
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed annulation of styrenes with α-bromoacetic acids†
Tung T. Nguyen *ab,
Bao H. T. Ngoab,
Huy X. Leab,
Linh N. P. Vuab,
Tuong A. Toab,
Anh N. Q. Phanab and
Nam T. S. Phan*ab
*ab,
Bao H. T. Ngoab,
Huy X. Leab,
Linh N. P. Vuab,
Tuong A. Toab,
Anh N. Q. Phanab and
Nam T. S. Phan*ab
aFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tungtn@hcmut.edu.vn; ptsnam@hcmut.edu.vn
bVietnam National University Ho Chi Minh City, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
First published on 29th January 2021
Abstract
We report a method for addition of α-bromophenylacetic acids to vinyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in styrenes to afford γ-lactones. Reactions employed a simple cobalt catalyst Co(NO3)2·6H2O in the presence of dipivaloylmethane (dpm) ligand. Many functionalities including halogen, ester, and nitro groups were compatible with reaction conditions. If α-bromoesters were used, vinylacetates were the major products.
C bonds in styrenes to afford γ-lactones. Reactions employed a simple cobalt catalyst Co(NO3)2·6H2O in the presence of dipivaloylmethane (dpm) ligand. Many functionalities including halogen, ester, and nitro groups were compatible with reaction conditions. If α-bromoesters were used, vinylacetates were the major products.
γ-Lactones are ubiquitously found in many natural products and bioactive molecules.1 Among the available synthetic methods to obtain these five-membered rings, those using transition metals for functionalization of unsaturated carbon–carbon bonds, such as alkenes and alkynes, with oxygen-containing nucleophiles have been substantially developed.2 Notable reports have used scarce noble metals and/or photoredox conditions.3 Some examples employing abundant first-row metals to obtain γ-lactones from α-bromoacetic acids or isosteric esters have been reported.4 A seminal method for copper(0)-promoted hydrocarboxylation of alkenes with α-iodocarboxylates was introduced by Metzger and Mahler.4a Following that, copper(I) salts have been developed for catalytic annulation of α-bromoacetamides with styrenes.4b,4c Methods for using low valent complexes of other non-noble metals such as those of Ni(0) or Fe(0) are known as well.4d,4e It should be noted that the reported transformations were presumably triggered by a homolytic cleavage of carbon–halogen bonds in α-halocarboxylates.4d We envisioned that such an activation could be obtained if a suitable cobalt complex is used.
Using cobalt adducts in the organic transformations that included alkyl halides was known.5 Most of the known examples involved the formation of cobalt(I) species, thus usually suffering from the presence of strong reductants and/or tailored ligands. Meanwhile, a direct elaboration of cobalt(II) complexes for incorporation of alkyl electrophiles is much rarer. It was hypothesized that a Co(II)/Co(III) catalytic cycle could be exploited since single-electron-transfer oxidation of Co(II) was feasible.6 Herein we report our attempts to annulate styrenes with α-bromophenylacetic acids for synthesis of γ-lactones using a simple cobalt(II) catalyst (Scheme 1). The method has allowed for the use of α-halocarboxylic acids that are not limited to those containing quaternary centers.4d,4e
 | ||
| Scheme 1 Metal-catalyzed synthesis of γ-lactones via activation of α-bromoacetic acids. | ||
We firstly investigated the coupling of diphenylethylene 1a and α-bromophenylacetic acid 2a to afford the γ-lactone 3aa. Brief results and control experiments are shown in Table 1.7 Optimization was carried out with respect to cobalt salt, ligand, base, and solvent. Using Co(acac)2 as catalyst for the annulation afforded 3aa in 69% yield (entry 1). If a cobalt(III) precursor was employed, a trace amount of the lactone was detected (entry 2). Since Co(OAc)2 was inferior to Co(acac)2 (entry 3), our hypothesis was that 1,3-diketone may play an important role in the coupling.8 Running the reaction with Co(NO3)2·6H2O catalyst and acetylacetone ligand (L1) furnished 3aa in a comparable yield with respect to that of Co(acac)2 (entry 4). A better yield of the product was observed if dipivaloylmethane (dpm) ligand (L2) was used (entry 5). Only 27% yield of 3aa was obtained in case of an electron-poor 1,3-diketone (L3, entry 6). An unsymmetrical ligand bearing a phenyl moiety (L4) was also attempted, furnishing the lactone 3aa in moderate yield (entry 7). Sodium carbonate was an inferior base to potassium carbonate (entry 8). To our surprise, adding a stoichiometric amount of DMF helped to increase the yield of 3aa (entry 9). 1,4-Dioxane could be used as the solvent for this coupling (entry 10). Notably, a small amount of by-product derived from 2a and CH3CN could be observed. Thus, pivalonitrile solvent was attempted to avoid such addition (entry 11), affording 91% yield of the lactone 3aa.
|
||||
|---|---|---|---|---|
| Entry | [Co] | Base | Solvent | Yield of 3aa (%) |
| a 1a (0.1 mmol), 2a (0.2 mmol), [Co] catalyst (0.02 mmol), ligand (0.05 mmol), base (0.1 mmol), solvent (1 mL), at 140 °C for 24 h. Yields of 3aa are GC yields using diphenyl ether internal standard.b L1 was used.c L2 was used.d L3 was used.e L4 was used.f DMF (0.1 mmol) was added. Abbreviation: acac = acetylacetonate. | ||||
| 1 | Co(acac)2 | K2CO3 | CH3CN | 69 |
| 2 | Co(acac)3 | K2CO3 | CH3CN | Trace |
| 3 | Co(OAc)2 | K2CO3 | CH3CN | 57 |
| 4b | Co(NO3)2·6H2O | K2CO3 | CH3CN | 70 |
| 5c | Co(NO3)2·6H2O | K2CO3 | CH3CN | 78 |
| 6d | Co(NO3)2·6H2O | K2CO3 | CH3CN | 27 |
| 7e | Co(NO3)2·6H2O | K2CO3 | CH3CN | 52 |
| 8c | Co(NO3)2·6H2O | Na2CO3 | CH3CN | 67 |
| 9c,f | Co(NO3)2·6H2O | K2CO3 | CH3CN | 84 |
| 10c,f | Co(NO3)2·6H2O | K2CO3 | 1,4-Dioxane | 75 |
| 11c,f | Co(NO3)2·6H2O | K2CO3 | (CH3)3CCN | 91 |
 |
||||
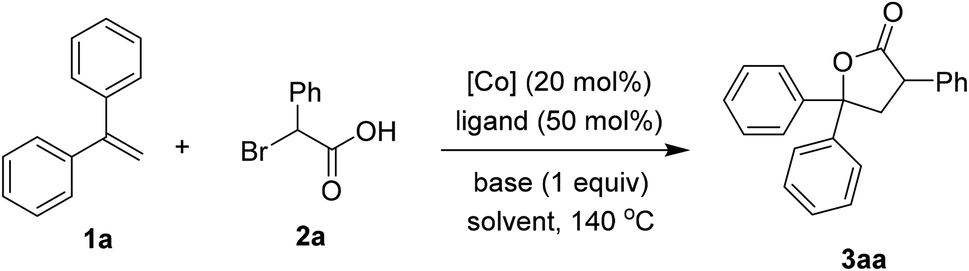
Scope of the reaction with respect to reagents containing CC bonds was next studied. The results are shown in Scheme 2. Notably, some products were obtained in excellent diastereoselectivities in comparison with two examples reported by Nishihara and Iwasaki.4e A triphenyl-substituted γ-lactone 3aa was isolated in 85% yield. The simplest styrene coupled with α-bromophenylacetic acid to furnish 3ba in 81% yield. Functionalities including bromo (3ca), chloro (3da), ester (3ea), and nitro (3fa, 3ga) groups were compatible with reaction conditions. Some vinyl arenes containing polyaromatics were also attempted. 2-Vinyl naphthalene was reacted to give the lactone 3ha in 88% yield. Cyclization of 9-vinylanthracene yielded a trace amount of the desired product, somewhat implying the importance of steric effect. Heterocycles, such as thiazole (3ia), were competent substrates. Other α-bromoarylacetic acids such as those containing chloro substituents could successfully annulate with CC bonds. Thus, the lactones (3hb, 3ac) were obtained in moderate to good yields. At this moment, very electron-poor styrenes (i.e. 3,5-bis(trifluoromethyl)styrene) or internal alkenes (i.e. indene) were not tolerant of reaction conditions.
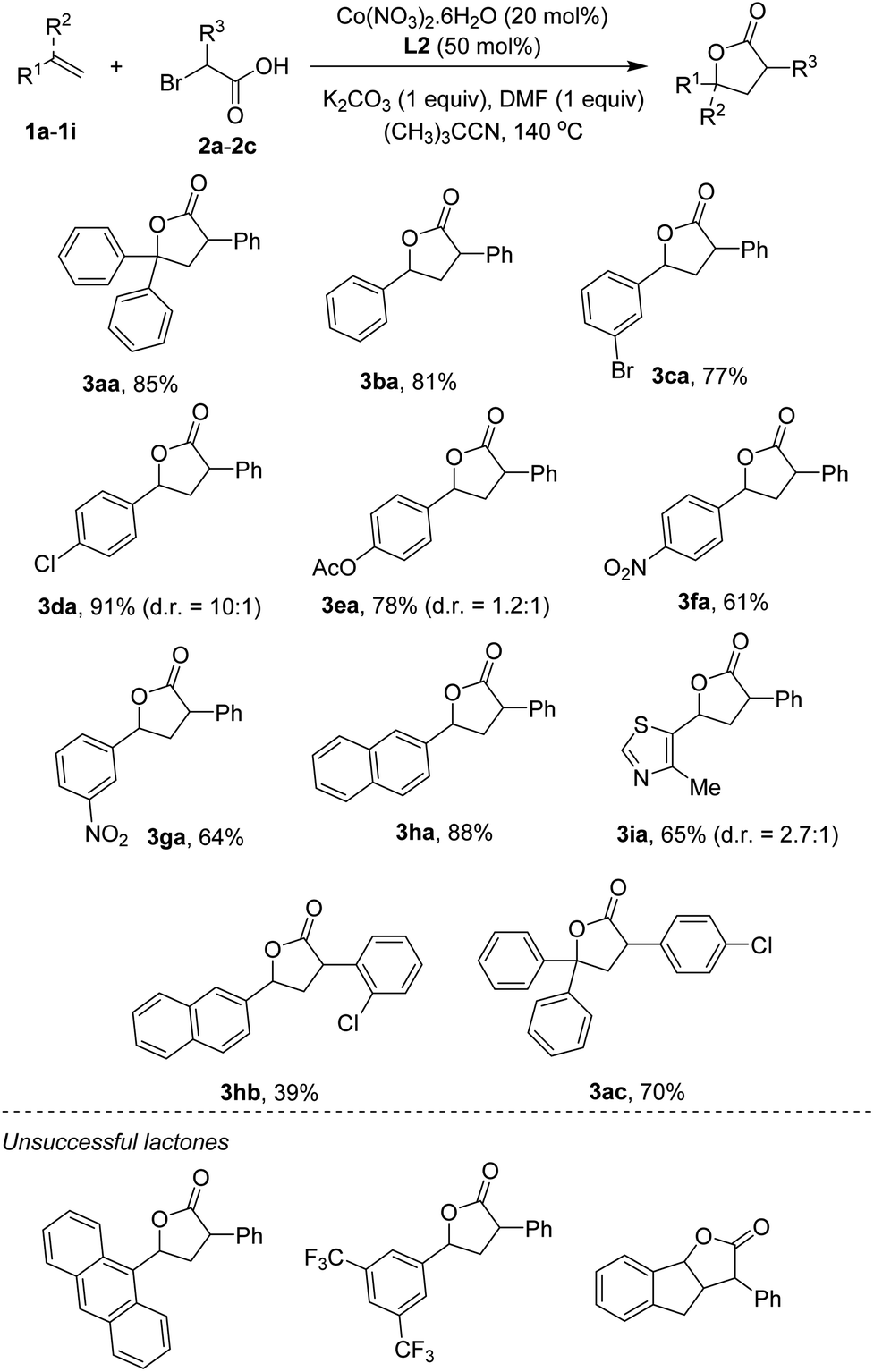 | ||
| Scheme 2 Annulation of styrenes and α-bromo arylacetic acids. Conditions: 1a–1j (0.5 mmol), 2a–2c (1.0 mmol), Co(NO3)2·6H2O (0.1 mmol), L2 (0.25 mmol), K2CO3 (0.5 mmol), DMF (0.5 mmol), (CH3)3CCN (5 mL), at 140 °C for 24 h. Yields are isolated yields. Diastereomeric ratios (d.r.) were determined using 1H NMR spectrum. | ||
To show the generality of our method, annulation of styrenes with α-bromoisobutyric acid 4 was attempted. The conditions were relatively milder than those of α-bromophenylacetic acids, presumably due to the presence of the quaternary carbon that help cyclization step. Scope of styrenes is presented in Scheme 3. An excellent yield of the lactone (5a) was obtained in case of diphenylethylene substrate. Reactions of molecules containing bromo (5c), ester (5e), and nitro (5f) substituents occurred without any difficulties. Similar to the iron-catalyzed method,4e 2-vinyl naphthalene was suitable for the annulation (5h). Vinyl heteroarenes were competent substrates (5i). Electron-rich arenes were also attempted, furnishing the lactones in good yields (5k, 5l).
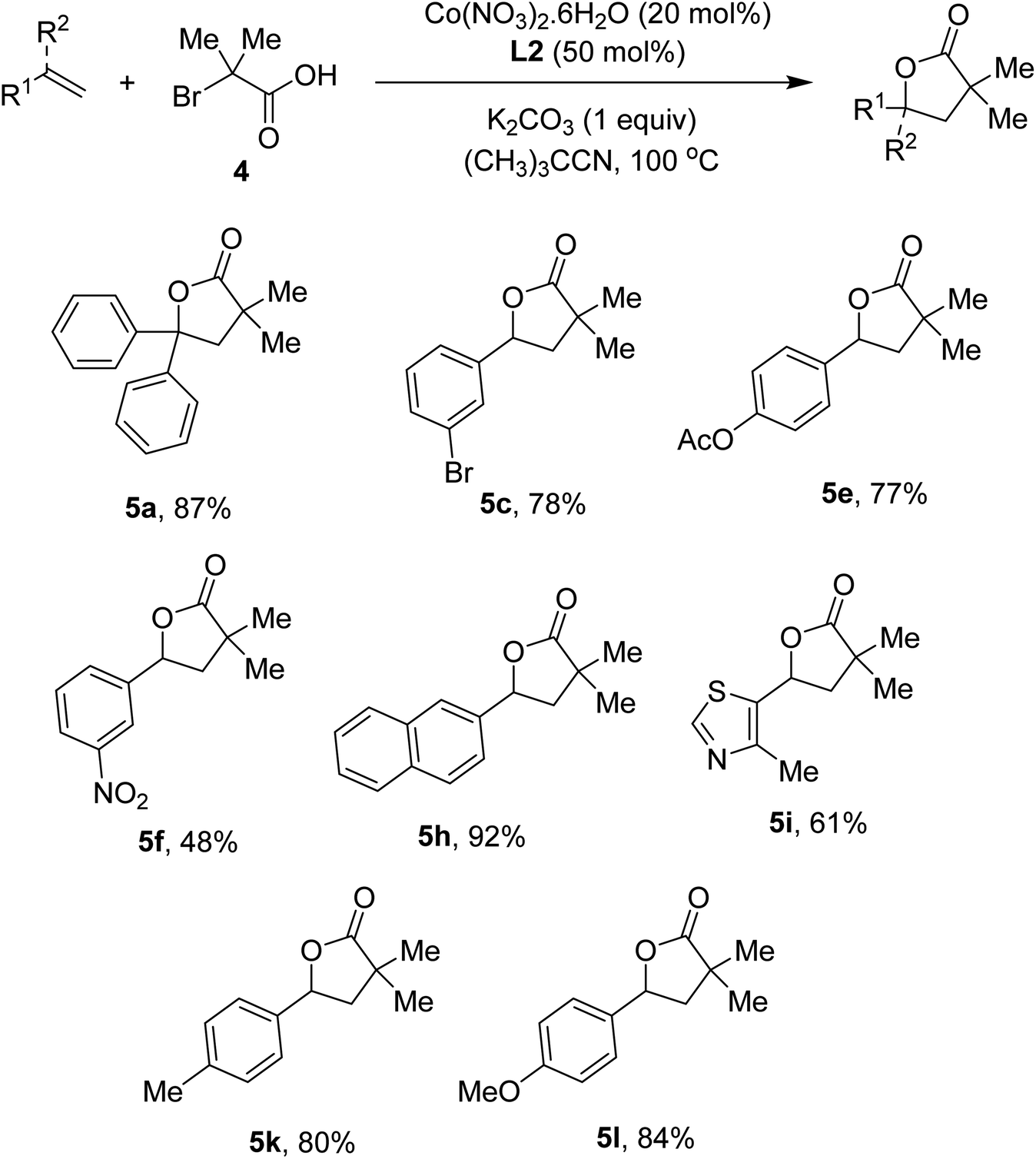 | ||
| Scheme 3 Annulation of styrenes and α-bromoisobutyric acid. Conditions: styrenes (0.5 mmol), 4 (1.0 mmol), Co(NO3)2·6H2O (0.1 mmol), L2 (0.25 mmol), K2CO3 (0.5 mmol), (CH3)3CCN (5 mL), at 100 °C for 24 h. Yields are isolated yields. | ||
Using α-bromoacetates to couple with styrenes could yield lactones.4d,4e However, only vinylacetates 7a–7c were obtained in our conditions if similar substrates were employed (Scheme 4). The results were indicative of a slow hydrolysis during reaction course. It should be noted that the presence of quaternary carbons in esters was crucial to afford the addition products.
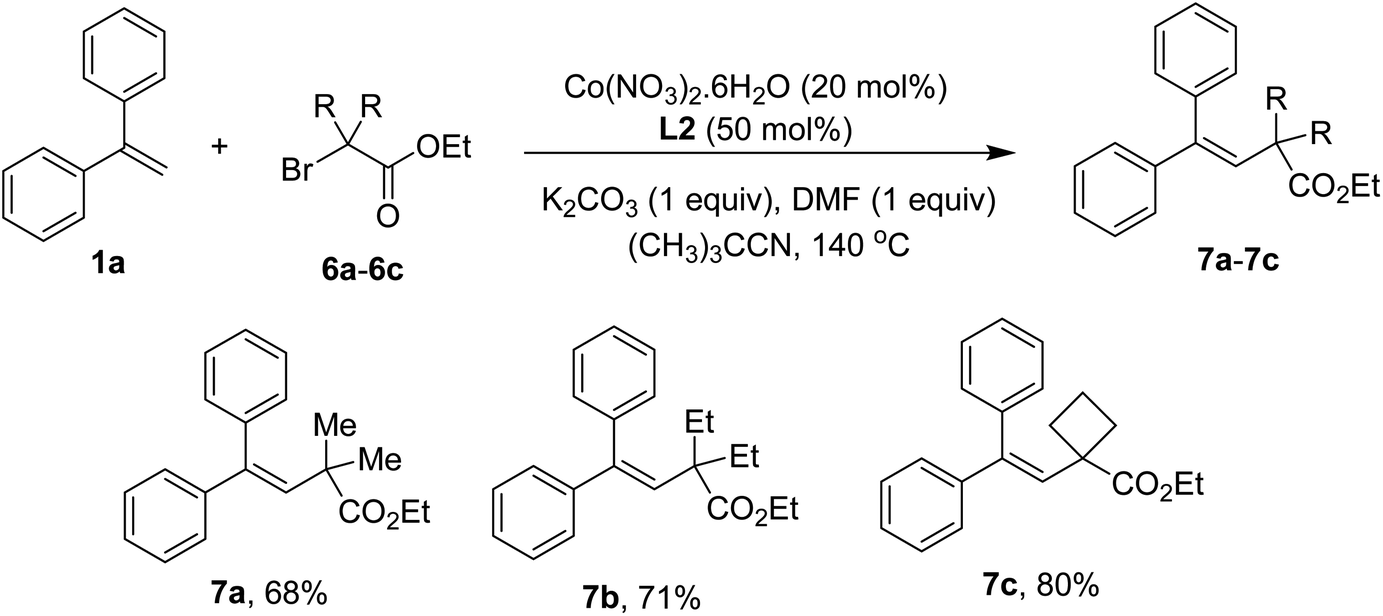 | ||
| Scheme 4 Scope of α-bromoacetates. Conditions: 1a (0.5 mmol), α-bromoacetates 6a–6c (1.0 mmol), Co(NO3)2·6H2O (0.1 mmol), L2 (0.25 mmol), K2CO3 (0.5 mmol), DMF (0.5 mmol), (CH3)3CCN (5 mL), at 140 °C for 24 h. Yields are isolated yields. | ||
Based on previous studies,4d,4e a plausible mechanism was proposed as that shown in Scheme 5. Homolytic cleavage of C(sp3)–Br bond in 2a would afford the benzyl radical 8. Radical addition to 1a generated 9 followed by an oxidation that could afford a benzylic cation 10. The resulting species underwent cyclization, in the presence of base, to eventually furnish the lactone 3aa. To support the hypothesis, some control experiments were carried out. Firstly, addition of radical quenchers such as TEMPO or BHT stopped the annulation, somewhat confirming the involvement of radical species during the reaction course. Secondly, running the reaction in absence of 1a gave an anhydride 11 (detected by GC-MS). Such the dead-end intermediate could be obtained via a homo-combination of the radical 8 followed by a dehydration.
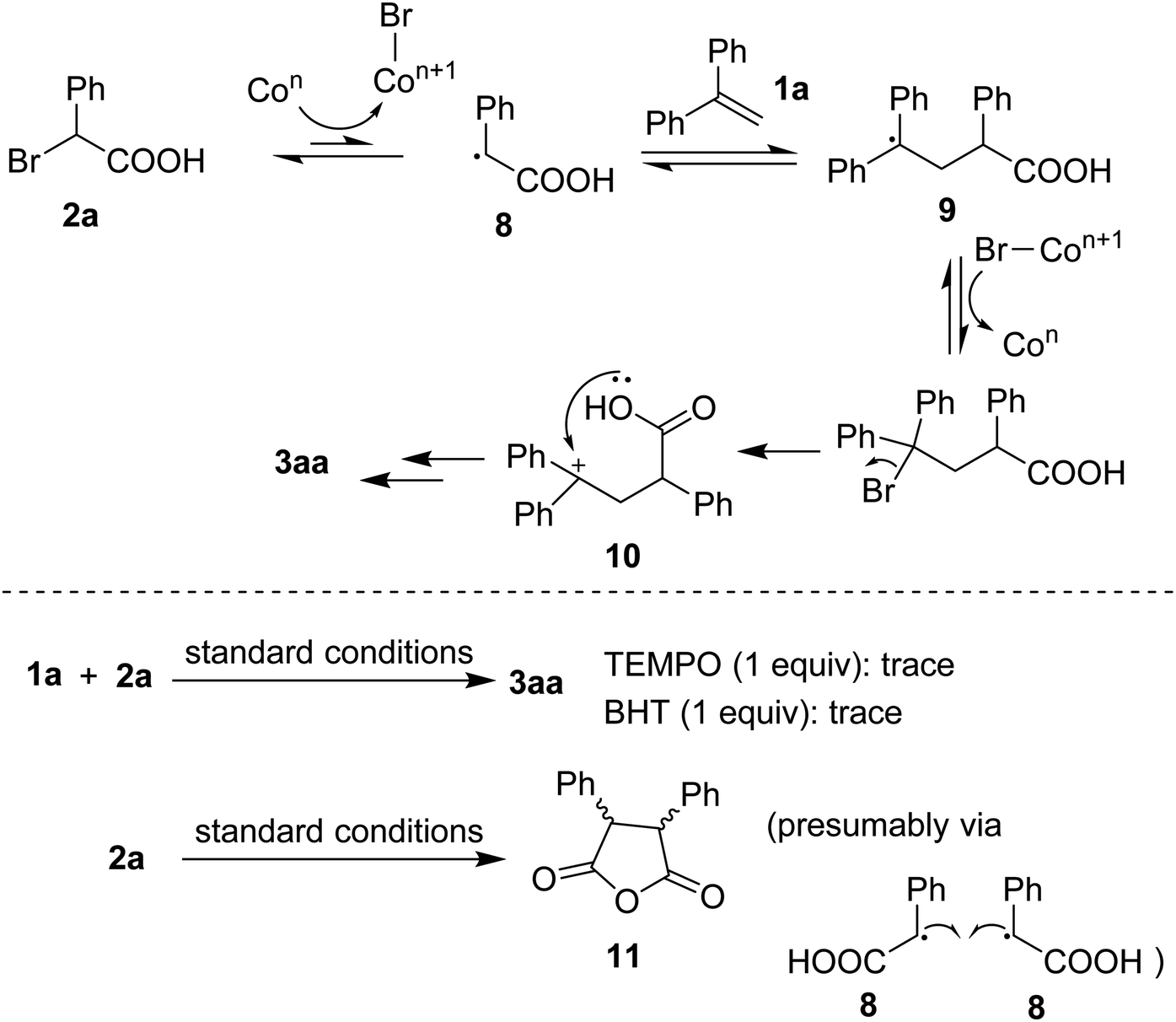 | ||
| Scheme 5 Mechanistic considerations. | ||
Conclusions
In conclusion, we have developed a general method to couple styrene CC bonds with α-bromoarylacetic acids to afford γ-lactones. The transformation employed a simple cobalt precursor Co(NO3)2·6H2O facilitated by dipivaloylmethane ligand. Reaction conditions were well compatible with a diversified array of functional groups. Preliminary control experiments supported a radical-involved mechanism that could triggered by the cobalt-catalyzed activation of carbon–bromide bond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Vietnam National Foundation for Science and Technology Development (NAFOSTED) for supporting this research (under grant number 104.01-2019.44).Notes and references
- (a) E. J. Kang and E. Lee, Chem. Rev., 2005, 105, 4348 CrossRef CAS PubMed; (b) G. Lin, S. S. K. Chan, H. S. Chung and S. L. Li, Stud. Nat. Prod. Chem, 2005, 32, 611 CAS.
- N. T. Patil, R. D. Kavthe and V. S. Shinde, Tetrahedron, 2012, 68, 8079 CrossRef CAS.
- (a) N. G. Kundu and M. Pal, J. Chem. Soc., Chem. Commun., 1993, 86 RSC; (b) H.-Y. Liao and C.-H. Cheng, J. Org. Chem., 1995, 60, 3711 CrossRef CAS; (c) R. C. Larock, M. J. Doty and X. Han, J. Org. Chem., 1999, 64, 8770 CrossRef CAS PubMed; (d) X.-J. Wei, D.-T. Yang, L. Wang, T. Song, L.-Z. Wu and Q. Liu, Org. Lett., 2013, 15, 6054 CrossRef CAS PubMed; (e) I. Triandafillidi, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2018, 20, 36 CrossRef CAS PubMed.
- (a) J. O. Metzger and R. Mahler, Angew. Chem., Int. Ed. Engl., 1995, 34, 902 CrossRef CAS; (b) T. Nishikata, K. Itonaga, N. Yamaguchi and M. Sumimoto, Org. Lett., 2017, 19, 2686 CrossRef CAS PubMed; (c) Y. Lv, W. Pu, S. Mao, X. Ren, Y. Wu and H. Cui, RSC Adv., 2017, 7, 41723 RSC; (d) D. Liu, S. Tang, H. Yi, C. Liu, X. Qi, Y. Lan and A. Lei, Chem. Eur. J., 2014, 20, 15605 CrossRef CAS PubMed; (e) M. Iwasaki, N. Miki, Y. Ikemoto, Y. Ura and Y. Nishihara, Org. Lett., 2018, 20, 3848 CrossRef CAS PubMed; (f) B. Wei, K.-W. Li, Y.-C. Wu, S.-Q. Tong and R.-J. Song, Synthesis, 2020, 52, 3855 CrossRef CAS.
- (a) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351 CrossRef PubMed; (b) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed.
- For recent examples, see: (a) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed; (b) S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493 CrossRef CAS.
- Please see the ESI† for full optimization studies.
- For recent examples of the cobalt complex ligated by dipivaloylmethane, see: (a) T. T. Nguyen, L. Grigorjeva and O. Daugulis, ACS Catal., 2016, 6, 551 CrossRef CAS PubMed; (b) L. Lukasevics, A. Cizikovs and L. Grigorjeva, Org. Lett., 2020, 22, 2720 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09588e |
| This journal is © The Royal Society of Chemistry 2021 |