DOI:
10.1039/D0RA10189C
(Paper)
RSC Adv., 2021,
11, 3972-3980
Synthesis and biological activity of squaramido-tethered bisbenzimidazoles as synthetic anion transporters†
Received
3rd December 2020
, Accepted 13th January 2021
First published on 20th January 2021
Abstract
A series of squaramido-tethered bisbenzimidazoles were synthesized from the reaction of diethyl squarate with substituted 2-aminomethylbenzimidazoles. These conjugates exhibit moderate binding affinity toward chloride anions. They are able to facilitate the transmembrane transport of chloride anions most probably via an anion exchange process, and tend to be more active at acidic pH than at physiological pH. The viability of these conjugates toward four selected solid tumor cell lines was evaluated using an MTT assay and the results suggest that some of these conjugates exhibit moderate cytotoxicity probably in an apoptotic fashion.
1. Introduction
During the past decades, substantial interest has been shown in the creation of small-molecule, organic compounds that are able to facilitate the transport of anions, in particular chloride anions across cellular phospholipid membranes.1–10 These so-called synthetic anion transporters may serve as useful research tools to clarify the structures and functions of natural anion channels and transporters. More interestingly, because anion transporters are able to perturb the chemical gradients within cells to trigger apoptosis, they may be developed as a novel class of chemotherapeutic agents for the treatment of cancers.11–20 In addition, they may also have high chemotherapeutic potentials for channelopathies such as cystic fibrosis (CF) by restoring the flux of anions through epithelial cell membranes in CF patients.20,21 As such, more small-molecule anion transporters with diverse structures and promising biological activity are in demand.1–10
In these aspects, squaramides and (benz)imidazoles represent two classes of ideal structural skeletons for the construction of superior anion transporters.22–40 This may be ascribed to their unique features, including ready availability, high stability and strong capability to complex anions mainly via hydrogen-bonding interactions. Some simple squaramides, such as trifluoromethylphenyl derivatives have been found to efficiently facilitate the transport of chloride anions across phospholipid bilayers.32 More interestingly, it has been demonstrated that these squaramide derivatives are able to trigger the apoptosis of cancer cell lines by deacidifying lysosomes, inhibiting Cathepsin B enzyme activity and disturbing autophagy.24 Replacement of the oxygen atoms in squaramides with sulfur atoms (to give thiosquaramides) leads to pH dependent anion transport.26,27 Such pH-regulating anion transporters may serve as attractive candidates for the development of anticancer drugs. Attachment of squaramido substituents to a rigid choloyl unit leads to extremely strong anion receptors that show promising anion transport.28 As for (benz)imidazole-based anion transporters, it has been reported that incorporation of (benz)imidazolyl or (benz)imidazolium groups into the structures of anion transporters may be favorable to the anion transport efficiency and biological activity.33–40 For example, Schmitzer et al. have described the strong antibacterial properties of some benzimidazolium-based anion transporters, which is a likely consequence of their ability to insert into the cellular membranes and alter the membrane permeability for chloride anions.38 We have also shown in our previous studies that 1,3-bis(benzimidazol-2-yl)benzene and its derivatives exhibit potent anion transport with promising anti-proliferative activity towards the selected solid tumor cell lines.41–43
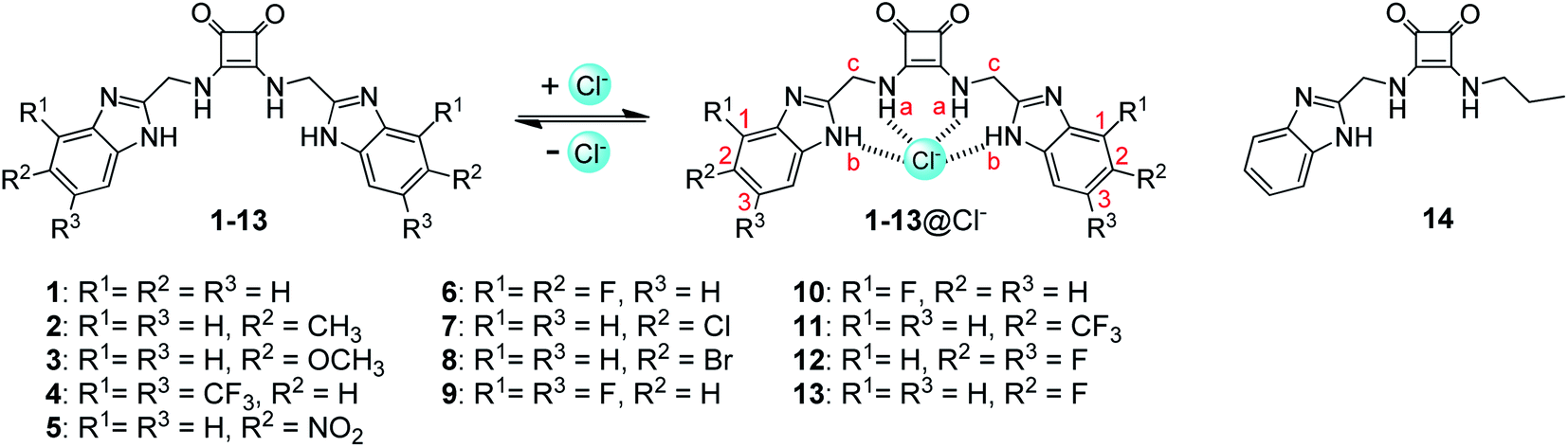
These studies have raised the feasibility that conjugation of squaramides with benzimidazoles may lead to a novel class of strong anion receptors. To test this hypothesis, we synthesized a squaramide-tethered bisbenzimidazole 1 (Fig. 1). In this conjugate, the two benzimidazolyl subunits are linked to the squaramido moiety via a CH2 unit so that the target molecule is flexible enough to complex anions through induced-fit, cooperative interactions with both the benzimidazolyl and squaramido NHs (Fig. 1).44–46 In addition, inspired by our previous findings that introduction of electron-withdrawing substituents onto the benzimidazolyl subunits of 1,3-bis(benzimidazol-2-yl)benzene is favorable to the anion transport,42,43 we modified the benzimidazolyl subunits of compound 1 with diverse substituents (to give compounds 2–13) with the aim at optimizing the anionophoric activity. For comparison, we also prepared compound 14 bearing only one benzimidazolyl subunit. Herein we report the synthesis of compounds 1–14 as well as their anion transport properties and anti-proliferative activity toward some solid tumor cell lines.
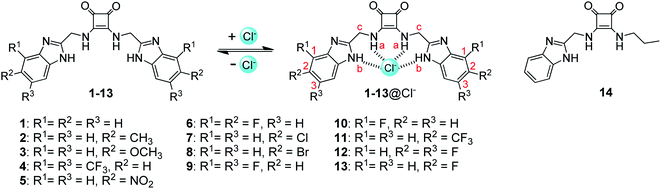 |
| | Fig. 1 Structures of compounds 1–14 and the interactions of compounds 1–13 with chloride anions. | |
2. Results and discussion
2.1 Synthesis
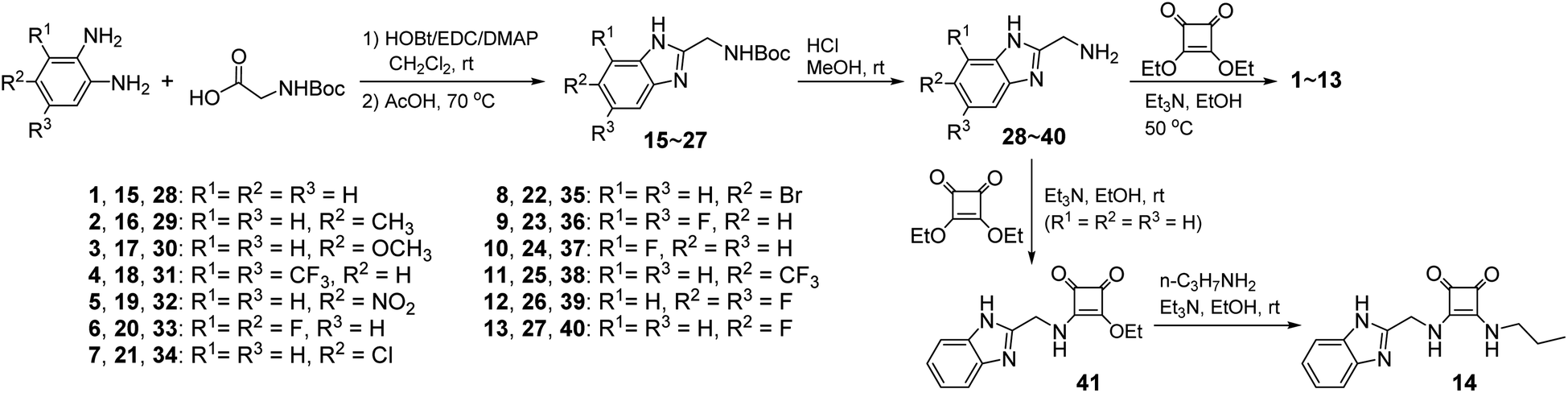
Compounds 1–14 were synthesized as shown in Scheme 1. Reaction of diethyl squarate with substituted 2-aminomethylbenzimidazoles 28–40 gave compounds 1–13. Reaction of compound 28 with excess diethyl squarate (to give compound 41) and subsequent reaction with n-propylamine afforded compound 14. Compounds 28–40 were prepared starting from the condensation of N-Boc-gly with substituted 1,2-diaminobenzene using the procedures previously reported.47,48 Compounds 1–14 were characterized by means of NMR (1H and 13C) and ESI MS (LR and HR) (see Experimental section and Fig. S1–S56†).
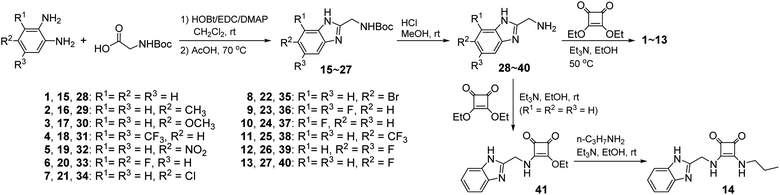 |
| | Scheme 1 Synthesis of compounds 1–14. | |
2.2 Anion recognition
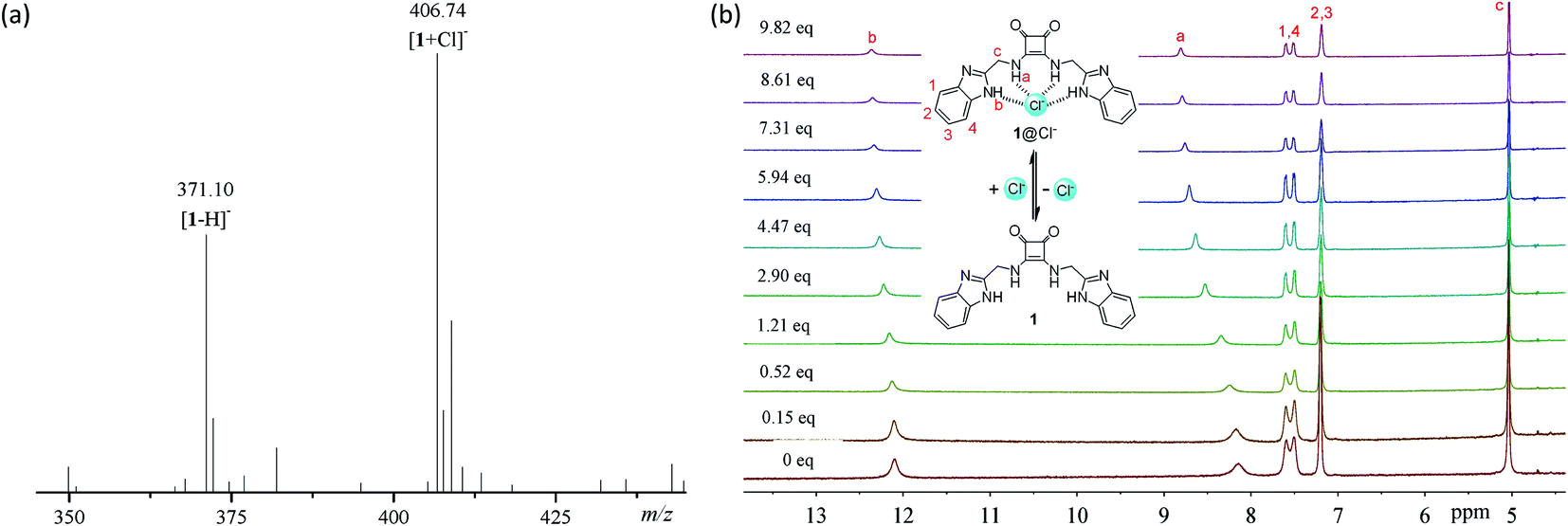
To explore the anion binding properties of compounds 1–14, we firstly measured the electrospray ionization (ESI) mass spectra of a mixture of compounds 1–14 with tetra(n-butyl)ammonium chloride (TBACl). As shown in Fig. 2a and S57 and S58,† in addition to the ion peaks from compounds 1–14 (for example, m/z = 371.17, [1 − H]−, Fig. 2a), new ion peaks that are assignable to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with chloride anions were observed (for example, m/z = 406.86, [1 + Cl]−, Fig. 2a). These observations suggest that compounds 1–14 are able to form stable 1:1 complexes with chloride anions.
:1 complexes with chloride anions were observed (for example, m/z = 406.86, [1 + Cl]−, Fig. 2a). These observations suggest that compounds 1–14 are able to form stable 1:1 complexes with chloride anions.
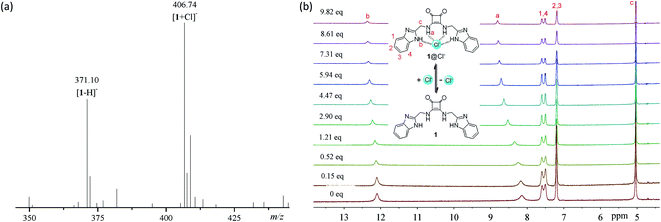 |
| | Fig. 2 (a) Negative ESI MS spectrum of compound 1 (1.0 × 10−3 M) mixed with TBACl (1.5 × 10−2 M) in 4:1 CH3CN-DMSO. (b) 1H NMR spectra of compound 1 (1.0 × 10−3 M) in the presence of TBACl of varying concentrations, in 4:1 CD3CN-DMSO-d6. | |
To clarify the binding sites and affinity of compounds 1–14 toward chloride anions, we carried out 1H NMR titrations with TBACl (Fig. 2b and S59–S71†). Upon the addition of TBACl, both the benzimidazolyl and squaramido NHs were gradually downfield shifted simultaneously, suggesting that they are involved in the recognition of chloride anions, probably via hydrogen bonding interactions (Fig. 1). To quantitatively characterize the affinity, we measured the association constants (Ka's) of compounds 1–14 with chloride anions, by nonlinear curve fitting the relationship between the downfield shifts of the squaramido NHs and the concentrations of TBACl based on a 1:1 model (Fig. S72 and S73†). The results are listed in Table 1 and suggest that compounds 1–13 exhibit moderate affinity toward chloride anions. Compound 14 is weaker than compounds 1–13, which suggests that a second benzimidazolyl subunit is necessary for achieving stronger anion recognition. Structural optimization of compound 1 with diverse substituents led to up to 10-fold increase in the binding affinity toward chloride anions, suggesting that the anion-binding affinity may be regulated by the substituents.
Table 1 Lipophilicity (clogP), anion binding affinity and chloride efflux efficiency of compounds 1–14
| Compound |
clogPa |
Anion bindingb |
Chloride efflux efficiencyc (EC50, mol%) |
| Ka (M−1) |
RA1d |
pH 7.0 |
pH 6.0 |
pH 5.0 |
pH 4.0 |
RA2e |
RA3f |
| Calculated using MarvinSketch (Version 6.1.0, Weighted Model, ChemAxon, MA). Estimated by means of 1H NMR titrations in CD3CN-DMSO-d6 (4/1, v/v). Measured by means of chloride ion selective electrode techniques, under the conditions of an intravesicular 500 mM NaCl solution (25 mM HEPES buffer) and extravesicular 500 mM NaNO3 solution (25 mM HEPES buffer). RA1 denotes the anion binding affinity of each compound relative to compound 14, that is, RA1 = Ka (each compound)/Ka (compound 14). RA2 denotes the chloride efflux efficiency of each compound at pH 7 relative to compound 1 at pH 7, that is, RA2 = EC50 (compound 1)/EC50 (each compound). RA3 denotes the chloride efflux efficiency of each compound at pH 4 relative to itself at pH 7, that is, RA3 = EC50 (pH 7)/EC50 (pH 4). |
| 1 |
1.52 |
(1.31 ± 0.25) × 102 |
1.9 |
16.7 ± 2.83 |
10.9 ± 1.10 |
9.12 ± 1.13 |
1.64 ± 0.50 |
1.0 |
10.2 |
| 2 |
2.55 |
(2.92 ± 0.32) × 102 |
4.3 |
3.78 ± 0.72 |
3.72 ± 0.65 |
1.93 ± 0.21 |
0.27 ± 0.09 |
4.4 |
14.0 |
| 3 |
1.21 |
(3.67 ± 0.67) × 102 |
5.5 |
6.67 ± 0.79 |
6.08 ± 0.77 |
3.74 ± 0.50 |
1.15 ± 0.48 |
2.5 |
5.8 |
| 4 |
5.03 |
(4.34 ± 0.49) × 102 |
6.4 |
50.4 ± 22.9 |
37.3 ± 11.1 |
26.7 ± 9.52 |
21.4 ± 10.6 |
0.3 |
2.4 |
| 5 |
1.40 |
(1.52 ± 0.91) × 102 |
2.3 |
1.84 ± 0.35 |
1.76 ± 0.16 |
1.38 ± 0.12 |
0.44 ± 0.07 |
9.1 |
4.2 |
| 6 |
2.09 |
(3.70 ± 0.22) × 102 |
5.5 |
6.41 ± 0.62 |
5.60 ± 0.47 |
2.80 ± 0.24 |
1.99 ± 0.63 |
2.6 |
3.2 |
| 7 |
2.73 |
(2.97 ± 0.83) × 102 |
4.4 |
31.5 ± 10.4 |
21.4 ± 3.58 |
15.4 ± 3.73 |
6.84 ± 2.98 |
0.5 |
4.6 |
| 8 |
3.06 |
(2.54 ± 0.18) × 102 |
3.8 |
47.8 ± 15.7 |
23.5 ± 2.89 |
16.7 ± 4.04 |
2.77 ± 1.03 |
0.3 |
17.3 |
| 9 |
2.09 |
(2.41 ± 0.30) × 102 |
3.6 |
58.0 ± 20.3 |
20.7 ± 2.72 |
11.0 ± 1.94 |
1.82 ± 0.57 |
0.3 |
31.9 |
| 10 |
1.81 |
(2.84 ± 0.26) × 102 |
4.2 |
23.3 ± 2.88 |
20.5 ± 4.52 |
10.15 ± 1.48 |
5.99 ± 2.31 |
0.7 |
3.9 |
| 11 |
3.28 |
(1.26 ± 0.34) × 103 |
18.7 |
48.9 ± 16.4 |
41.1 ± 14.0 |
30.5 ± 12.6 |
27.2 ± 23.4 |
0.3 |
1.8 |
| 12 |
2.09 |
(3.71 ± 0.33) × 102 |
5.5 |
28.0 ± 7.80 |
14.4 ± 2.30 |
10.00 ± 1.47 |
6.06 ± 0.80 |
0.6 |
4.6 |
| 13 |
1.81 |
(2.37 ± 0.32) × 102 |
3.5 |
13.9 ± 1.80 |
10.2 ± 1.16 |
6.10 ± 0.84 |
3.38 ± 0.79 |
1.2 |
4.1 |
| 14 |
1.32 |
67.3 ± 20.7 |
1.0 |
NA |
NA |
NA |
NA |
NA |
NA |
2.3 Anion transport
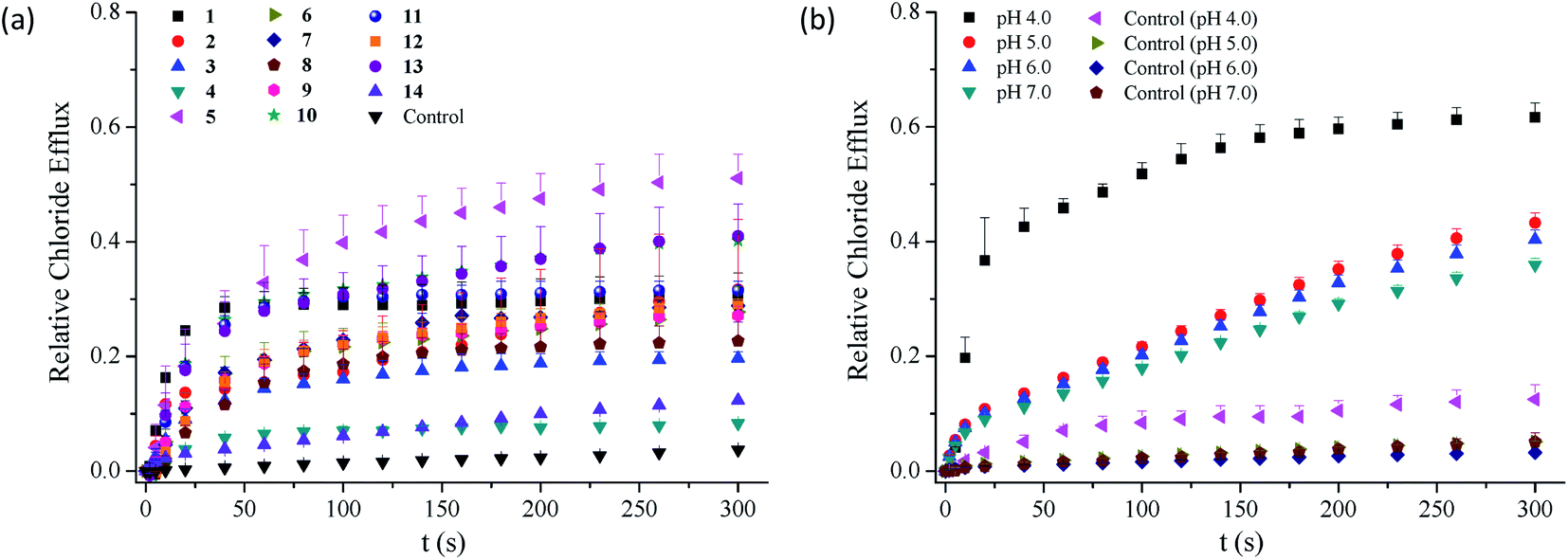
2.3.1 Chloride transport at physiological pH. As compounds 1–14 are able to complex chloride anions, they may be capable of facilitating the transport of chloride anions across phospholipid membranes. To address this, we measured the efflux of chloride anions out of egg-yolk L-α-phosphatidylcholine (EYPC) vesicles (100 nm, extrusion) using chloride ion selective electrode techniques (Fig. 3a and S74–S87†).49 The results suggest that the efflux of chloride anions across the liposomal membranes is facilitated in the presence of conjugates 1–13. Expectedly, compound 14 is not active in facilitating the transport of chloride anions (Fig. S87†). To quantitatively characterize the transport efficiency of compounds 1–13 toward chloride anions, we carried out concentration-dependent chloride efflux experiments and used a Hill equation to analyze the relationship between the relative chloride efflux at 260 s and the concentration of each compound. This analysis gives the EC50 value, the concentration that each compound needs to reach 50% of the maximum efflux.50
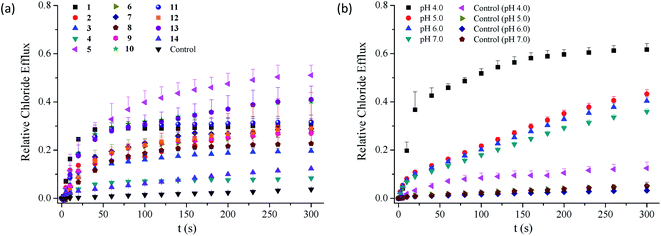 |
| | Fig. 3 (a) Relative efflux of chloride anions out of EYPC liposomes enhanced by compounds 1–14 (5 mol%) under the conditions of an intravesicular 500 mM NaCl solution in 25 mM HEPES buffer (pH 7.0) and extravesicular 500 mM NaNO3 in 25 mM HEPES buffer (pH 7.0). (b) Relative efflux of chloride anions out of EYPC liposomes enhanced by compound 1 (5 mol%) at pHs varying from 4.0 to 7.0, under the conditions of an intravesicular 500 mM NaCl solution in 25 mM HEPES buffer (pH 7.0) and extravesicular 500 mM NaNO3 in 25 mM HEPES buffer (pH 7.0). | |
As shown in Table 1, compounds 1–13 exhibit chloride transport efficiency with the EC50 values ranging from 1.84 mol% to 58.0 mol% at pH 7.0, suggesting that they are modest anion transporters. Calcein leakage experiments based on compounds 1–3 and 5–6 indicate that the transport of chloride anions across the membranes was triggered by the presence of these compounds (Fig. S88†).51 Careful analysis of the EC50 values afforded several observations on the structure–activity relationships. Firstly, the observation that compound 14 exhibits negligible anion transport activity, suggests that two benzimidazolyl subunits are required in the chloride transport. Secondly, the substituents exhibit obvious effects on the transport efficiency of compounds 1–13. Compared with compound 1, compounds 2, 3, 5, 6 and 13 exhibit 2.1–9.1-fold higher anion transport, whereas the other compounds are less active. This result suggests that the transport efficiency may be moderately regulated by the substituents. Thirdly, the highest chloride transport was observed in compound 5 bearing nitro groups on the benzimidazolyl subunits, ca. 9-fold more active than compound 1. The other conjugates bearing electron-withdrawing substituents, in particular trifluoromethyl groups (i.e., compounds 4 and 11) do not show higher activity than compound 1. These results, together with the finding that the conjugates bearing methoxy or methyl groups on the benzimidazolyl subunits exhibit higher chloride transport, suggest that introducing electron-withdrawing substituents on the benzimidazolyl subunits may not necessarily lead to an increase in the activity. This is apparently different from our the findings that electron-withdrawing groups on the benzimidazolyl subunits of 1,3-bis(benzimidazol-2-yl)benzene are favourable to the chloride transport.41–43
2.3.2 Effect of pH on the chloride transport. Literature reports show that benzimidazolyl derivatives exhibit pH dependent anion transport.52 This encouraged us to explore whether compounds 1–13 are able to facilitate the transport of chloride anions in a pH dependent fashion. To address this, we measured the efflux of chloride anions out of the EYPC liposomes at pHs varying from 4.0 to 7.0 (Fig. 3b and S74–S87,† Table 1). The EC50 value for each compound increases by 2 to 32 folds when the pH increases from 4 to 7, suggesting that all the conjugates tend to be more active at lower pHs. Interestingly, the pH-dependent increment in the activity is greater for the derivatives that are less active at physiological pH (e.g., compounds 8 and 9) than for those that are more active at physiological pH (e.g., compounds 2, 3, 6 and 13). It should be noted that compound 14 exhibits negligible activity in the pH range from 4 to 7.
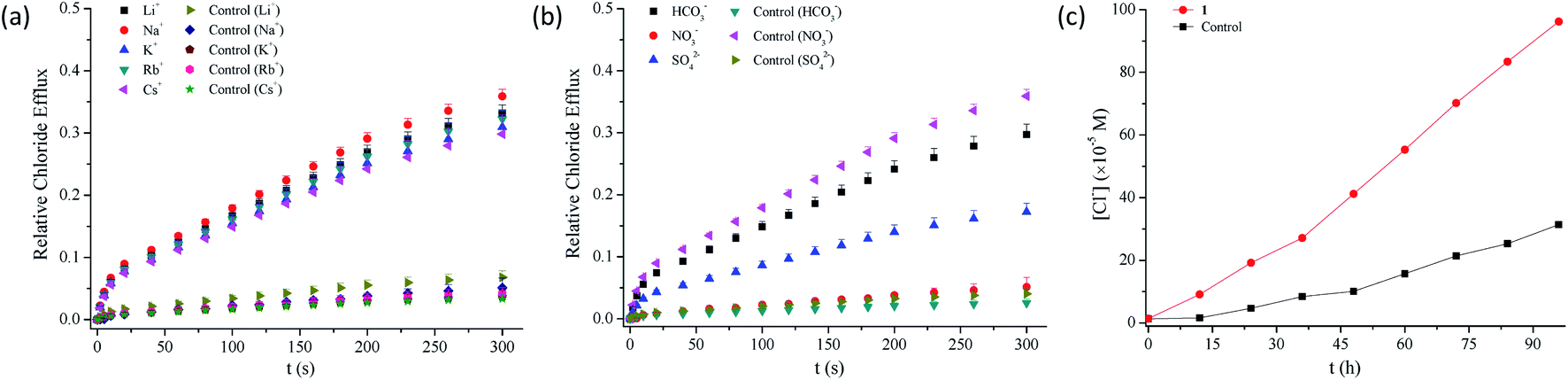
2.3.3 Probable mechanism of action of anion transport. To clarify the probable mechanism of action by which compounds 1–13 facilitate the transmembrane transport of chloride anions, we measured the efflux of chloride anions in the presence of different alkali metal ions or anions.53,54 As can be seen from Fig. 4a, b and S89–S92,† the efflux of chloride anions is regulated by the anions rather than by the metal ions, which suggests that an anion exchange process may be involved in the efflux of chloride anions. In addition, U-tube experiments show that these conjugates are able to facilitate the transport of chloride anions across bulky organic layers (Fig. 4c and S93†), suggesting that compounds 1–13 function primarily as mobile carriers.55
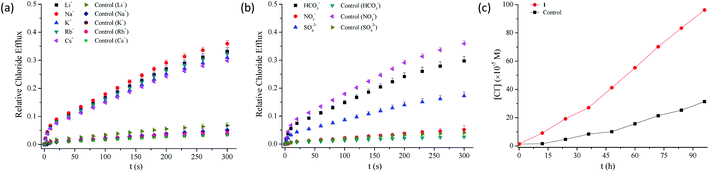 |
| | Fig. 4 (a) Relative chloride efflux out of EYPC liposomes containing Li+, Na+, K+, Rb+ or Cs+, enhanced by compound 1 (5 mol%), under the conditions of an intravesicular 500 mM MCl solution in 25 mM HEPES buffer (pH 7.0) and extravesicular 500 mM MNO3 in 25 mM HEPES buffer (pH 7.0) (M = Li, Na, K, Rb or Cs). (b) Relative chloride efflux out of EYPC liposomes, enhanced by compound 1 (5 mol%), under the conditions of an intravesicular 500 mM NaCl in 25 mM HEPES buffer (pH 7.0) and extravesicular 500 mM NaNO3, 500 mM NaHCO3 or 250 mM Na2SO4 in 25 mM HEPES buffer (pH 7.0). (c) Chloride transport across a bulky nitrobenzene membrane, promoted by compound 1 (1.0 mM) and detected by a chloride ion selective electrode in the receiving aqueous phase of U-tube, under the conditions of 500 mM NaNO3 (25 mM HEPES, pH 7.0) for the U-tube chloride receiver, 500 mM NaCl (25 mM HEPES, pH 7.0) for the U-tube chloride donor and 2 mM TBAPF6 in the nitrobenzene organic phase. | |
2.4 Cell viability
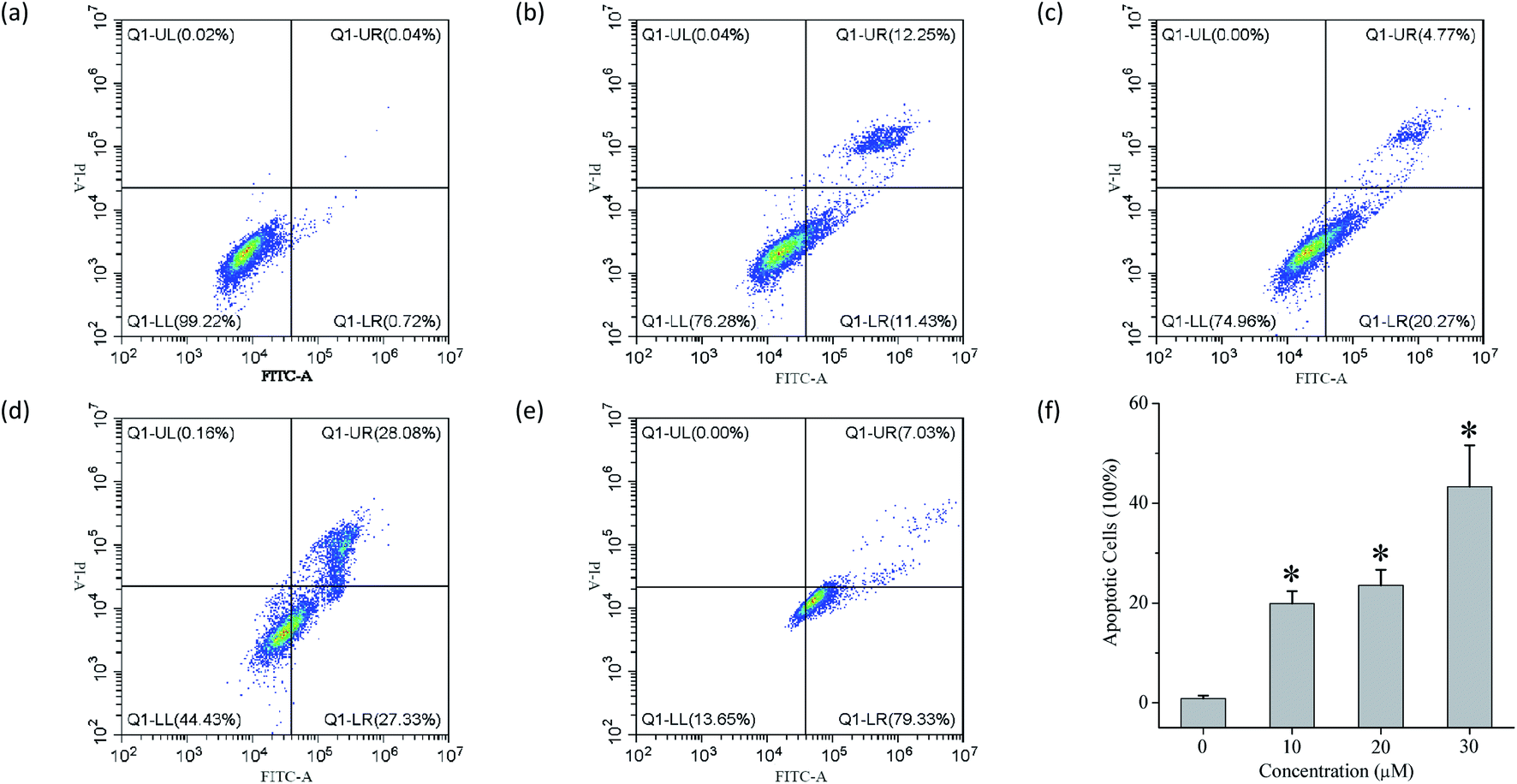
The ability of compounds 1–13 to facilitate the transport of anions across liposomal membranes encouraged us to investigate the biological effect of each compound on four selected solid cancer cell lines (that is, HeLa cervical cancer cells, A549 lung adenocarcinoma cells, MCF-7 breast cancer cells and HepG2 human liver cancer cells) and one normal LO2 cell line. Using a conventional MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra zolium bromide) assay, we measured the inhibitory activity of each compound at 50 μM toward those cell lines (Fig. S94†), and the IC50 values of those conjugates that exhibit higher than 50% inhibition at the above-mentioned concentrations (Fig. S94–S96† and Table 2). Doxorubicin was used as a positive drug. The results indicate that some of these conjugates, such as compounds 4, 5, 7, 8 and 11 exhibit moderate inhibitory activity toward the four cancer cells. It appears that these conjugates exhibit no obvious cytotoxic effect on the LO2 cell line. In addition, we used flow cytometry to investigate the probable mode of cell death triggered by compound 11 (Fig. 5). The results imply that compound 11 may cause cell death via an apoptotic fashion. More work will be carried out to clarify this.
Table 2 Inhibitory activity of compounds 4, 5, 7, 8, 11 and doxorubicin
| Compound |
Inhibitory activitya (IC50, μM) |
| HeLa |
A549 |
MCF-7 |
HepG2 |
| Calculated using GraphPad Prism v8.0. |
| 4 |
23.27 ± 2.19 |
>50 |
18.10 ± 3.19 |
18.74 ± 1.84 |
| 5 |
>50 |
>50 |
>50 |
12.62 ± 2.30 |
| 7 |
>50 |
37.31 ± 6.07 |
9.85 ± 1.72 |
16.09 ± 4.52 |
| 8 |
31.94 ± 13.36 |
>50 |
17.35 ± 5.45 |
14.41 ± 5.48 |
| 11 |
9.59 ± 0.56 |
>50 |
10.51 ± 2.57 |
12.81 ± 2.17 |
| Doxorubicin |
0.99 ± 0.16 |
1.99 ± 0.08 |
0.77 ± 0.10 |
1.75 ± 0.12 |
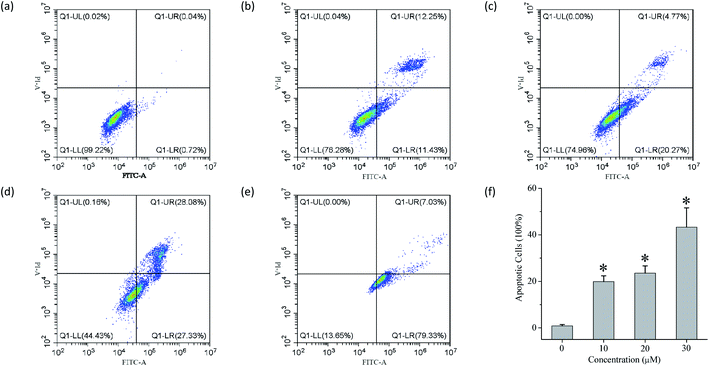 |
| | Fig. 5 Flow cytometry of (a) untreated HeLa cells and (b–e) HeLa cells treated with 11 ((b) 10 μM; (c) 20 μM; (d) 30 μM) and doxorubicin ((e) 0.03 μM) for 24 h and stained with Annexin V-FITC and PI solution. (f) The apoptosis rate of HeLa cells by compound 11 at different concentrations (mean ± s.d., n = 3, *P < 0.05, One-Way ANOVA). | |
3. Conclusion
In conclusion, we have successfully synthesized a series of squaramido-tethered bisbenzimidazoles and assessed their anion binding affinity, transport properties and biological activity. These conjugates are able to complex chloride anions, and facilitate the transport of chloride anions with moderate pH dependence and via an anion exchange process. In addition, some of these conjugates exhibit moderate anti-proliferative effects on the selected solid tumor cell lines, whereas without significant cytotoxicity toward the normal cell line. Further work will be carried out to optimize the structures of these conjugates so that promising pH-regulated anionophoric activity may be achieved. The outcomes will be reported in due course.
4. Experimental
4.1 Generals
The 1H and 13C NMR spectra were measured on a Bruker Avance AV 400 or 500 spectrometer, and the data were reported relative to the deuterium solvents. Waters UPLC/Quattro Premier XE and Bruker maXis 4G ESI-Q-TOF mass spectrometers were used to measure the LR and HR ESI-MS spectra, respectively. Analytical thin-layer chromatography (TLC) plates (silica gel, GF254) were detected by use of iodine and UV (254 or 365 nm). EYPC vesicles were prepared by extrusion through nuclepore track-etched polycarbonate membranes (100 nm, Whatman, Florham Park, New Jersey, USA) on an Avanti's Mini-Extruder (Avanti Polar Lipids, Inc., Alabaster, Alabama, USA). Chloride efflux was measured by using a Mettler-Toledo PerfectIon™ chloride ion selective electrode assembled with a Mettler-Toledo Seven Compact S220 ionometer.
EYPC and MTT were purchased from Sigma Chemical Co. (St Louis, USA). All the other chemicals and reagents were obtained from commercial sources and used without further purification. The experimental protocols for the measurement of anion recognition, anion transport and biological activity were included in the ESI.†
4.2 Synthesis of compounds 15–27
4.2.1 Compound 15. A solution of N-Boc-gly (805 mg, 4.59 mmol), EDC (1.77 g, 9.23 mmol), HOBt (936 mg, 6.92 mmol) and DMAP (30 mg, 0.24 mmol) in CH2Cl2 (40 mL) was stirred at room temperature for 10 min, and added drop wise to a solution of 1,2-diaminobenzene (500 mg, 4.62 mmol) in DMF (1.5 mL). The resulting mixture was stirred at room temperature for 3 d and concentrated under reduced pressure. The obtained residue was dissolved in EtOAc (20 mL) and washed subsequently with brine (20 mL × 2), saturated aqueous NH4Cl solution (20 mL × 2), saturated aqueous NaHCO3 solution (20 mL × 2) and brine (20 mL). The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The obtained residue was dissolved in acetic acid (5 mL). The resulting solution was heated at 70 °C for 10 h, and concentrated under reduced pressure. The obtained residue was dissolved in ether (20 mL) and filtered. The filtrate was concentrated under reduced pressure and the obtained residue was purified by chromatography on a silica gel column (petroleum ether/EtOAc, 2/1, v/v) to give compound 15 (515 mg, 45%) having 1H NMR (400 MHz, CD3OD) δ 7.51 (s, 2H), 7.20 (s, 2H), 4.48 (s, 2H), 1.47 (s, 9H) and negative ESI-MS: m/z 245.96 ([M − H]−).
4.2.2 Compound 16. Similar procedures as described for compound 15. 266 mg (25%). 1H NMR (400 MHz, CD3OD) δ 7.38 (d, J = 7.2 Hz, 1H), 7.30 (s, 1H), 7.03 (d, J = 7.6 Hz, 1H), 4.45 (s, 2H), 2.42 (s, 3H), 1.46 (s, 9H) and negative ESI-MS: m/z 259.83 ([M − H]−).
4.2.3 Compound 17. Similar procedures as described for compound 15. 458 mg (47%). 1H NMR (400 MHz, CD3OD) δ 7.38 (d, J = 8.8 Hz, 1H), 7.02 (s, 1H), 6.83 (d, J = 8.8 Hz, 1H), 4.44 (s, 2H), 3.81 (s, 3H), 1.46 (s, 9H) and negative ESI-MS: m/z 275.83 ([M − H]−).
4.2.4 Compound 18. Similar procedures as described for compound 15. 302 mg (38%). 1H NMR (400 MHz, CD3OD) δ 8.09 (s, 1H), 7.76 (s, 1H), 4.57 (s, 2H), 1.47 (s, 9H) and negative ESI-MS: m/z 381.87 ([M − H]−).
4.2.5 Compound 19. Similar procedures as described for compound 15. 287 mg (30%). 1H NMR (400 MHz, CD3OD) δ 8.46 (s, 1H), 8.17 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 7.1 Hz, 1H), 4.53 (s, 2H), 1.47 (s, 9H); negative ESI-MS: m/z 290.88 ([M − H]−) and negative HR-ESI-MS for C24H12F12N6O2 ([M − H]−) calcd: 291.10878; found: 291.10983.
4.2.6 Compound 20. Similar procedures as described for compound 15. 342 mg (34%). 1H NMR (400 MHz, CD3OD) δ 7.05 (d, J = 5.4 Hz, 1H), 6.84–6.80 (m, 1H), 4.47 (s, 2H), 1.47 (s, 9H) and negative ESI-MS: m/z 281.88 ([M − H]−).
4.2.7 Compound 21. Similar procedures as described for compound 15. 140 mg (14%). 1H NMR (CD3OD, 400 MHz) δ 7.51–7.46 (m, 2H), 7.19 (d, J = 8.5 Hz, 1H), 4.47 (s, 2H), 1.46 (s, 9H) and negative ESI-MS: m/z 280.12 ([M − H]−).
4.2.8 Compound 22. Similar procedures as described for compound 15. 221 mg (38%). 1H NMR (400 MHz, CD3OD) δ 7.67 (s, 1H), 7.42 (s, 1H), 7.32 (d, J = 8.4 Hz, 1H), 4.42 (s, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 158.5, 155.9, 129.1, 123.8, 81.0, 39.6, 28.9; negative ESI-MS: m/z 323.81 ([M − H]−) and negative HR-ESI-MS for C13H16BrN3O2 ([M − H]−) calcd: 324.03421; found: 324.03513.
4.2.9 Compound 23. Similar procedures as described for compound 15. 365 mg (37%). 1H NMR (400 MHz, CD3OD) δ 7.05 (d, J = 8.3 Hz, 1H), 6.81 (t, J = 10.3 Hz, 1H), 4.46 (s, 2H), 1.45 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 161.5 (d, J = 42.5 Hz), 159.1 (d, J = 63.4 Hz), 158.1, 156.0, 129.7, 98.7, 98.5, 98.4, 98.2, 80.8, 39.2, 28.7; negative ESI-MS: m/z 281.91 ([M − H]−) and negative HR-ESI-MS for C13H15F2N3O2 ([M − H]−) calcd: 282.10485; found: 282.10547.
4.2.10 Compound 24. Similar procedures as described for compound 15. 143 mg (23%). 1H NMR (400 MHz, CD3OD) δ 7.30 (d, J = 7.3 Hz, 1H), 7.19–7.14 (m, 1H), 6.92 (t, J = 9.2 Hz, 1H), 4.39 (s, 2H), 1.37 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 158.4, 155.0, 124.1 (d, J = 27.2 Hz), 108.4 (d, J = 69.1 Hz), 80.9, 39.6, 28.5; negative ESI-MS: m/z 263.93 ([M − H]−) and negative HR-ESI-MS for C13H16FN3O2 ([M − H]−) calcd: 264.11428; found: 264.11514.
4.2.11 Compound 25. Similar procedures as described for compound 15. 246 mg (28%). 1H NMR (400 MHz, CD3OD) δ 7.83 (s, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.49 (d, J = 8.3 Hz, 1H), 4.52 (s, 2H), 1.47 (s, 9H) and negative ESI-MS: m/z 314.08 ([M − H]−).
4.2.12 Compound 26. Similar procedures as described for compound 15. 159 mg (16%). 1H NMR (400 MHz, CD3OD) δ 7.29 (s, 2H), 4.36 (s, 2H), 1.37 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 158.4, 156.4, 150.3 (d, J = 61.6 Hz), 147.9 (d, J = 62.4 Hz), 132.4, 129.9, 106.2, 100.5, 80.9, 39.6, 28.7; negative ESI-MS: m/z 281.94 ([M − H]−) and negative HR-ESI-MS for C13H15F2N3O2 ([M − H]−) calcd: 282.10485; found: 282.10583.
4.2.13 Compound 27. Similar procedures as described for compound 15. 268 mg (29%). 1H NMR (400 MHz, CD3OD) δ 7.84 (s, 1H), 7.67 (d, J = 6.6 Hz, 1H), 7.50 (d, J = 6.7 Hz, 1H), 4.53 (s, 2H), 1.48 (s, 9H) and negative ESI-MS: m/z 263.93 ([M − H]−).
4.3 Synthesis of compounds 28–40
4.3.1 Compound 28. To a solution of compound 15 (60 mg) in CH3OH (3 mL) was added HCl aqueous solution (2 M, 3 mL). The resulting solution was stirred at room temperature for 1 h and concentrated under reduced pressure. The obtained residue was partitioned in ammonia solution (3 mL). The mixture was stirred at room temperature for 15 min and concentrated under reduced pressure. This procedure was repeated. Purification by preparative thin-layer chromatography (CH2Cl2/MeOH/NH3·H2O, 24/3/1, v/v/v) afforded compound 28 (39 mg, 45%). 1H NMR (500 MHz, CD3OD) δ 7.54 (dd, J = 4.8, 2.5 Hz, 2H), 7.22 (dd, J = 4.8, 2.5 Hz, 2H), 4.03 (s, 2H).
4.3.2 Compound 29. Similar procedures as described for compound 28. 40 mg (72%) from compound 16 (90 mg). 1H NMR (400 MHz, CD3OD) δ 7.40 (d, J = 8.2 Hz, 1H), 7.31 (s, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.01 (s, 2H), 2.44 (s, 3H).
4.3.3 Compound 30. Similar procedures as described for compound 28. 55 mg (71%) from compound 17 (122 mg). 1H NMR (400 MHz, CD3OD) δ 7.40 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 2.3 Hz, 1H), 6.85 (dd, J = 8.8, 2.4 Hz, 1H), 3.99 (s, 2H), 3.82 (s, 3H).
4.3.4 Compound 31. Similar procedures as described for compound 28. 69 mg (70%) from compound 18 (134 mg). 1H NMR (400 MHz, CD3OD) δ 8.06 (s, 1H), 7.72 (s, 1H), 4.11 (s, 2H).
4.3.5 Compound 32. Similar procedures as described for compound 28. 62 mg (73%) from compound 19 (129 mg). 1H NMR (400 MHz, CD3OD) δ 8.45 (d, J = 2.0 Hz, 1H), 8.16 (dd, J = 8.9, 2.0 Hz, 1H), 7.65 (d, J = 8.9 Hz, 1H), 4.09 (s, 2H).
4.3.6 Compound 33. Similar procedures as described for compound 28. 42 mg (75%) from compound 20 (86 mg). 1H NMR (400 MHz, CD3OD) δ 7.28–7.25 (m, 1H), 7.16–7.09 (m, 1H), 4.04 (s, 2H).
4.3.7 Compound 34. Similar procedures as described for compound 28. 72 mg (85%) from compound 21 (132 mg). 1H NMR (400 MHz, CD3OD) δ 7.68 (s, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.33 (dd, J = 8.5, 1.7 Hz, 1H), 4.04 (s, 2H).
4.3.8 Compound 35. Similar procedures as described for compound 28. 144 mg (68%) from compound 22 (307 mg). 1H NMR (400 MHz, CD3OD) δ 7.75 (d, J = 1.0 Hz, 1H), 7.51 (d, J = 6.9 Hz, 1H), 7.38 (dd, J = 6.8, 1.4 Hz, 1H), 4.33 (s, 2H).
4.3.9 Compound 36. Similar procedures as described for compound 28. 76 mg (70%) from compound 23 (167 mg). 1H NMR (400 MHz, CD3OD) δ 7.07 (dd, J = 8.4, 1.7 Hz, 1H), 6.82 (td, J = 10.4, 2.0 Hz, 1H), 4.01 (s, 2H).
4.3.10 Compound 37. Similar procedures as described for compound 28. 64 mg (73%) from compound 24 (140 mg). 1H NMR (400 MHz, CD3OD) δ 7.31 (d, J = 8.0 Hz, 1H), 7.17 (ddd, J = 8.0, 8.0, 4.8 Hz, 1H), 6.93 (dd, J = 10.8, 8.0 Hz, 1H), 4.04 (s, 2H).
4.3.11 Compound 38. Similar procedures as described for compound 28. 54 mg (72%) from compound 25 (110 mg). 1H NMR (400 MHz, CD3OD) δ 7.84 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 4.08 (s, 2H).
4.3.12 Compound 39. Similar procedures as described for compound 28. 70 mg (72%) from compound 26 (150 mg). 1H NMR (400 MHz, CD3OD) δ 7.45 (t, J = 8.8 Hz, 2H), 4.23 (s, 2H).
4.3.13 Compound 40. Similar procedures as described for compound 28. 79 mg (73%) from compound 27 (172 mg). 1H NMR (400 MHz, CD3OD) δ 7.50 (dd, J = 8.8, 4.7 Hz, 1H), 7.15 (dd, J = 9.1, 2.2 Hz, 1H), 7.00 (ddd, J = 9.3, 9.3, 2.4 Hz, 1H), 4.08 (s, 2H).
4.4 Synthesis of compound 41
A solution of compound 28 (40 mg, 0.27 mmol) and Et3N (300 μL) in EtOH (3 mL) was stirred at room temperature for 30 min. Then, a solution of diethyl squarate (55 mg, 0.32 mmol) in EtOH (270 μL) was added. The resulting solution was stirred at room temperature for 23 h. Purification by preparative thin-layer chromatography (CH2Cl2/CH3OH, 20/1, v/v) afforded compound 41 (44 mg, 71%) having 1H NMR (500 MHz, CD3OD) δ 7.54 (dd, J = 6.0, 3.2 Hz, 2H), 7.23 (dd, J = 6.0, 3.2 Hz, 2H), 5.07 (s, 2H), 3.59 (q, J = 7.1 Hz, 2H), 1.16 (t, J = 7.1 Hz, 3H); negative ESI-MS: m/z 270.09 ([M − H]−) and negative HR-ESI-MS for C14H13N3O3 ([M − H]−) calcd: 270.08542; found: 270.08732.
4.5 Synthesis of compounds 1–13
4.5.1 Compound 1. A solution of compound 28 (39 mg, 0.16 mmol) and Et3N (300 μL) in EtOH (3 mL) was stirred at 50 °C for 30 min. Then, a solution of diethyl squarate (7 mg, 0.042 mmol) in EtOH (100 μL) was added. The resulting solution was stirred at 50 °C for 54 h. The formed precipitates were collected through filtration under reduced pressure and washed with EtOH (20 mL × 5) to afford compound 1 (12 mg, 31%) having 1H NMR (400 MHz, DMSO-d6) δ 12.45 (s, 2H), 8.24 (s, 2H), 7.53 (s, 4H), 7.17 (s, 4H), 5.02 (s, 4H); selected 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.7, 151.5, 121.7, 41.4; negative ESI-MS: m/z 371.07 ([M − H]−) and negative HR-ESI-MS for C20H16N6O2 ([M − H]−) calcd: 371.12510; found: 371.12570.
4.5.2 Compound 2. Similar procedures as described for compound 1; 35 mg (79%) from compound 29 (40 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 2H), 8.18 (s, 2H), 7.44–7.26 (m, 4H), 6.99 (s, 2H), 4.99 (s, 4H), 2.39 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 183.0, 167.9, 151.4, 151.0, 143.3, 141.1, 134.7, 132.4, 131.7, 130.4, 123.8, 122.9, 118.2, 111.3, 41.5, 21.3; negative ESI-MS: m/z 399.06 ([M − H]−) and negative HR-ESI-MS for C22H20N6O2 ([M − H]−) calcd: 399.15640; found: 399.15680.
4.5.3 Compound 3. Similar procedures as described for compound 1; 43 mg (71%) from compound 30 (55 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 2H), 8.19 (s, 2H), 7.44 (s, 2H), 7.04 (s, 2H), 6.83 (d, J = 7.8 Hz, 2H), 5.00 (s, 4H), 3.79 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.7, 155.8, 154.8, 150.3, 137.3, 134.9, 128.5, 119.0, 111.6, 110.7, 101.3, 94.7, 55.4, 41.4; negative ESI-MS: m/z 431.01 ([M − H]−) and negative HR-ESI-MS for C22H20N6O4 ([M − H]−) calcd: 431.14622; found: 431.14667.
4.5.4 Compound 4. Similar procedures as described for compound 1; 33 mg (43%) from compound 31 (66 mg). 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 2H), 8.20 (s, 2H), 7.79 (s, 2H), 5.14 (s, 4H); 13C NMR (125 MHz, DMSO-d6) δ 183.2, 168.9, 167.6, 144.3, 127.0, 125.2, 125.1, 124.0, 123.1, 123.0, 121.1, 114.4 (q, J = 132.3 Hz), 111.2 (q, J = 120.8 Hz), 45.8; negative ESI-MS: m/z 642.89 ([M − H]−) and negative HR-ESI-MS for C24H12F12N6O2 ([M − H]−) calcd: 643.07463; found: 643.07489.
4.5.5 Compound 5. Similar procedures as described for compound 1; 45 mg (69%) from compound 32 (59 mg). 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 2H), 8.30 (s, 2H), 8.11 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 5.10 (s, 4H); selected 13C NMR (125 MHz, DMSO-d6) δ 183.1, 168.0, 156.8, 142.6, 117.9, 41.5; negative ESI-MS: m/z 460.99 ([M − H]−) and negative HR-ESI-MS for C20H14N8O6 ([M − H]−) calcd: 461.09525; found: 461.09579.
4.5.6 Compound 6. Similar procedures as described for compound 1; 30 mg (70%) from compound 33 (39 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 2H), 8.24 (s, 2H), 7.32–7.20 (m, 4H), 5.04 (s, 4H); selected 13C NMR (100 MHz, DMSO-d6) δ 183.5, 167.3, 154.5, 146.3 (d, J = 42.8 Hz), 144.0 (d, J = 40.8 Hz), 111.1, 99.4, 41.5; negative ESI-MS: m/z 443.02 ([M − H]−) and negative HR-ESI-MS for C20H12F4N6O2 ([M − H]−) calcd: 443.08741; found: 443.08801.
4.5.7 Compound 7. Similar procedures as described for compound 1; 52 mg (71%) from compound 34 (69 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.63 (s, 2H), 8.20 (s, 2H), 7.73 (s, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.31 (dd, J = 8.5, 1.7 Hz, 2H), 5.01 (s, 4H); selected 13C NMR (100 MHz, DMSO-d6) δ 182.6, 168.0, 153.1, 124.7, 114.2, 41.5; negative ESI-MS: m/z 438.89 ([M − H]−) and negative HR-ESI-MS for C20H14Cl2N6O2 ([M − H]−) calcd: 439.04715; found: 439.04749.
4.5.8 Compound 8. Similar procedures as described for compound 1; 96 mg (73%) from compound 35 (142 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.62 (s, 2H), 8.22 (s, 2H), 7.60–7.53 (m, 2H), 7.20 (dd, J = 8.5, 1.6 Hz, 2H), 5.02 (s, 4H); selected 13C NMR (100 MHz, DMSO-d6) δ 183.3, 168.0, 153.1, 126.4, 122.0, 41.5; negative ESI-MS: m/z 526.85 ([M − H]−) and negative HR-ESI-MS for C20H14Br2N6O2 ([M − H]−) calcd: 526.94612; found: 526.94629.
4.5.9 Compound 9. Similar procedures as described for compound 1; 58 mg (72%) from compound 36 (73 mg). 1H NMR (400 MHz, DMSO-d6) δ, 8.19 (s, 2H), 7.22 (dd, J = 8.6, 1.5 Hz, 2H), 7.04 (td, J = 10.7, 1.7 Hz, 2H), 5.02 (s, 4H); selected 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.8, 159.1 (d, J = 77.0 Hz), 156.8 (d, J = 55.7 Hz), 153.1, 97.5, 97.3, 97.2, 96.9, 41.3; negative ESI-MS: m/z 442.99 ([M − H]−) and negative HR-ESI-MS for C20H12F4N6O2 ([M − H]−) calcd: 443.08741; found: 443.08789.
4.5.10 Compound 10. Similar procedures as described for compound 1; 51 mg (73%) from compound 37 (61 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.87 (s, 2H), 8.22 (s, 2H), 7.35 (d, J = 7.6 Hz, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.16 (t, J = 8.0 Hz, 1H), 6.99 (t, J = 8.6 Hz, 2H), 5.05 (s, 4H); 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.7, 152.1, 137.1, 131.2, 122.8, 107.9, 106.8, 106.7, 41.3; negative ESI-MS: m/z 407.01 ([M − H]−) and negative HR-ESI-MS for C20H14F2N6O2 ([M − H]−) calcd: 407.10625; found: 407.10687.
4.5.11 Compound 11. Similar procedures as described for compound 1; 42 mg (74%) from compound 38 (52 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 2H), 8.24 (s, 2H), 7.90 (s, 2H), 7.73 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 5.08 (s, 4H); 13C NMR (125 MHz, DMSO-d6) δ 183.1, 167.9, 154.6, 129.1, 126.4, 123.7, 122.6 (q, J = 156.1 Hz), 121.0, 118.7, 41.5; negative ESI-MS: m/z 507.03 ([M − H]−) and negative HR-ESI-MS for C22H14F6N6O2 ([M − H]−) calcd: 507.09986; found: 507.10004.
4.5.12 Compound 12. Similar procedures as described for compound 1; 54 mg (71%) from compound 39 (67 mg). 1H NMR (400 MHz, DMSO-d6) δ 8.13 (s, 2H), 7.58 (t, J = 9.2 Hz, 4H), 4.99 (s, 4H); selected 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.8, 153.7, 147.8 (d, J = 76.5 Hz), 145.4 (d, J = 75.7 Hz), 41.4; negative ESI-MS: m/z 442.94 ([M − H]−) and negative HR-ESI-MS for C20H12F4N6O2 ([M − H]−) calcd: 443.08741; found: 443.08786.
4.5.13 Compound 13. Similar procedures as described for compound 1; 61 mg (72%) from compound 40 (76 mg). 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 2H), 8.17 (s, 2H), 7.54–7.51 (m, 2H), 7.34 (d, J = 8.1 Hz, 2H), 7.03 (t, J = 8.7 Hz, 2H), 5.01 (s, 4H); 13C NMR (125 MHz, DMSO-d6) δ 183.0, 167.8, 159.6, 157.3, 153.0, 110.0, 109.8, 41.1; negative ESI-MS: m/z 407.04 ([M − H]−) and negative HR-ESI-MS for C20H14F2N6O2 ([M − H]−) calcd: 407.10625; found: 407.10605.
4.6 Synthesis of compound 14
To a solution of compound 41 (40 mg, 0.15 mmol) in EtOH (2 mL) were added Et3N (200 μL) and 1-propylamine (9 mg, 0.16 mmol). The resulting mixture was stirred at room temperature for 23 h. The formed precipitates were collected through filtration under reduced pressure and washed with EtOH (20 mL × 5) to give compound 14 (34 mg, 79%) having 1H NMR (500 MHz, DMSO-d6) δ 12.48 (s, 1H), 7.97 (s, 1H), 7.58 (s, 1H), 7.48 (s, 1H), 7.17 (s, 2H), 5.00 (s, 2H), 3.50 (s, 2H), 1.54 (sext, J = 7.1 Hz, 2H), 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 183.0, 182.3, 168.4, 167.1, 151.6, 142.9, 134.0, 122.2, 121.4, 118.5, 111.4, 45.1, 41.1, 24.0, 11.2; negative ESI-MS: m/z 283.08 ([M − H]−) and negative HR-ESI-MS for C13H16N2O2 ([M − H]−) calcd: 283.11895, found: 283.11703.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21877057) and Jiangmen Program for Innovative Research Team (No. 2018630100180019806) is acknowledged.
Notes and references
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- X.-H. Yu, X.-Q. Hong, Q.-C. Mao and W.-H. Chen, Eur. J. Med. Chem., 2019, 184, 111782 CrossRef CAS.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- Z. Li and W.-H. Chen, Mini-Rev. Med. Chem., 2017, 17, 1398–1405 CAS.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS.
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC.
- N. Busschaert and A. P. Gale, Angew. Chem., Int. Ed., 2013, 52, 1374–1382 CrossRef CAS.
- G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824–2833 CrossRef CAS.
- P. A. Gale, R. Pérez-Tomás and R. Quesada, Acc. Chem. Res., 2013, 46, 2801–2813 CrossRef CAS.
- F. De Riccardis, I. Izzo, D. Montesarchio and P. Tecilla, Acc. Chem. Res., 2013, 46, 2781–2790 CrossRef CAS.
- S. K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. V. Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS.
- C. R. Elie, G. David and A. R. Schmitzer, J. Med. Chem., 2015, 58, 2358–2366 CrossRef CAS.
- W.-H. Chen, X.-B. Shao, R. Moellering, C. Wennersten and S. L. Regen, Bioconjugate Chem., 2006, 17, 1582–1591 CrossRef CAS.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS.
- T. Saha, M. S. Hossain, D. Saha, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 7558–7567 CrossRef CAS.
- V. Soto-Cerrato, P. Manuel-Manresa, E. Hernando, S. Calabuig-Farinas, A. Martínez-Romero, V. Fernandez-Duenas, K. Sahlholm, T. Knopfel, M. García-Valverde, A. M. Rodilla, E. Jantus-Lewintre, R. Farras, F. Ciruela, R. Perez-Tomas and R. Quesada, J. Am. Chem. Soc., 2015, 137, 15892–15898 CrossRef CAS.
- E. Hernando, V. Soto-Cerrato, S. Cortes-Arroyo, R. Pérez-Tomás and R. Quesada, Org. Biomol. Chem., 2014, 12, 1771–1778 RSC.
- S. J. Moore, M. Wenzel, M. E. Light, R. Morley, S. J. Bradberry, P. Gomez-Iglesias, V. Soto-Cerrato, R. Pérez-Tomás and P. A. Gale, Chem. Sci., 2012, 3, 2501–2509 RSC.
- S. J. Moore, C. J. E. Haynes, J. Gonzalez, J. L. Sutton, S. J. Brooks, M. E. Light, J. Herniman, G. J. Langley, V. Soto-Cerrato, R. Perez-Tomas, I. Marques, P. J. Costa, V. Felix and P. A. Gale, Chem. Sci., 2013, 4, 103–117 RSC.
- H. Li, H. Valkenier, A. G. Thorne, C. M. Dias, J. A. Cooper, M. Kieffer, N. Busschaert, P. A. Gale, D. N. Sheppard and A. P. Davis, Chem. Sci., 2019, 10, 9663–9672 RSC.
- X.-J. Cai, Z. Li and W.-H. Chen, Mini-Rev. Org. Chem., 2018, 15, 148–156 CrossRef CAS.
- X.-H. Yu, X.-J. Cai, X.-Q. Hong, K. Y. Tam, K. Zhang and W.-H. Chen, Future Med. Chem., 2019, 11, 1091–1106 CrossRef CAS.
- X. Bao, X. Wu, S. N. Berry, E. N. W. Howe, Y.-T. Chang and P. A. Gale, Chem. Commun., 2018, 54, 1363–1366 RSC.
- N. Busschaert, S. H. Park, K. H. Beak, Y. P. Choi, J. H. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannids, I. Marques, V. Felix, W. Namkung, J. L. Sesseler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS.
- X.-J. Cai, Z. Li and W.-H. Chen, Bioorg. Med. Chem. Lett., 2017, 27, 1999–2002 CrossRef CAS.
- X. Wu, L. W. Judd, E. N. W. Howe, A. M. Withecombe, V. Soto-Cerrato, H. Li, N. Busschaert, H. Valkenier, R. Perez-Tomas, D. N. Sheppard, Y.-B. Jiang, A. P. Davis and P. A. Gale, Chem, 2016, 1, 127–146 CAS.
- R. B. P. Elmes, N. Busschaert, D. D. Czech, P. A. Gale and K. A. Jolliffe, Chem. Commun., 2015, 51, 10107–10110 RSC.
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS.
- Z. Li, L.-Q. Deng, J.-X. Chen, C.-Q. Zhou and W.-H. Chen, Org. Biomol. Chem., 2015, 13, 11761–11769 RSC.
- N. Busschaert, R. B. P. Elmes, D. D. Czech, X. Wu, I. L. Kirby, E. M. Peck, K. D. Hendzel, S. K. Shaw, B. Chan, B. D. Smith, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 3617–3626 RSC.
- L.-Q. Deng, Y.-M. Lu, C.-Q. Zhou, J.-X. Chen, B. Wang and W.-H. Chen, Bioorg. Med. Chem. Lett., 2014, 24, 2859–2862 CrossRef CAS.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS.
- Z. Li, L.-Q. Deng, Y. Chen, T. Wu and W.-H. Chen, Bioorg. Med. Chem. Lett., 2016, 26, 3665–3668 CrossRef CAS.
- C.-C. Peng, Z. Li, L.-Q. Deng, Z.-F. Ke and W.-H. Chen, Bioorg. Med. Chem. Lett., 2016, 26, 2442–2445 CrossRef CAS.
- L. Gonzalez-Mendoza, B. Altava, M. I. Burguete, J. Escorihuela, E. Hernando, S. V. Luis, R. Quesada and C. Vicent, RSC Adv., 2015, 5, 34415–34423 RSC.
- J. Gravel and A. R. Schmitzer, Supramol. Chem., 2015, 27, 364–371 CrossRef CAS.
- J. Kempf, N. Noujeim and A. R. Schmitzer, RSC Adv., 2014, 4, 42293–42299 RSC.
- C. R. Elie, G. David and A. R. Schmitzer, J. Med. Chem., 2015, 58, 2358–2366 CrossRef CAS.
- C.-R. Elie, M. Charbonneau and A. R. Schmitzer, RSC Med. Chem., 2012, 3, 1231–1234 RSC.
- C.-R. Elie, N. Noujeim, C. Pardin and A. R. Schmitzer, Chem. Commun., 2011, 47, 1788–1790 RSC.
- X.-H. Yu, X.-Q. Hong and W.-H. Chen, Org. Biomol. Chem., 2019, 17, 1558–1571 RSC.
- X.-H. Yu, C.-C. Peng, X.-X. Sun and W.-H. Chen, Eur. J. Med. Chem., 2018, 152, 115–125 CrossRef CAS.
- C.-C. Peng, M.-J. Zhang, X.-X. Sun, X.-J. Cai, Y. Chen and W.-H. Chen, Org. Biomol. Chem., 2016, 14, 8232–8236 RSC.
- C. Montoya, R. Cervantes and J. Tiburcio, Tetrahedron Lett., 2015, 56, 6177–6182 CrossRef CAS.
- J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS.
- L. Yin, W.-R. Xu, Z.-G. Wang, D.-T. Zhang, J. Jia, Y.-Q. Ge, Y. Li and J.-W. Wang, Arkivoc, 2010,(9), 196–205 Search PubMed.
- M. Bibian, R. J. Rahaim, J. Y. Choi, Y. Noguchi, S. Schurer, W. Chen, S. Nakanishi, K. Licht, L. H. Rosenberg, L. Li, Y. Feng, M. D. Cameron, D. R. Duckett, J. L. Cleveland and W. R. Roush, Bioorg. Med. Chem. Lett., 2013, 23, 4374–4380 CrossRef CAS.
- J. T. Davis, P. A. Gale, O. A. Okunola, P. Prados, J. C. Iglesias-Sanchez, T. Torroba and R. Quesada, Nat. Chem., 2009, 1, 138–144 CrossRef.
- S. Bhosale and S. Matile, Chirality, 2006, 18, 849–856 CrossRef CAS.
- S. Dutta, B. G. Watson, S. Mattoo and J.-C. Rochet, Bio-Protoc., 2020, 10, e3690 Search PubMed.
- S.-P. Zheng, J.-J. Jiang, A. V. D. Lee and M. Barboiu, Angew. Chem., Int. Ed., 2020, 132, 19082–19088 CrossRef.
- P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS.
- Z. Li, X.-H. Yu, Y. Chen, D.-Q. Yuan and W.-H. Chen, J. Org. Chem., 2017, 82, 13368–13375 CrossRef CAS.
- C. J. E. Haynes, N. Busschaert, I. L. Kirby, J. Herniman, M. E. Light, N. J. Wells, I. Marques, V. Félix and P. A. Gale, Org. Biomol. Chem., 2014, 12, 62–72 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization and experimental data for the anion recognition, anion transport and biological activity of each compound. See DOI: 10.1039/d0ra10189c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac
*ac