
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Me2NH2)10[H2-Dodecatungstate] polymorphs: dodecatungstate cages embedded in a variable dimethylammonium cation + water of crystallization matrix†
György Lendvay a,
Eszter Majzikab,
Laura Bereczkia,
Attila Domjána,
László Trifa,
István E. Sajóc,
Fernanda Paiva Franguelli*ab,
Attila Farkasd,
Szilvia Kléberta,
Petra Bombicza,
Csaba Németha,
Imre Miklós Szilágyib and
László Kótai*ae
a,
Eszter Majzikab,
Laura Bereczkia,
Attila Domjána,
László Trifa,
István E. Sajóc,
Fernanda Paiva Franguelli*ab,
Attila Farkasd,
Szilvia Kléberta,
Petra Bombicza,
Csaba Németha,
Imre Miklós Szilágyib and
László Kótai*ae
aInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Budapest, H-1117, Hungary. E-mail: kotai.laszlo@ttk.mta.hu
bUniversity of Technology and Economics, Department of Inorganic and Analytical Chemistry, Budapest, H-1111, Hungary
cUniversity of Pécs, János Szentágothai Research Centre, Pécs, H-7624, Hungary
dUniversity of Technology and Economics, Department of Organic Chemistry, Budapest, H-1111, Hungary
eDeuton-X Ltd., Selmeci u. 89, Érd, H-2030, Hungary
First published on 19th January 2021
Abstract
Two polymorphs and a solvatomorph of a new dimethylammonium polytungstate—decakis(dimethylammonium) dihydrogendodecatungstate, (Me2NH2)10(W12O42)·nH2O (n = 10 or 11)—have been synthesized. Their structures were characterized by single-crystal X-ray diffraction and solid-phase NMR methods. The shape of the dodecatungstate anions is essentially the same in all three structures, their interaction with the cations and water of crystallization, however, is remarkably variable, because the latter forms different hydrogen-bonded networks, and provides a highly versatile matrix. Accordingly, the N–H⋯O and C–H⋯O hydrogen bonds are positioned in each crystal lattice in a variety of environments, characteristic to the structure, which can be distinguished by solid-state 1H-CRAMPS, 13C, 15N CP MAS and 1H–13C heteronuclear correlation NMR. Thermogravimetry of the solvatomorphs also reflect the difference and multiformity of the environment of the water molecules in the different crystal lattices. The major factors behind the variability of the matrix are the ability of ammonium cations to form two hydrogen bonds and the rigidity of the polyoxometalate anion cage. The positions of the oxygen atoms in the latter are favourable for the formation of bifurcated and trifurcated cation–anion hydrogen bonds, some which are so durable that they persist after the crystals are dissolved in water, forming ion associates even in dilute solutions. The H atom involved in furcated hydrogen bonds cannot be exchanged by deuterium when the compound is dissolved in D2O. An obvious consequence of the versatility of the matrix is the propensity of these compounds to form multiple polymorphs.
Introduction
Polyoxometalates (POMs) represent one of the most extensively studied classes of compounds of anionic metal oxide structures. The interest in them is unceasing because their structure is tunable, offering access to a virtually unmatched range of physical properties.1–4 Systematic studies on the classical and representative group of the family, polyoxotungstates (POTs) have established standards for the bottom-up assembly of cluster-based crystalline materials which, thanks to their appealing electronic and molecular properties exhibit excellent performance in a wide range of applications such as catalysis, medicine or materials science.5–8Combination of alkylammonium cations with polyoxotungstate anions yields POTs with a variety of properties, offering new special applications.9 One of the factors influencing the thermal behavior of POTs is the amount of water of crystallization in their crystal lattice. In the crystals of alkali-polytungstates the multiple negative charge of the tungstate clusters is only locally screened by the alkali anions, and a large number of water molecules is needed to keep the structure stable.10–12 The salt of the ammonium ion, whose size is close to that of the K+ ion differs from the potassium salt and has more solvatomorphs, as many as five.13–17 The difference between the crystal-building properties of potassium and ammonium ions is reflected in the variation of the number of water of crystallization molecules in the triclinic/monoclinic solid solutions of the potassium and ammonium salts, in which there are 9.7, 9.5, 8.5 and 4.0 water molecules, corresponding to decreasing potassium/ammonium ion ratio.12 This shows that some property of the ammonium cation increases the propensity of the salt to have multiple solvatomorphs. A candidate for this property is the ability of the cation to form hydrogen bonds.
The properties of the polyoxotungstate anion may also play a role in determining the water of crystallization content and the chances of polymorphism. The main factors determining the chemical nature and abundance of species in tungstate solutions are the pH and the temperature.18,19 Acidification leads to polycondensation and formation of various condensed polytungstates up to as high nuclearity as H12W36O12012−, in which some hydrogen atoms are not exchangeable by deuterium in solution.20,21 In neutral or slightly alkaline tungstate solutions paratungstate A (heptatungstate, W7O246−) ions are in equilibrium with the condensed paratungstate B (dihydrogen-dodecatungstate, H2W12O4210−) ions, the former being the dominant species.22–24
Polytungstates salts formed by alkyl-substituted ammonium ions also display polymorphism and solvatomorphism, which offers the possibility of investigating the factors governing the formation of solvatomorphs. What makes these systems unique is the variability of the tungstate–alkylammonium ion interaction: the ability of the cation to form more than one hydrogen bond and the large number of hydrogen-bond acceptor sites of the tungstate anions allow them to form many different spatial arrangements, which appear as different polymorphs. The presence of water of crystallization increases further the number of possible arrangements of the cations and water molecules between the tungstate cages.
Among the alkylammonium ions investigated so far, the easily formed Keggin-type salts of dimethylammonium and tetramethylammonium ion were found to form multiple solvatomorphs.25,26 Synthesis and structure investigation of novel alkylammonium tungstates can provide more detailed information on the role of cation–anion hydrogen bonding. When selecting the possible anions and cations for synthesis and structure investigation, advantageous features are simplicity and purity of the compound. The dimethylammonium cation has some favorable properties that may be utilized in understanding the role of anion–cation hydrogen bonds: it is simple, can be a donor in two (and not more) N–H–X hydrogen bonds, and the methyl groups are small, so that the chance for complications due to steric effects is minimal. Since the anion studied so far, the Keggin-type dodecatungstate has been found to produce impure crystals, these salts are not appropriate for investigating the hydrogen bonds, thus, compounds that form pure crystals is desirable.
The purpose of this work is to synthesize some new alkylammonium polytungstates and demonstrate the remarkable structural versatility of crystal structure that can be achieved when large and rigid polytungstate cages are connected to cations capable of forming hydrogen bonds in the presence of water of crystallization molecules. The cation and the water molecules surround the large anions and can form a versatile network of hydrogen bonds; in other words, the anions are embedded in a highly variable matrix. Different stable arrangements (and water content) of the same matrix manifest in the formation of polymorphs and solvatomorphs. In the following, we first describe the synthesis and constitution of a new alkylammonium polyoxotungstate, dimethylammonium dihydrogendodecatungstate. Then the results of single-crystal XRD structure determinations are presented, with an emphasis of the number and type of hydrogen bonds and on the variability of the structure of the cation–water of crystallization framework surrounding the polytungstate anions. This will be followed by a summary of the consequences of the peculiarities of the structure involving cations strongly bound to the polytungstate anions, manifested in the thermal behaviour, solid-state NMR spectra, presence of strongly bound ion associates in solution and inefficient solution-phase deuterium exchange.
Results
Synthesis and composition of decakis(dimethylammonium) dihydrogendodecatungstate
The methods used for the preparation of decakis(dimethylammonium) dihydrogendodecatungstate are described in ESI, Section 1.† To avoid contaminating effects, instead of using an external base, the pH was varied by starting from a high-concentration aqueous solution of dimethylamine, and letting the amine evaporate. After dissolving WO3 in such a solution, the pH of the dimethylamine solution of was set to be neutral or slightly basic to ensure the presence of heptatungstates in the solution, in order to allow the simultaneous formation of new dodecatungstate-ion clusters and the bonds connecting them to the cations, which we think is an important factor enabling the build-up of the species that can form the crystalline material. The precipitation of a solid phase was initiated by adding organic solvents like ethanol and keeping the temperature as low as possible to avoid the condensation into Keggin structures.27,28In particular, WO3 was dissolved in a large excess of 40% aq. dimethylamine (under basic conditions), and the product was let to crystallize. At room temperature in 24 h we obtained triclinic crystals (hereafter referred to as modification 1T) very well soluble in water. The same single-phase product was obtained at 50 °C in 2 h. Another modification was found when we attempted to re-crystallize compound 1T from aqueous solution and, instead of the single crystals of 1T, we obtained triclinic crystals with a different constitution, which will be referred to as 2T.
The determination of the constitution of compound 1T by chemical methods involved several steps. Elemental analysis yielded C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N:W:H = 20:10:12:90 atomic ratio for this compound, i.e., the cation:tungsten ratio can be 5:6 or 10:12. The former can be excluded since no hexatungstate ion can have 5 negative charges. Accordingly, the product is decakis-(dimethylammonium) dodecatungstate, (Me2NH2)10H2W12O42. The hydrogen content found by elemental analysis in compound 1T is higher than what is expected for the anhydrous salt (20:10:12:82). This indicates that, as expected, the compound contains water of crystallization. The accuracy of the elemental analysis allows each of n = 9, 10 or 11H2O molecules per anhydrous polytungstate molecule, because the molecular weight of the latter (Manhydrous = 3340.98 g mol−1) so much larger than that of H2O, that the weight measurement cannot reliably distinguish the three possibilities. The standard way of measuring the water content, Karl-Fischer titration is known to give incorrect results for dodecatungstates, because a part of water of crystallization proved to be too strongly bound and not to be liberated under the standard measurement conditions.29 Another option is the thermogravimetry-mass spectrometry (TG-MS) method, according to which the crystals are subjected to thermal decomposition, and the mass of the water leaving crystals is determined. Our TG-MS experiments showed that when the crystals of 1T are heated, initially pure H2O departs, but the represents only a small fraction of the water content, and its bulk is released simultaneously with dimethylamine, preventing the direct determination of the mass of water of crystallization (for details, see ESI, Sections 2 and 3†). This problem has been circumvented utilizing the observation that if the thermal decomposition of 1T is conducted in air all the way to completion, the final product is pure WO3. The latter was identified by powder X-ray diffraction (ESI, Section 2†).30 Since the final product is a well-defined, pure compound with known composition, and the amount of dimethylamine is known from the stoichiometric formula, then from the total mass loss one can estimate the mass of the water of crystallization. This way we obtained n = 9.62 for the number of H2O molecules per anhydrous dodecatungstate salt. As a test of this method, an additional estimate was obtained from the comparison of the theoretical density, ρcell, obtained from the cell volume determined by powder X-ray diffraction and the pycnometric density of the crystals (ESI, Sections 3 and 10†). The former can be obtained by assuming that the volume of the unit cell, Vcell, contains Z anhydrous dimethylammonium dodecatungstate molecules. In the pycnometric density, representing the real crystal, in addition to the anhydrous molecule, the mass of the n water molecules is also included. Thus, the difference of the two densities provides the number of the water molecules associated with an anhydrous dodecatungstate salt molecule. From Vcell = 1916 ± 16 Å3, with the assumption that Z is unity and considering that the smaller volumes in the uncertainty range (around 1908 Å3) characterize better the crystals with the smallest number of defects than the larger ones, we obtained ρcell = 2.9077 ± 0.0120 g cm−3. Pycnometry yielded ρpyc = 3.0678 g cm−3, from which the approximate number of the water of crystallization molecules is n = 10.21 ± 0.82. This value and the n = 9.62 obtained by TG-MS bracket the integer n = 10. Accordingly, the formula of the hydrated salt is (Me2NH2)10H2W12O42·10H2O. The TG-based and the density-based methods mutually validate each other. For compound 2T, the TG experiments also showed that initially water is released, this time at much lower temperatures (120 vs. 150 °C), but again, the majority of water leaves simultaneously with dimethylamine. The complete thermal decomposition in air analogous to that described for 1T, yielded again the same final product, pure WO3, and from the mass loss the water content was found to correspond to n = 10.72. Thus, the formula of compound 2T can be estimated to be (Me2NH2)10H2W12O42·11H2O. The comparison of the formulas shows that 1T and 2T are solvatomorphs.
:N:W:H = 20:10:12:90 atomic ratio for this compound, i.e., the cation:tungsten ratio can be 5:6 or 10:12. The former can be excluded since no hexatungstate ion can have 5 negative charges. Accordingly, the product is decakis-(dimethylammonium) dodecatungstate, (Me2NH2)10H2W12O42. The hydrogen content found by elemental analysis in compound 1T is higher than what is expected for the anhydrous salt (20:10:12:82). This indicates that, as expected, the compound contains water of crystallization. The accuracy of the elemental analysis allows each of n = 9, 10 or 11H2O molecules per anhydrous polytungstate molecule, because the molecular weight of the latter (Manhydrous = 3340.98 g mol−1) so much larger than that of H2O, that the weight measurement cannot reliably distinguish the three possibilities. The standard way of measuring the water content, Karl-Fischer titration is known to give incorrect results for dodecatungstates, because a part of water of crystallization proved to be too strongly bound and not to be liberated under the standard measurement conditions.29 Another option is the thermogravimetry-mass spectrometry (TG-MS) method, according to which the crystals are subjected to thermal decomposition, and the mass of the water leaving crystals is determined. Our TG-MS experiments showed that when the crystals of 1T are heated, initially pure H2O departs, but the represents only a small fraction of the water content, and its bulk is released simultaneously with dimethylamine, preventing the direct determination of the mass of water of crystallization (for details, see ESI, Sections 2 and 3†). This problem has been circumvented utilizing the observation that if the thermal decomposition of 1T is conducted in air all the way to completion, the final product is pure WO3. The latter was identified by powder X-ray diffraction (ESI, Section 2†).30 Since the final product is a well-defined, pure compound with known composition, and the amount of dimethylamine is known from the stoichiometric formula, then from the total mass loss one can estimate the mass of the water of crystallization. This way we obtained n = 9.62 for the number of H2O molecules per anhydrous dodecatungstate salt. As a test of this method, an additional estimate was obtained from the comparison of the theoretical density, ρcell, obtained from the cell volume determined by powder X-ray diffraction and the pycnometric density of the crystals (ESI, Sections 3 and 10†). The former can be obtained by assuming that the volume of the unit cell, Vcell, contains Z anhydrous dimethylammonium dodecatungstate molecules. In the pycnometric density, representing the real crystal, in addition to the anhydrous molecule, the mass of the n water molecules is also included. Thus, the difference of the two densities provides the number of the water molecules associated with an anhydrous dodecatungstate salt molecule. From Vcell = 1916 ± 16 Å3, with the assumption that Z is unity and considering that the smaller volumes in the uncertainty range (around 1908 Å3) characterize better the crystals with the smallest number of defects than the larger ones, we obtained ρcell = 2.9077 ± 0.0120 g cm−3. Pycnometry yielded ρpyc = 3.0678 g cm−3, from which the approximate number of the water of crystallization molecules is n = 10.21 ± 0.82. This value and the n = 9.62 obtained by TG-MS bracket the integer n = 10. Accordingly, the formula of the hydrated salt is (Me2NH2)10H2W12O42·10H2O. The TG-based and the density-based methods mutually validate each other. For compound 2T, the TG experiments also showed that initially water is released, this time at much lower temperatures (120 vs. 150 °C), but again, the majority of water leaves simultaneously with dimethylamine. The complete thermal decomposition in air analogous to that described for 1T, yielded again the same final product, pure WO3, and from the mass loss the water content was found to correspond to n = 10.72. Thus, the formula of compound 2T can be estimated to be (Me2NH2)10H2W12O42·11H2O. The comparison of the formulas shows that 1T and 2T are solvatomorphs.
Crystallographic features of the modifications of (Me2NH2)10H2W12O42
The solvatomorphs of decakis(dimethylammonium) dihydrogendodecatungstate were both found to crystallize in triclinic forms. While polymorphism and solvatomorphism is not rare among dodecatungstates with univalent cations, the factors giving rise to the phenomenon have not yet been understood.11–17 In our compounds the cation, dimethylammonium is relatively simple; its N atom can form hydrogen bonds but not more than two. The structure determination and analysis offer us a chance to understand the origin of polymorphism and solvatomorphism in dodecatungstate salts involving cations that can form hydrogen bonds.Single crystals of 1T (colourless needles) were grown from 96% ethanol at room temperature in months while those of 2T (colourless platelets) from aqueous solution of 1T in a week. As described in the next section, based on the diversity of the matrix formed by the dimethylammonium cations and water molecules, we expected the existence of additional modifications. The search for them was successful: a repeated attempt to crystallize the freshly synthesized compound from 96% ethanol allowed us to observe the formation of a small amount of monoclinic crystals in addition to those of 1T. We denote this modification 1M, because it proved to be a polymorph of 1T. In the following we compare the crystal structures of the three modifications. The major parameters characterizing them are shown in Table 1. All other data, including crystal data and details of structure determination and refinement, the atomic coordinates and equivalent isotropic and anisotropic displacement parameters, anisotropic displacement parameters, bond lengths and angles and the intermolecular interactions is collected in the ESI Section 4.†
| Empirical formula | C20H102N10O52W12 | C20H104N10O53W12 | C20H102N10O52W12 |
| Label | 1T | 2T | 1M |
| Formula weight | 3483.01 | 3515.14 | 3499.14 |
| Temperature | 150(2) | 153(2) | 143(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
C2/c |
| Unit cell dimensions | a = 11.4037(5) Å | a = 13.5917(3) Å | a = 14.7682(3) Å |
| b = 13.7428(7) Å | b = 16.6057(4) Å | b = 24.7368(4) Å | |
| c = 13.9665(8) Å | c = 18.6449(4) Å | c = 21.1511(4) Å | |
| α = 63.686(4)° | α = 90.344(6)° | α = 90° | |
| β = 79.005(6)° | β = 105.743(7)° | β = 108.924(8)° | |
| γ = 69.227(5)° | γ = 112.051(8)° | γ = 90° | |
| Volume | 1832.99(18) Å3 | 3726.5(3) Å3 | 7309.2(4) Å3 |
| Z | 1 | 2 | 4 |
| Density (calculated) | 3.190 mg m−3 | 3.145 mg m−3 | 3.200 mg m−3 |
In the three polytungstate crystal structures, relatively large residual electron density can be found mainly in the neighborhood of the large tungsten atoms that give level A alerts when checking the cif files. In the structures, no void volumes can be found. These residual electron densities correspond to the imperfect fitting of the structure model in some very high electron density areas. Generally, large residual electron density is characteristic to polytungstate crystal structures.
Compound 1T crystallizes in the triclinic system, in the P space group. The asymmetric unit contains one half of a dihydrododecatungstate(10-) anion, with an inversion center inside, four entire and two half dimethylammonium cations (some of the nitrogen atoms being disordered over two positions) and five water molecules.
Solvatomorph 2T crystallizes in the triclinic system, in the same P space group as 1T. The asymmetric unit contains two half dihydrododecatungstate(10-) anions, ten dimethylammonium cations and eleven water molecules.
The composition of 1M is identical to that of 1T, thus it is a polymorphic modification of 1T. It crystallizes in the monoclinic system, in the C2/c space group. The asymmetric unit contains one half dihydrododecatungstate(10-) anion, five dimethylammonium cations and five water molecules.
The structural features of dihydrogendodecatungstate anions in compounds 1T, 2T and 1M are very similar. The coordination geometry around each tungsten atom is octahedral. The cage consists of two trigonal and two linear units of three edge-sharing WO6 octahedra. The polytungstate cages are arranged according to slightly different patterns in the three modifications, and the dimethylammonium cations and the water molecules are located in the open spaces between the cages. The [H2W12O42]10− cages taken from the three structures are almost perfectly superimposable. The positions of the dimethylammonium cations were found to be similar but by far not identical in the different crystal forms. One of the basic features determining the crystal structure is that the dihydrododecatungstate(10-) anions are extended and have a large negative charge. Instead of being connected by cations in strictly defined positions, the anions are embedded in what is best described as a matrix composed of dimethylammonium ions and water molecules (see Fig. 1 for the lattice of 1M, the structure of which is slightly different in the crystal modifications).
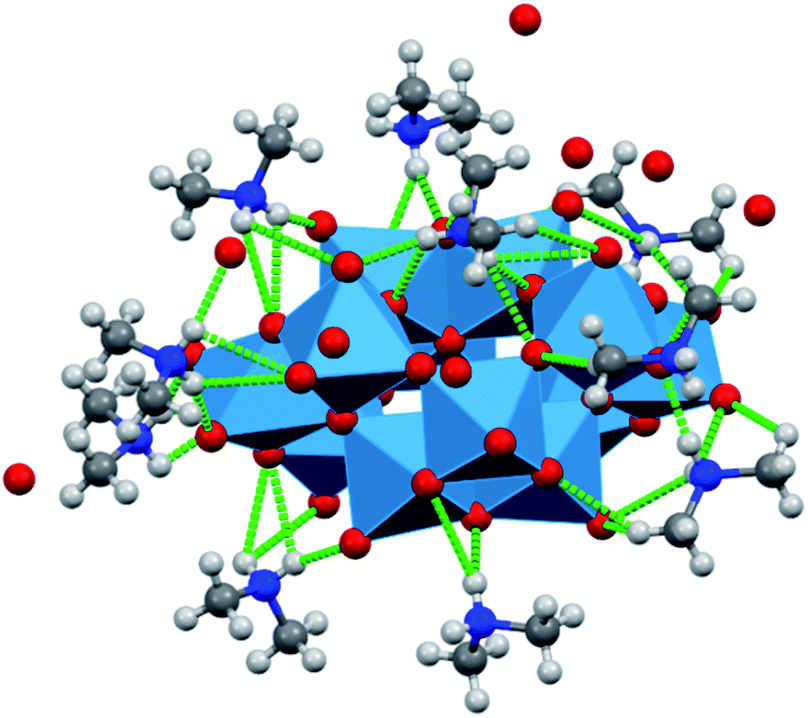 | ||
| Fig. 1 Hydrogen bonding in the crystal of 1M. | ||
The largest difference between the lattices and thus in the networks of hydrogen bonds of the three compounds is the number and the arrangement of the water molecules, which is associated with a variation of the relative position and alignment of the tungstate cages. These observations indicate that the matrix consists of cation and water acts like a versatile medium and foreshadows the possibility of the formation of further crystal modifications. While similar hydrogen-bond networks between the organic cation, water and polyoxometalate cations have been observed for some heteropolytungstates31 and heteropolymoybdates,32 the versatility we demonstrate below has not been mentioned.
A property characterizing the interrelations between the constituents is the number of N–H⋯O and C–H⋯O hydrogen bonds involving the ammonium cations and the water molecules in the crystal lattices of the three modifications (Table 2).
| H-bond type | 1T | 2T | 1M |
|---|---|---|---|
| N–H⋯O(cage) | 28 | 31 | 28 |
| N–H⋯O(water) | 0 | 0 | 4 |
| C–H⋯O(cage) | 16 | 14 | 20 |
| C–H⋯O(water) | 2 | 2 | 0 |
| O(water)–H⋯O(cage) | 10 | 8 | 14 |
| O(water)–H⋯O(water) | 10 | 14 | 18 |
In Table 3 the bonding pattern is demonstrated (ESI, Section 5†). As an extension to the widely used criteria33 to include weaker and furcated hydrogen bonds, two bridgehead atoms (including not only O and N but also C) are considered to be bonded by a hydrogen bond if their distance is smaller than the sum of their van der Waals radii + 0.2 Å. The results of the solid-state NMR investigation, the reduced conductivity and limited H/D exchange in the aqueous solutions of our compounds (see the next section), which prove the existence of furcated hydrogen bonds, justify the choice for this criterion.
| Hydrogen bond type | 1T | 2T | 1M |
|---|---|---|---|
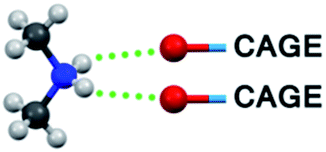 |
4 (N1, N4, N1′, N4′) | 2 (N6, N7) | 4 (N2, N5, N2′, N5′) |
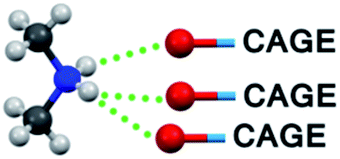 |
2 (N3, N3′) | 6 (N1, N2, N3, N4, N8, N10) | — |
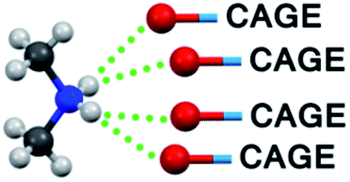 |
— | 1 (N9) | 2 (N1, N1′) |
 |
— | — | 4 (N3, N4, N3′, N4′) |
 |
— | 1 (N5) | — |
 |
2 (N2, N2′) | — | — |
 |
2 (N5, N6, N5′, N6′) | — | — |
In each structure the largest number of hydrogen bonds is formed by the 10 dimethylammonium cations. These include N–H⋯O and C–H⋯O bonds connecting the ammonium ions to the polytungstate anion. In addition, in 1M four N–H⋯O(water) hydrogen bonds, and in both 1T and 2T, two C–H⋯O(water) bonds have also been observed. The number of N–H⋯O(cage) hydrogen bonds is similar in the three modifications, between 28 and 31, but their distribution is quite different (see below). The methyl groups of the cations also participate in C–H⋯O(cage) bonds with the same or a neighboring anion; the number of such interactions is between 14 and 20 in the three structures. There are large differences in the number of water–tungstate cage and water–water interactions in the three structures: 10 of both in 1T, 8 and 14 in 2T and 14 and 8 in 1M.
Table 3 shows that both H atoms on the N atoms of every ammonium ion are connected to some of the O atoms of the tungstate anion, in some cases to two different anions. These bonds are regular N–H⋯O(cage) bonds in many cases, but the majority of them are furcated, the H atom being connected to two or three nearby electronegative acceptors. The geometrical factor that makes this possible is that on the “surface” of the polytungstate anion, the oxygen atoms are relatively close to each other, so that the cations can easily find some favorable binding sites. In the three structures all combinations can be observed, from one monofurcated and one bifurcated up to two trifurcated hydrogen bonds originating from the same N atom. The feasibility of the formation of many different regular hydrogen bonds accompanied by branched ones renders enormous versatility to the network. Branched hydrogen bonds are generally thermodynamically weaker than a regular one, and they cannot be expected to provide the rigidity offered by a regular hydrogen bond in the gas phase. However, they are assisted by the electrostatic interaction between the ions and by the multitude of forces acting on the dimethylammonium cations from different directions.
If, for example, a N atom of a cation is anchored to four cage oxygen atoms by two bifurcated hydrogen bonds, the N–H⋯O interactions exert a significant geometrical constraint, fixing the cation in a favorable orientation. If the methyl groups are also clamped to one or more oxygens of the cage (or even those of a neighboring one), the sideways motion of the cation can also be completely hindered, stabilizing the given structure. And, as Table 4 shows, this does happen: there are numerous C–H⋯O interactions providing additional constraint at the methyl ends of the cation.
| C–H⋯O bond type | 1T | 2T | 1M |
|---|---|---|---|
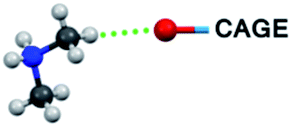 |
8 (C1, C2, C4, C6) | 10 (C2, C3, C10, C11, C12, C14, C15, C16, C19, C20) | 16 (C1, C2, C3, C4, C5, C6, C7, C10) |
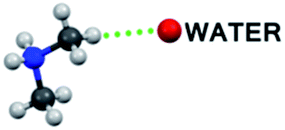 |
— | 2 (C1, C13) | — |
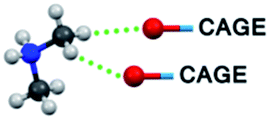 |
4 (C5) | 2 (C4) | 4 (C9) |
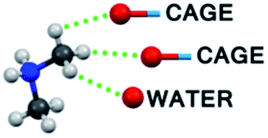 |
6 (C8) | — | — |
The methyl groups of the dimethylammonium ions form C–H⋯O hydrogen bonds not only with the same anion as the N atom, but also with a neighboring cage. The hydrogen-bond network involving the water molecules further stabilizes the structure. Snapshots of this network are shown in Fig. 2 and 3 for 1T and 2T, respectively. The water molecules tend to assemble in the spaces between the cages, forming “clusters”. Most of the water molecules of the clusters are bonded to the oxygen atoms of the anions and to each other (ESI, Section 6†).
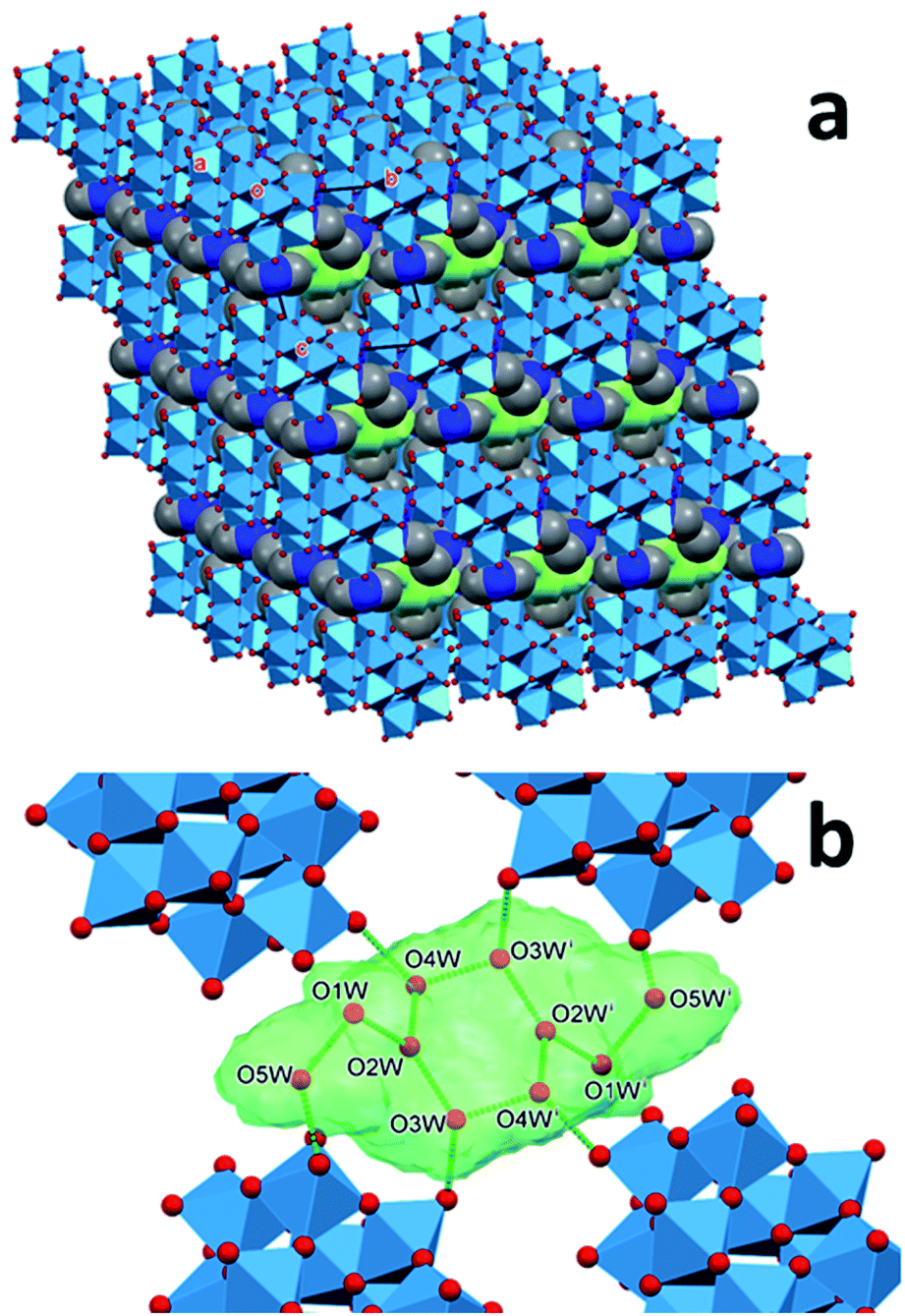 | ||
| Fig. 2 Water islands (green) in the structure of 1T. Light blue octahedrons represent WO4 units. Green sticks refer to the hydrogen bonds connecting the oxygen atoms (red dots) of the water molecules assembled in a cluster (green) in the structure of 1T (dimethylammonium ions are omitted for clarity, symmetry code: (i) 1 − x,1 − y,1 − z). | ||
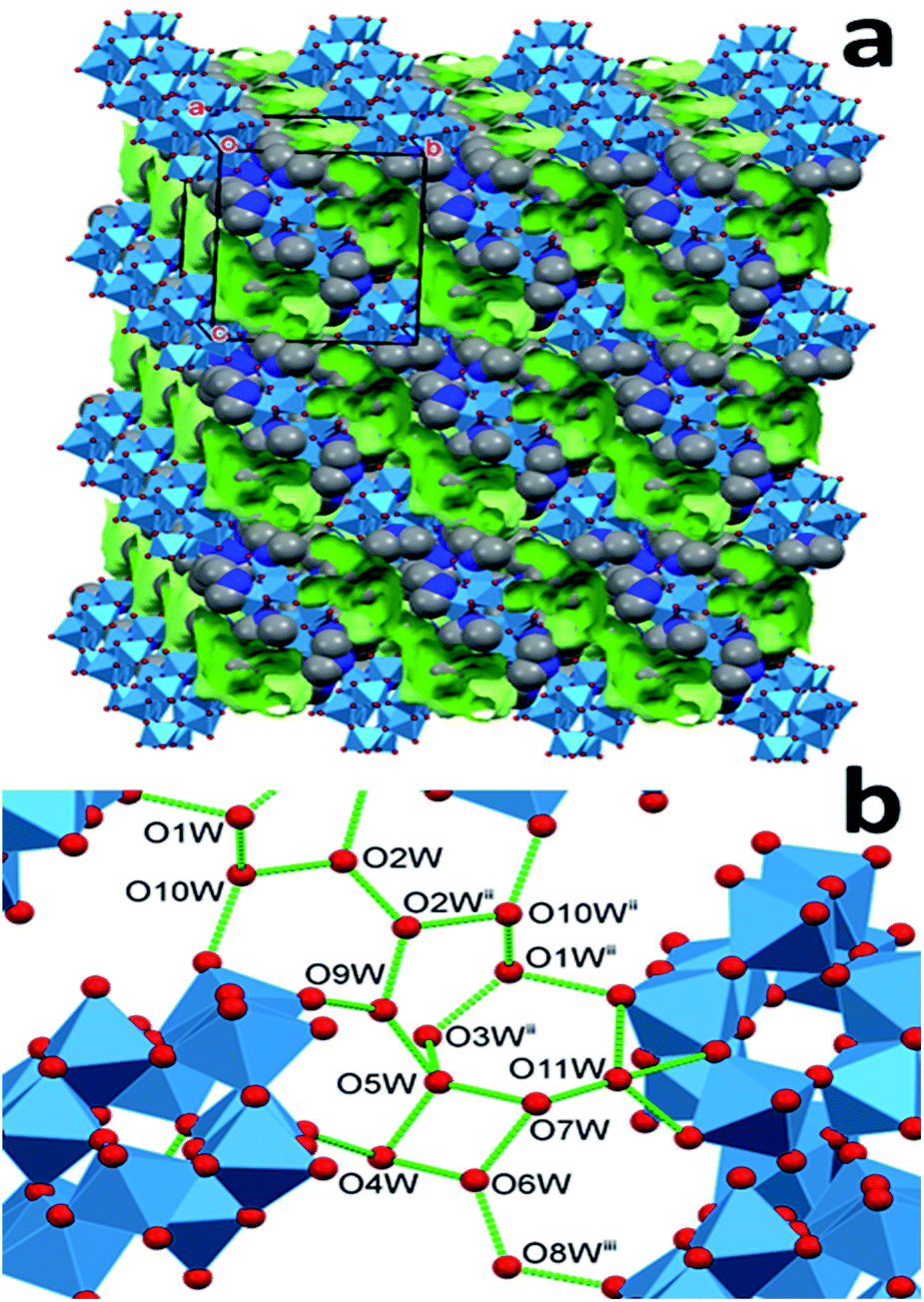 | ||
| Fig. 3 The network of hydrogen bonds (green sticks) connecting the O atoms of the water molecules (red dots) in the structure of 2T (dimethylammonium ions are omitted for clarity, symmetry codes: (ii) 2 − x,−y,1 − z; (iii) 1 − x,1 − y,−z). | ||
1T crystallizes in the triclinic system, in the P space group. The asymmetric unit contains one half of a dihydrododecatungstate(10-) anion, four entire and two half dimethylammonium cations (nitrogens disordered over two positions) and five water molecules. An inversion centre is situated at the centre of the [H2W12O42]10− anion. 2T crystallizes in the triclinic system, in the same P space group as 1T. The asymmetric unit contains two half dihydrododecatungstate(10-) anions, ten dimethylammonium cations and eleven water molecules. 2T is a solvatomorphic modification of 1T since it contains one more water molecule per one dihydrododecatungstate(10-). At the centre of both [H2W12O42]10− polyanions in the asymmetric unit, an inversion centre can be found. 1M crystallizes in the monoclinic system, in the C2/c space group (Fig. 5). The asymmetric unit contains one half dihydrododecatungstate(10-) anion, five dimethylammonium cations and five water molecules. 1M is a polymorphic modification of 1T. The [H2W12O42]10− polyanion contains a twofold symmetry axis but it is not centrosymmetric.
In the lattice of 1T all disk-shaped cages are parallel and the plane of each is at an angle to each crystallographic axis. The cages are surrounded by 9 water molecules and 20 dimethylammonium ions. The water molecules form a cluster in the body center of a parallelepiped formed by eight tungstate cages. There is a planar six-membered ring in the center of each cluster, extended by two arms on its opposite ends (resembling the p-xylene molecule). The two water molecules at the branching points are bonded only to other water molecules (O2W in Fig. 2). These are probably the most mobile constituents of the lattice. The clusters occupy 11.8% of the unit cell, and form chains flanked by ammonium ions. The intra- and interchain distances of the clusters are at least 5.44 Å long, and a dimethylammonium ion is between the closest cluster tails. The tungstate layers formed this way are held together by some disordered O⋯H–N–H⋯O bonds whose N atoms are randomly closer to one or the other layer (Fig. 4).
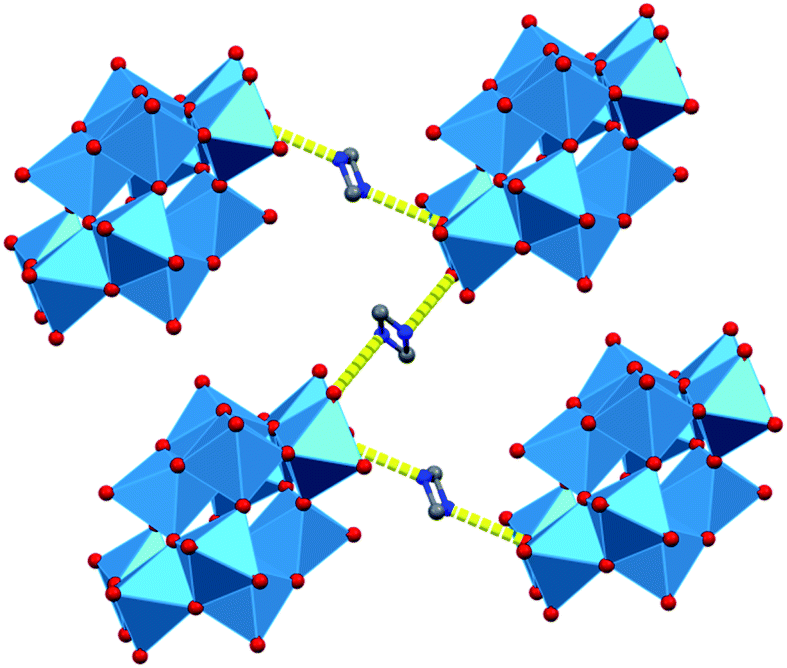 | ||
| Fig. 4 Position and hydrogen bonding of the two disordered ammonium ions in 1T. | ||
In the solvatomorph 2T (Fig. 3) the cages are not parallel, instead, they are alternately almost perpendicular to each other. The cages are bound together by three cations between each pair. The water molecules form long clusters in this lattice, too, which occupy 14.6% of the unit cell volume. The molecules of these clusters are bonded to the cages only at 8 points, and 14 hydrogen bonds connect the water molecules to each other. Probably this is what lends the lowest density to 2T among the three structures. In line with this, there are three water molecules connected only to other water molecules and not to the cages. These are probably the most weakly bound among all water molecules in the three structures. The clusters form quasi-continuous water channels in this solvatomorph, similarly to 1M.
Discussion
In the synthesized modifications of decakis(dimethyl-ammonium) dihydrododecatungstates the shapes of the tungstate cages are very similar. The arrangement of the cations and the water of crystallization molecules shows larger variety. The polytungstate cores can “float” in the matrix formed by the cations and water molecules. Although this matrix is not fluid, it is “flexible” in the sense that the ensemble can be stabilized in multiple arrangements by different combinations of type, number or multiplicity of hydrogen bonds. These spatial arrangements are energetically almost equivalent, and can be expected to be separated by low potential barriers, which makes the matrix highly variable. The rigid tungstate cages are embedded into this matrix as the seeds are in a watermelon: the arrangement of the seeds in different melons is similar but can be somewhat different. It is reasonable to assume that there are further possibilities for the tungstate “seeds” to be arranged in the ammonium cation + water flesh of a melon. There is little doubt that several other stable arrangements of the matrix are possible and can give rise to additional polymorphs and solvatomorphs. As we detailed in the previous section, this assumption prompted us to look for some more polymorphs which led us to find a polymorph of 1T. The structural properties of this modification confirm our expectation of structural diversity of the dimethylammonium ion + water matrix. In the lattice of 1M, the cages are also parallel, similarly to 1T. They are covered by 12 cations and 12 water molecules. The cages forming the same plane are connected by two cations along one and by three along the second in-plane axis of the cage, and those lying in neighboring planes by two cations. A unique cation is within 3.5 Å from three cages, indicating hydrogen-bond interaction with all three of them (an N–H⋯O and two C–H⋯O interactions), thus stabilizing the lattice. There are several contact points where water molecules also bind together the cages.The water of crystallization molecules form elongated clusters, and are hardly separated (the smallest distance between neighbouring clusters is 4.62 Å), forming almost continuous channels. The clusters occupy only 9.3% of the cell volume. The reason for their large density is probably that they are connected to the cages by as many as 14 hydrogen bonds. The other properties of this modification are listed together with those of 1T and 2T in Tables 3 and 4. We note that formation of water clusters has been reported for acidic sodium dodecatungstates,34 where they connect the cationic and anionic constituents. The main difference between those clusters and the ones we observe is that the sodium cations do not participate in hydrogen bonds, in particular, not in multifurcated ones. Thus, in contrast to dimethyl-ammonium cations, they do not support the formation of polymorphs, because they cannot induce structural versatility.
Manifestation of the structural properties in the chemical properties of the solvatomorphs.
Thermal decomposition
The cations and the water molecules are the most loosely bound in structure 2T, with three water molecules being bound to other water molecules only, like in bulk water. The consequence is that this modification releases water molecules already at low temperatures (120 °C).30 The arrangement of the latter into long channels also facilitates the water removal. Since the water channels are intertwined with the cation network, the departure of water molecules also disrupts the latter, so that the lattice collapses at relatively low temperatures, and water and dimethylamine is released simultaneously. The lattice of 1T is more tightly ordered than that of 2T. The water molecules are involved in stronger bonds, mostly to the tungstate cages, and, as a result, they start to leave the crystals only above 150 °C.30 One can conjecture that the water removal from 1M also starts at relatively high temperatures because the water molecules forming the clusters in this lattice are also bound to the cage oxygen atoms by relatively strong bonds. In the TG-MS experiments30 on 1T and 2T we have seen that the elimination of dimethylamine extends to high temperatures, and in inert atmosphere auto-oxidation takes place. This means that in (probably all three) poly/solvatomorphs a part of the individual cations remain strongly bound to the tungstate anions after the removal of water and the relatively weakly bound amine molecules. These are probably the ones that remain associated with the anion even in solution (see below).IR and NMR spectra
The cations and the molecules of the water of crystallization assume numerous relative spatial positions and are connected by a large variety of hydrogen bonds, including bifurcated and trifurcated ones. We recorded the IR and Raman spectra of the compounds and performed their complete assignment. From this we learned that the O–H stretch, H–O–H bend and the N–H stretch frequencies are really shifted towards lower wave numbers with respect to their gas-phase positions, indicating that the hydrogen atoms of these bonds are donated into hydrogen bonds.35–37 The peaks are also smeared out and their separability remains limited even after complete deuteration. From this one can conclude that there are many kinds of N–H⋯O and O–H⋯(O,N,C) hydrogen bonds with different strength. Very probably, the furcated hydrogen bonds, whose stretching frequencies in crystals are known to vary according to their type36 also contribute to the widening of the spectral lines. However, just because of the smearing of the lines, vibrational spectroscopy, although it hints at the structural variability, does not display any characteristic features reflecting the variability of the structures.Solid-state NMR spectroscopy offers better chances for characterizing the environments of different kinds of atoms forming the cation + water matrix (ESI, Section 7†). The solid-state 15N CP-MAS NMR spectrum of 1T indicates that the N atoms occur in two different neighborhoods (with δ = 29.2 and 26.6 ppm). In 2T, on the other hand, all N atoms are in very similar environment (δ = 26.5 ppm). Since the shifts around 26.5 ppm are common in the two solvatomorphs, it sounds reasonable to assume that their positions are similar in both lattices. These are probably the cations lying on one anion cage. On the other hand, the N atom with the larger shift in 1T is definitely in a distinct environment with no counterpart in 2T. This is the cation forming a disordered O⋯H–N–H⋯O bridge between two tungstate cage layers in the lattice.
The 13C CP-MAS NMR spectrum of 1T contains six (δ = 38.9, 36.7, 36.0, 35.3, 34.8 and 33.9 ppm), while that of 2T only five signals (δ = 36.4, 35.9, 35.2, 35.6 and 33.5 ppm). The lower five chemical shifts of 1T match very well those of 2T, which again can be assigned to the dimethylammonium cations lying on the surfaces of the tungstate anions and connected to them by one or two C–H⋯O bonds in addition to the N–H⋯O bond(s). The numerous chemical shifts correspond to carbon atoms of dimethylammonium cations in slightly different positions. The signal with the largest chemical shift in the spectrum of 1T (δ = 38.9 ppm) represents a methyl group that is in a significantly different neighborhood compared with the rest. It is reasonable to assume that the corresponding carbon atoms are associated with the N atoms with a separate 15N signal, which we have assigned to the ammonium ions bridging the tungstate layers. The highest peak in both spectra is the one at about 35.3 ppm. This peak probably corresponds to the methyl carbon position that is most common in both lattices. These are those methyl groups of dimethylammonium ions, which are connected to a tungstate oxygen by one C–H⋯O bond. There is not enough information to make a one-to-one connection between the rest of the peaks and the C–H⋯O bridges listed in Table 4.
The solid-state 1H{15N} CRAMPS NMR spectra of both 1T and 2T contain two signals corresponding to H2O molecules, and one to NH2 and one to CH3. This indicates that two types of environments of H2O are different enough to be distinguished by the method. The chemical shifts of the two signals corresponding to water molecules as well as the one corresponding to NH2 are remarkably different in the two lattices (water signals at 5.7 and 3.4 ppm in 1T and 6.9 and 5.7 ppm in 2T as well as NH2 signals at 2.63 vs. 3.15 ppm in 1T and 2T, respectively), while the CH3 signals are much closer in the two solvatomorphs (2.28 and 2.39 ppm). This indicates that in the two lattices the strengths of the hydrogen-bond interactions involving the H atoms of the more electronegative atoms is significantly larger than those of the C–H⋯O bonds. This is consistent with the observation seen in the 13C CP-MAS spectrum: the strengths of the C–H⋯O interactions is almost the same in the two solvatomorphs, which supports the assumption that they correspond to the cations lying on the surface of the tungstate cage. The information from the CRAMPS NMR spectra seems not to provide enough detail to make more structural associations.
The 1H–13C HETCOR NMR spectra of structures 1T and 2T (Fig. 6) characterize further the properties of C–H⋯O type interactions in the two lattices. Along the horizontal axis the distribution of the peaks seen in the 13C CP-MAS spectra are reproduced with lower resolution. In the spectrum of 1T the peak at about 39 ppm associated with the cation forming the disordered O⋯H–N–H⋯O bonds remains distinct. Along the vertical axis the four different peaks observed in the 1H{15N} CRAMPS spectrum also appear, indicating that the carbon atoms with these chemical shifts are magnetically associated with protons in (at least) four different environments. The five peaks in the 13C CP-MAS spectrum of 1T in the 33–36 ppm range, assigned to the cations lying on the surface of the tungstate cage merge into a wide a peak in which two maxima are still distinguishable. The carbon associated with the higher peak is interacting with four, the lower with at least two but probably three, and the shoulder at higher 13C chemical shifts with two protons. This shows that the environments of the methyl groups of the dimethylammonium cations lying on the surface of the anion is different. Unfortunately, no further connection can be made with the crystal structure. In the 1H–13C HETCOR NMR spectrum of 2T the five peaks the 13C CP-MAS spectrum merge into one structureless peak, which along the vertical axis is split only into two, indicating that here the cations laid on the anion surface are less distinguishable.
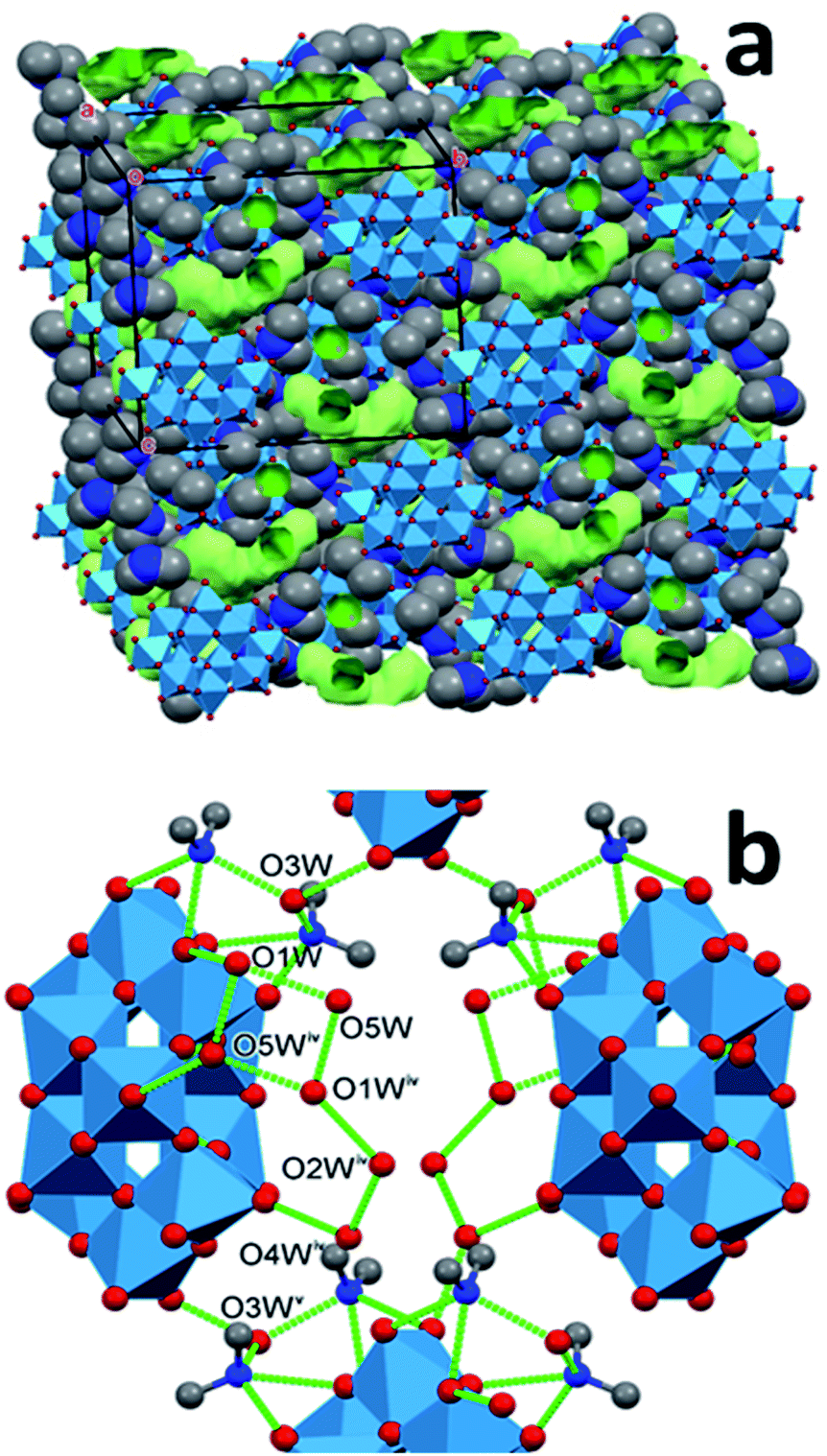 | ||
| Fig. 5 Hydrogen bonded water clusters in the structure of 1M (symmetry codes: (iv) 3/2 − x,1/2 − y,−z, (v) 1/2 + x,1/2 + y,z). | ||
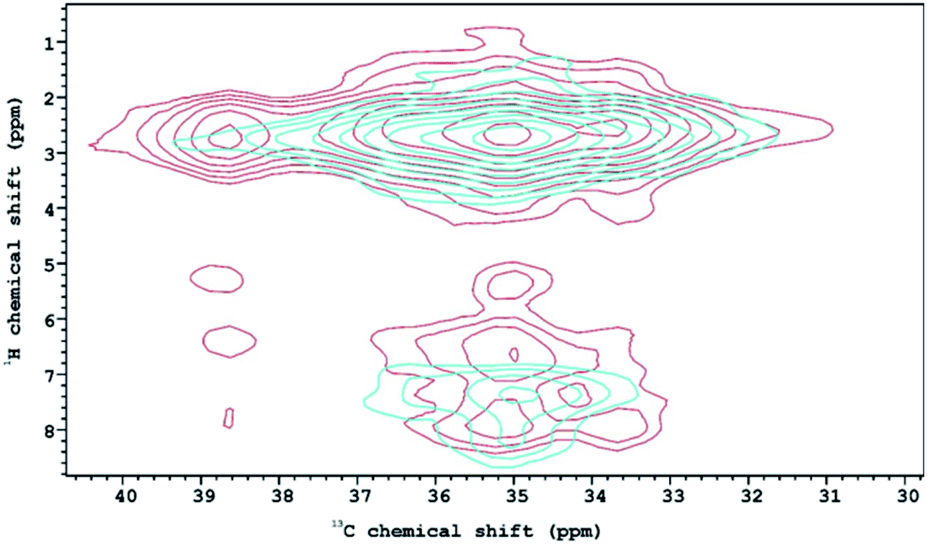 | ||
| Fig. 6 13C–1H HETCOR NMR spectra of solvatomorphs 1T (blue) and 2T (red) at room temperature. | ||
The interpretation of the solid-sate NMR spectra shows that this method is sensitive enough to reflect the diversity of the properties of the cation + water matrix, including the large variety of regular and furcated hydrogen bonds. Thus, this is a method that can be recommended for quick distinction of the matrices of different poly/solvatomorphs and for the detection of the variability of matrices involving a network of hydrogen bonds.
Formation of ion associates in solution
The features of the crystal structure and the autooxidation of the dimethylammonium ions that stay connected to the anions up to high temperatures indicate that the binding of the cations to the large and highly charged anion is persistent. Structural information from the single-crystal X-ray diffraction shows that the hydrogen bonds connecting the nitrogen atoms of dimethylammonium ion to the anion, in addition to bridging oppositely charged ions, are often multifurcated. The anions are wrapped into a dialkylammonium ion layer in the crystal lattices and are somewhat separated from each other by the water clusters. This explains that the crystals are soluble in water. One can also expect that the dialkylammonium ion–anion bonds remain in force after dissolution, so that some of the cations of the “wrapping” stay tied to the anion, forming ion associates. To check this possibility, we measured the conductivity of the dimethylammonium-dihydrododecatungstate solutions at different concentrations. At these same concentrations we also determined the conductivities of solutions of the analogous salt of sodium, in which only electrostatic forces support associate formation. The details are presented in Section 8 of ESI† (where the hitherto unknown specific conductivity of the dihydrododecatungstate(10-) ion that we measured is also reported). We found that the conductivity of the solutions of the dimethylammonium salt is much smaller than that of the sodium analog, and the quantitative comparison showed that the degree of formation of ion associates is 14 or 15 times larger in the dimethylammonium salt as compared with the sodium analog. This means that the hydrogen bonds connecting the ammonium cation to the cation are rather durable and can really keep at least some of the cations tied to the anion even in aqueous solution.Limited solution-phase H/D exchange in heavy water
If the cation–anion hydrogen bonds are so persistent that they keep the ions associated even in aqueous solution, then one can expect further observable consequences. Since the N–H⋯O bonds are furcated in the crystal lattice, one can expect them to remain furcated in solution. The hydrogen atoms involved in multifurcated hydrogen bonds are immobilized, in the sense that they cannot easily dissociate in the form of protons as in regular H-bonds, because two or more bonds should brake simultaneously. If ionic dissociation is not possible, then the hydrogen atoms cannot be easily substituted by deuterium after dissolving the crystal in heavy water, which is the common way of deuterating compounds involving mobile hydrogen atoms. To check this possibility, we attempted to deuterate dimethylammonium dihydrogen-dodecatungstate by placing 1T in heavy water. After dissolution, the water of crystallization molecules are released and their protium atoms can be completely exchanged by deuterium, so only some of the furcated N–H–X bonds can resist deuteration. The amount of the hydrogen atoms replaced by D was determined by 1H NMR spectroscopy, based on the fact that each substituted H atom will appear in the form of an HDO molecule. From the comparison of the area of the HDO signal to that of the six un-exchangeable H atoms in the methyl groups of each dimethylammonium cation (for details, see Section 9 of ESI†), we found that only 25% of the possible 20 (N–)H atoms were replaced by D. This means that on average, 15H atoms are deactivated by being “buried” in multifurcated hydrogen bonds. In other words, on average 7 or 8 of the 10 cations remain associated to the polytungstate anion.The poor efficiency of deuterium exchange has also been seen when we deuterated the compound for IR spectroscopy: an aggressive procedure involving four successive recrystallizations from D2O was needed to remove all exchangeable hydrogen atoms from the salt. The covalently bound methyl hydrogens remained intact as they should, and the two H atoms that are inside the tungstate cage also resisted even the rather aggressive conditions.38,39
Conclusions
We have synthesized three polymorphs and solvatomorphs of decakis(dimethylammonium) dodecatungstate and determined their crystal structure by single-crystal XRD. The thermal decomposition as well as the IR and solid-state NMR spectra and some other properties of two of the crystal modifications have also been characterized, with the purpose of understanding the properties of the dodecatungstate – cation/water interaction. According to single-crystal XRD structure determination, in the crystals of these salts the polytungstate clusters are embedded in a matrix of alkylammonium cations and water of crystallization molecules, stabilized in different arrangements by a variable network of regular and branching hydrogen bonds. The ability of the R2NH2+ cations to form two hydrogen bonds is key to the formation of these networks. The structures resemble the arrangement of the seeds of a watermelon (the polytungstate cages) embedded in a flesh with various arrangements of filaments (the hydrogen-bond network of the dimethylammonium + water matrix).A remarkable observation is that some of the hydrogen bonds binding the ammonium cations to the tungstate anions are persistent and remain active even after dissolution in water, keeping the ions associated and causing poor deuterium exchange. The reason for the durability of the hydrogen bonds is that there are many of them and some are furcated, the same hydrogen atom being connected to two or three different hydrogen-bond acceptors in the lattice. Branched hydrogen bonds are generally not strong thermodynamically;33 the distances of the bridgehead atoms are larger than in regular hydrogen bonds and the XHY bond angle is far from the optimal 180°. Since there are several cations connected to the rigid anion with relatively weak but numerous bonds, it is natural that there are different spatial arrangements corresponding to local potential minima whose energies are close to each other. This provides considerable structural diversity to the geometrical structures, i.e. polymorphism. In fact, based on the properties of solvatomorphs 1T and 2T, we predicted the existence of polymorphs and found a third modification, 1M. The conditions favorable for the variability of crystal structures of POM salts are that the polyoxo anion is rigid with a multifaceted geometry and that the positions of its oxygen atoms are favorable for the cation to form two or more hydrogen bonds with them. These bonds can be furcated because of the proximity of several acceptors. While no characteristic features were observed in the vibrational spectra of these compounds, solid-state NMR spectroscopy has proved to be a tool appropriate for the detection the structural variability of polytungstates of cations that can form hydrogen bonds.
POMs formed by alkylated ammonium ions with other polyoxometalate anions are also good candidates for generation of numerous polymorphs. The advantage of the alkylated ammonium cations is that they are volatile and can be removed by thermal treatment. The decomposition of the polymorphs takes place in significantly different steps and different temperatures, and complete decomposition can produce the same metal oxide with different morphology. As a manifestation of this, we observed that the final decomposition product of our compounds can be stoichiometric WO3 or an oxygen-deficient oxide corresponding to the WO2.93 formula.
Overall, our study demonstrates that the variability of the interaction of rigid and relatively bulky polyoxometalate anions with alkylated ammonium ions and molecules of water of crystallization is due to the numerous possible arrangements and furcation patterns of hydrogen bonds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research within projects No. VEKOP-2.3.2-16-2017-00013 and GINOP-2.2.1-15-2017-00084 was supported by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. N. V. M. and P. B. are grateful for grants by the Hungarian Scientific Research Found (K-124544 and K-115762). The K-124212 and an NRDI TNN_16 123631 grants are also acknowledged. I. M. S. thanks the Hungarian Academy of Sciences for a János Bolyai Research Fellowship and acknowledges the ÚNKP-18-4-BME-238 grant supported by the New National Excellence Program of the Ministry of Human Capacities, Hungary. An NRDI K 124212 and an NRDI TNN_16 123631 grants are acknowledged. The research reported in this paper was supported by the BME Nanotechnology and Materials Science TKP2020 IE grant of NKFIH Hungary (BME IE-NAT TKP2020).References
- (a) H. L. Li, C. Lian, L. J. Chen, J. W. Zhao and G. Y. Yang, Two Ce3+-Substituted Selenotungstates Regulated by N,N-Dimethylethanolamine and Dimethylamine Hydrochloride, Inorg. Chem., 2019, 58, 8442–8450 CrossRef CAS; (b) N. I. Gumerova and A. Rompel, Synthesis, structures and applications of electron-rich polyoxometalates, Nat. Rev. Chem., 2018, 2, 1–20 CrossRef; (c) F. Xie, X. Li, Y. Li, X. Jiang, Q. Rui and J. Sha, Assembly of polyoxometalate-templated metal-organic framework with effective peroxidase-like catalytic activity, J. Coord. Chem., 2019, 72, 279–282 CrossRef.
- (a) Y. J. Liu, M. T. Jin, L. J. Chen and J. W. Zhao, Recent advances in isopolyoxotungstates and their derivatives, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1202–1221 CrossRef CAS; (b) S. U. Khan, M. Akhtar, F. U. Khan, J. Peng, A. Hussain, H. Shi, J. Du, G. Yan and Y. Li, Polyoxometalates decorated with metal-organic moieties as new molecular photo- and electro-catalysts, J. Coord. Chem., 2018, 71, 2604–2621 CrossRef CAS; (c) S. Chen and X. Xu, The loading of polyoxometalates compound on a biomass derived N-doped mesoporous carbon matrix, a composite material for electrolytical energy storage, J. Coord. Chem., 2018, 71, 3035–3044 CrossRef CAS.
- D. M. Neelam, A. Burg, D. Shamir, Y. Albo, X. Li, Y. Wang, K. Zhou, Y. Wang, T. Han and J. Sha, Polyoxometalates entrapped in sol-gel matrices as electron exchange columns and catalysts for the reductive de-halogenation of halo-organic acids in water, J. Coord. Chem., 2018, 71, 468–482 CrossRef.
- J. Wang, H. Zhou, M. Ge, Y. Wang, Y. Zhang and H. Lu, Synthesis and peroxidase-like mimic study in H2O2 detection of a stable polyoxometalate-pillared coordination polymer, J. Coord. Chem., 2018, 71, 3127–3138 CrossRef CAS.
- T. Yamase, Isopoly and Heteropoly Compounds of Molybdenum, Tungsten, and Vanadium, Eur. J. Inorg. Chem., 2019,(3–4), 343–345 CrossRef CAS.
- A. Y. Olenin, P. G. Mingalev and G. V. Lisichkin, Partial Catalytic Oxidation of Alcohols: Catalysts Based on Metals and Metal Coordination Compounds (a Review), Pet. Chem., 2018, 58, 577–592 CrossRef CAS.
- K. Shakeela and G. R. Rao, Thermoreversible, Hydrophobic Ionic Liquids of Keggin-type Polyanions and Their Application for the Removal of Metal Ions from Water, ACS Appl. Nano Mater., 2018, 1, 4642–4651 CrossRef CAS.
- A. Singh and G. Kumar, Advancement in catalysts for transesterification in the production of biodiesel: a review, J. Biochem. Technol., 2018, 9, 17–27 Search PubMed.
- J. Ichihara and Y. Sasaki, Participation of new active species in epoxidation with cetylpyridinium dodecatungstate/FAp/urea–H2O2 system, Catal. Today, 2006, 117, 120–125 CrossRef CAS.
- S. J. Liu, Q. Y. Chen, P. M. Zhang and M. N. Li, Study on thermal decomposition of sodium paratungstate, Na10H2W12O42.27H2O, Acta Phys. Sin., 1998, 14, 821–825 CAS.
- A. Chrissafidou, J. Fuchs, H. Hartl and R. Palm, Kristallisation und Strukturuntersuchung von Alkali-Parawolframaten/Crystallization and Structure Determination of Alkaline Metal-Paratungstates, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 217–222 CAS.
- H. J. Lunk, M. Fait, B. Ziemer, J. Fuchs and H. Hartl, Formation of Heterotypic Substitutional Solid Solutions (NH4)10-xKx[H2W12O42]·nH2O in the Ammonium Paratungstate ‘Z’/Potassium Paratungstate ‘Z’ System, Z. Anorg. Allg. Chem., 1999, 625, 673–680 CrossRef CAS.
- H. d'Amour and R. Allmann, Die Kristallstruktur des Ammoniumparawolframat-tetrahydrats (NH4)10[H2W12O42]·4H2O, Z. Kristallogr., 1972, 136, 23–47 CrossRef.
- R. Allmann, Die Struktur des Ammoniumparawolframates (NH4)10[H2W12O42].10H2O, Acta Crystallogr., 1971, 27, 1393–1404 CrossRef CAS.
- M. Fait, Synthese, Charakterisierung sowie thermisches Verhalten von Ammonium/Kalium-Parawolframat-Mischkristallen, Doctoral Dissertation, Humboldt University, Berlin, Germany, 1995 Search PubMed.
- J. W. van Put, T. W. Verkroost and E. J. Sonneveld, X-Ray Powder Diffraction Data and Unit Cells of Ammonium Paratungstate Tetrahydrate, Powder Diffr., 1990, 5, 167–169 CrossRef CAS.
- J. W. van Put, G. J. Witkamp and G. M. van Rosmalen, Formation of ammonium paratungstate tetra- and hexahydrate. I: Stability, Hydrometallurgy, 1993, 34, 187–201 CrossRef CAS.
- J. J. Cruywagen, Protonation, oligomerization, and condensation reactions of vanadate(V), molybdate (VI) and tungstate(VI), Adv. Inorg. Chem., 2000, 49, 127–182 CrossRef CAS.
- R. I. Maksimovskaya and K. G. Burtseva, 17O and 183W NMR studies of the paratungstate anions in aqueous solutions, Polyhedron, 1985, 4, 1559–1562 CrossRef CAS.
- D. L. Long, H. Abbas, P. Kögerler and L. Cronin, High, “Celtic-Ring” Isopolyoxotungstate, [H12W36O120]12- That Captures Trace Potassium Ions, J. Am. Chem. Soc., 2004, 126, 13880–13881 CrossRef CAS.
- S. Liu, Q. Chen, P. Zhang and S. Li, Raman spectral study on isopolytungstates in aqueous solutions, Trans. Nonferrous Met. Soc. China, 1998, 8, 688–692 CAS.
- L. Fan, J. Cao and C. Hu, What can electrospray mass spectrometry of paratungstates in an equilibrating mixture tell us?, RSC Adv., 2015, 5, 83377–83382 RSC.
- J. Fuchs and E. P. Flindt, Preparation and Structure Investigation of Polytungstates- Contribution to the Paratungstate-A Problem, Z. Naturforsch., 1979, 34, 412–422 Search PubMed.
- P. Haufe, Raman-Spectrophotometric, Determination of the Tungstate Anion and Its Isopolyanions in Aqueous Systems, Fresenius' Z. Anal. Chem., 1982, 310, 388–391 CrossRef CAS.
- P. Zavalij, J. Guo, M. S. Whittingham, R. A. Jacobson, V. Pecharsky, C. K. Bucher and S.-J. Hwu, Keggin Cluster Formation by Hydrothermal Reaction of Tungsten Trioxide with Methyl Substituted Ammonium: The Crystal Structure of Two Novel Compounds, [NH2(CH3)2]6H2W12O40.4H2O and [N(CH3)4]6H2W12O40·2H2O, J. Solid State Chem., 1996, 123, 83–92 CrossRef CAS.
- J. Chojnacka, E. Hodorowicz and K. Stadnicka, Analytical and crystal data for the reaction product of tungstate (WO42-) with an organic base, Rocz. Chem., 1977, 51, 1593–1596 CAS; E. Hodorowitz, S. Sagnowski and A. Samotus, Synthesis and characterization of isopolytungsates with tetramethylammonium cation, Proceedings of the 8th Conference on Coordination Chemistry, 1980, pp. 121–122 Search PubMed.
- G. M. Rozantsev and O. I. Sazonova, Thermodynamic Parameters of Interconversions of Isopolyanions in Solutions of Tungsten(VI), Russ. J. Coord. Chem., 2005, 31, 552–558 CrossRef CAS.
- O. I. Sazonova, G. M. Rozantsev and Y. V. Kholin, State of tungsten(VI) ions in aqueous solutions, Zh. Neorg. Khim., 1998, 43, 1894–1899 CAS.
- F. B. Sherman and V. A. Klimova, Determination of water in some aqua-and heteropolytungstates, Russ. Chem. Bull., 1971, 20, 1040–1041 CrossRef.
- L. Trif, G. Lendvay, F. P. Franguelli, E. Majzik and I. M. Szilágyi, Thermal analysis of solvatomorphic decakis(dimethylammonium) dihydrododecatungstate hydrates, J. Therm. Anal. Calorim., 2020 DOI:10.1007/s10973-020-10494-4.
- T. Bouallegui, A. Harchani, N. Dege, A. Haddad and B. Ayed, Synthesis, characterization, Hirschfield surface and theoretical properties of a new non-centrosymmetric inorganic/organic material: (C7H12N2)7[β-SbMo6O24]2·8H2O, J. Mol. Struct., 2018, 1166, 195–201 CrossRef CAS.
- Y.-F. Li, D. G. Hubble, R. G. Miller, H.-Y. Zhao, W.-P. Pan, S. Parkin and B. Yan, Synthesis and characterization of two novel organic-inorganic hybrid solids from Keggin ions and metal coordination complexes, Polyhedron, 2010, 29, 3324–3328 CrossRef CAS.
- E. S. Feldblum and I. T. Arkin, Strength of a bifurcated H bond, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4085–4090 CrossRef CAS; I. Rozas, I. Alkorta and J. Elguero, Bifurcated Hydrogen Bonds: Three-Centered Interactions, J. Phys. Chem. A, 1998, 102, 9925–9932 CrossRef.
- K. V. Redrup and M. T. Weller, Hydrothermal routes to new sodium hydrogen polytungstates, Dalton Trans., 2009, 23, 4468–4472 RSC.
- I. E. Sajó, L. P. Bakos, I. M. Szilágyi, G. Lendvay, J. Magyari, M. Mohai, Á. Szegedi, A. Farkas, A. Jánosity, S. Klébert and L. Kótai, Unexpected Sequential NH3/H2O Solid/Gas Phase Ligand Exchange and Quasi-Intramolecular Self-Protonation Yield [NH4Cu(OH)MoO4], a Photocatalyst Misidentified before as (NH4)2Cu(MoO4)2, Inorg. Chem., 2018, 57, 13679–13692 CrossRef.
- E. Majzik, F. P. Franguelli, G. Lendvay, L. Trif, S. Klebert, A. Farkas, I. M. Szilagyi and L. Kotai, Deuteration and Vibrational spectroscopy of dimethylammonium paratungstate-B hydrates, Z. Anorg. Allg. Chem., 2020 DOI:10.1002/zaac.202000283.
- G. Novodárszki, H. E. Solt, G. Lendvay, R. M. Mihályi, A. Vikár, F. Lónyi, J. Hancsók and J. Valyon, Hydroconversion mechanism of biomass-derived γ-valerolactone, Catal. Today, 2019, 336, 50–62 CrossRef.
- M. Fait, D. Heidemann and H. J. Lunk, Characterization of the protons in polycrystalline paratungstates using 1H MAS NMR investigations, Z. Anorg. Allg. Chem., 1999, 625, 530–538 CrossRef CAS.
- H. T. Evans and E. Prince, Location of internal hydrogen atoms in the paradodecatungstate polyanion by neutron diffraction, J. Am. Chem. Soc., 1983, 105, 4838–4839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Materials and synthetic methods. (2) Instrumental analyses. (3) Determination of the water content. (4) Single crystal data and characterization. (5) Hydrogen bonding in the three crystal forms. (6) Water channels or islands in the three structures. (7) Solid state NMR studies. (8) Conductivity studies. (9) Limited solution-phase H/D exchange in heavy water. (10) Powder X-ray diffraction and indexing. (11) Thermogravimetry. CCDC file numbers of 1T, 2T and 1M are 1998540, 1998541 and 1998542 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09997j |
| This journal is © The Royal Society of Chemistry 2021 |