.-H. Yu, Nano Energy, 2019, 55, 226–233 CrossRef CAS.
- T. Sun, B. B. Tian, J. Lu and C. L. Su, J. Mater. Chem. A, 2017, 5, 18933–18950 RSC.
- M. Q. Wang, C. Ye, M. Wang, T. H. Li, Y.-N. Yu and S. J. Bao, Energy Storage Mater., 2018, 11, 112–117 CrossRef.
- J. L. Long, R. Li and X. L. Gou, Catal. Commun., 2017, 95, 31–35 CrossRef CAS.
- H. J. Meng, X. H Chen, T. L. Gong, H. R. Liu, Y. M. Liu, H. Li and Y. M. Zhang, ChemCatChem, 2019, 11, 6015–6021 CrossRef CAS.
- X. Liu, L. Wang, P. Yu, C. G. Tian, F. F. Sun, J. Y. Ma, W. Li and H. G. Fu, Angew. Chem., Int. Ed., 2018, 57, 16166–16170 CrossRef CAS PubMed.
- P. W. Cai, Y. Hong, S. Q. Ci and Z. H. Wen, Nanoscale, 2016, 8, 20048–20055 RSC.
- J. Deng, L. Yu, D. H. Deng, X. Q. Chen, F. Yang and X. H. Bao, J. Mater. Chem. A, 2013, 1, 14868–14873 RSC.
- H. Hu, Y. H. Xie, F. M. D. Kazim, K. G. Qu, M. Li, Z. K. Xu and Z. H. Yang, Sustainable Energy Fuels, 2020, 4, 5188–5194 RSC.
- A. L. Shen, Y. Q. Zou, Q. Wang, R. A. W. Dryfe, X. B. Huang, S. Dou, L. M. Dai and S. Y. Wang, Angew. Chem., Int. Ed., 2014, 53, 10804–10808 CrossRef CAS PubMed.
- I. Y. Jeon, H. J. Choi, M. J. Ju, I. T. Choi, K. Lim, J. Ko, H. K. Kim, J. C. Kim, J. J. Lee, D. Shin, S. M. Jung, J. M. Seo, M. J. Kim, N. Park, L. Dai and J. B. Baek, Sci. Rep., 2013, 3, 2260 CrossRef PubMed.
- T. Sharifi, G. Z. Hu, X. E. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904–8912 CrossRef CAS PubMed.
- S. Chen, J. Y. Bi, Y. Zhao, L. J. Yang, C. Zhang, Y. W. Ma, Q. Wu, X. Z. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed , 5646.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1–5 Search PubMed.
- H. Yin, C. Z. Zhang, F. Liu and Y. L. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- Y. Y. Wen, C. Ma, Z. T. Wei, X. X. Zhu and Z. X. Li, RSC Adv., 2019, 9, 13424–13430 RSC.
- T. Thanh-Nhan, H. Y. Lee, J. D. Park, T. H. Kang, B. J. Lee and J. S. Yu, ACS Appl. Energy Mater., 2020, 3, 6310–6322 Search PubMed.
- Y. J. Qian, Z. Y. Chen, S. F. Huang and D. X. Li, Integr. Ferroelectr., 2020, 209, 68–75 CrossRef CAS.
- M. T. Lu, Y. W. Li, Y. H. Wu, H. Xu, J. K. Gao and S. Q. Xu, ChemNanoMat, 2020, 6, 1496–1501 CrossRef CAS.
- H. S. Liang, H. Xing, M. Qin and H. J. Wu, Composites, Part A, 2020, 135, 105959 CrossRef CAS.
- T. Dong, X. Zhang, P. Wang, H.-S. Chen and P. Yang, Carbon, 2019, 149, 222–233 CrossRef CAS.
- G. L. Wen, H. J. Niu, J. J. Feng, X. L. Luo, X. X. Weng and A. J. Wang, J. Colloid Interface Sci., 2020, 569, 277–285 CrossRef CAS PubMed.
- L. J. Yang, S. Z. Feng, G. C. Xu, B. Wei and L. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 5462–5475 CrossRef CAS.
- Y. T. Xu, B. L. Chen, J. Nie and G. P. Ma, Nanoscale, 2018, 10, 17021–17029 RSC.
- Z. Liu, B. Tang, X. C. Gu, H. Liu and L. G. Feng, Chem. Eng. J., 2020, 395 Search PubMed.
- W. Y. Gou, J. H. Bian, M. K. Zhang, Z. M. Xia, Y. X. Liu, Y. D. Yang, Q. C. Dong, J. Y. Li and Y. Q. Qu, Carbon, 2019, 155, 545–552 CrossRef CAS.
- A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08768h |
| This journal is © The Royal Society of Chemistry 2021 |