
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePseudo-octahedral nickel(II) complexes of strongly absorbing benzannulated pincer-type amido ligands: ligand-based redox and non-Aufbau electronic behaviour†
Jason D. Braun a,
Issiah B. Lozadaa,
Michael Shepitb,
Johan van Lieropbc and
David E. Herbert*ac
a,
Issiah B. Lozadaa,
Michael Shepitb,
Johan van Lieropbc and
David E. Herbert*ac
aDepartment of Chemistry, University of Manitoba, 144 Dysart Road, Winnipeg, Manitoba, Canada. E-mail: david.herbert@umanitoba.ca
bDepartment of Physics and Astronomy, University of Manitoba, 31A Sifton Rd, Winnipeg, Manitoba, Canada
cManitoba Institute for Materials, University of Manitoba, 20 Sifton Rd, Winnipeg, MB, Canada
First published on 18th January 2021
Abstract
The synthesis, structures and electronic characterization of three strongly coloured, pseudo-octahedral Ni(II) complexes supported by redox-active diarylamido ligands featuring benzannulated N-heterocyclic donor arms are reported. The S = 1 paramagnets each present two singly occupied molecular orbitals (SOMOs) identified as metal-based by density functional theory (DFT), consistent with solid-state and solution magnetism measurements. Upon applying oxidative potentials, non-Aufbau behaviour leads to the appearance of intense and well-defined absorption features extending into the near IR (NIR). The attribution of these features to the onset of aminyl radical character through ligand-based redox is corroborated by exceptionally strong intervalence charge-transfer (IVCT) transitions ascribed to electronic communication between two Namido moieties across a Ni(II) bridge.
Introduction
Redox ‘non-innocent’ ligands have long been a topic of interest owing to their ability to imbue metal coordination complexes (MCCs) with unconventional physical properties1,2 and reactivity.3,4 Of the myriad redox-active motifs explored, the inclusion of heteroatoms with lone pairs strategically within a ligand framework is a particularly useful design principle for introducing reservoirs of electron-density accessible through ligand-based electron transfer. For example, redox-active biquinone O^N^O ligands have yielded transition metal complexes of unusual oxidation states,5 often boasting impressive absorption profiles.6 While these tridentate ligands contain central amido donors, their redox-active nature centres on the catecholate/semiquinonate couple. In the context of MCCs, nitrogen-based amido lone pairs can be oxidized to give metal-coordinated aminyl radicals and represent what is by now a common example of redox ‘non-innocence’.7 Metal-coordination of aminyl radicals has been exploited to access substrate-based radical-like reactivity8,9 and interesting electronic/spectroscopic properties10 derived from mixed-valence character, including strengthened absorption in the near IR (NIR).11 Flanking aryl groups have a further stabilizing effect, and diarylamido architectures have been widely explored in this context.12,13With respect to finding spectroscopic markers of ligand-based radicals in transition metal coordination complexes, the ability of MCCs of redox-active ligands to strongly absorb visible and NIR light can be beneficial.14 Benzannulation is a common strategy for boosting the absorptive cross-sections of organic ligands and their coordination complexes.15 We have investigated the incorporation of benzannulated phenanthridine (3,4-benzoquinoline) moieties in diarylamido ligands.16 In addition to producing Pt MCCs that combine strong absorption and red-shifted phosphorescence,17,18 neutral Fe-based MCCs comprised of two such ligands show strong panchromatic absorption and nanosecond charge-transfer lifetimes in their neutral forms, along with a rich redox profile.19 Similarly strong but narrower absorption bands can also be seen with isoelectronic Co(III) analogues, as well as congeners of redox-silent Zn(II) and Ga(III) ions.20 Together with a Ni(II) variant, this series presents a gradient of dπ–pπ mixing between filled metal d and amido N(2p) orbitals. Despite this gradient in orbital character, however, the redox chemistry of the MCCs based on metals other than iron is uniformly ligand-centred, with strong intervalence charge-transfer (IVCT) bands observed in the NIR upon oxidation (Fig. 1).
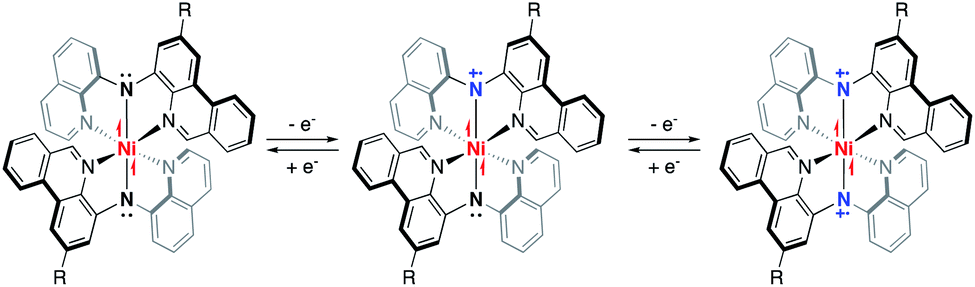 | ||
| Fig. 1 Redox-related series of paramagnetic Ni(II) complexes of phenanthridine/quinoline containing diarylamido pincer-type ligands. | ||
Compared with other common air-stable transition metal oxidation states, chromophores based on MCCs bearing d8 Ni(II) ions outside of bis-dithiolenes21 are relatively rare,22,23 especially those displaying strong NIR absorptions.24 In a notable example, a pseudo-octahedral Ni(II) complex of the non-innocent N,N′-bis(3,5-di-tert-butyl-2-hydroxy-phenyl)-1,2-phenylenediamine ligand was recently shown to exhibit very strong absorptive cross-sections well into the NIR25 thanks to the redox-active nature of the ligand. Noting the strengthened absorptive properties enabled by our readily synthesized benzannulated diarylamido ligands, we report here a series of three pseudo-octahedral nickel complexes of phenanthridine/quinoline-based diarlyamido ligands and describe the impact that ligand substitution has on their electronic structures and stabilities upon oxidation. We find that a degree of covalency to the Ni–Namido interaction strengthens the IVCT features observed. This work is meant to contribute examples of strongly absorbing MCCs to the growing literature describing electronic communication between organic fragments mediated by metal bridges.26,27
Results and discussion
Compared with quinoline and acridine, the coordination chemistry of phenanthridine-containing multidentate ligand scaffolds is much less developed.28 We previously described the preparation of (4-amino)phenanthridines amenable for incorporation into multidentate ligands, including those bearing methyl,16 tert-butyl (tBu; 1a)19 and trifluoromethyl (1b)29 substituents in the 2-position of the heterocycle. Here, we extend this methodology to include installation of a halide (Cl; 1c). Briefly, tandem Suzuki coupling/condensation reaction of 2-formylphenyl boronic acid and (2-iodo-6-nitro-4-chloro)aniline affords 1c in 65% yield (Scheme 1a). Reduction of (4-nitro-2-chloro)phenanthridine 1c to (4-amino-2-chloro)phenanthridine 1e was accomplished in a facile manner using zinc and a hydrazine/formate mixture (analogous to the preparation of 1d19). Pd-catalyzed C–N coupling of 1e with commercially available (8-bromo)quinoline gave the desired proligand ClLH in 90% isolated yield. The preparation of all three proligands RLH is summarized in Scheme 1b. Metalation with Ni(II) was achieved in refluxing methanol using NiCl2·6H2O and an exogenous base in the same manner as reported for (tBuL)2Ni.20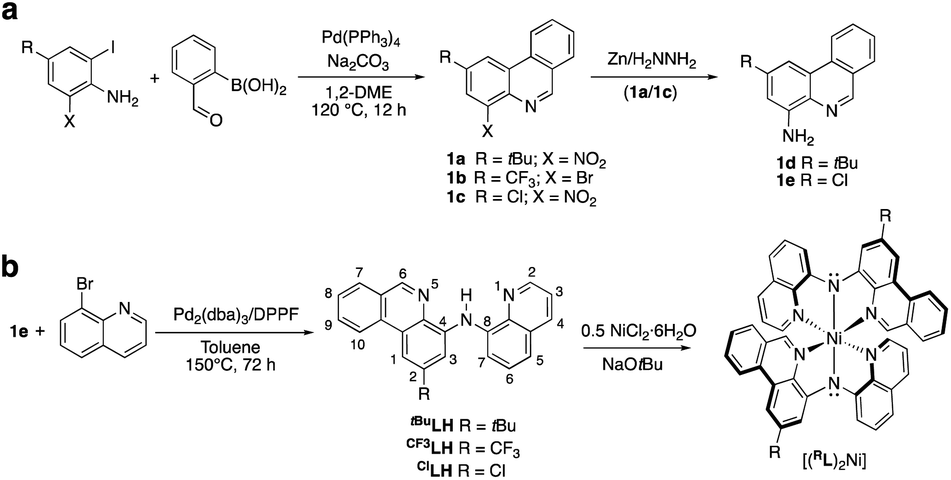 | ||
| Scheme 1 Synthesis of (a) 1a–e; and (b) proligands RLH and their (RL)2Ni complexes. See noted references for conditions for preparation of 1a, 1b, 1d, tBuLH, CF3LH and (tBuL)2Ni. The IUPAC numbering system for quinolines and phenanthridines is illustrated for RLH. | ||
The resulting complexes are deep red. All are paramagnetic and in solution present broad 1H NMR spectra covering an expanded chemical shift range of 0–45 ppm (Fig. S1 and S2†). Evans' method NMR experiments (Fig. S3 and S4†) are consistent with two unpaired electrons [μeff = 2.90 μB (tBuL)2Ni,20 2.68 μB (CF3L)2Ni, 2.97 μB (ClL)2Ni]. Solid-state SQUID magnetometry agrees with this assignment [μeff = 2.99(3) μB (tBuL)2Ni, 3.02(3) μB (CF3L)2Ni, 2.82(3) μB (ClL)2Ni; Fig. S5–S7†]. The molecular formulae of the 20-electron complexes were corroborated by high-resolution mass spectrometry (HR-MS). For the new complexes, (CF3L)2Ni and (ClL)2Ni, crystals suitable for X-ray diffraction were obtained by slow evaporation of pentane solutions. The solid-state structures show the metal in a pseudo-octahedral coordination environment with two Namido donors trans to each other (Fig. 2; see Table S1† for a summary of bond distances and angles). As expected, the shortest Ni–N contacts involve the anionic Namido nitrogens and are very similar among the three complexes ranging from 2.00–2.02 Å. Contacts between the metal and the N-heterocyclic N donor atoms are also very similar, spanning only a narrow range from 2.06–2.09 Å. In comparison, the 18-electron d6 analogue (tBuL)2Fe exhibits a tighter coordination environment with M–Namido bond lengths of ∼1.90 Å and M–Nphen/quin bond lengths of ∼1.90–1.97 Å,19 correlating with trends in ionic radii.30
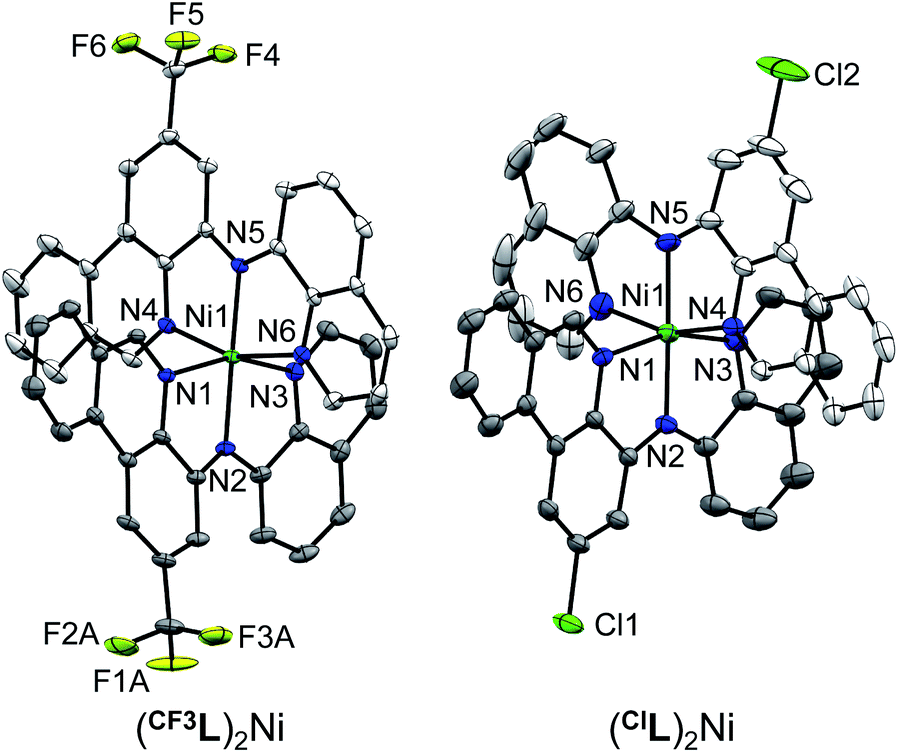 | ||
| Fig. 2 Solid-state structures of (CF3L)2Ni and (ClL)2Ni with thermal ellipsoids shown at 30% probability levels. Carbon atom labels, hydrogen atoms, disordered component to one CF3 group and co-crystallized solvent molecules have been omitted for clarity. | ||
The electronic structure of the Ni complexes was probed by electrochemical techniques using cyclic voltammetry (CV) and differential pulse voltammetry (DPV; Fig. 3). The precise onset of the broad, irreversible reduction events for the complexes is difficult to assign, but the observation of cathodic events past ∼−2.5 V vs. ferrocene/ferrocenium (FcH0/+) is consistent with ligand-based reduction, specifically at the phenanthridinyl arm.19 The oxidative chemistry of all three complexes is also similar within the series, though the identity of the substituent in the 2-position has a notable influence on both the potential and reversibility of the oxidation. These two effects do not trend together, however, as the oxidation events for (ClL)2Ni are the least reversible, though not the most anodically shifted (Table 1). As befits the electronic influence of the three substituents, the oxidation potentials shift to more positive values as the substitution on the ligand changes from electron-releasing (tBu, Eox1 = −0.23 V vs. FcH0/+) to increasingly electron-withdrawing (Cl, Eox1 = −0.19 V vs. FcH0/+; CF3, Eox1 = −0.05 V vs. FcH0/+) in accordance with their respective meta Hammett substituent constants31 (tBu = −0.10; Cl = +0.37; CF3 = +0.43). These anodic events are consistent with the formation of aminyl radical character at the Namido nitrogen atoms, stabilized to some degree by ligation to the metal.12
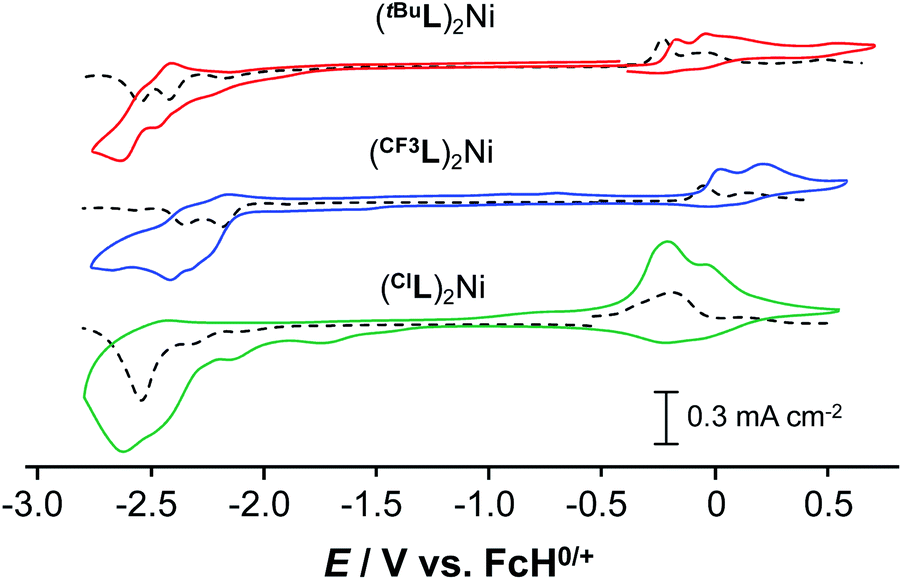 | ||
| Fig. 3 Cyclic voltammograms (—) and differential pulse voltammograms (---) of (RL)2Ni in CH3CN with 0.10 M [nBu4N][PF6] as the supporting electrolyte, using a glassy carbon working electrode. CV scan rates were 100 mV s−1. Potentials are listed vs. the FcH0/+ redox couple (FcH = ferrocene). | ||
| Compound | [(tBuL)2Ni]0/1+/2+ | [(CF3L)2Ni]0/1+/2+ | [(ClL)2Ni]0/1+/2+ |
|---|---|---|---|
| a Oxidation potentials starting from (RL)2Ni vs. Fc/FcH0/+.b Eox2 − Eox1. | |||
| Eox1a | −0.23 | −0.05 | −0.19 |
| Eox2a | −0.04 | +0.15 | +0.11 |
| ΔE1°b | 0.19 | 0.20 | 0.30 |
| λmax (RL)2Ni | 519 (28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370) 370) |
508 (28890) |
503 (27170) |
| λmax [(RL)2Ni]+ | 1802 (15740) |
1729 (14400) |
1751 (4970) |
| λmax [(RL)2Ni]2+ | 683 (101430) |
663 (59170) |
689 (19030) |
Electronic absorption spectra of the complexes in CH3CN solution are shown in Fig. 4. The deep red complexes absorb strongly peaking near 500 nm (ε = ∼30000 M−1 cm−1), with marked shifts as a function of ligand substitution. The lowest-energy peak of (tBuL)2Ni is the most red-shifted (Table 1), with the electron-releasing tBu group destabilizing the HOMO through electronic effects. In comparison, the lowest energy absorptions of (CF3L)2Ni and (ClL)2Ni are present at slightly higher energy. For these latter complexes, unlike the oxidation potential, the peak position and band edge does not track exactly with the electron-withdrawing character derived from Hammett substituent constants;31 the λmax for (ClL)2Ni is more blue-shifted than that of (CF3L)2Ni. The overall effect is inverted from what is seen for (RL)2Fe (R = CF3, tBu).19 Time-dependent DFT (TDDFT) reveals subtle functional group effects. Exchanging an electron-releasing (R = tBu) substituent for an electron withdrawing one (R = CF3, Cl), a decrease in the number of electronic excitations (fosc > 0.05) are observed within the lowest energy absorption band (E < 2.8 eV; tBu = 6,20 CF3 = 5; Cl = 4). Furthermore, configuration interactions of multiple contributing (>10%) particle-hole pairs in a given state decrease when electron-withdrawing substituents are present. These effects seem to be the origin of the discrepancy between the experimental λmax of (ClL)2Ni and (CF3L)2Ni, and their respective Hammett substituent constants. Compared with the d8 complexes investigated here, the phenanthridine substituent most strongly influences the LUMO energy in d6 Fe(II) complexes and (CF3L)2Fe (bearing the more strongly electron-withdrawing substituent on the heterocycle) absorbs to lower energy than (tBuL)2Fe.
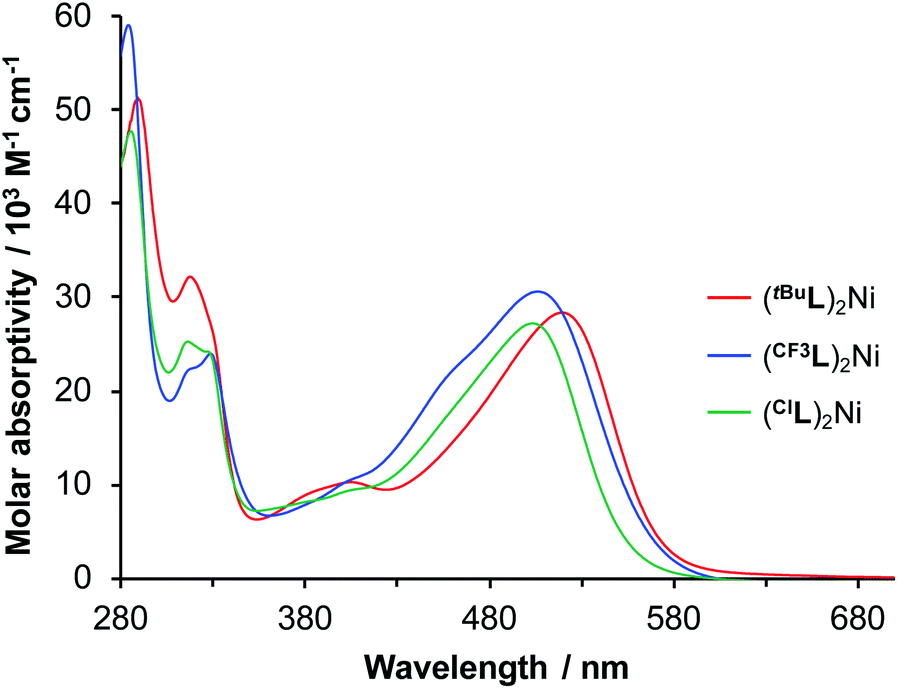 | ||
| Fig. 4 UV-vis absorption spectra of (RL)2Ni in CH3CN. | ||
Density functional theory (DFT) optimization of (CF3L)2Ni and (ClL)2Ni reproduced the structural metrics observed in the solid-state using the same basis set and method reported for (tBuL)2Ni20 (Table S2;† SMD-O3LYP/6-31+G(d,p)). Consistent with the observed trends in the Ni–N distances in the solid-state, DFT predicts shorter Ni–Namido distances compared to the Ni–Nphen/Ni–Nquin separations, with minimal differences between the Ni–Nphen and Ni–Nquin values. The electronic structures optimized in the triplet spin-state corroborate the Evans' method and solid-state magnetometry results. For both (CF3L)2Ni and (ClL)2Ni, spin density plots show localization of the unpaired electrons at the metal (Fig. S8†). Accordingly, the two singly occupied molecular orbitals (SOMOs) are predicted by DFT to be metal-based, primarily of dz2 and dx2–y2 character, with fragment contributions to the respective orbitals by Ni of 80% and 84%, respectively (Table S3†), as befits a pseudo-octahedral ligand field and a d8 Ni(II) ion.32
An orbital energy level diagram showing the two SOMOs, the next three highest energy fully occupied MOs, and the lowest unoccupied molecular orbitals (LUMOs) of (CF3L)2Ni is shown in Fig. 5. The LUMO and LUMO+1 are both low-lying ligand-based π* orbitals with significant contributions from the (H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nphen subunit of the phenanthridinyl arm of the benzannulated ligand framework (26% for the LUMO; see Table S3† for population analysis). Thus, RL retains its strong electron-accepting character. In related Ni(II) complexes of pyrazolyl diarylamido ligands27 the metal centre was described as an isolated Ni(II) ion sitting within an N6 coordination environment, only interacting weakly with filled ligand π-type orbitals. While the filled–filled interactions between the metal dπ and amido pπ-type orbitals is not as strong as for the analogous Fe(II) complexes19 there a non-negligible contribution to the SOMO from the Namido units, and metal participation in the next highest-lying filled MOs (labelled nN,amπ in Fig. 5; see Table S3† for population analysis). Electron-withdrawing substituents (R = CF3, Cl) on the phenanthridinyl arms stabilize the energy of the Namido orbitals relative to the electron releasing tBu-substituent but their characters are unperturbed, again evident from the population analysis. These are consistent with the experimental redox potentials (Table 1) and assigned aminyl radical character.
Nphen subunit of the phenanthridinyl arm of the benzannulated ligand framework (26% for the LUMO; see Table S3† for population analysis). Thus, RL retains its strong electron-accepting character. In related Ni(II) complexes of pyrazolyl diarylamido ligands27 the metal centre was described as an isolated Ni(II) ion sitting within an N6 coordination environment, only interacting weakly with filled ligand π-type orbitals. While the filled–filled interactions between the metal dπ and amido pπ-type orbitals is not as strong as for the analogous Fe(II) complexes19 there a non-negligible contribution to the SOMO from the Namido units, and metal participation in the next highest-lying filled MOs (labelled nN,amπ in Fig. 5; see Table S3† for population analysis). Electron-withdrawing substituents (R = CF3, Cl) on the phenanthridinyl arms stabilize the energy of the Namido orbitals relative to the electron releasing tBu-substituent but their characters are unperturbed, again evident from the population analysis. These are consistent with the experimental redox potentials (Table 1) and assigned aminyl radical character.
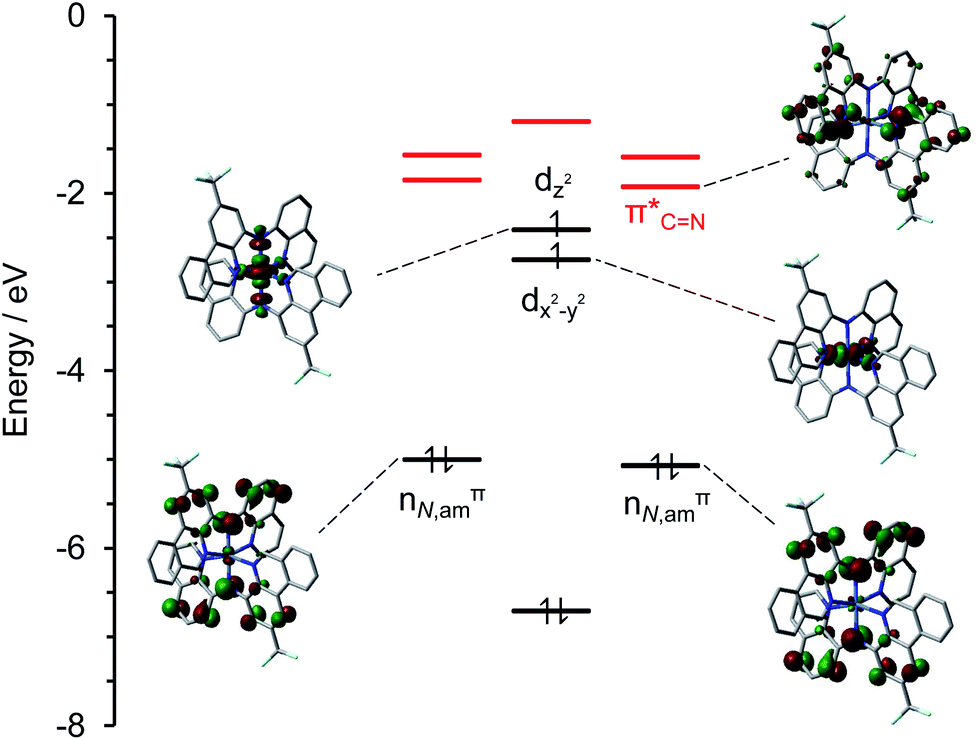 | ||
| Fig. 5 Selected ground-state restricted open-shell molecular orbital isosurfaces (isovalue = 0.04) and energy diagram for (CF3L)2Ni (SMD-roPBE0/6-31+G(d,p)//SMD-uO3LYP/6-31+G(d,p)). Vacant, acceptor ligand-based π* orbitals are highlighted in red. Fragment contributions to selected MOs are tabulated in Table S3.† | ||
The MCCs were next characterized using spectroelectrochemistry to ascertain the spectroscopic response to oxidation. Upon single-electron oxidation (Fig. 6a, c and e), the lowest energy absorption band for each of (RL)2Ni at ∼500 nm attenuates rapidly, with the growth of a new peak at ∼700 nm and a considerably broader, lower energy NIR absorption at ∼1800 nm (FWHM ∼ 3000 cm−1), typical of oxidized metal-bridged diarylamines.33 The strong absorptivity values (ε = ∼16000 M−1 cm−1 for [(tBuL)2Ni]+ and [(CF3L)2Ni]+, ∼5000 M−1 cm−1 for [(ClL)2Ni]+) and position of this new absorption are consistent with an intervalence charge-transfer (IVCT) transition from one Namido lone pair to the aminyl radical on the opposing ligand, mediated through the Ni metal centre.11 A structurally distinct square-planar Ni(II) dye with a similarly intense but narrower absorption at ∼1000 nm has been described.24 That complex also contained a ligand-based radical, localized on a 3,5-(dimedonyl)azadiisoindomethene scaffold. For that system, reduction of the paramagnetic complex to a neutral anion brought about the appearance of an even stronger absorption at 748 nm and disappearance of the NIR absorption at 1008 nm. The NIR peaks attenuated upon oxidation. Here, the second oxidation of (RL)2Ni to [(RL)2Ni]2+ results in the disappearance of the low-energy peak but continued growth of the strong peak at ∼700 nm (Fig. 6b, d and f). This is consistent with two completely oxidized Namido fragments, and a blue colour could be seen in the spectroelectrochemical cell. Compared with (tBuL)2Ni and (CF3L)2Ni, (ClL)2Ni initially appears to follow the same trend, however, each new peak observed is much smaller in intensity. Also, particulate matter could be observed deposited in the cell following the experiment. The degradation of (ClL)2Ni upon oxidation is the likely cause of the difference in peak intensity and corroborates the electrochemical irreversibility seen in the CV of (ClL)2Ni compared to (tBuL)2Ni and (CF3L)2Ni.
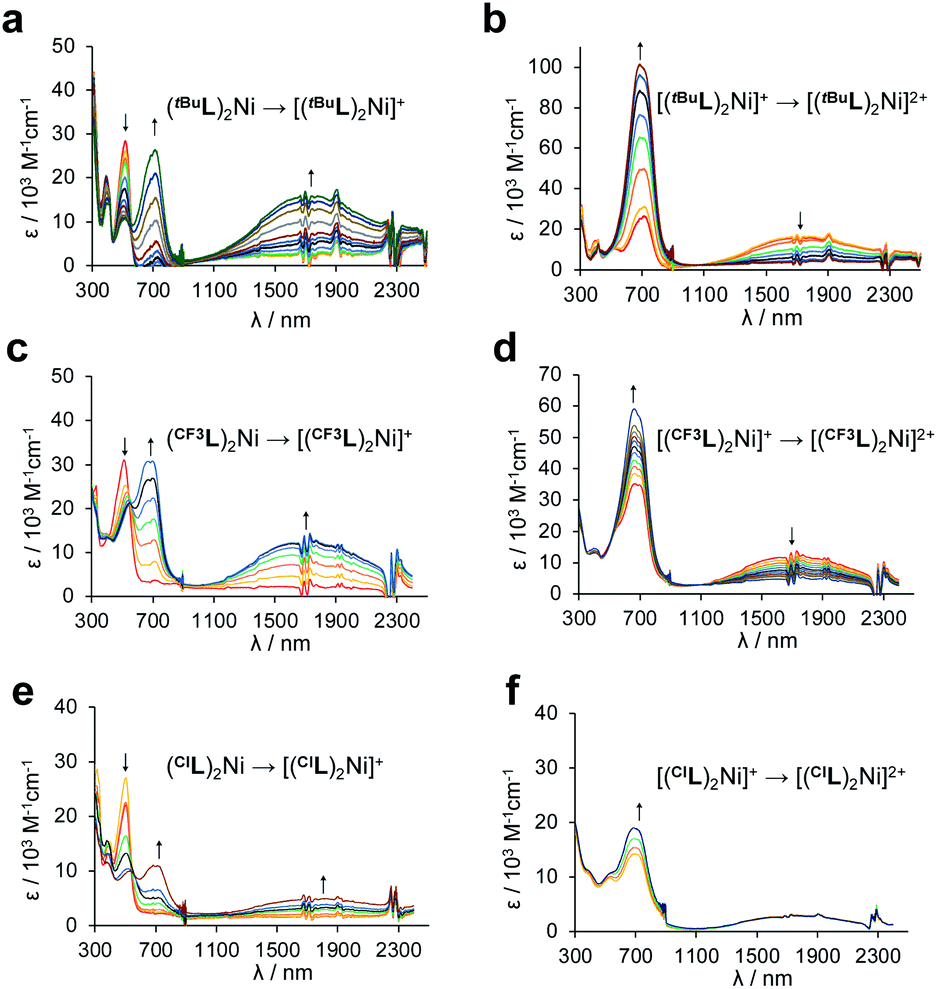 | ||
| Fig. 6 UV-Vis/NIR absorption spectra 0.3 M [nBu4N][PF6] CH3CN solution of (a) (tBuL)2Ni, oxidative potentials applied from −0.1 to 0.35 V; (b) (tBuL)2Ni, oxidative potentials applied from 0.35 to 1.4 V; (c) (CF3L)2Ni, oxidative potentials applied from −0.3 to 0.8 V; (d) (CF3L)2Ni, oxidative potentials applied from 0.8 to 1.4 V; (e) (ClL)2Ni, oxidative potentials applied from −0.1 to 0.7 V; and (f) (ClL)2Ni, oxidative potentials applied from 0.7 to 1.3 V. All potentials referenced to Ag/Ag+. | ||
The electrochemical and spectroelectrochemical data together reveal a degree of metal-mediated electronic communication between the Namido fragments in the three complexes. Observation of a second separated, though irreversible, oxidation in the CV, and the presence of IVCT bands, support this claim and negate the competing possibility of coulombic effects.27 Similar observations have recently been made of Ni complexes supported by sterically hindered salen ligands,34 and more comparably, for pseudo-octahedral Ga(III) complexes of di(2-3R-pyrazolyl)-p-arylamine ligands.26 Upon exchanging Ga(III) for Ni(II) in that ligand environment, the electronic communication is significantly enhanced owing to strengthened (although still relatively weak) dπ–pπ interactions between Ni d orbitals and amido p orbitals.27 A similar degree of mixing appears to operate in [(RL)2Ni]0/1+/2+, with strong IVCT features observed for [(RL)2Ni]+.
Accordingly, quantitative analysis of the electronic relationship between the electroactive Namido centres can be carried out using classical Marcus35 theory along with Hush36 relations (Table 2; see equations in ESI†). Comproportionation constants (Kcom) gleaned from the electrochemical data suggest that all three complexes exhibit Robin-Day Class II behavior.37 Spectroelectrochemical data supports a Class IIB assignment for (CF3L)2Ni (Hab = 2432 cm−1), very similar to that reported for (tBuL)2Ni (Table 2).20 Though (ClL)2Ni has a larger Kcom value (1.33 × 105) than (tBuL)2Ni or (CF3L)2Ni, likely due to exhibiting the most irreversible oxidations, IVCT analysis suggests electronic communication in the Robin-Day Class IIA regime (Hab = 1703 cm−1; Table 2). However, in light of the aforementioned decomposition of (ClL)2Ni under conditions of spectroelectrochemical analysis, Class IIB assignment may be more appropriate. Comparing these complexes to the homoleptic Ga and Ni complexes of pyrazolyl diarlyamido ligands mentioned above,26,27 ε values nearing 20 times greater are observed in the NIR region of the absorption spectra.
| Compound | [(tBuL)2Ni]+ | [(CF3L)2Ni]+ | [(ClL)2Ni]+ |
|---|---|---|---|
| a Value for extinction coefficient may be artificially low due to decomposition. See text for full discussion of implications for Class IIB vs. IIA assignment.b Distance (Å) between electroactive Namido centres.c Average value from DFT-optimized structure of [(tBuL)2Ni]+ [S = 3/2; SMD-uPBE0/6-31+G(d,p)]. Due to the similarities of the other complexes to [(tBuL)2Ni]+, DFT was not run for these complexes. Note: very small differences were observed between calculated bond distances of [(tBuL)2Ni] and [(tBuL)2Ni]+.d Kcom = e(ΔEF/RT), T = 295 K; kB = 0.695 cm−1 K−1; h = 3.36 × 10−11 cm−1 s; T = 295 K. | |||
| EOP = λ (cm−1) | 5549 | 5784 | 5711 |
| εmax (M−1 cm−1) | 15740 |
14400 |
4970a |
| Δν1/2 (cm−1) | 3051 | 2781 | 3998 |
| Δν1/2 HTL (cm−1) | 3580 | 3627 | 3604 |
| θ = Δν1/2/Δν1/2 (HTL) | 0.852 | 0.767 | 1.109 |
| d (Å)b | 4.055c | 4.055c | 4.055c |
| Hab (cm−1) | 2609 | 2432 | 1703 |
| α = 2Hab/λ | 0.940 | 0.841 | 0.596 |
| ΔG* (cm−1) | 4.936 | 36.58 | 232.6 |
| ket (s−1) | 2.065 × 1015 | 1.505 × 1015 | 2.859 × 1014 |
| Kcomd | 1.76 × 103 | 2.61 × 103 | 1.33 × 105 |
| Class IIB | Class IIB | Class IIBa | |
The MO diagram, spin-density maps and magnetism are all consistent with a paramagnetic d8 Ni(II) ion in an octahedral ligand field. Yet, the spectroelectrochemical response suggests formation of a ligand-based radical, supported by the covalency implied by the strong IVCT features for the oxidized species. Similar non-Aufbau behaviour has been reported for vanadium porphyrinate and phthalocyanines,38 as well as for closely related pseudo-octahedral Ni complexes,27 where the minimization of the spin-pairing energy favours removal of an electron from a lower-lying Namido lone-pair orbital rather than from a higher energy metal-based SOMO. Accordingly, time-dependent DFT (TDDFT) simulations (Fig. S9–S12†) identify three to five electronic transitions with significant oscillator strengths (fosc > 0.05; Tables S6 and S7†) in the lowest energy band (E < 3 eV) of the experimental UV-vis spectra as ligand-to-ligand charge-transfer (LLCT) from the Namido lone pair to the LUMO, rather than involving the metal-based SOMOs, further supporting the non-Aufbau assignment. In all cases, the acceptor orbital (LUMO) is mainly localized on the phenanthridine imine-like CN unit.
Conclusions
The use of a benzannulated N^N−^N tridentate diarylamido ligand has afforded deeply coloured pseudo-octahedral d8 Ni complexes that exhibit non-Aufbau behaviour in their electronic structures. Introducing highly energetic Namido-based lone pairs into the frontier orbital manifold gives rise to several ligand-derived transitions with significantly large epsilon values, ascribable to the extended π system offered by the phenanthridine/quinoline donors.Incorporating the synthetically tunable phenanthridine moiety into this ligand scaffold furthermore allowed us to study the impact of ligand substitution on the electrochemical and absorptive properties of these complexes and their oxidized analogues. We find that although the electronic properties of the neutral complexes are quite similar, they differ upon oxidation in terms of oxidative potential and the stability of the radical species evidenced by spectroelectrochemical data. The exceptionally strong IVCT bands observed in (tBuL)2Ni and (CF3L)2Ni are on the order of 20× more intense than previous examples of electronic communication of oxidized diarylamido N(2p) lone pairs across paramagnetic Ni bridges27 in an octahedral geometry, revealing the impact of the benzannulated ligand framework.
Experimental
Materials
Unless otherwise specified, air-sensitive manipulations were carried either in a N2 filled glove box or using standard Schlenk techniques under Ar. 2-Formylphenyl boronic acid, N-iodosuccinimide (AK Scientific); Pd(PPh3)4; Pd2(dba)3, (1,1′-diphenylphosphino)ferrocene (dppf), sodium tert-butoxide (NaOtBu), hydrazine hydrate (Sigma Aldrich), 4-chloro-2-nitronaniline (Combi-Blocks), Na2CO3, zinc, formic acid, and NiCl2·6H2O (Alfa Aesar) were used as purchased.Organic solvents were dried and distilled using appropriate drying agents prior to use. 1- and 2D NMR spectra were recorded on Bruker Avance 300 MHz or Bruker Avance – III 500 MHz spectrometers. 1H and 13C{1H} NMR spectra were referenced to residual solvent peaks. Elemental analyses were performed at the University of Manitoba using a PerkinElmer 2400 Series II CHNS/O Elemental Analyzer. tBuLH and CF3LH were synthesized according to literature procedures.19
Instrumentation and methods
For electrochemical analysis, 5–10 mg of each compound investigated was dissolved in 15 mL of 0.1 M [nBu4N][PF6] in CH3CN and purged with Ar for 20 minutes before analysis. All electrochemical experiments were conducted under inert (Ar) atmosphere using a CHI 760c bipotentiostat, a 3 mm diameter glassy carbon working electrode, a Ag/Ag+ quasi-non-aqueous reference electrode separated by a Vycor tip, and a Pt wire counter electrode. Cyclic voltammetry (CV) experiments were conducted using scan rates of 50–800 mV s−1. Differential pulse voltammetry (DPV) experiments were also conducted, using a 5 mV increment, 50 mV amplitude, 0.1 s pulse width, 0.0167 s sample width, and 0.5 s pulse period. Upon completion of all CV and DPV analyses, ferrocene (FcH) was added to the solution as an internal standard, with all potentials reported versus the FcH0/+ redox couple. Magnetic measurements were performed using a Quantum Design MPMS-XL SQUID magnetometer. Samples were mounted in NMR Suprasil quartz tubes. Susceptibility was measured on cooling from 300–2 K, in an applied field of 0.1 T. After reaching 2 K, magnetization measurements were performed from 0–5 T. Magnetic moments (μeff) were obtained by fitting 1/χ using a modified Curie–Weiss law, with a subtraction performed on χ accounting for the diamagnetic components arising from the ligands using Pascal's method.39Synthesis
040), 317 (22310), 329 (24010), 407 (sh), 455 (sh), 508 nm (28890 M−1 cm−1). μeff (Evans' method) = 2.68 μB. MS (ESI-TOF/MS, m/z) calcd for C46H26F6N6Ni [M]+, 834.1471; found 834.1482.600), 316 (25220), 406 (sh), 503 nm (27170 M−1 cm−1). μeff (Evans' method) = 2.97 μB. MS (ESI-TOF/MS, m/z) calcd for C44H26Cl2N6Ni [M]+, 766.0944; found 766.0963.X-ray crystallography
X-ray crystal structure data was collected in each case from a multi-faceted crystal of suitable size and quality selected from a representative sample of crystals of the same habit using an optical microscope. The crystals were mounted on MiTiGen loops and data collection carried out in a cold stream of nitrogen (150 K; Bruker D8 QUEST ECO; Mo Kα radiation). All diffractometer manipulations were carried out using Bruker APEX3 software.41 Structure solution and refinement was carried out using XS, XT and XL software, embedded within Olex2.42 For each structure, the absence of additional symmetry was confirmed using ADDSYM incorporated in the PLATON program.43![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 11.9342(6) Å, b = 14.6616(7) Å, c = 16.6289(8) Å, α = 113.683(2)°, β = 90.763(2)°, γ = 113.819(2)°, V = 2383.5(2) Å3; Z = 2, ρcalcd = 1.365 g cm−3; crystal dimensions 0.270 × 0.150 × 0.110 mm; 2θmax = 55.058°; 68060 reflections, 10937 independent (Rint = 0.0580), intrinsic phasing; absorption coeff (μ = 0.476 mm−1), absorption correction semi-empirical from equivalents (SADABS); refinement (against Fo2) with SHELXTL V6.1, 654 parameters, 0 restraints, R1 = 0.0511 (I > 2σ) and wR2 = 0.1191 (all data), Goof = 1.046, residual electron density 0.72/−0.66 Å−3.087.1(16) Å3; Z = 16, ρcalcd = 1.269 g cm−3; crystal dimensions 0.270 × 0.180 × 0.140 mm; 2θmax = 50.000°; 183624 reflections, 7058 independent (Rint = 0.0960), intrinsic phasing; absorption coeff (μ = 0.653 mm−1), absorption correction semi-empirical from equivalents (SADABS); refinement (against Fo2) with SHELXTL V6.1, 478 parameters, 0 restraints, R1 = 0.0592 (I > 2σ) and wR2 = 0.1390 (all data), Goof = 1.040, residual electron density 0.65/−0.84 Å−3. Several pentane solvent molecules were found to be disordered and could not be modeled successfully. The SQUEEZE protocol embedded in PLATON software was used to mask four solvent voids of 899 Å3 containing 215 e− each.
; a = 11.9342(6) Å, b = 14.6616(7) Å, c = 16.6289(8) Å, α = 113.683(2)°, β = 90.763(2)°, γ = 113.819(2)°, V = 2383.5(2) Å3; Z = 2, ρcalcd = 1.365 g cm−3; crystal dimensions 0.270 × 0.150 × 0.110 mm; 2θmax = 55.058°; 68060 reflections, 10937 independent (Rint = 0.0580), intrinsic phasing; absorption coeff (μ = 0.476 mm−1), absorption correction semi-empirical from equivalents (SADABS); refinement (against Fo2) with SHELXTL V6.1, 654 parameters, 0 restraints, R1 = 0.0511 (I > 2σ) and wR2 = 0.1191 (all data), Goof = 1.046, residual electron density 0.72/−0.66 Å−3.087.1(16) Å3; Z = 16, ρcalcd = 1.269 g cm−3; crystal dimensions 0.270 × 0.180 × 0.140 mm; 2θmax = 50.000°; 183624 reflections, 7058 independent (Rint = 0.0960), intrinsic phasing; absorption coeff (μ = 0.653 mm−1), absorption correction semi-empirical from equivalents (SADABS); refinement (against Fo2) with SHELXTL V6.1, 478 parameters, 0 restraints, R1 = 0.0592 (I > 2σ) and wR2 = 0.1390 (all data), Goof = 1.040, residual electron density 0.65/−0.84 Å−3. Several pentane solvent molecules were found to be disordered and could not be modeled successfully. The SQUEEZE protocol embedded in PLATON software was used to mask four solvent voids of 899 Å3 containing 215 e− each.Computational methods
All calculations were performed using Gaussian 16, Rev. B01 (ref. 44) with unrestricted Kohn–Sham DFT (UKS), unless otherwise mentioned, and the solvent-model based on density (SMD, solvent = CH3CN) to account for implicit-solvent effects.45 Ground triplet state geometries for (CF3L)2Ni and (ClL)2Ni were optimized with the O3LYP functional46 and 6-31+G(d,p) basis sets on all atoms.47–51 Restricted open-shell DFT (ROKS) single point calculation, with the PBE0 functional52 and the 6-31+G(d,p) basis set on all atoms, at the optimized triplet ground state geometry of (CF3L)2Ni was performed to maximally pair the α- and β-electrons and determine the relative energies of the MOs. Time-dependent DFT (TDDFT) calculations were carried out with the same PBE0 functional and the 6-31+G(d,p) basis set on all atoms on (CF3L)2Ni and (ClL)2Ni. Ground state molecular orbital (isosurface value = 0.04) and spin density isosurfaces (isosurface value = 0.004) were generated using Gaussview,53 while electron-hole density maps of select excited states (isosurface value = 0.002) were generated using Multiwfn.54 TDDFT calculated vertical excitations (f > 0.05) and simulated spectra (FWHM = 3000 cm−1) were generated with GaussSum.55Author contributions
JDB and DEH conceived of the project. JDB conducted all experimental work. IBL carried out computational analysis. MS and JvL conducted magnetism experiments. JDB, IBL and DEH analysed all data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Sciences Engineering Research Council of Canada for a Discovery Grant to D. E. H. (RGPIN-2014-03733) and J. v. L. (NSERC RGPIN-2018-05012); the Canadian Foundation for Innovation and Research Manitoba for an award in support of an X-ray diffractometer (CFI #32146); the University of Manitoba for the Bert & Lee Friesen Graduate Scholarship (I. B. L.) and GETS/SEGS support (J. D. B., I. B. L.); and Compute Canada for access to computational resources. Prof. Viktor N. Nemykin is thanked for access to spectroelectrochemical equipment and helpful discussions.References
- P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- S. K. Larsen and C. G. Pierpont, J. Am. Chem. Soc., 1988, 110, 1827–1832 CrossRef CAS.
- E. P. Ivakhnenko, A. G. Starikov, V. I. Minkin, K. A. Lyssenko, M. Yu. Antipin, V. I. Simakov, M. S. Korobov, G. S. Borodkin and P. A. Knyazev, Inorg. Chem., 2011, 50, 7022–7032 CrossRef CAS.
- R. Gross and W. Kaim, Inorg. Chem., 1987, 26, 3596–3600 CrossRef CAS.
- T. Büttner, J. Geier, G. Frison, J. Harmer, C. Calle, A. Schweiger, H. Schönberg and H. Grützmacher, Science, 2005, 307, 235–238 CrossRef.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Dalton Trans., 2007, 1552–1566 RSC.
- W. Kaim, Coord. Chem. Rev., 2019, 255, 2503–2513 CrossRef.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS.
- A. Rajput, A. K. Sharma, S. K. Barman, D. Koley, M. Steinert and R. Mukherjee, Inorg. Chem., 2014, 53, 36–48 CrossRef CAS.
- T. Storr and R. Mukherjee, Inorg. Chem., 2018, 57, 9577–9579 CrossRef CAS.
- K. Hanson, L. Roskop, P. I. Djurovich, F. Zahariev, M. S. Gordon and M. E. Thompson, J. Am. Chem. Soc., 2010, 132, 16247–16255 CrossRef CAS.
- P. Mandapati, P. K. Giesbrecht, R. L. Davis and D. E. Herbert, Inorg. Chem., 2017, 56, 3674–3685 CrossRef CAS.
- P. Mandapati, J. D. Braun, C. Killeen, R. L. Davis, J. A. G. Williams and D. E. Herbert, Inorg. Chem., 2019, 58, 14808–14817 CrossRef CAS.
- P. Mandapati, J. D. Braun, I. B. Lozada, J. A. G. Williams and D. E. Herbert, Inorg. Chem., 2020, 59, 12504–12517 CrossRef CAS.
- J. D. Braun, I. B. Lozada, C. Kolodziej, C. Burda, K. M. E. Newman, J. van Lierop, R. L. Davis and D. E. Herbert, Nat. Chem., 2019, 11, 1144–1150 CrossRef CAS.
- J. D. Braun, I. B. Lozada and D. E. Herbert, Inorg. Chem., 2020, 59, 17746–17757 CrossRef CAS.
- B. Garreau-de Bonneval, K. I. M.-C. Ching, F. Alary, T.-T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- N. D. Fenton and M. Gerloch, Inorg. Chem., 1990, 29, 3726–3733 CrossRef CAS.
- M. V. Andreocci, G. Mattogno, R. Zanoni, P. Giannoccaro and G. Vasapollo, Inorg. Chim. Acta, 1982, 63, 225–231 CrossRef CAS.
- E. A. Makarova, Y. V. Zatsikha, K. M. E. Newman, V. K. Paidi, V. A. Beletsky, J. van Lierop, E. A. Lukyanets and V. N. Nemykin, Inorg. Chem., 2017, 56, 6052–6055 CrossRef CAS.
- E. Salojärvi, A. Peuronen, M. Lahtinen, H. Huhtinen, L. S. Vlasenko, M. Lastusaari and A. Lehtonen, Molecules, 2020, 25, 2531 CrossRef.
- B. J. Liddle, S. Wanniarachchi, J. S. Hewage, S. V. Lindeman, B. Bennett and J. R. Gardinier, Inorg. Chem., 2012, 51, 12720–12728 CrossRef CAS.
- J. S. Hewage, S. Wanniarachchi, T. J. Morin, B. J. Liddle, M. Banaszynski, S. V. Lindeman, B. Bennett and J. R. Gardinier, Inorg. Chem., 2014, 53, 10070–10084 CrossRef CAS.
- R. Mondal, P. K. Giesbrecht and D. E. Herbert, Polyhedron, 2016, 108, 156–162 CrossRef CAS.
- P. Mandapati, J. D. Braun, B. K. Sidhu, G. Wilson and D. E. Herbert, Organometallics, 2020, 39, 1989–1997 CrossRef CAS.
- A. F. Wells, Structural Inorganic Chemistry, Clarendon Press; Oxford University Press, Oxford, 5th edn, 1984 Search PubMed.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
- H. Basch, A. Viste and H. B. Gray, J. Chem. Phys., 1966, 44, 10–19 CrossRef CAS.
- M. Parthey and M. Kaupp, Chem. Soc. Rev., 2014, 43, 5067–5088 RSC.
- F. Thomas, Dalton Trans., 2016, 45, 10866–10877 RSC.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391–444 CAS.
- P. Zanello, F. Fabrizi de Biani and C. Nervi, in Inorganic Electrochemistry, Royal Society of Chemistry, Cambridge, 2007, pp. 159–216 Search PubMed.
- B. L. Westcott, N. E. Gruhn, L. J. Michelsen and D. L. Lichtenberger, J. Am. Chem. Soc., 2000, 122, 8083–8084 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- W. Dohle, A. Staubitz and P. Knochel, Chem.–Eur. J., 2003, 9, 5323–5331 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- A. J. Cohen and N. C. Handy, Mol. Phys., 2001, 99, 607–615 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Multi-nuclear NMR and HR-MS spectra; magnetism plots, DFT and TD-DFT tables and plots, and crystallographic information files containing all X-ray data. CCDC 2044311 and 2044312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10761a |
| This journal is © The Royal Society of Chemistry 2021 |