
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVesicular systems for dermal and transdermal drug delivery
Claire Richard,
Stéphanie Cassel and
Muriel Blanzat*
and
Muriel Blanzat*
Laboratoire des Interactions Moléculaires et Réactivité Chimique et Photochimique, UMR 5623, Université Paul Sabatier, 31062 Toulouse Cedex 4, France. E-mail: blanzat@chimie.ups-tlse.fr
First published on 23rd December 2020
Abstract
Dermal/transdermal drug delivery continues to grow in importance as a means of enhancing treatment activity while reducing toxicity by avoiding the systemic absorption of the drug. At the same time, this has led to the adjustment of a wide diversity of drug carriers. This paper begins with a review of the skin, including its structure and the parameters that influence drug diffusion, followed by strategies to improve dermal drug delivery. Of the multitude of existing carriers, we will focus on the most advanced vectors in dermal/transdermal delivery, and in particular, on vesicular systems. This review will present the state of the art as well as the new trends in this domain. Through the description of these systems, we will try to obtain information on the ideal properties that the carrier must have in order to improve the cutaneous and transcutaneous penetration of the drug.
1 Introduction
Since its introduction in the late 1970s, dermal/transdermal drug delivery has been seen as a novel and pain-free route of administering drug treatments, thanks in large part to its non-invasive nature. It reduces the risk of drug overdoses associated with oral administration or injection, while also allowing a satisfactory therapeutic efficacy by avoiding the drug's early metabolization by the liver.1 It is thus well adapted to long-term treatments, as in chronic pain therapies. Dermal administration using transdermal patches has already had a significant influence on a wide variety of therapeutic drugs, particularly in pain treatment2 or for hormonal therapy.3 However, it is still far from being systematically used, as human skin is a complex organ with a protective function,4 which limits the ability of many molecules that have adequate physicochemical characteristics and pharmacokinetic/pharmacodynamic properties from diffusing through the skin.5Many novel techniques to overcome this limitation have thus been developed to improve and control the transport of drugs through the skin.5–7 Technologies used to modify the barrier properties of the stratum corneum can be divided into passive or chemical methods (chemical permeation enhancers,8 drug delivery systems9–12) and active or physical methods (microneedles,13 ultrasounds,14 electroporation,15 or iontophoresis16).
For the past twenty years, most chemical methods developed have focused on the use of skin penetration enhancers to temporarily modify the integrity of the skin barrier by increasing its permeability or by fluidizing its lipid phase.17 However, this strategy suffers from its tendency to irritate the skin.
Drug delivery systems offer a gentler alternative to facilitate passive dermal passage.7,18,19 Such systems were found capable of increasing the residence time of the drug in the stratum corneum and epidermis, while controlling its systemic absorption.
2. The skin: a biological barrier
2.1. Composition of the skin
The skin spreads to a total thickness of 0.5 to 5 mm depending on the body region. It consists of different parallel layers, each with specific natures and functions (Fig. 1A). It is separated into three distinct functional strata, from the deepest to the most superficial: the hypodermis, the dermis, and the epidermis.20 Various types of skin appendages pass through it: the pilosebaceous follicles, which ensure hair growth as well as the sebum secretion, and the sweat glands, which allow water evacuation as a means of maintaining the body temperature.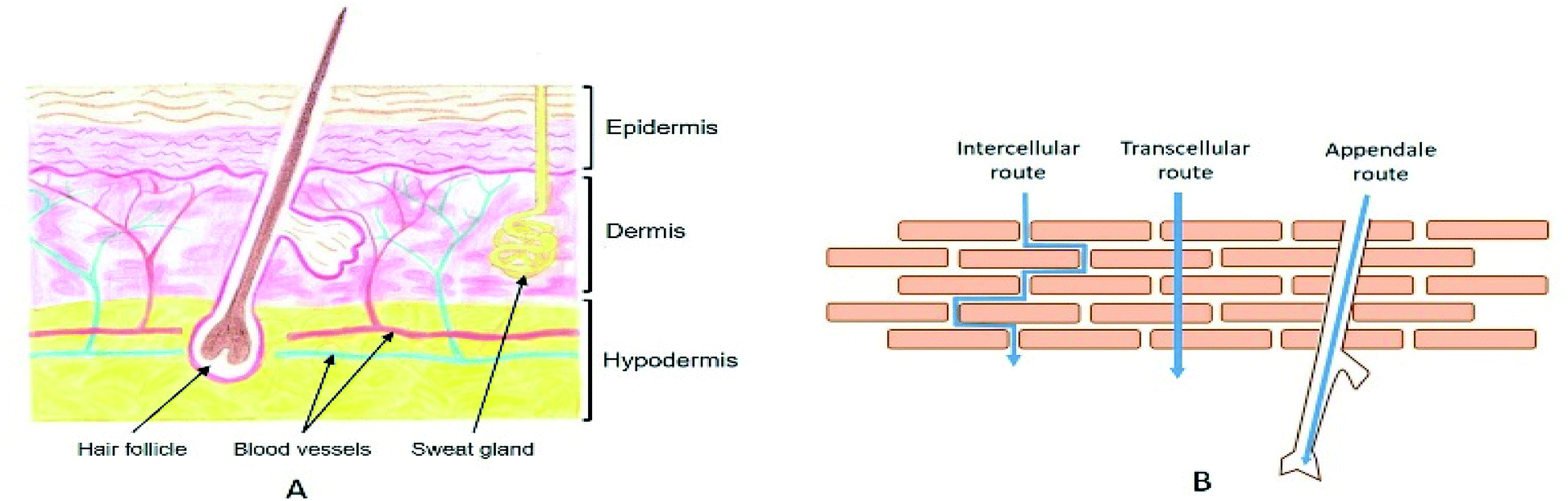 | ||
| Fig. 1 (A) Schematic representation of a skin cross section. Drawing by Valentine Turrin. (B) Illustration of the three different routes for stratum corneum crossing. | ||
The stratum corneum (SC) is the superficial portion of the epidermis. While only 10 μm thick, it plays a determining role in the body. It is a unique biomembrane that serves a barrier function for the skin. It is therefore absolutely essential to understand the SC's structure in order to be able to cross it.
The SC is made of corneocytes surrounded by a lipidic cement following the “brick-mortar” principle, which gives the skin its mechanical resistance. It consists of 5 to 20 layers of “bricks”, the corneocytes, which are dead cells made of keratin and filaggrin filaments. The corneocytes are organized in clusters, separated by channels of a few microns. The cells are held together by a protein structure, called corneodesmosome, which give the corneocytes a high structural stability. It is the cleavage of these proteins, under the action of proteases, which is at the origin of the phenomenon of desquamation of the first layers, by separating the cells of the network. Then, these “bricks” are included in an extracellular lipid matrix that forces their alignment. This lipidic matrix is mainly composed of ceramides, free fatty acids, and cholesterol.21–23 The combination of the different lipids leads to their self-association in multilayers, formed by the repetition of lipophilic and hydrophilic planes parallel to the cells' surface. These multilayers play a critical role in the barrier function that makes the SC waterproof. However, the elucidation of their complex structure, facilitated in recent years by analytical techniques, remains a challenge for the scientific world. Variability of the composition and behavior phase within the thickness of the SC makes the task all the more arduous.
2.2. Routes for the stratum corneum crossing
When a drug is applied to the surface of the skin, the first and hardest step consists in crossing the SC. There are three major routes to go through the upper layers (Fig. 1B). The first one consists of using the different annexes that cross the skin to the dermis, also known as the transannexial/appendale route. The other two routes cross the SC, either through the corneocytes (transcellular route), or between corneocyte cells through the lipidic cement (intercellular route).It is noteworthy that penetration of external molecules into the skin takes place simultaneously by these three parallel penetration pathways. However, their relative contribution can be largely dependent on the physicochemical characteristics of the drug. Knowing that it is generally impossible to select its characteristics according to the intended application, it is necessary to develop strategies that will promote skin transport.
2.3. Physicochemical parameters of skin diffusion
Skin presents a number of obstacles to drug transport. However, it is not impassable and some molecules can diffuse passively. It is therefore important to understand the absorption process and the characteristics that allow a substance to penetrate.Thus, each stage of penetration has its own kinetics and depends on many factors (Fig. 2).
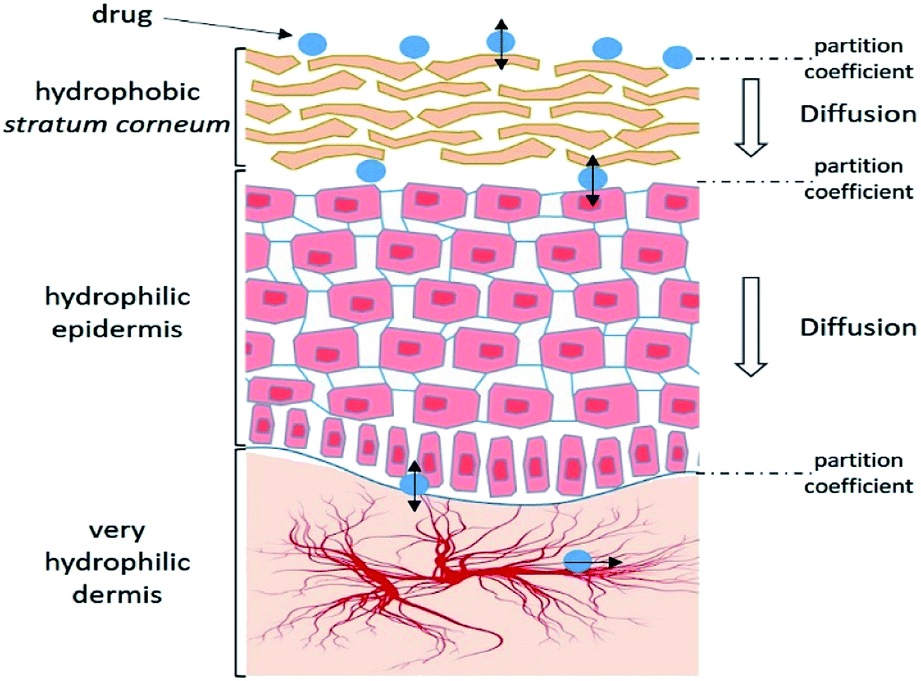 | ||
| Fig. 2 Schematic representation of the different steps of the cutaneous absorption mechanism. Adapted from ref. 25. | ||
The absorption of external molecules is mainly controlled by passive diffusion. So, the stationary state of diffusion can be defined by a simplified Fick's law:
| J = DkΔc/h | (1) |
Thus, the combinations are infinite. The study of the effects of these different parameters makes it possible to define the ideal physicochemical parameters of a permeant.20,27
2.3.2.1 Molecular size. The diffusion coefficients are defined by the Stokes–Einstein equation. They are inversely proportional to the radius of the diffuser. Thus, the smaller the penetrating molecule, the more its absorption is facilitated. It is generally accepted that it must not exceed a mass of 500 Da.
2.3.2.2 Affinities. The drug must be able to diffuse from its vehicle into the SC—an extremely hydrophobic environment—and then into the viable epidermis that contains 70% water. Therefore, the drug must present a hydrophilic/hydrophobic balance, which corresponds to an n-octanol/water partition coefficient (log
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) between 1 and 3.28
P) between 1 and 3.28
2.3.2.3 Solubility. The solubility in the SC intercellular lipids is influenced by the drug melting point, which should ideally be below 200 °C. Moreover, solubility in water is defined by the drug's ability to form hydrogen bonds. All of these settings help classify good permeants.29
2.3.2.4 Ionization. Penetration also depends on the degree of ionization of the species. Due to their low log
P, ionic molecules have in general a lower permeability coefficient than the associated nonionic species. It is therefore more advantageous to use the acid form or the free base. However, the charged molecule can instead increase its solubility in hydrophilic tissues. It is then possible to play on the skin pH gradient to maximize absorption.If the drug does not meet all of these criteria, which is the most common case, it is necessary to find ways to modify its skin penetration profile.
2.4. Strategies to improve dermal drug delivery
Before designing a strategy to improve drug dermal penetration, it is first necessary to identify the real target of the active molecule. In certain formulations such as insect repellents or sunscreens, the drug has to be effective in the SC. In other applications, such as with anti-inflammatory, antibiotic, or anesthetic drugs, the desired effect needs to take place in the viable epidermis. In all cases, we will speak of dermal transport. However, when the drug is supposed to cross all layers of the skin to reach the vascular network of the dermis and be distributed by the blood circulation, we will speak of transdermal delivery. This distinction makes all the difference as to the strategies to adopt to bring the molecule to its target.30Three distinct approaches exist for improving dermal penetration. First, some strategies are based on a simple improvement in passive permeation, mostly using permeation enhancers;31 second, the integrity of the SC can be damaged32 using physical techniques (microneedles,13 electroporation,15 iontophoresis16 or sonophoresis14) in order to reduce the barrier effect of the SC.
These strategies are well adapted for transdermal administration.33 But the last and least damaging strategy, which does not permanently impair the barrier properties, consists in using nanovectors to temporarily improve the dermal drug penetration. In this review, we will focus on dermal delivery systems with nanovectors and especially vesicles.
3 Dermal drug delivery with vesicular systems
As previously mentioned, nanovectors are of great interest in the dermal delivery field because they are mostly non-toxic and offer control on drug bioavailability.34 Moreover, they can be used both as permeation enhancers and as local containers for the sustained delivery of drugs via the skin.35 They can also increase the drug residence time in the stratum corneum and epidermis, while reducing its systemic absorption.36,37Nanovectors are colloidal systems whose size is less than 1 μm.38 By enclosing the drug, these vectors make it possible to increase its stability, to pass the biological barriers to reach its therapeutic target, but also to protect the body against its possible toxicity. While research was focused a few years ago on oral and parenteral administration, more studies are now widely focused on topical applications. Of the multitude of existing systems, we will focus, in a non-exhaustive manner, on the most advanced vectors in dermal/transdermal delivery and in particular on vesicular systems (systems in which the active ingredient is enclosed in a cavity delimited by a membrane). A number of matrix systems are also developed to enhance dermal penetration,39 but we will not discuss them here. Generally, matrix systems include those systems made of three-dimensional networks formed by polymers,12,40 surfactants or dendrimers, and in which active principles are trapped, as for instance emulsions,41,42 hydrogels,43 dendrimers,44 nanospheres,45,46 and solid lipid particles.47–49
Vesicles are defined as colloidal soft matter vectors containing an aqueous or an oily heart, separated from the external environment by a membrane, made of phospholipids, polymers, or surfactants.12 Depending on its affinities, the drug can be dispersed in the internal cavity and/or be included in the membrane. In addition, the carrying capacity of vesicles is generally greater than that of matrix systems, making it a vector of choice for limiting the quantity of exogenous molecules in the body. Moreover, by varying the nature of the membrane to obtain the appropriate physicochemical properties, vesicles could in particular succeed in crossing the skin barrier to improve the distribution of the encapsulated drug in the deep layers. In fact, the drug delivery ability of vesicles is largely influenced by their physicochemical characteristics, particularly those of the membrane. We will therefore describe the different types of vesicles according to the family of molecules that comprise them.
3.1. Nanocapsules and polymersomes
Nanocapsules are colloidal particles of less than 1 μm in size. They are made of a very thin polymer shell, surrounding a liquid core (Fig. 3). The medium in the cavity is oily in most cases (nanocapsules), but there are specific preparation methods for obtaining a hydrophilic core (polymersomes).50 In both systems, the active molecule is encapsulated as a liquid, a solid, or even as a molecular dispersion. The nanocapsules can also be obtained by the layer-by-layer method, giving rise to polyelectrolyte nanocapsules.51,52 In this case, oppositely charged polyelectrolytes are adsorbed at the surface of a template which is finally dissolved.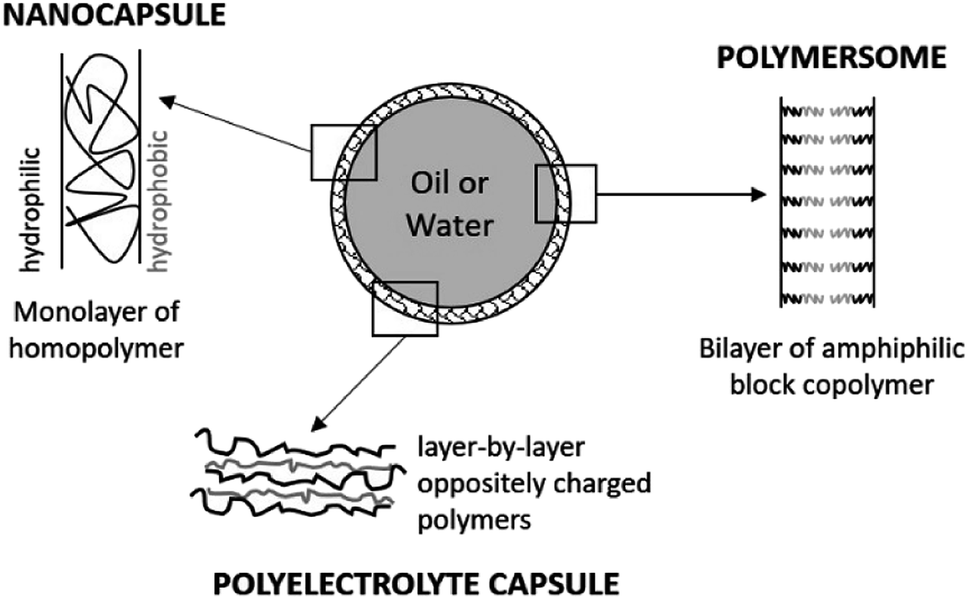 | ||
| Fig. 3 Structure of nanocapsules and polymersomes. | ||
Ideally, the polymer used should be biodegradable and non-toxic, whether natural or synthetic. Literature reports the wide use of poly (ε-caprolactones) and poly (D, L-lactides) for dermal applications of nanocapsules.53–55 The core is generally made of triglycerides with the dispersed drug. It is also possible to find sorbitan monostearate, or even other additives providing an intrinsic effect (turmeric oil for its antibacterial and antioxidant action, for example).
Nanocapsules are most often stabilized by poly(ethylene glycol) derivatives, such as poloxamers, phospholipids, and possibly poly(vinyl alcohol).56,57 To avoid the stabilizers, it is possible to form nanocapsules based on block copolymers. Their outwardly pointing hydrophilic parts provide the necessary hydrophilicity and hindrance to the surface of the particle.58
Two different types of preparation methods can be used. If the polymer is not preformed, the nanocapsules are synthesized by interfacial polymerization. This method requires the prior formation of a nanoemulsion, which serves as a template for the future nanoparticles. If the polymer is preformed, the nanocapsules are obtained by precipitation on the surface of oil droplets. For this, the polymer must be insoluble both in water and in the oily core.58
The potential of nanocapsules for dermatological or cosmetic uses was evaluated in the early 1990s. Their first application in a commercial product was developed by L'Oréal in 1995, to encapsulate active ingredients such as vitamins A, C, E and beta-carotene.59
Studies on this subject have multiplied for the last ten years. After topical application, nanocapsules have been shown to form a thin film on the skin due to the evaporation of water. This film ensures prolonged delivery and acts as a reservoir of drug for the skin. A positive surface charge of the particles can promote bioadhesiveness and increase the phenomenon.58
These properties are particularly interesting in the field of sun protection. For this application, UV screens should be kept in the first layers of the SC to absorb radiation and prevent skin damage. Alvarez-Romá et al.60 were the first to encapsulate the lipophilic dioctyl methoxycinnamate (OMC) sunscreen into nanocapsules. The nanoencapsulation has shown an improvement in skin retention and an absence of penetration of organic sunscreens such as OMC or benzophenone-3, or even TiO2. Being isolated from potential reactive species in their environment, their photostability and their ability to block UV were also optimized.61 Other applications for nanocapsules include disinfection with the encapsulation of chlorhexidine and the local delivery of indomethacin or minoxidil.62
3.2. Liposomes
The most historically studied type of vesicles is the liposome.63 It is made of phospholipids which spontaneously self-associate in flat bilayers. When dispersed in water, liposomes form vesicles enclosing an aqueous core (Fig. 4).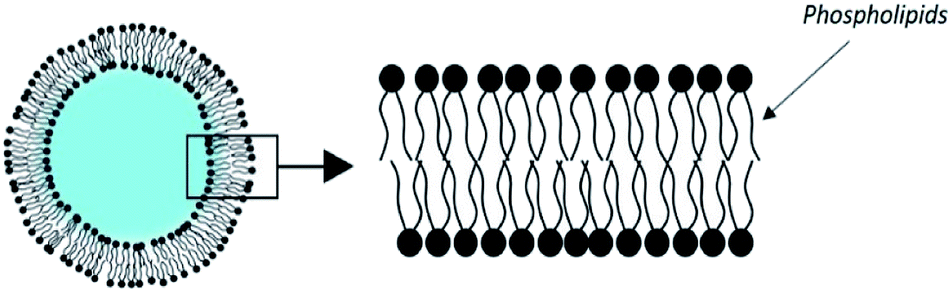 | ||
| Fig. 4 Structure of a liposome. | ||
Among the variety of existing phospholipids, phosphatidyl-cholines are most commonly used for the production of liposomes. Most often cholesterol is added to it. It does not intrinsically form a bilayer, but the cholesterol intercalates between the phospholipid molecules. The defects it causes make the membrane more rigid, which reduces the permeability to water-soluble molecules. It also increases the stability of objects in the presence of biological fluids.64
There are many methods for preparing liposomes, but they generally follow three main steps:
(i) dissolution of the phospholipids and possibly hydrophobic drug in an organic solvent, which is then evaporated;
(ii) redispersion, most often by sonication, in the aqueous phase possibly containing the water-soluble drugs.
(iii) Purification of the resulting liposomes, generally to reduce their polydispersity.65
The size of the liposomes can vary from 25 nm to 2.5 μm and can contain one or more concentric phospholipid bilayers. Depending on the method of preparation, it is possible to obtain multilamellar vesicles (MLV), small or large unilamellar vesicles (SUV or LUV), and even multivesicular vesicles containing several liposomes within a single bilayer.66
Of interest to researchers around the world since the 1970s, these systems have naturally found applications in topical administration. Thus, MLVs are already used to encapsulate heparin, sodium diclofenac, and iodides within their aqueous heart, for dermal delivery applications.38 However, the ability of liposomes to exceed the first layers of the SC is largely questioned, which makes them unsuitable for transdermal transport.63 According to the various studies carried out, it would seem that the thermodynamic state of the membrane is of the greatest importance for the passage of this biological barrier. Microscopic observation of skins treated with rigid vesicles, whose carbon chains are in the gel state, has shown their presence only on the surface. However, the use of vesicles with fluid and elastic bilayers results in the presence of the encapsulated fluorescent probe in the extracellular matrix and in the deeper layers. This result confirms that skin penetration is greater with flexible vesicles.67 Therefore, researchers have developed a large number of liposome derivatives to obtain such properties.68
3.3. Ethosomes
Adding up to 50% of an alcohol to the aqueous phase of liposomes has shown a substantial improvement in dermal transport. This concept was elaborated by Touitou et al. in the late 1990s under the name of ethosome (Fig. 5).69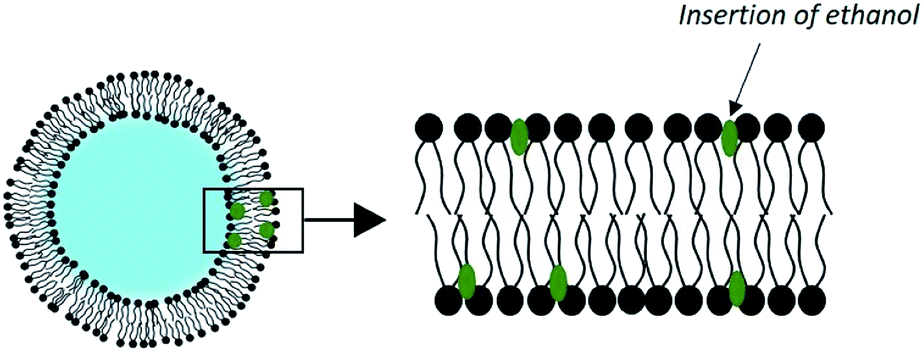 | ||
| Fig. 5 Structure of an ethosome. | ||
Their method of preparation is simpler than for conventional liposomes. It is sufficient to dissolve the phospholipids and the drug at 30 °C in ethanol with vigorous stirring and then to add the water isothermally. No solvent removal step is necessary.
Numerous studies have demonstrated ethosomes' capacity to vectorize both hydrophilic and hydrophobic drugs for topical applications. In addition, they have the advantage of being efficient, regardless of the state of hydration of the skin. Therefore, they can be used in occlusive conditions.70
This effectiveness can be explained by the synergy existing between ethanol and the phospholipid vesicles. First, ethanol is intrinsically a penetration enhancer and induces a local alteration of the extracellular matrix of the SC. Second, ethanol has an effect on the physicochemical characteristics of ethosomes. It induces a negative surface charge and a reduction in the size of vesicles, as observed in a study on the encapsulation of trihexyphenidyl HCl. The ethosomes had an average diameter of 109 nm, compared to 324 nm for the corresponding liposomes.71 In addition, the insertion of ethanol into the membrane of the vesicles allows for disruption of the phospholipids' order, which lowers the phase transition of their carbon chains. The result is vesicles with very fluid and easily deformable membranes, which slide through the disturbed SC to release their charge in the deep layers of the skin.72 This delivery system is already commercialized in various anti-cellulite products.73 The performance of skin drug delivery systems such as ethosomes has motivated the development of other types of vesicles taking advantage of the synergistic effect of chemical penetration promoters. They are called PEV for “Penetration Enhancer-containing Vesicles”. Studied compounds include oleic acid, limonene, propylene glycol, glycerol, and transcutol.74
3.4. Transfersomes
Since 1992, Cevc and his collaborators have developed liposomes which they qualify as ultra-deformable vesicles.75 Patented under the name of transfersomes, these are phospholipid vesicles to which is added an adjuvant called ‘edge activator’ (EA), a surfactant or a single-chain lipid, up to a limit of 25% (Fig. 6).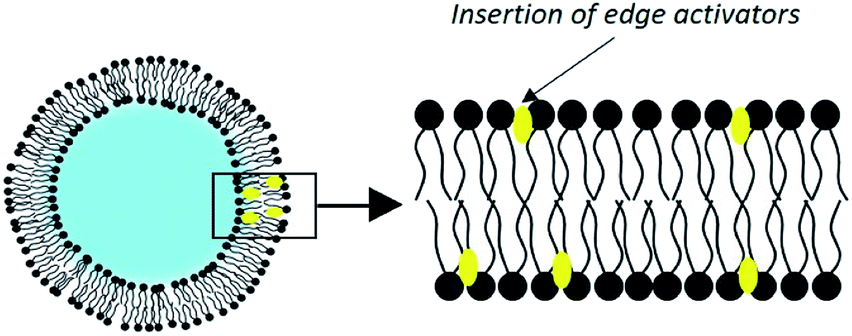 | ||
| Fig. 6 Structure of a transfersome. | ||
By far the most widely used edge activator is sodium cholate, but similar properties are seen with deoxycholate as well as with some Spans® and Tweens®. These molecules have a single fatty chain and form objects with a large radius of curvature.70 Their introduction into the liposomes creates defects in the organization of the membranes. These discontinuities in the order of phospholipids make the bilayer less rigid.
Edge activators give transfersomes their ultra-deformable properties. Indeed, during a destabilization event, the edge activators accumulate at the pressure points of the vesicle because of their affinity for curved configurations. Changing the shape of the liposome therefore requires less energy. Deformability is maximum when the membrane attempts to optimize its local composition in response to an anisotropic external stress.76
Numerous studies have demonstrated the excellent performance of this type of flexible vesicles in the dermal and transdermal delivery of drugs.63 Thanks to its flexibility, its inventors affirm that the transfersome is capable of flattening to pass through the fine pores of the SC. However, this requires an osmotic gradient, which are non-occlusive conditions, to serve as a driving force.
The hydrophilicity of phospholipids pushes them to avoid dry environments. So in order to remain swollen after skin application and vehicle evaporation, the vesicles must follow the local hydration gradient, navigating towards the deeper layers of the skin.77 This hypothesis is corroborated by the reduction in their performance if the application is occlusive, that is to say with an increased hydration of SC.78 Thus, transfersomes have been used successfully for the transdermal delivery of certain model drugs, such as for transdermal immunization.79
3.5. Niosomes
Niosomes have a structure similar to liposomes but their membranes are made of one or more nonionic surfactants and cholesterol (Fig. 7). | ||
| Fig. 7 Structure of a niosome. | ||
They have the advantage of being able to encapsulate a wide variety of hydrophilic and lipophilic drugs, while being much cheaper and more stable than liposomes. In fact, surfactants are generally less expensive than pure phospholipids and are more resistant to hydrolytic degradation.74
Niosomes serve as a reservoir system and the release kinetics can be modified by changing their composition. For pharmaceutical applications, the surfactants chosen should be biocompatible, biodegradable, non-immunogenic, and non-carcinogenic. They include polyglycerol alkyl ethers, glucosyldialkyl ethers, crown ethers or some Tween®, but the most commonly used are Brij® and Spans®.80 Cholesterol or its derivatives are generally included in a 1:1 molar ratio, as a membrane stabilizer that prevents aggregation. Stabilization can be further increased by adding charged molecules of the niosome composition. These additives may provide electrostatic repulsion between objects.
The physicochemical properties of the surfactants used in the composition of niosomes, and in particular the hydrophilic lipophilic balance (HLB), are very important. They determine the niosome's size, encapsulation efficiency, and drug release kinetics and can also influence the level of interactions between the vesicles and the skin.81 Most of the preparation methods involve the hydration of the surfactants mixture at high temperature, followed by a reduction in the size of the vesicles, which is initially the order of a micron. The use of sonication or extrusion results in vesicles around 100–200 nm diameter and can reach 50–100 nm diameter with high pressure homogenization or microfluidization. However, if the smaller particles are more likely to penetrate the SC, it is at the expense of stability and encapsulation capacity.82
Different studies show that niosomes induce an increase in the residence time of the drug in the epidermis, and improve their penetration while reducing systemic absorption. However, direct permeation of niosomes in the viable epidermis is limited. They are mainly located in the SC where they act as a penetration enhancer.
The predominant mechanism would be an increase in the fluidity of intercellular lipids, which reduces the barrier effect of the epidermis. It has also been suggested that niosomes dissociate or merge in the SC to form weakly bound aggregates which could penetrate more deeply.83 Furthermore, their ability to promote drug permeation has been attributed to their flexibility. When the cholesterol composition is modified to increase the fluidity of the membrane, a better retention of the drug is observed.82
Many researchers have studied the potential of niosomes as innovative vectors for percutaneous drug delivery. The variety of drugs used is very large, including 5-fluorouracil, aceclofenac, tretinoin, and estradiol.84 The addition of ethanol to the composition results in the formation of niosomes qualified as elastic because they have a fluid membrane. The encapsulation of diclofenac diethylammonium,85 papain,86 and sodium sulfadiazine87 show the ability of this type of vector to increase the flow of drug through the skin for an ethanol content up to 20%. Despite the abundant research on niosomes for topical delivery of drugs, few studies have led to patents and commercial products. However, we note the development of niosomal formulations of anti-aging active ingredients by Lancôme and L’Oréal. Niosomal gels of urea and griseofulvin have also reached the clinical trial stage.84
3.6. Catanionic vesicles
As an alternative to liposomes, catanionic vesicles are obtained by spontaneous self-assembly of catanionic surfactants (Fig. 8) in the absence of applied external forces.88–90 Catanionic surfactants are amphiphilic systems resulting from the mixing of oppositely charged surfactants in water in which the inorganic counter-ions associated to the amphiphiles are eliminated. These are obtained in few steps with different simple methods: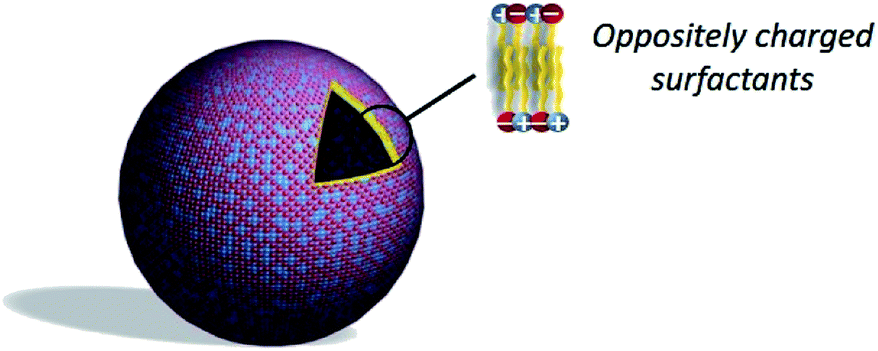 | ||
| Fig. 8 Structure of a catanionic vesicle. | ||
(i) extraction of the catanionic surfactant in an organic solvent, the salts remaining in the aqueous phase;
(ii) precipitation of the catanionic surfactant out of a supersaturated aqueous solution of the oppositely charged surfactants, with the salts remaining in the aqueous phase;
(iii) ion exchange, which consists of changing the cationic surfactant into its hydroxylated form using an ion exchange resin;91
(iv) proton exchange between equimolar quantities of an amphiphilic acid and an amphiphilic amine in water.
In this last procedure no residual salts are generated.90,92
These catanionic surfactants are known for their ability to spontaneously form vesicles in water,88,93,94 but they can precipitate when they are prepared in an equimolar ratio of both surfactants.95 Catanionic vesicles are thus generally formed with an excess of either positive or negative charge, as a loss of solvation of the polar head is observed with the ion-pairing. However, the use of sugar-based amphiphiles can enhance the water solubility of catanionic mixtures90,92,96 and therefore lead to vesicles in a large domain of concentration for equimolar systems without any excess of cationic or anionic surfactant.
Catanionic vesicles show good stability and dispersion properties in aqueous phase89,92 that make them potential candidates for drug delivery.12 But despite their many advantages, relatively little work is dedicated to the design of catanionic vesicles for dermal drug delivery. Ethosome-like catanionic vesicles were developed with commonly used decyltrimethylammonium and dodecyl sulfate and ethanol as a cosolvent.97 The potential application in dermal delivery of these ethosome-like catanionic vesicles was validated with the encapsulation of α-tocopherol.98
Catanionic vesicles made of biomimetic lactose-derived amphiphiles have also proved capable of delivering various hydrophobic drugs in the living epidermis of the skin.99 As previously observed in literature with transfersomes or ethosomes, the deformability and/or the fluidity of vesicular vectors can strongly improve dermal or transdermal drug delivery. In the present case, the catanionic vesicles are fluid at skin temperature, thus improving dermal penetration and drug delivery in general.100,101 The incorporation of catanionic vesicles in gels for dermal administration was also studied,102–104 showing the great stability of the vesicles and the input of the gels for an improved sustained delivery.102
Like niosomes, catanionic vesicles can be built from a large variety of surfactants. In particular, one of the partner of the ion-pairing can be an amphiphilic drug, which then participates to its own formulation. For instance, this strategy was successfully validated on antihistamines associated to SDS105 or fatty acids,106 and with non-steroidal anti-inflammatory drugs (NSAID) associated with sugar-derived surfactants,10 resulting in an improved anti-inflammatory activity of the NSAID in vivo, thanks to a sustained diffusion through the skin.107
4 Conclusions
Despite the different and widespread proof of their activity, the mode of action of the various vesicular systems is still the subject of controversy today. Indeed, the mechanisms by which the vectors promote the drug penetration in the epidermis are relatively difficult to investigate, particularly in vivo. Two main modes of action are proposed. The first hypothesis is that the vesicle remains intact and serves as a vehicle for the drug to deliver enter the skin. And even if it does not reach the dermis, it would serve as a reservoir within the epidermis.The second hypothesis is that the vesicle, initially protecting the drug, only acts as a promoter of penetration through the SC. Thus, the components of the vector disturb the lipid ultrastructure by dissolving into the extracellular matrix. The diffusion of active molecules, which cross along the different skin layers, is improved.
On the one hand, it seems that this second mechanism is adopted by the vesicles in the fluid state. In fact, changes in the deep layers of the SC have been observed for fluid liposomes as well as for niosomes. Their components (lipids or nonionic surfactants) have probably succeeded in penetrating these layers by altering the lipidic lamellar structure. On the other hand, vesicles in the gel state do not even penetrate the skin. They have been shown to adsorb and fuse on the skin's surface.67
Despite their undeniable qualities, the interactions of ultra-deformable vesicles with the skin is one of the most debated questions in dermal research. To date, the exact mode of action and the nature of transport in this type of vesicles is still not fully understood. Researchers working on transfersomes advocate that these vesicles remain intact while crossing the skin to the bloodstream.108 However, other experts refute this hypothesis.109 Evidence for changes in the lipid structure of the SC after treatment with transfersomes has been provided by Duangjit et al.110 However, nothing indicates a permeation of the vesicles without alteration of their structure. Different works suggest a sharing in the SC followed by a release of the drug at the junction of the viable epidermis.63 The multiplicity of parameters in these studies – composition and preparation of vesicles, administration, different skin types – probably explains the contradictions of results. Thus, the degree of permeation of intact vesicles through the SC, while also their sharing in the viable epidermis, remains to be determined in detail.79
As we have seen, the different types of vesicles can have opposite skin penetration capacities depending on their physicochemical characteristics. Nanocapsules or rigid liposomes do not seem to penetrate the SC but merge on the surface of the skin. On the contrary, flexible vesicles such as transfersomes, ethosomes, or catanionic vesicles promote the dermal diffusion of the encapsulated drug. Thus, these two families of carriers can find applications in different suitable fields. The liposomes and nanocapsules can be used as reservoir systems for the prolonged dermal delivery of drugs in which no systemic effect is desired. Deformable or fluid vesicles can be considered for dermal and transdermal diffusion of active substances of all polarities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CNRS, MENESR and Toulouse University are acknowledged for institutional funding.References
- S. M. Pond and T. N. Tozer, Clin. Pharmacokinet., 1984, 9, 1–25 CrossRef CAS.
- L. Peng, H. Y. Zheng and Y. Dai, Braz. J. Med. Biol. Res., 2019, 52(11), e8567–71 CrossRef CAS.
- N. W. Kopper, J. Gudeman and D. J. Thompson, Drug Des., Dev. Ther., 2009, 2, 193–202 Search PubMed.
- S. H. Lee, S. K. Jeong and S. K. Ahn, Yonsei Med. J., 2006, 47, 293–306 CrossRef CAS.
- M. R. Prausnitz, S. Mitragotri and R. Langer, Nat. Rev. Drug Discovery, 2004, 3, 115–124 CrossRef CAS.
- K. S. Paudel, M. Milewski, C. L. Swadley, N. K. Brogden, P. Ghosh and A. L. Stinchcomb, Ther. Delivery, 2010, 1, 109–131 CrossRef CAS.
- P. Carter, B. Narasimhan and Q. Wang, Int. J. Pharm., 2019, 555, 49–62 CrossRef CAS.
- B. W. Barry, Nat. Biotechnol., 2004, 22, 165–167 CrossRef CAS.
- H. Schreier and J. Bouwstra, J. Controlled Release, 1994, 30, 1–15 CrossRef CAS.
- S. Consola, M. Blanzat, E. Perez, J.-C. Garrigues, P. Bordat and I. Rico-Lattes, Chem. – Eur. J., 2007, 13, 3039–3047 CrossRef CAS.
- M. Paulsson and K. Edsman, J. Pharm. Sci., 2001, 90, 1216–1225 CrossRef CAS.
- E. Soussan, S. Cassel, M. Blanzat and I. Rico-Lattes, Angew. Chem., Int. Ed., 2009, 48, 274–288 CrossRef CAS.
- K. Cheung and D. B. Das, Drug Delivery, 2016, 23, 2338–2354 CrossRef CAS.
- D. Park, H. Park, J. Seo and S. Lee, Ultrasonics, 2014, 54, 56–65 CrossRef CAS.
- K. Ita, Pharmaceutics, 2016, 8, 9 CrossRef.
- D. R. Kalaria, S. Dubey and Y. N. Kalia, in Transdermal and topical drug delivery: principles and practice, ed. H. A. E. Benson and A. C. Watkinson, Wiley, Hoboken, NJ, 2012, pp. 67–83 Search PubMed.
- V. R. Sinha and M. P. Kaur, Drug Dev. Ind. Pharm., 2000, 26, 1131–1140 CrossRef CAS.
- C. Sinico, M. Manconi, M. Peppi, F. Lai, D. Valenti and A. M. Fadda, J. Controlled Release, 2005, 103, 123–136 CrossRef CAS.
- M. J. Choi and H. I. Maibach, Skin Pharmacol. Physiol., 2005, 18, 209–219 CrossRef CAS.
- H. A. E. Benson, in Transdermal and topical drug delivery: principles and practice, ed. A. C. Watkinson and H. A. E. Benson, Wiley, Hoboken, NJ, 2012, pp. 3–22 Search PubMed.
- A. V. Rawlings and P. J. Matts, J. Invest. Dermatol., 2005, 124, 1099–1110 CrossRef CAS.
- R. K. Freinkel and T. N. Traczyk, J. Invest. Dermatol., 1985, 85, 295–298 CrossRef CAS.
- P. M. Elias, J. Invest. Dermatol., 2012, 132, 2131–2133 CrossRef CAS.
- C. Lafforgue and J.-P. Marty, in Annales de dermatologie et de Vénéréologie, Elsevier Masson, 2007, vol. 134, pp. 18–23 Search PubMed.
- A. Cortial, PhD Thesis, Université Claude Bernard-Lyon I, 2015.
- J. Hadgraft, Int. J. Pharm., 2001, 224, 1–18 CrossRef CAS.
- M. E. Lane, P. Santos, A. C. Watkinson and J. Hadgraft, in Transdermal and topical drug delivery: principles and practice, ed. H. A. E. Benson and A. C. Watkinson, Wiley, Hoboken, NJ, 2012, pp. 23–42 Search PubMed.
- J. Hadgraft, Eur. J. Pharm. Biopharm., 2004, 58, 291–299 CrossRef CAS.
- B. M. Magnusson, W. J. Pugh and M. S. Roberts, Pharm. Res., 2004, 21, 1047–1054 CrossRef CAS.
- J. Hadgraft, in Modified-release drug delivery technology, ed. M. J. Rathbone, J. Hadgraft and M. S. Roberts, Marcel Dekker, New York, 2003, pp. 471–479 Search PubMed.
- T. Haque and M. M. U. Talukder, Adv. Pharm. Bull., 2018, 8, 169–179 CrossRef CAS.
- A. Alkilani, M. T. McCrudden and R. Donnelly, Pharmaceutics, 2015, 7, 438–470 CrossRef CAS.
- N. Akhtar, V. Singh, M. Yusuf and R. A. Khan, Biomed. Eng. Biomed. Tech, 2020, 65(3), 243–272 Search PubMed.
- R. Agarwal, O. P. Katare and S. P. Vyas, Int. J. Pharm., 2001, 228, 43–52 CrossRef CAS.
- P. Karande and S. Mitragotri, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 2362–2373 CrossRef CAS.
- R. R. Boinpally, S. L. Zhou, S. Poondru, G. Devraj and B. R. Jasti, Eur. J. Pharm. Biopharm., 2003, 56, 389–392 CrossRef CAS.
- C. Puglia, D. Trombetta, V. Venuti, A. Saija and F. Bonina, J. Pharm. Pharmacol., 2004, 56, 1225–1232 CrossRef CAS.
- R. H. H. Neubert, Eur. J. Pharm. Biopharm., 2011, 77, 1–2 CrossRef CAS.
- S. Pappinen and A. Urtti, in Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers, ed. N. Dragicevic and H. I. Maibach, Springer, 2016, pp. 253–262 Search PubMed.
- Z. Davoudi and Q. Wang, in Imaging Technologies and Transdermal Delivery in Skin Disorders, John Wiley & Sons, Ltd, 2019, pp. 147–168 Search PubMed.
- C. Nastiti, T. Ponto, E. Abd, J. Grice, H. Benson and M. Roberts, Pharmaceutics, 2017, 9, 37 CrossRef.
- N. Salim, N. Ahmad, S. H. Musa, R. Hashim, T. F. Tadros and M. Basri, RSC Adv., 2016, 6, 6234–6250 RSC.
- I. P. Harrison and F. Spada, Pharmaceutics, 2018, 10, 71–83 CrossRef.
- M. Sun, A. Fan, Z. Wang and Y. Zhao, Soft Matter, 2012, 8, 4301 RSC.
- T. Ramezanli, B. E. Kilfoyle, Z. Zhang and B. B. Michniak-Kohn, Int. J. Pharm., 2017, 516, 196–203 CrossRef CAS.
- J. Akbuga, S. Ozbas-Turan and C. Ekentok, in Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers, ed. N. Dragicevic and H. I. Maibach, Springer, 2016, pp. 337–351 Search PubMed.
- Y. Zhai and G. Zhai, J. Controlled Release, 2014, 193, 90–99 CrossRef CAS.
- A. Garcês, M. H. Amaral, J. M. Sousa Lobo and A. C. Silva, Eur. J. Pharm. Sci., 2018, 112, 159–167 CrossRef.
- Y. Duan, A. Dhar, C. Patel, M. Khimani, S. Neogi, P. Sharma, N. Siva Kumar and R. L. Vekariya, RSC Adv., 2020, 10, 26777–26791 RSC.
- D. Sarkar, J. El-Khoury, S. T. Lopina and J. Hu, Macromolecules, 2005, 38, 8603–8605 CrossRef CAS.
- G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Möhwald, Polym. Adv. Technol., 1998, 9, 759–767 CrossRef CAS.
- G. Cheng, C. Yin, H. Tu, S. Jiang, Q. Wang, X. Zhou, X. Xing, C. Xie, X. Shi, Y. Du, H. Deng and Z. Li, ACS Nano, 2019, 13, 6372–6382 CrossRef CAS.
- A. T. Rangari, Asian J. Biomed. Pharm. Sci., 2015, 05, 05–12 CrossRef CAS.
- H. H. Joo, H. Y. Lee, Y. S. Guan and J.-C. Kim, J. Ind. Eng. Chem., 2008, 14, 608–613 CrossRef CAS.
- Z. Teixeira, C. A. Dreiss, M. J. Lawrence, R. K. Heenan, D. Machado, G. Z. Justo, S. S. Guterres and N. Durán, J. Colloid Interface Sci., 2012, 382, 36–47 CrossRef CAS.
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Int. J. Pharm., 2010, 385, 113–142 CrossRef CAS.
- Y. Zhan, W. Zeng, G. Jiang, Q. Wang, X. Shi, Z. Zhou, H. Deng and Y. Du, J. Appl. Polym. Sci., 2015, 132, 41496 CrossRef.
- F. S. Poletto, R. C. R. Beck, S. S. Guterres and A. R. Pohlmann, in Nanocosmetics and Nanomedicines, Springer, Berlin, Heidelberg, 2011, pp. 49–68 Search PubMed.
- J. A. Schwarz, C. I. Contescu and K. Putyera, Dekker Encyclopedia of Nanoscience and Nanotechnology, CRC Press, 2004 Search PubMed.
- R. Alvarez-Román, G. Barré, R. H. Guy and H. Fessi, Eur. J. Pharm. Biopharm., 2001, 52, 191–195 CrossRef.
- C. B. Detoni, K. Paese, R. C. Beck, A. R. Pohlmann and S. S. Guterres, in Nanocosmetics and Nanomedicines, Springer, 2011, pp. 333–362 Search PubMed.
- S. S. Guterres, M. P. Alves and A. R. Pohlmann, Drug Target Insights, 2007, 2, 117739280700200000 CrossRef.
- M. Sala, R. Diab, A. Elaissari and H. Fessi, Int. J. Pharm., 2018, 535, 1–17 CrossRef CAS.
- K. Banerjee, S. Banerjee and M. Mandal, in Biological and Pharmaceutical Applications of Nanomaterials, ed. P. Prokopovich, CRC Press, 2015, pp. 53–100 Search PubMed.
- H. Anwekar, S. Patel and A. K. Singhai, Int. J. Pharm. Life Sci., 2011, 945–951 CAS.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef.
- J. A. Bouwstra and P. L. Honeywell-Nguyen, Adv. Drug Delivery Rev., 2002, 54, S41–S55 CrossRef CAS.
- S. Siler-Marinkovic, in Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers, ed. N. Dragicevic and H. I. Maibach, Springer, 2016, pp. 15–38 Search PubMed.
- E. Touitou, N. Dayan, L. Bergelson, B. Godin and M. Eliaz, J. Controlled Release, 2000, 65, 403–418 CrossRef CAS.
- M. M. A. Elsayed, O. Y. Abdallah, V. F. Naggar and N. M. Khalafallah, Int. J. Pharm., 2007, 332, 1–16 CrossRef CAS.
- N. Dayan and E. Touitou, Biomaterials, 2000, 21, 1879–1885 CrossRef CAS.
- E. Touitou and B. Godin, J. Drug Delivery Sci. Technol., 2007, 17, 303–308 CrossRef CAS.
- N. A. Pratima and T. Shailee, Int. J. Res. Pharm. Sci., 2012, 2, 1–20 Search PubMed.
- H. Abdelkader, A. W. G. Alani and R. G. Alany, Drug Delivery, 2014, 21, 87–100 CrossRef CAS.
- G. Cevc and G. Blume, Biochim. Biophys. Acta, Biomembr., 1992, 1104, 226–232 CrossRef CAS.
- A. Kumar, K. Pathak and V. Bali, Drug Discovery Today, 2012, 17, 1233–1241 CrossRef CAS.
- G. Cevc and A. Chopra, in Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers, ed. N. Dragicevic and H. I. Maibach, Springer, 2016, pp. 39–59 Search PubMed.
- D. I. J. Morrow, P. A. McCarron, A. D. Woolfson and R. F. Donnelly, Open Drug Delivery J., 2007, 1, 36–59 CrossRef CAS.
- P. L. Honeywell-Nguyen and J. A. Bouwstra, Drug Discovery Today: Technol., 2005, 2, 67–74 CrossRef CAS.
- S. Kamboj, V. Saini, N. Maggon, S. Bala and V. Jhawat, Int. J. Drug Delivery, 2013, 5, 121 Search PubMed.
- Y. Rahimpour and H. Hamishehkar, in Recent Advances in Novel Drug Carrier Systems, ed. A. D. Sezer, InTech, 2012 Search PubMed.
- R. Muzzalupo, in Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers, ed. N. Dragicevic and H. I. Maibach, Springer, 2016, pp. 39–59 Search PubMed.
- J.-Y. Fang, C.-T. Hong, W.-T. Chiu and Y.-Y. Wang, Int. J. Pharm., 2001, 219, 61–72 CrossRef CAS.
- R. Muzzalupo and L. Tavano, Res. Rep. Transdermal Drug Delivery, 2015, 4, 23–33 CrossRef CAS.
- A. Manosroi, P. Jantrawut and J. Manosroi, Int. J. Pharm., 2008, 360, 156–163 CrossRef CAS.
- A. Manosroi, C. Chankhampan, W. Manosroi and J. Manosroi, Eur. J. Pharm. Sci., 2013, 48, 474–483 CrossRef CAS.
- R. Muzzalupo, L. Tavano, F. Lai and N. Picci, Colloids Surf., B, 2014, 123, 207–212 CrossRef CAS.
- E. W. Kaler, A. K. Murthy, B. E. Rodriguez and J. A. N. Zasadzinski, Sci. New Ser., 1989, 245, 1371–1374 CAS.
- E. F. Marques, O. Regev, A. Khan and B. Lindman, Adv. Colloid Interface Sci., 2003, 100–102, 83–104 CrossRef CAS.
- M. Blanzat, E. Perez, I. Rico-Lattes, D. Prome, J. C. Prome and A. Lattes, Langmuir, 1999, 15, 6163–6169 CrossRef CAS.
- H. Fukuda, K. Kawata, H. Okuda and S. L. Regen, J. Am. Chem. Soc., 1990, 112, 1635–1637 CrossRef CAS.
- E. Soussan, C. Mille, M. Blanzat, P. Bordat and I. Rico-Lattes, Langmuir, 2008, 24, 2326–2330 CrossRef CAS.
- E. Soussan, A. Pasc-Banu, S. Consola, T. Labrot, E. Perez, M. Blanzat, R. Oda, C. Vidal and I. Rico-Lattes, ChemPhysChem, 2005, 6, 2492–2494 CrossRef CAS.
- D. Vivares, E. Soussan, M. Blanzat and I. Rico-Lattes, Langmuir, 2008, 24, 9260–9267 CrossRef CAS.
- E. F. Marques, O. Regev, A. Khan, M. da Graça Miguel and B. Lindman, J. Phys. Chem. B, 1999, 103, 8353–8363 CrossRef CAS.
- F. M. Menger, W. H. Binder and J. S. Keiper, Langmuir, 1997, 13, 3247–3250 CrossRef CAS.
- C.-W. Chiu, C.-H. Chang and Y.-M. Yang, Soft Matter, 2013, 9, 7628–7636 RSC.
- Y.-S. Liu, C.-F. Wen and Y.-M. Yang, J. Taiwan Inst. Chem. Eng., 2012, 43, 830–838 CrossRef CAS.
- C. Richard, PhD Thesis, Université Toulouse 3, 2018.
- C. Mauroy, P. Castagnos, J. Orio, M.-C. Blache, I. Rico-Lattes, J. Teissié, M.-P. Rols and M. Blanzat, Mol. Pharm., 2015, 12, 103–110 CrossRef CAS.
- C. Mauroy, P. Castagnos, M.-C. Blache, J. Teissié, I. Rico-Lattes, M.-P. Rols and M. Blanzat, Chem. Commun., 2012, 48, 6648 RSC.
- N. Dew, K. Edsman and E. Björk, J. Pharm. Pharmacol., 2011, 63, 1265–1273 CrossRef CAS.
- C. Bize, J.-C. Garrigues, J.-P. Corbet, I. Rico-Lattes and M. Blanzat, ChemPhysChem, 2013, 14, 1126–1131 CrossRef CAS.
- C. Richard, E. Souloumiac, J. Jestin, M. Blanzat and S. Cassel, Colloids Surf., A, 2018, 558, 373–383 CrossRef CAS.
- N. Dew, K. Edwards, J. Eriksson, K. Edsman and E. Björk, Colloids Surf., B, 2012, 89, 53–60 CrossRef CAS.
- N. Dew, T. Bramer and K. Edsman, J. Colloid Interface Sci., 2008, 323, 386–394 CrossRef CAS.
- F. Roig, M. Blanzat, C. Solans, J. Esquena and M. J. Garcia-Celma, Colloids Surf., B, 2018, 164, 218–223 CrossRef CAS.
- G. Cevc, A. Schätzlein and H. Richardsen, Biochim. Biophys. Acta, Biomembr., 2002, 1564, 21–30 CrossRef CAS.
- G. M. El Maghraby, B. W. Barry and A. C. Williams, Eur. J. Pharm. Sci., 2008, 34, 203–222 CrossRef CAS.
- S. Duangjit, P. Opanasopit, T. Rojanarata, Y. Obata, K. Takayama, T. Ngawhirunpat and B. Pamornpathomkul, Int. J. Nanomed., 2014, 9(1), 2005–2007 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |