
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemoval of microplastics via tannic acid-mediated coagulation and in vitro impact assessment†
Jun Woo Park,
Su Jin Lee,
Dae Youn Hwang and
Sungbaek Seo *
*
Department of Biomaterials Science (BK21 FOUR Program), College of Natural Resources and Life Science, Life and Industry Convergence Research Institute, Pusan National University, Miryang 50463, Republic of Korea. E-mail: sbseo81@pusan.ac.kr
First published on 18th January 2021
Abstract
Microplastics are distributed in oceans worldwide, and the negative effects of microplastics on the environment and human health are increasing. Generally, three methods are employed to remove microplastics: filtration, biological degradation, and coagulation. Of these methods, filtration is the most commonly used but depends on the filter size or degree of microplastic coagulation. Although Fe- or Al-salts are generally used for coagulation via electrostatic interactions between metal ion and microplastics, their microplastic removal efficiency is less than 40%, and the smaller the size of microplastics, the lower is the removal efficiency. In order to improve the removal efficiency, metal–phenolic coordinate bonds were newly utilized for microplastic coagulation. Plant-derived tannic acid contributed to interfacial bridging between the microplastics and Fe3+. Using 0.5 μm polystyrene beads as model microplastics, a removal efficiency of more than 90% within 5 min was achieved. Since microplastics mostly accumulate in the gut of animals, rat intestine IEC18 cells exposed to purified water from the microbead suspension were risk assessed, revealing that water purified using the coagulation-based method reduced oxidative stress and inflammatory cytokines to levels similar to those in cells exposed to water without microbeads.
1. Introduction
With the growing use of plastics, the accumulation of plastic waste has considerably increased.1–3 Exposure of plastics to photo-oxidation and mechanical collision of plastics, results in the generation of microplastics (MPs), i.e., plastics smaller than 5 mm. In recent decades, MPs have become a serious issue because of the scale of their accumulation and their ability to carry toxic heavy metals or organic compounds4,5 without weathering or degradation. The toxicological effect of MPs has been primarily investigated in marine organisms6–8 however, humans are affected by the MPs via the food chain. In vitro and in vivo studies revealed that MPs from the bloodstream could penetrate the capillaries, resulting in toxicity or development of endocrine system disorders.9–13Several approaches have been employed to remove MPs present in the environment; these include filtration of MPs using a membrane,14–16 biological degradation of MPs by microorganisms,17,18 sequestration of MPs using magnetic nanoparticles19,20 or metal–organic frameworks,21 and coagulation—which creates larger clusters of suspended MPs—using coagulants and subsequent ultrafiltration.22–25 Although membrane filtration is a commonly used method for removing pollutants from water, isolation of small-sized (<20 μm) MPs is challenging. The biological degradation of MPs is an environmentally friendly approach, but the degradation kinetics is too slow, for example, less than 10 wt% loss of MPs occurred after 40 days.18 The removal of MPs using functional nanoparticles requires the use of potentially toxic chemicals. For coagulating small-sized MPs, coagulants such as Fe- or Al-based salts, are required in large amounts to achieve substantial MPs removal efficiency. In recent studies, MPs were identified in even purified drinking water upon using one of these techniques.26,27 Therefore, qualitative and efficient (additive- and time-saving) methods that employ nontoxic components are necessary for the purification of water containing MPs.
In nature, numerous strong coordinate bonds exist; these include the bonds between metal ions and phenolic molecules. Coordinate bonds are responsible for the unique behaviors or the functionalities encountered in natural systems. For instance, the byssal plaque of marine mussels contains an interfacial polyphenolic protein that contributes to the strong adhesion to the rock surface in environments containing metal ions via the formation of metal–phenolic coordinates.28,29 Researchers have used coordinate bonds to formulate interfacial nanolayers in water-in-oil particles for live cell encapsulation30 and development of nanocarriers for chemotherapeutic drugs.31 Inspired by the coordinate bonds existing in nature, we attempted to leverage the strong coordinate bonds between surface phenolic MPs and ferric ions (Fe3+) to enable the coagulation of MPs. In our previous study, a high degree of interspecific interaction-driven aggregation was observed between surface phenolic liposomes and Fe3+.32 Furthermore, few studies have investigated MPs-associated human health risks—and those of residual MPs after MPs removal. There is disagreement regarding how MPs affect viability and toxicity in various cell types. Therefore, systematic studies pertaining to MPs- and residual MPs-associated risk assessment are required.
In this study, the coagulation of MPs was demonstrated by using nontoxic plant-derived tannic acid. The formation of metal–phenolic coordinate bonds was used to initiate the coagulation of the MPs. 0.5–125 μm sized beads of polystyrene (PS) or polyethylene (PE) were used as model MPs (Fig. 1). After MPs coagulation, the removal efficiency of MPs was quantified using a fluorescence reader and optical/fluorescence microscopy. Epithelial cells of the gut and liver were used to observe how the coagulation-based MPs removal improves cell viability, inflammation, and cytokine level, as the gut is the dominant tissue affected by MPs accumulation.
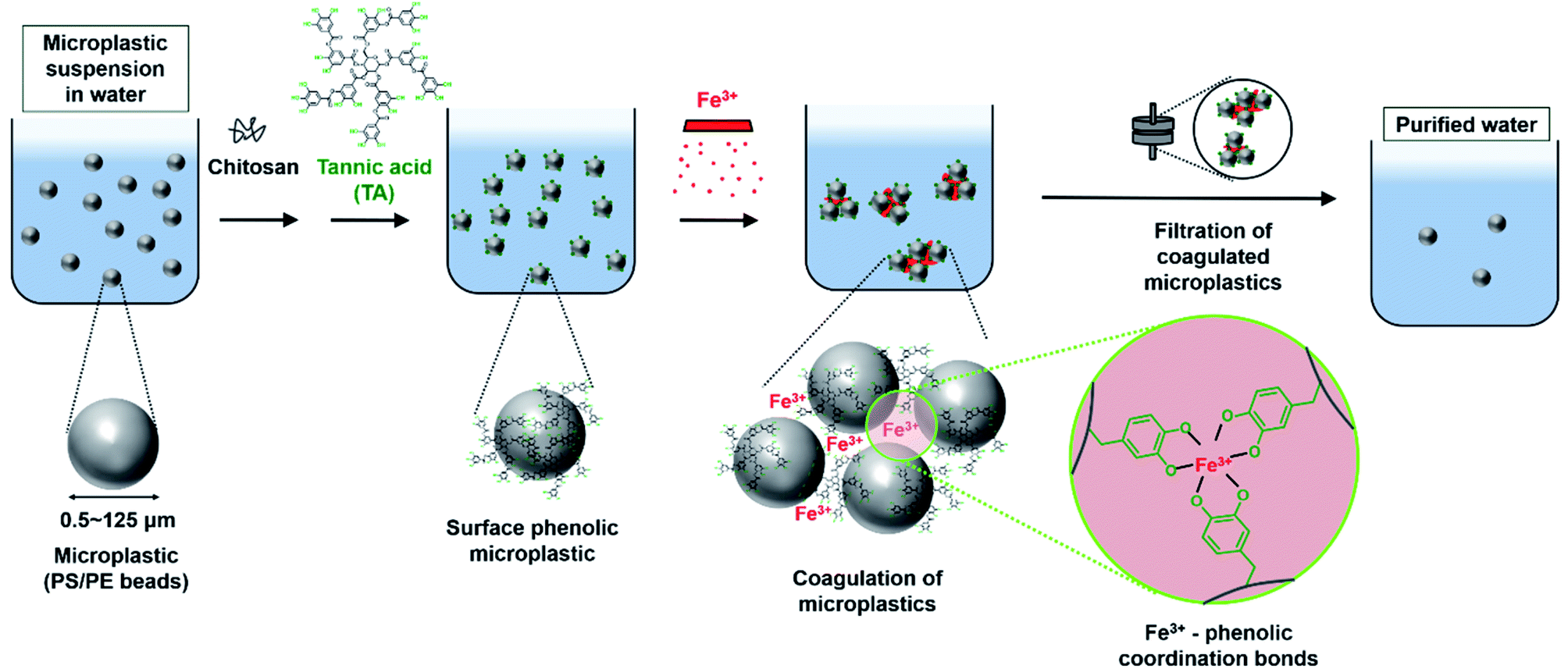 | ||
| Fig. 1 Schematic illustration of purification of microplastics (0.5–125 μm) suspension in water. Polystyrene (PS)/polyethylene (PE) beads are used as a model of the microplastics. The surface of the microplastics is treated with chitosan and tannic acid to form surface phenolic microplastics. Then, Fe3+ triggers the coagulation of the microplastics by Fe3+-phenolic coordinate bonds. The coagulated microplastics are filtered for the removal of microplastics, results in purified water. | ||
2. Experimental details
2.1. Materials
Tannic acid (TA), PS beads (mean particle size, 0.5 μm; fluorescent λex = ∼520 nm and λem = ∼540 nm), sodium chloride, and chitosan (molecular weight = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–190000 Da), were purchased from Sigma Aldrich (USA). PE beads (45–53 μm; red-colored; 106–125 μm; fluorescent λex = 414 nm and λem = 515 nm) were purchased from Cospheric (USA). Iron(III) chloride anhydrous, PS bead (90 μm) aqueous suspension, and all other chemical reagents were obtained from Thermo Fisher Scientific (USA). Humic acid was purchased from Wako (USA). Calcium chloride was purchased from OCI (Korea).
000–190000 Da), were purchased from Sigma Aldrich (USA). PE beads (45–53 μm; red-colored; 106–125 μm; fluorescent λex = 414 nm and λem = 515 nm) were purchased from Cospheric (USA). Iron(III) chloride anhydrous, PS bead (90 μm) aqueous suspension, and all other chemical reagents were obtained from Thermo Fisher Scientific (USA). Humic acid was purchased from Wako (USA). Calcium chloride was purchased from OCI (Korea).
2.2. Surface modification of PS/PE beads
A 0.01 wt% aqueous solution of PS or PE beads (1 mL) was mixed with 1 wt% aqueous solution of chitosan (4 mL) on a shaker (KS 130, IKA) for 24 h. The mixed solution was centrifuged (Micro Prime Centrifuge, Centurion Scientific) at 10000 rpm (1411 × g) for 5 min. The supernatant was removed and the beads were redispersed in distilled water (DI water, 1 mL). To this solution, 0.06 mM of TA aqueous solution (0.5 mL) was added, and the mixture was shaken for 2 h. The mixed solution was centrifuged at 10000 rpm (1411 × g) for 5 min. The supernatant was removed and the beads were redispersed in DI water (1.5 mL).
The size and zeta potential of the beads based on the surface modification were measured using a Zetasizer (Nano ZS90, Malvern Instruments). Scanning electron microscopy/energy-dispersive X-ray spectroscopy (SEM/EDX spectroscopy, JSM-7900F, JEOL) was used to analyze the surface morphology and surface elements of the beads according to the surface modification.
2.3. Coagulation of PS/PE beads and filtration of coagulated beads
For the coagulating the beads, 3 mM of FeCl3 (0.5 mL) was added to the solution (1.5 mL) of surface-modified beads and shaken for 5 min. Coagulation was monitored using optical/fluorescence microscopy (Eclipse TS100, Nikon).The coagulated beads were filtered using a filter paper (qualitative filter paper, Grade 1, Whatman) with a pore size of 11 μm. The removal efficiency of 0.01 wt% fluorescent PS beads (0.5 μm) was calculated using the following equation:

The fluorescence intensity of the beads was measured using a fluorescence reader (Spectramax i5D, Molecular Devices). Bead removal efficiency according to various reaction times (5–120 min), concentration of beads (0.01–0.1 mg mL−1), volume of bead aqueous solutions (1–100 mL), pH-dependent (pH 6–8), actual water conditions including inorganic ions (NaCl, CaCl2), natural organic material-rich water conditions with humic acidwere calculated. As control coagulants, an aqueous solution of Fe-salts (FeCl3) and Al-salts (AlCl3) was mixed with non-treated beads. Morning burst facial scrub (Clean&Clear) was used as a practical sample representing MPs. The fine particles were separated from the facial scrubs filtered using filter paper and rinsed with DI water.
2.4. In vitro tests
000 rpm for 5 min, the protein concentrations were determined using a SMARTTM Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific Inc.). Proteins were separated on 4–20% SDS–PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) gels after running for 2 h, and then transferred to nitrocellulose membranes at 40 V for 2 h. Individual membranes were incubated overnight at 4 °C with the following primary antibodies: anti-iNOS (Thermo Fisher Scientific, Inc.), anti-COX-2 (Cell Signaling Technology, Danvers, MA, USA), and anti-β-actin antibody (Cell Signaling Technology, Inc.). The membranes were subsequently washed with a buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 0.05% Tween 20) and incubated with 1:2000 diluted horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Invitrogen) at 23 ± 2 °C for 1 h. Blots were developed using the Amersham ECL Select Western Blotting detection reagent (GE Healthcare, Little Chalfont, UK). Chemiluminescence signals from specific bands were detected using FluorChemi®FC2 (Alpha Innotech, Co., San Leandro, CA, USA).For IEC18 cells. NF-κB, sense primer 5′-CCTGTAGCCCACGTCGTAGC-3′, antisense primer 5′-TTGACCTCAGCGCTGACTTG-3′; TNF-α, sense primer 5′-CCTGT AGCCC ACGTC GTAGC-3′, antisense primer 5′-TTGAC CTCAG CGCTG ACTTG-3′; IL-6, sense primer 5′-TTGGG ACTGA TGTTG TTGAC A-3′, antisense primer 5′-TCATC GCTGT TGATA CAATC AGA-3′, IL-1 β, sense primer 5′-AGG CTT CCT TGT GCA AGT GT-3′, antisense primer 5′-TGA GTG ACA CTG CCT TCC TG-3′; β-actin sense and antisense primers 5′-TGGAA TCCTG TGGCA TCCAT GAAAC-3′ and 5′-TAAAA CGCAG CTCAG TAACA GTCCG-3′.
For HepG2 cells. NF-κB, NF-B, sense primer 5′-GTG AGG TCA CTC TAA CGT ATG CAA CAG G-3′, antisense primer 5′-CTC CAC CAC ATC TTC CTG CTT AGT G-3′; TNF-α, sense primer 5′-CTC TTC TGC CTG CTG CAC TTT G-3′; antisense primer 5′-ATG GGC TAC AGG CTT GTC ACT C-3′; IL-6, sense primer 5′-GAA CTC CTT CTC CAC AAG TAA GTG C-3′, antisense primer 5′-CTC CTC ATT GAA TCC AGA TTG GAA GCA TC-3′, IL-1 β, sense primer 5′-CAC AGA CCT TCC AGG AGA ATG ACC TGA G-3′, antisense primer 5′-CTG CTT GAG AGG TGC TGA TGT ACC AG-3′, β-actin, sense and antisense primers 5′-TGG AAT CCT GTG GCA TCC ATG AAA C-3′, 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′.
qPCR was performed for 40 cycles using the following sequence: denaturation at 95 °C for 15 s, followed by annealing and extension at 70 °C for 60 s. Fluorescence intensity was measured at the end of the extension phase of each cycle. Threshold value for the fluorescence intensities of all samples was set manually. The reaction cycle at which the PCR products exceeded this fluorescence intensity threshold during the exponential phase of PCR amplification was considered, as the threshold cycle (Ct). Expression of the target gene was quantified relative to that of the housekeeping gene β-actin, based on a comparison of the Cts at constant fluorescence intensity, as per the Livak and Schmittgen's method.
2.5. Statistical analysis
Significance between the groups was analyzed using a one-way analysis of variance (ANOVA) (SPSS for Windows, Release 10.10, Standard Version, Chicago, IL, USA) followed by Tukey's post hoc t-test for multiple comparisons. All values are presented as the mean ± standard deviation, and a p-value (p < 0.05) was considered to be significant.3. Results and discussion
PS, PE, and polypropylene-based MPs are pervasive in the environment and have been mostly studied for removal of MPs.33 The average size of plastic fragments in the Korean coastal waters was approximately 200 μm;34 beads smaller than 200 μm have primarily been studied for their toxicological and pathological effects in vitro and in vivo.9,12,13,35 Hence, we focused on demonstrating coagulation-based MP removal using PS and PE beads in the range of <200 μm as models.First, we modified the surface of PS beads (90 μm) using chitosan and TA. The change in the surface morphology and chemical elements present on the surface of PS beads based on the surface modification were observed by SEM/EDX spectroscopy (Fig. 2a–d). Untreated PS beads (PS beads) exhibited a smooth surface with a diameter of ∼86.6 μm (Fig. 2a). When chitosan was used to treat PS beads (PS beads/chitosan), the diameter increased by ∼3 μm, and a thin layer of chitosan was formed on the surface (Fig. 2b). The diameter increased by ∼0.2 μm, and the surface became rough upon treating the PS beads/chitosan (PS beads/chitosan–TA or surface phenolic beads) with TA (Fig. 2c). SEM/EDX spectral analysis of the beads revealed that changes in the C element (91.08 atomic%) and O element (1.22 atomic%) of PS beads/chitosan were minimal compared with those in the C element (88.77 atomic%) and O element (2.68 atomic%) of PS beads because of the thin layering of chitosan on relatively large-sized PS beads. The O element of PS beads/chitosan–TA (26.05 atomic%) was considerably increased after TA treatment because of the relatively sufficient O element of TA (Fig. 2d). The change in surface charge of PS beads according to modification was monitored using zeta potential measurements. The zeta potential of untreated PS bead was −28.7 ± 1.7 mV owing to the presence of anionic sulfate groups on the beads but was changed to 28.0 ± 4.3 mV after treatment with positively charged chitosan, and then became −2.6 ± 0.4 mV after TA treatment (Fig. 2e). We suggest that the oppositely charged molecules, chitosan and TA, were attracted to the surface of beads in a layer-by-layer manner. Based on these characterizations, chitosan and TA were successfully layered on the surface of the PS beads.
 | ||
| Fig. 2 Scanning electron microscopy images of (a) PS beads (90 μm), (b) the surface-modified PS beads with chitosan (PS beads/chitosan), and (c) beads further treated with tannic acid (PS beads/chitosan–TA). The square regions (yellow) of the surface of the beads were magnified in the next row. (d) Atomic percentage of the surface of the beads according to the modification. (e) Zeta potential of PS beads according to the modification. | ||
To observe effect of pH on the beads and surface treatment, zeta potential of the PS beads was checked in range of pH 2–12 (Fig. S1†). At pH 2, zeta potential of untreated PS bead was −0.1 ± 1.2 mV, but was changed to 0.2 ± 0.2 mV after treatment with positively charged chitosan, and then became 2.3 ± 0.1 mV after TA treatment. We believe that the untreated PS bead is neutralized at the acidic conditions, minimize the change of zeta potential after treatment with chitosan or TA. In range of pH 6–8 (pH of drinking water), trend of zeta potential values according to surface treatment was similar to the trend in DI water shown in Fig. 2e.
To validate whether the surface treatments were successful with other types of MPs, two different-sized PE beads (106–125 μm, 45–53 μm) were used (Fig. S2 and S3†). The PE beads had a relatively smooth surface (Fig. S2a and S3a†), a thin and rougher layer was formed after chitosan treatment (Fig. S2b and S3b†), and the surface became much rougher after TA treatment (Fig. S2c and S3c†). Elemental analysis of different sized PE beads revealed that the C element in PE beads/chitosan increased after treatment of the PE beads with chitosan, and the O element in PE beads/chitosan–TA increased after treatment of the PE beads/chitosan with TA (Fig. S2d and S3d†). This trend of change in surface elements was similarly observed in case of the surface treatment of PS beads (90 μm). The zeta potential of untreated PE beads (106–125 μm, 45–53 μm) was −18.8 ± 0.4 mV and −19.2 ± 2.1 mV, respectively. After treatment with chitosan, the values increased to 67.3 ± 6.4 mV and 50.0 ± 2.4 mV, respectively. Then, the zeta potential of the beads became 6.0 ± 0.4 mV and 11.4 ± 0.3 mV, respectively, after TA treatment (Fig. S2e and S3e†). This trend of change in the zeta potential was in agreement with the change observed during the surface treatment of PS beads (90 μm).
On the surface of the phenolic PS beads (90 μm), Fe3+ markedly triggered bead precipitation within 5 min (Fig. 3a and ESI Movie 1†). Optical microscopy images revealed that untreated PS beads in the dispersion phase coagulated with the beads in the precipitates (Fig. 3b and c). As a control experiment, untreated PS beads (90 μm) were not coagulated or aggregated upon introducing Fe3+ (Fig. S4†). To investigate how TA and Fe3+ contribute to the coagulation and to identify the contributing bonds involved in coagulation, optical microscopy and UV-Vis spectroscopy were used with various concentrations of TA and Fe3+ (Fig. 3d, S5 and S6†). As the concentration of TA increased, coagulation of the beads likely occurred (Fig. 3d) because of the formation of TA–Fe3+ coordinate complexes. The coordinate complex between TA and Fe3+ was visibly formed at TA concentrations (≥0.016 mM) (Fig. S5a†). However, in the presence of PS beads, coordinate complex-driven coagulation could occur even at lower concentrations of TA (≥0.004 mM) (Fig. S5b†). We suggest that a stronger interaction among the surface phenolic groups existed rather than that existed in the presence of coordinate complexes between TA and Fe3+. Additionally, the formation of Fe3+–TA coordinate bonds was identified by UV-Vis absorption spectra (Fig. S6†). Unlike the Fe3+ aqueous solution, a new peak appeared at approximately 650 nm in the TA–Fe3+ solution, indicating characteristic metal–phenolic coordinate bonds.36–38
 | ||
| Fig. 3 (a) Photographs of a solution of PS beads (90 μm) and PS beads after adding Fe3+ to the surface phenolic beads. Optical microscopic images of (b) the untreated PS beads and (c) the PS beads after adding Fe3+, at 200× magnification. (d) Optical microscopic images of PS beads after adding Fe3+ on the surface treatment of various concentrations of tannic acid (TA), at 40× magnification. | ||
To prove the ability of the surface phenolic treatment to coagulate smaller-sized PS beads, PS beads (0.5 μm, fluorescent) were treated with chitosan and TA as described herein. After chitosan and TA treatment, the difference in surface morphology and surface elements was observed by SEM (Fig. 4a–c). Surface elemental analysis revealed that the O element (40.02 atomic%) of PS beads/chitosan increased compared with the O element (19.77 atomic%) of untreated PS beads (Fig. 4d). The dramatic increase in the O element is believed to be caused by the layering of relatively O-sufficient chitosan onto the smaller-sized PS beads. The change in the average size of the PS beads increased from 596 nm to 1324 nm after chitosan treatment and became 1910 nm after TA treatment (Fig. 4e). The zeta potential of the PS beads was negatively charged, being −35.9 ± 0.2 mV, 49.2 ± 2.6 mV after chitosan treatment, and 19.3 ± 0.1 mV after TA treatment (Fig. 4f). The trend of the change in the zeta potential was the same as that observed for the PS beads (90 μm) according to surface modification. These results confirmed that the smaller-sized PS beads (0.5 μm) could be treated with chitosan and TA as well.
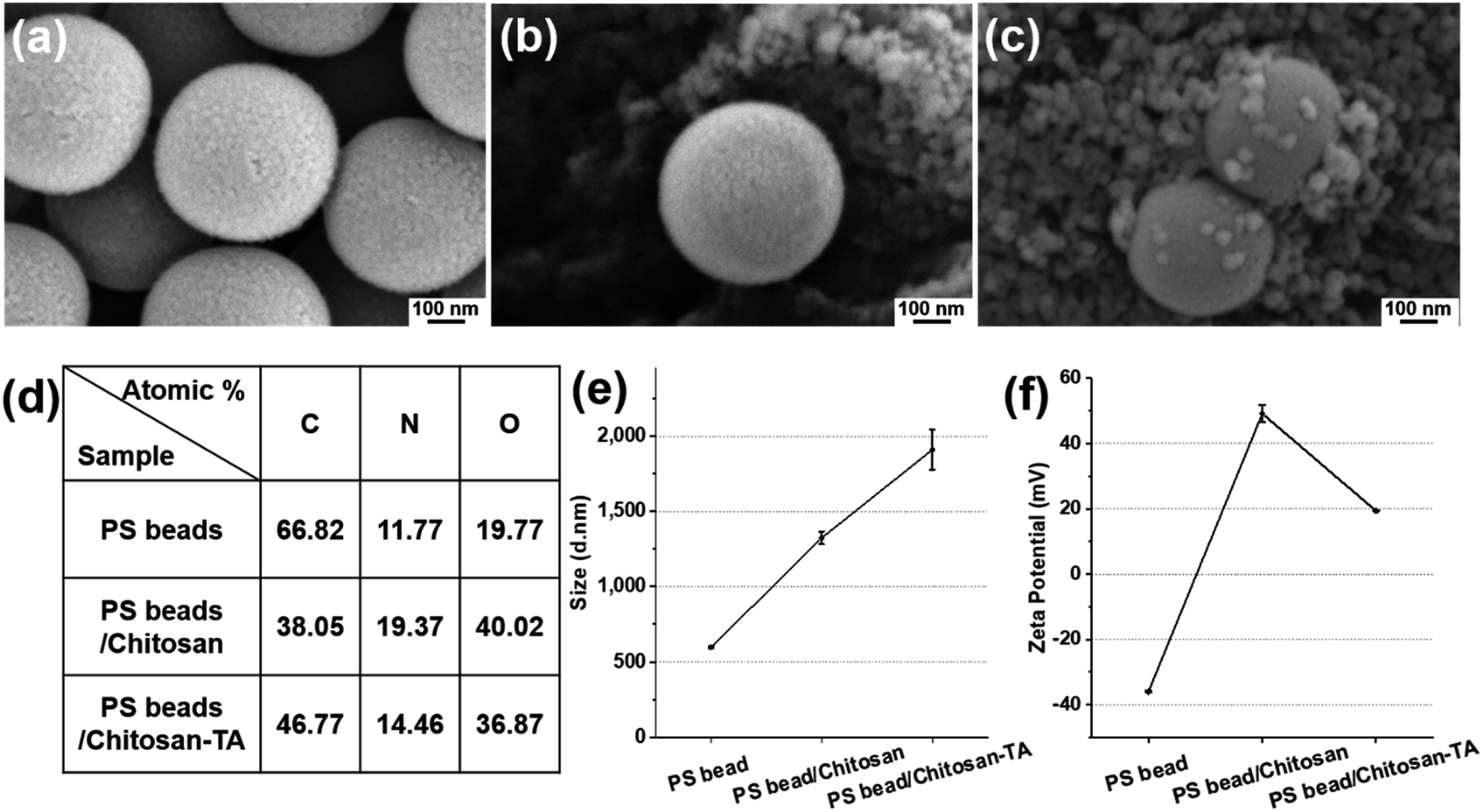 | ||
| Fig. 4 Scanning electron microscopy images of (a) PS beads (0.5 μm), (b) surface modified PS beads with chitosan (PS beads/chitosan), and (c) beads further treated with tannic acid (PS beads/chitosan–TA). (d) Atomic percentage of the surface of the beads according to the modification. (e) Size distribution of PS beads according to the modification. (f) Zeta potential of PS beads according to the modification. | ||
To observe effect of pH on the beads and surface treatment, zeta potential of the PS beads was checked in range of pH 2–12 (Fig. S7†). At pH 2, zeta potential of untreated PS bead was 0.9 ± 0.8 mV, but was changed to 0.3 ± 0.1 mV after chitosan treatment, and then became 5.6 ± 0.4 mV after TA treatment. Similar to the trend of zeta potential in PS beads (90 μm), in range of pH 6–8 (pH of drinking water), zeta potential values according to surface treatment was resembling to the trend in DI water shown in Fig. 4f.
Upon using the 0.5 μm sized fluorescent PS beads, the phase behavior of untreated beads, coagulated beads, and filtered beads in solution was observable by the naked eye (Fig. 5a) and fluorescence microscopic images (Fig. 5b–e). The untreated beads in solution (#1) presented bright fluorescence as individual beads (Fig. 5b). Once the beads were coagulated (#2), the supernatant of the bead suspension did not show fluorescence. In contrast, the fluorescent beads showed clustered/coagulated in the precipitates (Fig. 5c and d). After filtration of the coagulated suspension (#3), no noticeable fluorescent beads were observed in the solution (Fig. 5e). We hypothesized that the 0.5 μm sized individual PS beads coagulated and became clustered via TA–Fe3+ coordinate bonds; then, the coagulated PS beads were trapped in the filter paper (pore size of 11 μm).
 | ||
| Fig. 5 (a) Photographs of fluorescent PS beads (0.5 μm) according to treatment. #1: untreated beads. #2: after adding Fe3+ to the surface of phenolic beads. #3: after filtration of the coagulated beads using filter paper (11 μm of pore size). Fluorescence microscopic images of (b) sample #1, (c and d) supernatant and precipitated parts of sample #2, and (e) sample #3 at 200× magnification. (f) Reaction time dependent-removal efficiency of PS beads. The removal efficiency was calculated from the ratio of measured fluorescent intensity [1 − (fluorescent intensity of #3/fluorescent intensity of #1) × 100] of the beads. (g) Concentration of PS bead-dependent removal efficiency. (h) Sample volume-dependent removal efficiency of PS beads. Inset: photographs of filter paper of trapped coagulated beads. (i) Photographs of microplastic beads and filtered microplastic beads in cleanser before/after coagulation. | ||
The removal efficiency of MPs after purification is an important criterion in terms of time-savings, treated coagulant-savings, and purification capability over a wide range of volume scales. Herein, we conducted a quantitative analysis of the removal efficiency depending on the purification duration (or reaction time), the concentration of coagulants and MPs, and the volume of samples using fluorescent microbeads.
The removal efficiency of our coagulation-based purification of beads was compared with that of commonly used purification of MPs using coagulants, for example, Fe- and Al-salts.22,39,40 In the removal efficiency of the PS beads (0.5 μm) depending on the reaction time, the coagulation-based purification was as high as 97% compared with the efficiency of Fe-salts at 50–60% and Al-salts at 45–57% (Fig. 5f). Regarding removal efficiency according to the concentration of PS beads, the efficiency of Fe-salts and Al-salts was 52–75% and 29–57%, respectively. The coagulation-based purification used in this study was as high as 90–97% (Fig. 5g) indicating that the purification covered from high to low concentration low (0.1–0.01 mg mL−1) of MPs. At the concentration of PS beads (0.01 mg mL−1), removal efficiency of coagulation-based purification was as high as 99% compared with that of Fe-salts at 50–63% and Al-salts at 37–55% in various concentration (0.3, 0.6, 1.5 mM) of treated metal ion of coagulants (Fig. S8a†). Moreover, the removal efficiency of coagulation-based purification had a smaller standard deviation than did the Fe- and Al-salts, indicating the outstanding reproducibility and accuracy of the purification method. Regarding the removal efficiency of beads according to volume scale (1–100 mL), the coagulation-based purification efficiency was 97–99%, with a small error range, and the coagulation on the membrane was confirmed by photographs (Fig. 5h and inset). To demonstrate a practical sample of MPs, facial scrub was used to observe the successful coagulation of the original microbeads (Fig. 5i). The removal efficiency was preserved as high as 96–99% in range of pH 6–8 (pH of general drinking water), in actual water conditions including inorganic ions (NaCl, CaCl2), and even in natural organic material-rich water conditions with humic acid (Fig. S8b–d†).
Additionally, we demonstrated the coagulation of two different sized PE beads as a model of MPs. The green fluorescent beads (106–125 μm) were coagulated as precipitates by the naked eye and fluorescence microscopic images (Fig. S9a–c†). The red-colored exfoliating PE beads (45–53 μm) were precipitated by the coagulation step (Fig. S9d†). The dispersed beads were coagulated by the coordination complex (Fig. S9e and f†).
Furthermore, the effects of coagulation-based purification were examined in normal epithelial cells (IEC18 cells) derived from rat intestines because most of the PS beads accumulated in the gut of animals.9,41,42 Cell viability was maintained at a constant level in the purified PS-treated group compared with the untreated group, although a slight decrease in this level was detected in the Mid and High PS-treated groups (Fig. 6a and S10a†). The results of the present study on the cytotoxicity of PS were very similar to those of previous studies. In previous studies, some degree of cytotoxicity was detected after PS treatment. Carboxylated PS (20–100 nm), PS particles (0.1–5 μm), and PS MPs (4 μm) showed cytotoxic effects in THP-1 cells, CaCo-2 cells, and BEAS-2B cells.43–45 Additionally, oxidative stress and inflammatory responses in IEC18 cells were significantly decreased after purified PS treatment. The levels of ROS, iNOS, and COX-2 were higher in the high PS-treated group compared with the untreated group. However, these levels in the purified PS-treated group were remarkably recovered to those of the untreated group (Fig. 6b, c and S10b†). A similar pattern was observed in the expression levels of inflammatory cytokines including TNFα, IL-1, β and NF-κB, although IL-6 showed a different pattern (Fig. 6d and S10c†). These results were consistent with previous results showing inflammatory responses after PS treatment. Oxidative stress and inflammatory response were enhanced in PS particle-treated Caco-2 cells, PS MP-treated BEAS-2 cells, and PS nanoparticle-treated Hs27 cells, whereas the expression levels of inflammatory cytokines were increased in carboxylated PS-treated U937 cells and PS nanoparticle-treated A549 cells.43,44,46–48 Therefore, the above results suggest that coagulation-based purification can contribute to reducing oxidative stress and inflammatory response in IEC18 cells. Moreover, similar effects of coagulation-based purification in ICE18 cells were detected in the liver cell line (HepG2 cells), although the expression level of IL-6 mRNA showed a different pattern (Fig. S11 and S12†).
 | ||
| Fig. 6 In vitro tests of (a) IEC18 cell viability, (b) reactive oxygen species level, (c) inflammation, and (d) cytokine tests of PS beads (0.5 μm) treatment. Sample of purified PS beads was compared with low (0.01 mg mL−1), middle (0.05 mg mL−1), and high (0.1 mg mL−1) concentration of non-treated PS beads in these tests. | ||
4. Conclusions
We demonstrated that the coagulation of PS or PE beads using plant-derived tannic acid occurred within 5 min, and more than 95% of the 0.5 μm PS beads were removed by filtration. Application of this method improved the removal efficiency of MPs compared to that of commonly used coagulants, such as Fe- and Al-salts, from 40–60% at the same concentration of metal ions. The low to high volume scale samples and practical sample (microbeads in a facial cleanser) were shown to induce the coagulation of MPs and were almost completely purified by filtration. Furthermore, in vitro tests indicated that the purified water obtained from coagulation-based microbead removal was not cytotoxic and reduced oxidative stress and inflammatory cytokines in rat intestine IEC18 cells to levels similar to those in cells exposed to water without microbeads.Author contributions
The manuscript was written with contributions from all the authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP: Ministry of Science, ICT & Future Planning) (NRF-2018R1D1A1B07050070) and the BK21 FOUR Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Korea.References
- R. C. Thompson, Lost at Sea: Where Is All the Plastic?, Science, 2004, 304, 838 CrossRef CAS.
- J. Brahney, M. Hallerud, E. Heim, M. Hahnenberger and S. Sukumaran, Plastic Rain in Protected Areas of the United States, Science, 2020, 368, 1257–1260 CrossRef CAS.
- N. Evangeliou, H. Grythe, Z. Klimont, C. Heyes, S. Eckhardt, S. Lopez-Aparicio and A. Stohl, Atmospheric Transport is a Major Pathway of Microplastics to Remote Regions, Nat. Commun., 2020, 11, 3381 CrossRef CAS.
- K. Mizukawa, H. Takada, M. Ito, Y. B. Geok, J. Hosoda, R. Yamashita, M. Saha, S. Suzuki, C. Miguez and J. Frias, et al., Monitoring of a Wide Range of Organic Micropollutants on the Portuguese Coast using Plastic Resin Pellets, Mar. Pollut. Bull., 2013, 70, 296–302 CrossRef CAS.
- K. Ashton, L. Holmes and A. Turner, Association of Metals with Plastic Production Pellets in the Marine Environment, Mar. Pollut. Bull., 2010, 60, 2050–2055 CrossRef CAS.
- N. von Moos, P. Burkhardt-Holm and A. Köhler, Uptake and Effects of Microplastics on Cells and Tissue of the Blue Mussel Mytilus edulis L. after an Experimental Exposure, Environ. Sci. Technol., 2012, 46, 11327–11335 CrossRef CAS.
- D. X. Oh, E. Prajatelistia, S.-W. Ju, H. Jeong Kim, S.-J. Baek, H. Joon Cha, S. Ho Jun, J.-S. Ahn and D. Soo Hwang, A Rapid, Efficient and Facile Solution for Dental Hypersensitivity: The Tannin–Iron Complex, Sci. Rep., 2015, 5, 10884 CrossRef CAS.
- F. Ribeiro, E. D. Okoffo, J. W. O'Brien, S. Fraissinet-Tachet, S. O'Brien, M. Gallen, S. Samanipour, S. Kaserzon, J. F. Mueller and T. Galloway, et al., Quantitative Analysis of Selected Plastics in High-Commercial-Value Australian Seafood by Pyrolysis Gas Chromatography Mass Spectrometry, Environ. Sci. Technol., 2020, 54, 9408–9417 CrossRef CAS.
- Y. Deng, Y. Zhang, B. Lemos and H. Ren, Tissue Accumulation of Microplastics in Mice and Biomarker Responses suggest Widespread Health Risks of Exposure, Sci. Rep., 2017, 7, 46687 CrossRef.
- Y. He, J. Li, J. Chen, X. Miao, G. Li, Q. He, H. Xu, H. Li and Y. Wei, Cytotoxic Effects of Polystyrene Nanoplastics with Different Surface Functionalization on Human HepG2 Cells, Sci. Total Environ., 2020, 723, 138180 CrossRef CAS.
- M. Hesler, L. Aengenheister, B. Ellinger, R. Drexel, S. Straskraba, C. Jost, S. Wagner, F. Meier, H. von Briesen and C. Büchel, et al., Multi-Endpoint Toxicological Assessment of Polystyrene Nano- and Microparticles in Different Biological Models in Vitro, Toxicol. In Vitro, 2019, 61, 104610 CrossRef CAS.
- L. Lu, Z. Wan, T. Luo, Z. Fu and Y. Jin, Polystyrene Microplastics induce Gut Microbiota Dysbiosis and Hepatic Lipid Metabolism Disorder in Mice, Sci. Total Environ., 2018, 631–632, 449–458 CrossRef CAS.
- C. Q. Y. Yong, S. Valiyaveetill and B. L. Tang, Toxicity of Microplastics and Nanoplastics in Mammalian Systems, Int. J. Environ. Res. Public Health, 2020, 17, 1509 CrossRef CAS.
- T. Poerio, E. Piacentini and R. Mazzei, Membrane Processes for Microplastic Removal, Molecules, 2019, 24, 4148 CrossRef CAS.
- M. R. Michielssen, E. R. Michielssen, J. Ni and M. B. Duhaime, Fate of Microplastics and Other Small Anthropogenic Litter (SAL) in Wastewater Treatment Plants Depends on Unit Processes Employed, Environ. Sci.: Water Res. Technol., 2016, 2, 1064–1073 RSC.
- M. Lares, M. C. Ncibi, M. Sillanpää and M. Sillanpää, Occurrence, Identification and Removal of Microplastic Particles and Fibers in Conventional Activated Sludge Process and Advanced MBR Technology, Water Res., 2018, 133, 236–246 CrossRef CAS.
- A. Paço, K. Duarte, J. P. da Costa, P. S. M. Santos, R. Pereira, M. E. Pereira, A. C. Freitas, A. C. Duarte and T. A. P. Rocha-Santos, Biodegradation of Polyethylene Microplastics by The Marine Fungus Zalerion Maritimum, Sci. Total Environ., 2017, 586, 10–15 CrossRef.
- H. S. Auta, C. U. Emenike and S. H. Fauziah, Screening of Bacillus Strains Isolated from Mangrove Ecosystems in Peninsular Malaysia for Microplastic Degradation, Environ. Pollut., 2017, 231, 1552–1559 CrossRef CAS.
- A. Misra, C. Zambrzycki, G. Kloker, A. Kotyrba, M. H. Anjass, I. Franco Castillo, S. G. Mitchell, R. Güttel and C. Streb, Water Purification and Microplastics Removal Using Magnetic Polyoxometalate-Supported Ionic Liquid Phases (magPOM-SILPs), Angew. Chem., Int. Ed., 2020, 59, 1601–1605 CrossRef CAS.
- J. Grbic, B. Nguyen, E. Guo, J. B. You, D. Sinton and C. M. Rochman, Magnetic Extraction of Microplastics from Environmental Samples, Environ. Sci. Technol. Lett., 2019, 6, 68–72 CrossRef CAS.
- Y.-J. Chen, Y. Chen, C. Miao, Y.-R. Wang, G.-K. Gao, R.-X. Yang, H.-J. Zhu, J.-H. Wang, S.-L. Li and Y.-Q. Lan, Metal–Organic Framework-Based Foams for Efficient Microplastics Removal, J. Mater. Chem. A, 2020, 8, 14644–14652 RSC.
- B. Ma, W. Xue, C. Hu, H. Liu, J. Qu and L. Li, Characteristics of Microplastic Removal via Coagulation and Ultrafiltration during Drinking Water Treatment, Chem. Eng. J., 2019, 359, 159–167 CrossRef CAS.
- K. E. Lee, N. Morad, T. T. Teng and B. T. Poh, Development, Characterization and the Application of Hybrid Materials in Coagulation/Flocculation of Wastewater: A review, Chem. Eng. J., 2012, 203, 370–386 CrossRef CAS.
- N. Shirasaki, T. Matsushita, Y. Matsui and T. Marubayashi, Effect of Aluminum Hydrolyte Species on Human Enterovirus Removal from Water during the Coagulation Process, Chem. Eng. J., 2016, 284, 786–793 CrossRef CAS.
- B. Ma, W. Li, R. Liu, G. Liu, J. Sun, H. Liu, J. Qu and W. van der Meer, Multiple Dynamic Al-Based Floc Layers on Ultrafiltration Membrane Surfaces for Humic Acid and Reservoir Water Fouling Reduction, Water Res., 2018, 139, 291–300 CrossRef CAS.
- D. Elkhatib and V. Oyanedel-Craver, A Critical Review of Extraction and Identification Methods of Microplastics in Wastewater and Drinking Water, Environ. Sci. Technol., 2020, 54, 7037–7049 CrossRef CAS.
- Z. Zainuddin, Syuhada Study of Analysis Method on Microplastic Identification in Bottled Drinking Water, Macromol. Symp., 2020, 391, 1900195 CrossRef CAS.
- H. Zeng, D. S. Hwang, J. N. Israelachvili and J. H. Waite, Strong Reversible Fe3+-Mediated Bridging between Dopa-Containing Protein Films in Water, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12850–12853 CrossRef CAS.
- E. Prajatelistia, S.-W. Ju, N. D. Sanandiya, S. H. Jun, J.-S. Ahn and D. S. Hwang, Tunicate-Inspired Gallic Acid/Metal Ion Complex for Instant and Efficient Treatment of Dentin Hypersensitivity, Adv. Healthcare Mater., 2016, 5, 919–927 CrossRef CAS.
- B. J. Kim, S. Han, K.-B. Lee and I. S. Choi, Biphasic Supramolecular Self-Assembly of Ferric Ions and Tannic Acid across Interfaces for Nanofilm Formation, Adv. Mater., 2017, 29, 1700784 CrossRef.
- L. Shan, G. Gao, W. Wang, W. Tang, Z. Wang, Z. Yang, W. Fan, G. Zhu, K. Zhai and O. Jacobson, et al., Self-Assembled Green Tea Polyphenol-Based Coordination Nanomaterials to Improve Chemotherapy Efficacy by Inhibition of Carbonyl Reductase 1, Biomaterials, 2019, 210, 62–69 CrossRef CAS.
- K. H. Park, S. Y. Yang, B.-S. An, D. Y. Hwang, J. H. Lee, H. S. Kim and S. Seo, Metal Ion-Mediated Interliposomal Aggregation of Polydiacetylene Liposomes Incorporating a Phenolic Lipid, J. Nanosci. Nanotechnol., 2019, 19, 3755–3761 CrossRef CAS.
- S. A. Carr, J. Liu and A. G. Tesoro, Transport and Fate of Microplastic Particles in Wastewater Treatment Plants, Water Res., 2016, 91, 174–182 CrossRef CAS.
- Y. K. Song, S. H. Hong, S. Eo, M. Jang, G. M. Han, A. Isobe and W. J. Shim, Horizontal and Vertical Distribution of Microplastics in Korean Coastal Waters, Environ. Sci. Technol., 2018, 52, 12188–12197 CrossRef CAS.
- M. Hesler, L. Aengenheister, B. Ellinger, R. Drexel, S. Straskraba, C. Jost, S. Wagner, F. Meier, H. von Briesen and C. Büchel, et al., Multi-Endpoint Toxicological Assessment of Polystyrene Nano- and Microparticles in Different Biological Models in Vitro, Toxicol. In Vitro, 2019, 61, 104610 CrossRef CAS.
- C. Zhang, Z. Chen, Z. Xia, R. Z. Waldman, S.-L. Wu, H.-C. Yang and S. B. Darling, Ferric Tannate Photothermal Material for Efficient Water Distillation, Environ. Sci.: Water Res. Technol., 2020, 6, 911–915 RSC.
- H. Xu, J. Nishida, W. Ma, H. Wu, M. Kobayashi, H. Otsuka and A. Takahara, Competition between Oxidation and Coordination in Cross-Linking of Polystyrene Copolymer Containing Catechol Groups, ACS Macro Lett., 2012, 1, 457–460 CrossRef CAS.
- T. Liu, M. Zhang, W. Liu, X. Zeng, X. Song, X. Yang, X. Zhang and J. Feng, Metal Ion/Tannic Acid Assembly as a Versatile Photothermal Platform in Engineering Multimodal Nanotheranostics for Advanced Applications, ACS Nano, 2018, 12, 3917–3927 CrossRef CAS.
- M. Lapointe, J. M. Farner, L. M. Hernandez and N. Tufenkji, Understanding and Improving Microplastic Removal during Water Treatment: Impact of Coagulation and Flocculation, Environ. Sci. Technol., 2020, 54, 8719–8727 CrossRef CAS.
- B. Ma, W. Xue, Y. Ding, C. Hu, H. Liu and J. Qu, Removal Characteristics of Microplastics by Fe-Based Coagulants during Drinking Water Treatment, J. Environ. Sci., 2019, 78, 267–275 CrossRef.
- Y. Jin, L. Lu, W. Tu, T. Luo and Z. Fu, Impacts of Polystyrene Microplastic on the Gut Barrier, Microbiota and Metabolism of Mice, Sci. Total Environ., 2019, 649, 308–317 CrossRef CAS.
- Y.-F. Yang, C.-Y. Chen, T.-H. Lu and C.-M. Liao, Toxicity-Based Toxicokinetic/Toxicodynamic Assessment for Bioaccumulation of Polystyrene Microplastics in Mice, J. Hazard. Mater., 2019, 366, 703–713 CrossRef CAS.
- B. Prietl, C. Meindl, E. Roblegg, T. R. Pieber, G. Lanzer and E. Fröhlich, Nano-Sized and Micro-Sized Polystyrene Particles Affect Phagocyte Function, Cell Biol. Toxicol., 2014, 30, 1–16 CrossRef CAS.
- B. Wu, X. Wu, S. Liu, Z. Wang and L. Chen, Size-Dependent Effects of Polystyrene Microplastics on Cytotoxicity and Efflux Pump Inhibition in Human Caco-2 Cells, Chemosphere, 2019, 221, 333–341 CrossRef CAS.
- C.-D. Dong, C.-W. Chen, Y.-C. Chen, H.-H. Chen, J.-S. Lee and C.-H. Lin, Polystyrene Microplastic Particles: In Vitro Pulmonary Toxicity Assessment, J. Hazard. Mater., 2020, 385, 121575 CrossRef CAS.
- C.-D. Dong, C.-W. Chen, Y.-C. Chen, H.-H. Chen, J.-S. Lee and C.-H. Lin, Polystyrene Microplastic Particles: In Vitro Pulmonary Toxicity Assessment, J. Hazard. Mater., 2020, 385, 121575 CrossRef CAS.
- A. Poma, G. Vecchiotti, S. Colafarina, O. Zarivi, M. Aloisi, L. Arrizza, G. Chichiriccò and P. Di Carlo, Vitro Genotoxicity of Polystyrene Nanoparticles on the Human Fibroblast Hs27 Cell Line, Nanomaterials, 2019, 9, 1299 CrossRef CAS.
- M. Xu, G. Halimu, Q. Zhang, Y. Song, X. Fu, Y. Li, Y. Li and H. Zhang, Internalization and Toxicity: A Preliminary Study of Effects of Nanoplastic Particles on Human Lung Epithelial Cell, Sci. Total Environ., 2019, 694, 133794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09645h |
| This journal is © The Royal Society of Chemistry 2021 |