
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the biological activity of polyoxometalate–peptide nano-fibrils by spacer design†
Valeria Tagliavini a,
Claudia Honischb,
Simona Serratìc,
Amalia Azzaritid,
Marcella Bonchioa,
Paolo Ruzza*b and
Mauro Carraro*a
a,
Claudia Honischb,
Simona Serratìc,
Amalia Azzaritid,
Marcella Bonchioa,
Paolo Ruzza*b and
Mauro Carraro*a
aDepartment of Chemical Sciences, Institute on Membrane Technology of CNR, University of Padova, Padova, Italy. E-mail: mauro.carraro@unipd.it
bInstitute of Biomolecular Chemistry of CNR, Padova Unit, Padova, Italy. E-mail: paolo.ruzza@unipd.it
cNanotechnology Laboratory, IRCCS Istituto Tumori “Giovanni Paolo II”, Viale Orazio Flacco, 65, 70124 Bari, Italy
dExperimental Pharmacology Laboratory, IRCCS Istituto Tumori “Giovanni Paolo II”, Viale Orazio Flacco, 65, 70124 Bari, Italy
First published on 26th January 2021
Abstract
Polyoxometalates (POMs) and peptides can be conjugated to yield novel bio-hybrids with potential application as nanodrugs. However, the observed POM-induced folding of the peptide prevents its availability towards biological targets. An Anderson–Evans POM was functionalized with a bombesin analog peptide and engineered by adding a tailored hydrophilic and anionic spacer between the two moieties, to make the targeting sequence more accessible and enable an unprecedented cancer cell recognition capability.
Introduction
Polyoxometalates (POMs) are early transition metal oxoclusters that, by virtue of their nanodimensions, charge, and rigidity, can establish multiple interactions with biological macromolecules such as proteins, enzymes and peptides.1 In addition, ionomer capacity and proton coupled electron transfer (PCET) behaviour impart a unique biological activity, with potential applications as anti-bacterials, -virals, and -tumorals.2 Although the biological applications of inorganic POMs are often hampered by some drawbacks, such as scarce selectivity, low hydrolytic stability and difficult cell penetration, the possibility to endow them with organic pendants may allow better bioavailability and higher stability to be obtained.3 Moreover, new useful functionalities (e.g. for tracking or delivery) can be introduced.4 Nevertheless, the potential of hybrid POMs for cell targeting is yet undisclosed, and only a few bio-conjugates are known.5–7Among hybrid POMs, the Anderson–Evans [XMo6O24]3− polyoxomolybdates are disc-shaped polyanions of approximately D3d symmetry, with average dimensions of 8.6 × 8.6 × 2.7 Å.8 Being oxidatively and hydrolytically stable, they are suitable for biomedical applications. As for other polyoxomolybdates, their anticancer activity is likely to follow an interaction mechanism which affects ATP production and induces cell apoptosis. Moreover, their derivative decorated with two tris(hydroxymethyl)aminomethane (TRIS) molecules (Fig. 1, POM 3) can be easily synthetized in situ, resulting available for further functionalization through its amino-groups.9 Rompel and co-workers reported the interaction of aromatic Anderson–Evans derivatives with human or bovine serum albumin, showing that electrostatic, hydrophobic, or π–π interactions can be established with the protein.10 Wang and co-workers reported the functionalization of this POM with bioactive molecules, such as bile acids, resulting in an improved cytotoxicity with increased selectivity against cancer cells, thanks to the possibility to target the overexpressed farnesoid X receptor.6 Different Anderson–Evans POM–peptide conjugates were also described, obtained either by reaction with pre-synthesized peptides or by incorporation of the POM as an unnatural amino acid during step-wise peptide synthesis.7 In these hybrids, the strong interplay between the two domains was highlighted by the marked effect of the POM on the peptide secondary structure, which evolves from a random coil to α-helix conformation.
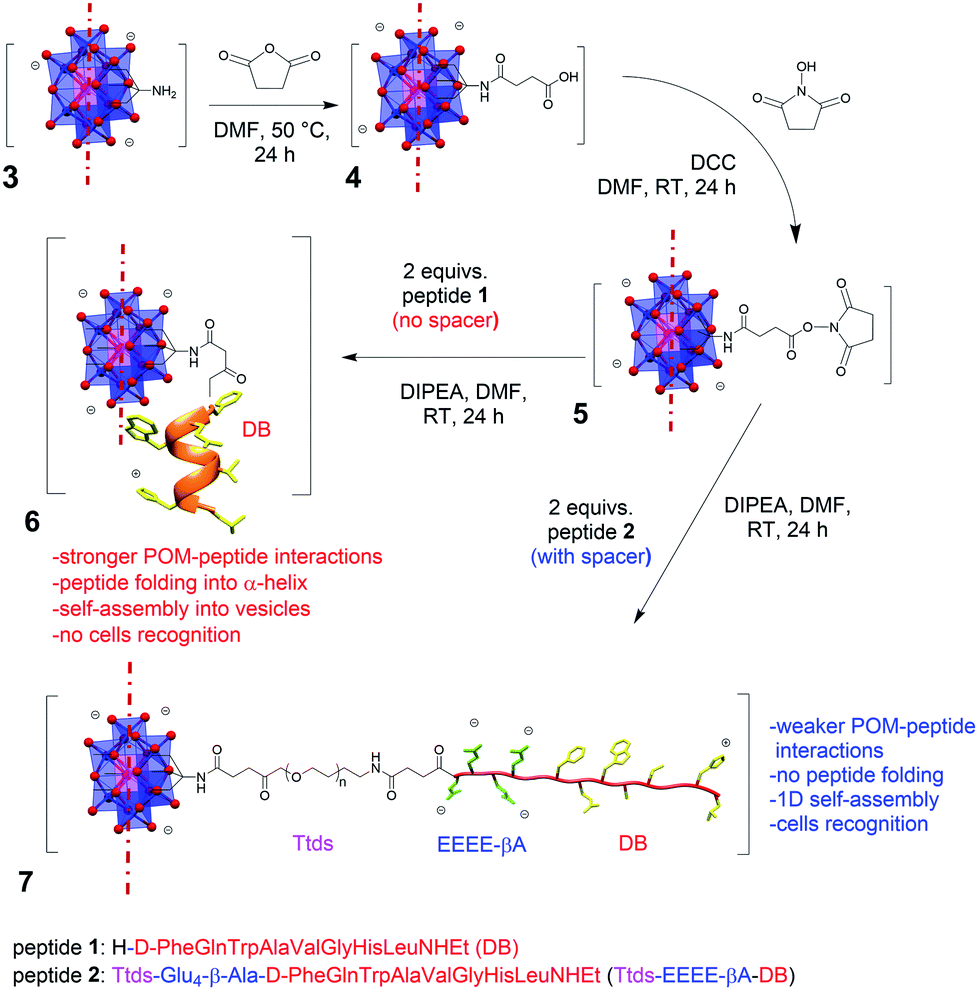 | ||
| Fig. 1 Reaction scheme to obtain POMs 6 and 7, showing the different impact of the spacer on the peptide secondary structure (evolving into α-helix for 6 and to random coil for 7). For graphical reasons, only one side of the bis-functionalized POM is shown. Countercations (TBA) are also omitted. DCC = N,N′-dicyclohexylcarbodiimide; DIPEA = N,N-diisopropylethylamine. | ||
In our previous research work, an Anderson–Evans POM was functionalized with demobensin-1 (DB) (1) to obtain POM 6 (Fig. 1).11 DB is a bombesin antagonist peptide that shows binding affinity for the gastrin-releasing peptide receptor (GRPr) overexpressed in different tumors, including prostatic carcinoma, breast cancer, and gastrointestinal stromal tumors, and it was successfully employed to improve the targeting ability of different nanomaterials.12 Preliminary in vitro results, however, demonstrated that the peptide loses its targeting ability once conjugated with the POM unit.11 The large size and the delocalized charge on POM surface can indeed foster competitive interactions with polar and positively charged residues of the peptide chain.1 As a result, change of secondary structure and self-assembly into vesicles were observed, with a strong impact on DB accessibility.11
Herein, we show the preparation of a polyoxomolybdate–demobensin-1 (DB) conjugate, implemented with a tailored hydrophilic/anionic spacer. Beside introducing a longer distance, the spacer guarantees electrostatic repulsion between POM and grafted peptide chains, thus allowing the peptide to retain its initial conformation and, in turn, its accessibility towards intermolecular interactions. As a matter of fact, the hybrid POM bearing the unfolded peptide displays supramolecular self-assembly into fibrillar nanostructures and enhanced cancer cells recognition capability. To the best of our knowledge, this is the first example of a POM–peptide conjugate with enhanced biological activity.
Results and discussion
Synthesis of the POM bio-conjugate and characterization
The spacer, consisting of the small PEG-like chain, Ttds (trioxatridecan-succinamic acid), linked to the anionic GluGluGluGluβ-Ala (EEEE-βA) sequence, was added to DB, to obtain peptide 2 (Fig. 1).Ttds improves water solubility, while inducing a better stability and decreasing the immune response;13 the short anionic sequence, instead, provided by the four glutamic residues, fosters charge repulsion to the negatively charged POM (an additional β-Ala residue is useful to avoid proximity effects with the DB). After the synthesis of the peptide 2 by manual solid phase peptide synthesis (SPPS, see ESI†), the strategy chosen to obtain the final hybrid involved the functionalization of the POM–TRIS cluster (3) with succinic anhydride, to form compound 4, and the subsequent activation with N,N′-dicyclohexylcarbodiimide and N-hydroxysuccinimide (DCC/NHS) of the succinic carboxyl group, to obtain compound 5 (Fig. 1).7,11 The final product (7) was obtained by reaction of compound 5 with the N-terminal amino group of peptide 2.
Each synthetic step was followed and confirmed via FT-IR, ESI-MS and 1H NMR (Fig. S.15–S.35†). The grafting of the peptide, through amidation of the POM-NHS 5, was assessed by ESI-MS analysis, where the signal at m/z = 1689.9 corresponds to the tri-charged POM (calcd m/z for [MnMo6O18{(OCH2)3CNHCO(CH2)2CO(Ttds-EEEEβA-fQWAVGHL-NHEt)}2]3− = 1689.3) (Fig. S.31†). The preservation of the inorganic scaffold, throughout the multistep synthesis, was assured by the presence of the typical Mo–O–Mo and Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands in the 1000–500 cm−1 FT-IR region (Fig. S.15, S.18, S.21, and S.30†). The infrared spectroscopy also highlights the appearance of the peptide bonds (CO stretching band at 1649 cm−1 and N–H stretching at 1531 cm−1) and the loss of the CO signals of NHS group (1815 and 1739 cm−1, Fig. S.30†).
O stretching bands in the 1000–500 cm−1 FT-IR region (Fig. S.15, S.18, S.21, and S.30†). The infrared spectroscopy also highlights the appearance of the peptide bonds (CO stretching band at 1649 cm−1 and N–H stretching at 1531 cm−1) and the loss of the CO signals of NHS group (1815 and 1739 cm−1, Fig. S.30†).
Spectroscopic investigations of the peptide–POM interaction
In order to evaluate the impact of the spacer on the POM–peptide interactions, the secondary structure of the peptide constructs was investigated by 2D 1H NMR, circular dichroism (CD) and fluorescence spectroscopies, upon comparison of 7 with the peptide 2 and with the parent compounds 1 and 6.11Due to the limited solubility in water of the hybrid 7, the NMR spectra were recorded in d6-DMSO. 2D homonuclear NMR experiments (COSY and TOCSY, Fig. S.10 and S.32†) allowed the sequential resonance assignment of both peptide 2 and its POM construct 7, for which the bis-functionalization with C2 symmetry was also confirmed. In Fig. 2, the overlap of COSY NMR spectra, in the region of NH amide and CHα resonances for compounds 1, 6, and 7, is reported, being evident the bigger displacement of signals belonging the peptide 1 anchored onto POM 6 with respect to POM 7, in particular for Gln, Trp, Ala, Val residues.
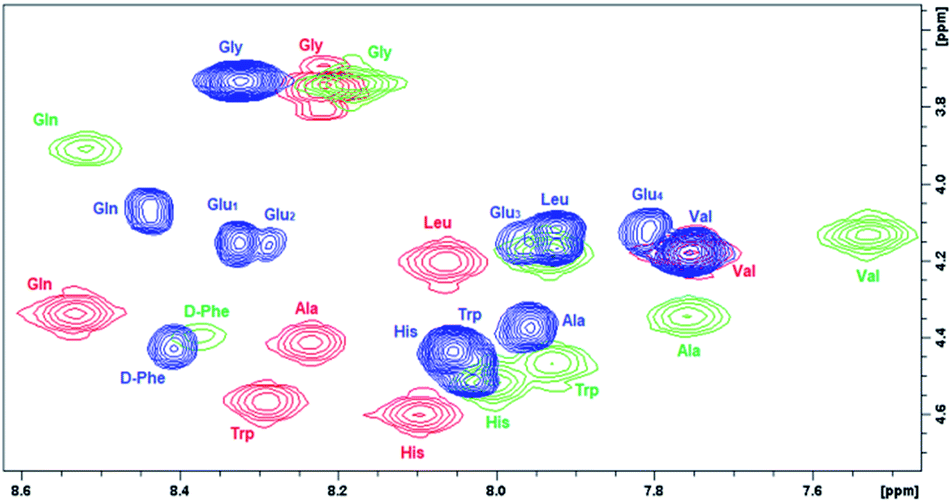 | ||
| Fig. 2 Comparison of chemical shift (400 MHz COSY spectra in d6-DMSO) for NH–CHα cross peaks belonging to free and POM-grafted DB (peptide 1 (red), POM 6 (green), POM 7 (blue)). | ||
To further highlight the beneficial effect of the spacer, the differences in chemical shift for 2 and 7, 1 and 2, 1 and 6 (Fig. S.37†) were calculated. As previously observed for the POM–DB peptide hybrid (6), most NH and/or CHα resonances are subjected to a upfield shift with respect to 1 (up to ca. 0.4 ppm, Fig. S.37c†), due to the shielding effect of the Mn-containing paramagnetic polyoxoanion, while a much lower difference between the resonance values of DB in peptide (2) and in its POM construct (7) was detected upon introduction of the spacer (<0.15 ppm, Fig. S.37a†). By comparing the NH and CHα resonances of the two DB chains, an upfield shift of both these resonances was also detected for peptide 2 in comparison to the parent peptide 1, suggesting that the negative spacer itself may has also an impact on the peptide conformation (Fig. S.37b†). Noteworthy, the diagnostic CHα, N(i, i + 4) cross peak between the CHα of the Gln (δ = 3.9 ppm) and the NH of the Gly (δ = 8.2 ppm), suggesting the presence of an α-helix conformation for POM–DB peptide hybrid (6),11 was not detected in the ROESY spectrum of the conjugate 7 (Fig. S.26 and S.32†).
The secondary structure of both spacer–peptide (2) and its POM construct (7) was then monitored by CD spectroscopy at increasing percentages (10–100% v/v) of trifluoroethanol (TFE).14 The lower polarity of TFE allows the evaluation of the influence of a membrane-like environment on the peptide conformation. Useful information on the bioactive conformation of a peptide that interacts with a receptor placed on the cell surface can thus be provided (Fig. S.38 and S.40†).
In 80% TFE solution (Fig. 3), the introduction of the POM moiety did not influence significantly the general shape of the CD spectrum, preserving the bands described for the spacer–DB peptide 2 alone, although with a decreased intensity.
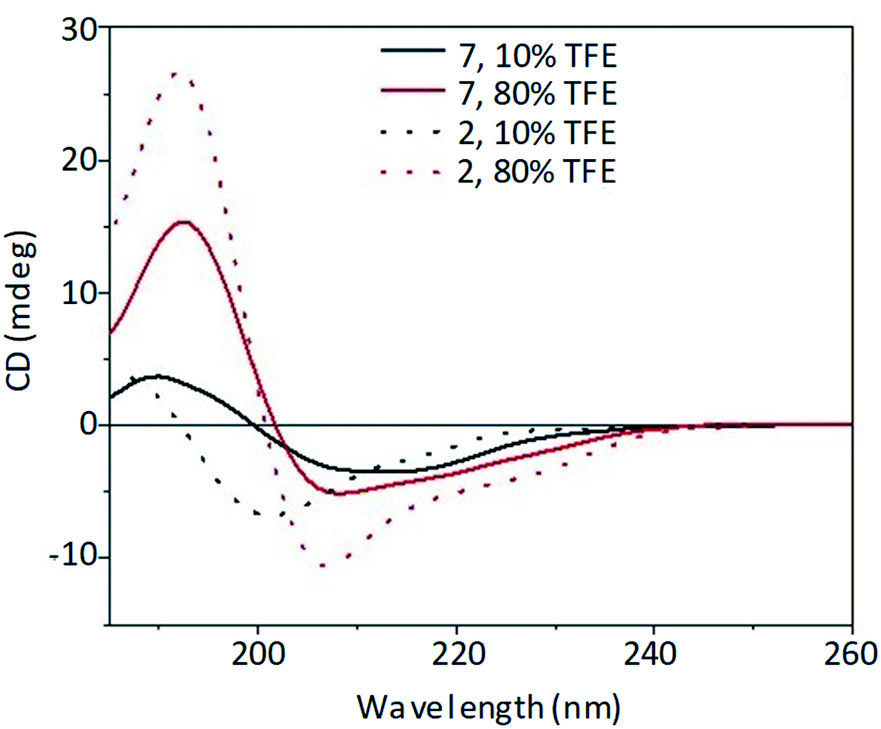 | ||
Fig. 3 Far-UV CD spectra of peptide 2 (53 μM, dot line) and POM–(peptide)2 7 (26.5 μM, solid line) at different TFE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water percentage (10% black line, 80% red line). :water percentage (10% black line, 80% red line). | ||
The CD spectra of both spacer–DB (2) peptide and its POM construct (7) are, indeed, characterized by a strong positive band at 192 nm and a negative band at 207 nm with a shoulder at about 224 nm suggesting the presence of an ordered secondary structure, likely arising from β-sheet signal overlapped with a contribution due to the induced chirality on the POM, which absorb in the same wavelength range of the peptide (Fig. S.14 and S.36†). On the other hand, at low percentages of the fluorinated alcohol (10% v/v) the peptide (2) adopted preferentially an unordered conformation (Fig. 3), similarly to that reported for the DB peptide 1,10 characterized by a positive band at about 185 nm and a negative band at 200 nm. In this case, the introduction of the POM moiety fosters a red-shift of the bands.
The influence of the POM moiety on the secondary structure of the receptor-binding sequence (Trp–Ala–Val–Gly–His–Leu) was investigated by near-UV CD and fluorescence studies. The highly environment-sensitive La transition of the Trp residue, observable in this spectral region, is indeed useful to evaluate the impact of the POM on the Trp surroundings. As shown by comparing Fig. S.34† (for POM 7) and Fig. S.12† (for peptide 2), the presence of the POM modifies the near-UV CD spectra, suggesting the presence of interactions between POM and peptide in the region under evaluation. However, in agreement with the CD observation, the impact is much lower in 80% v/v TFE in water.
Fluorescence studies were then performed at low TFE concentration (10% TFE), in order to exploit the higher sensitivity of Trp to the aqueous environment. In such conditions, Trp emission can be detected at 350 nm, typical of residues exposed to a hydrophilic medium. Trp fluorescence emission in the hybrid 7 (Fig. S.35†) is lower than that of the corresponding peptide 2 (Fig. S.13†), as a result of ca. 50% quenching of peptide luminescence by the POM, likely because of an energy transfer mechanism.15 Nevertheless, fluorescence quenching of the peptide is much higher for 6 (>95% from 1 to 6) providing a further evidence of reduced interaction between cluster 7 and peptide 2.
Successively, fluorescence quenching experiment were performed adding I− as quencher. Upon stepwise addition of KI, compound 1 shows a dynamic quenching in agreement with the Stern–Volmer equation (KSV = 6 M−1, Fig. S.41†) while its hybrid 6 shows a completely different behaviour, with 1.4 folds increase of fluorescence (Fig. S.43†). This suggests the partial disruption of the interactions between POM surface and peptide by the negative ion, with partial recovery of the starting peptide fluorescence. Quenching experiments performed on spacer–peptide 2 and its POM hybrid 7 showed, instead, similar dynamic quenching pattern with KSV = 3 M−1 values (Fig. S.42 and S.44†), indicating again the weaker interactions between POM and spacer–peptide around the Trp region.
Self-assembly of the hybrid POM and morphology
The higher availability of the peptide, in POM 7, is expected to modify the self-assembly behaviour of the hybrid. Supramolecular interactions were monitored by dynamic light scattering (DLS), with conditions narrowing physiological-like parameters (0.2 mM water solutions, pH 7.1, 3% DMSO). When the functionalized POMs were analyzed, 6 formed particles with diameter around 60 nm (Fig. S.48†), while 7 formed aggregates with dimensions of about 300 nm, as a result of a different interplay of intermolecular forces (Fig. S.49†). The addition of 0.5 equivalents of the inorganic cluster 4 (particles of 80 nm, Fig. S.45†) to a solution of 2 (hydrodynamic diameter hovering at 70–80 nm, Fig. S.46†), showed that weak supramolecular interactions can also occur between non-bonded moieties, since the size of the resulting particles doubled reaching 160 nm (Fig. S.47†). In this case, however, a less defined scattering signal was observed. Moreover, neither fluorescence (Fig. S.42†) nor CD (Fig. S.39†) in 10% v/v TFE/water gave evidence of any particular impact of the POM on peptide properties when they are not conjugated.TEM analyses were recorded in similar conditions as those used for DLS (0.2 mM water solutions, 3% DMSO). The role played by the spacer, as highlighted by scattering data, is better visible in terms of size and shape of the resulting assemblies. In the absence of a spacer, vesicle-like particles hovering at around 50 nm are observed for 6 (Fig. 4a and S.50†), while compound 7 forms bigger elongated structures with fiber-like shape (1 μm × 14 nm, Fig. 4b). Since elongated structures can also be formed by the POM–TRIS (Fig. S.51†), this behaviour suggests that, when the peptide is not folded on the POM, the latter subunit can still drive the resulting morphology. As previously observed for related Strandberg-type polyoxomolybdates, indeed, a network of hydrogen bonds can be established between protonated amino groups and the anionic polyoxygenated inorganic surface, with supramolecular self-assembly into fibrils.16
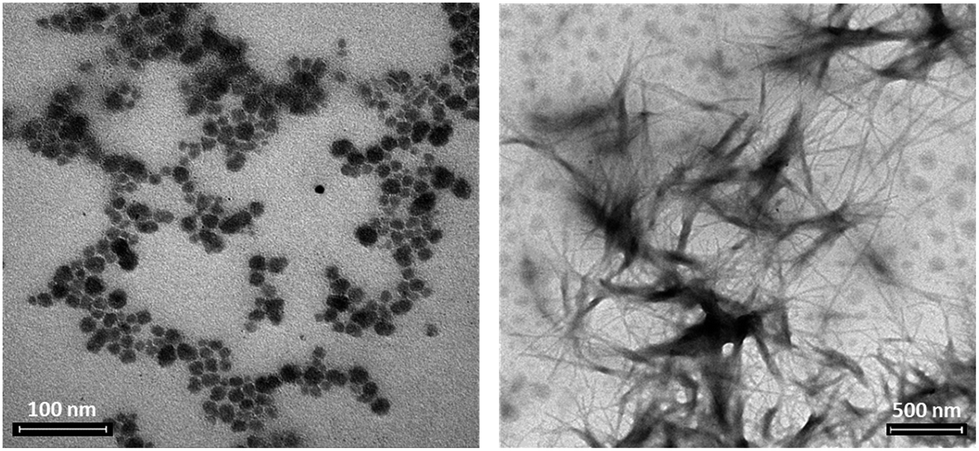 | ||
| Fig. 4 TEM images collected from 0.2 mM solutions in H2O with 3% DMSO of 7, scale bar of 100 nm (a) and 6, scale bar 500 nm (b). | ||
Interestingly, compound 7 forms a gel in DMF, although with shorter fibrils (500 × 14 nm, Fig. S.52†) than those observed in water. On the other hand, the peptide-free POM cannot form a gel, suggesting that the additional intermolecular interaction established by the unfolded peptide are relevant to enable the activity as organogelator. The lower polarity of the organic solvent might, indeed, (i) favour the interactions between chains also at the level of Ttds moiety, (ii) allow a higher degree of protonation of the tetraglutamic spacer, (iii) further stabilize switterionic-type hydrogen bonding,16 leading to well organized elongated structures with efficient solvent entrapment ability.
According to the intermolecular forces which come into play between POMs and peptides, a plethora of structures were reported in the literature.17 The 1D shape was previously reported for non-covalent POM–peptides assemblies, by using cationic peptides18 or peptides decorated with azobenzene side chains,19 where their β-sheet state drives the alignment into fibers. On the other hand, Anderson–Evans conjugated with peptides of different length were shown to form invariably hollow, spherical supramolecular structures.20
Our results highlight the possibility to modify the morphology of the assembly by using tailored peptide chains.
Cytotoxicity investigation
Concerning the biological activity, the peptide endowed by the spacer (compound 2) exhibits a moderate anticancer activity with 70% retention of viability of HeLa cells after 24 h (150 μM solution). HeLa cells display a moderate bombesin receptor overexpression. However, as mentioned above, previous studies reported no recognition capability for 6, with no improvement of cytotoxicity after POM functionalisation with the bombesin analogue: both compound 3 and 6 gave indeed similar results.11 Here, we report greater cell viability inhibition for the hybrid decorated with the additional spacer. Compound 7 reaches a remarkable cancer cells inhibition with a residual viability of 53% for a 150 μM concentration after 24 h (IC50 = 95 μM, Fig. S.53†) against 68% for unfunctionalized POM–TRIS cluster 3, (IC50 = 134 μM, Fig. S.54†). The viability further decreased at 33% after 48 h, confirming a better biological activity for POM 7 (13% more efficient than 3). In Table S1† are reported relevant literature data for a comparison of the activity of polyoxomolybdate bio-hybrids.2a,21 Even if the activity is not the best, the compound is still among most active ones, which display IC50 values in the range 10–100 μM.Experimental
Materials and methods
Samples were analyzed by a Z-Sizer Nano-S instrument (Malvern) as 0.2 mM solutions in H2O with 3% DMSO. In all cases, three measurements were collected at 25 °C.All CD measurements were performed at 25 °C using a nitrogen flushed Jasco J-1500 spectropolarimeter (Jasco, Easton, MD, USA) equipped with a thermostated cell holder. Each CD spectrum was the average of 9 scans, collected at a scan rate of 50 nm min−1 in a 1.0 mm path length quartz cuvette (200 μL).
NMR spectra were run on a Bruker DMX-600 instrument (Bruker Corp., Billerica, MA, USA), operating a 599.90 MHz for 1H, at 298 K. For the two-dimensional (2D) experiments, pulse programs of the standard Bruker library were used. With the exception of COSY experiment, all 2D experiments were acquired in the phase-sensitive mode, with quadrature detection in both dimensions, by use of the time proportional phase increments.
Fluorescence data were collected at 25 °C for 15 μM solutions in TFE/water, with an Agilent Cary Eclipse Fluorescence Spectrophotometer in the range between 300–550 nm with excitation at 290 nm, medium scanning speed, 5 accumulations, 5 excitation and emission slit with a 1 cm path cuvette.
FTIR measurements were carried out on a Nicolet 5700 FT-IR instrument by preparing KBr pellets. Analysis of IR spectra was performed on OMNIC software.
Preparative HPLC system (Shimadzu, Tokyo, Japan) was equipped with LC-8A pumps, SCL-8A controller and SPD-6A spectrophotometric detector, and were performed using a linear gradient (eluent A: 0.05% TFA in H2O; eluent B: 0.05% TFA in 9:1 v/v CH3CN–H2O; 22% B for 3 min; from 22% to 35% B in 40 min, flow 12 mL min−1, λ 216 nm), and a Vydac C18 column (300 Å, 10 μm, 250 × 22 mm).
TEM measurements were obtained with a FEI Tecnai G2 transmission electron microscope (Thermo Fisher Scientific, Waltham, MA, USA), operating at an excitation voltage of 100 kV.
Cytotoxicity/viability tests were performed as 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) assay. For the experiments we used HeLa cervical cancer cells.
Synthetic procedures
The peptide 2 was synthesized by manual solid phase using Fmoc-based synthetic strategy. ESI MS (+) in MeOH: m/z = 937.9 (calcd 938.0) for [C86H130N20O27]2+.For the synthesis of POM-NHS 7 (17.63 mg, 0.0056 mmol) and peptide 2 (20.99 mg, 0.0112 mmol) were dissolved in DMF (700 μL) and DIPEA (14.93 μL) was added. The reaction mixture was stirred at room temperature for one day and the resulting solution was stored for crystallization under diethyl ether vapour for one day. The pale orange product was washed with diethyl ether and dried for one day. Yield: 30.07 mg, 92.1%.
FT IR (KBr, cm−1): 3287 (s, br), 3080 (w), 2961 (w), 2935 (w), 2874 (w), 1670 (s), 1547 (m), 1457 (w), 1384 (w), 1032 (s), 942 (w), 920 (m), 902 (w), 747 (w), 670 (s), 562 (w), 470 (w). ESI-MS (−) (CH3CN): m/z 1688.9 ([M-3TBA]3−) (calcd 1689.3 m/z). UV-VIS (CH3CN): λmax = 221 nm. Shoulder at 281 nm. ελ221 = 67200 cm−1 M−1.
Conclusions
In conclusion, although the grafting of a targeting peptide sequence to a polyanionic cluster can strongly affect the conformation of the aminoacidic backbone, the introduction of a tailored spacer can allow to minimize such competing interferences. As a result, the novel bio-hybrid displays (i) moderate change of the secondary structure, especially in less hydrophilic (membrane-like) environment, (ii) decreased on-POM folding of the chains, leading to higher availability of receptor-binding region and (iii) no tendency to self-assemble into spherical vesicles, turning instead in the formation of a fibrous soft material. Finally, an enhancement of cytotoxicity toward cancer cells was obtained with respect to the POM precursor. These data provide a basis to rationally develop efficient POM-based nanodrugs.Author contributions
V. T. and C. H. made the synthesis and the characterization of the hybrid. S. S. and A. A. helped with methodology and formal analysis of biological tests. M. B. contributed to the design and discussion of the experiments, P. R. and M. C. planned the research, analysed the data and wrote the manuscript.Conflicts of interest
None of the authors has any conflict to declare.Acknowledgements
We thank Youssef Kerdi and Xhao Zhue for preliminary experiments. Financial support from Department of Chemical Sciences at University of Padova (Project P-DiSC #11NExuS_BIRD2019-UNIPD) and MIUR (PRIN Nanoredox, Prot. 2017PBXPN4) is gratefully acknowledged.Notes and references
- (a) M. Arefian, M. Mirzaei, H. Eshtiagh-Hosseini and A. Frontera, Dalton Trans., 2017, 46, 6812–6829 RSC; (b) M. Zhao, X. Chen, G. Chi, D. Shuai, L. Wang, B. Chen and J. Li, Inorg. Chem. Front., 2020, 7, 4320–4332 RSC; (c) L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92–99 CrossRef CAS.
- (a) A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS; (b) A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS; (b) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC; (c) Y. -F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC.
- (a) V. A. Zamolo, G. Modugno, E. Lubian, A. Cazzolaro, F. Mancin, L. Giotta, D. Mastrogiacomo, L. Valli, A. Saccani, S. Krol, M. Bonchio and M. Carraro, Front. Chem., 2018, 6, 1–10 CrossRef; (b) G. Modugno, E. Fabbretti, A. Dalle Vedove, T. Da Ros, C. Maccato, H. S. Hosseini, M. Bonchio and M. Carraro, Eur. J. Inorg. Chem., 2018, 4955–4961 CrossRef CAS; (c) D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS; (d) G. Geisberger, E. B. Gyenge, D. Hinger, P. Bösiger, C. Maake and G. R. Patzke, Dalton Trans., 2013, 42, 9914–9920 RSC.
- M. Ortiz, A. M. Debela, M. Svobodova, S. Thorimbert, D. Lesage, R. B. Cole, B. Hasenknopf and C. K. O'Sullivan, Chem.–Eur. J., 2017, 23, 10597–10603 CrossRef CAS.
- H. K. Yang, Y. X. Cheng, M. M. Su, Y. Xiao, M. B. Hu, W. Wang and Q. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 1462–1466 CrossRef CAS.
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef CAS.
- (a) J. S. Anderson, Nature, 1937, 140, 850 CrossRef CAS; (b) H. T. Evans Jr, J. Am. Chem. Soc., 1948, 70, 1291–1292 CrossRef.
- (a) B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081–1087 CrossRef CAS; (b) A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS.
- E. Al-Sayed, A. Blazevic, A. Roller and A. Rompel, Chem.–Eur. J., 2015, 21, 17800–17807 CrossRef CAS.
- D. Ventura, A. Calderan, C. Honisch, S. Krol, S. Serratì, M. Bonchio, M. Carraro and P. Ruzza, Pept. Sci., 2018, 110, 1–11 Search PubMed.
- F. Cuttitta, D. N. Carney, J. Mulshine, T. W. Moody, J. Fedorko, A. Fischler and J. D. Minna, Nature, 1985, 316, 823–826 CrossRef CAS.
- G. Pasut, A. Guiotto and F. M. Veronese, Expert Opin. Ther. Pat., 2004, 14, 859–894 CrossRef CAS.
- M. Buck, Q. Rev. Biophys., 1998, 31, 297–355 CrossRef CAS.
- G. Zhang, B. Keita, J. C. Brochon, P. de Oliveira, L. Nadjo, C. T. Craescu and S. Miron, J. Phys. Chem. B, 2007, 111, 1809–1814 CrossRef CAS.
- M. Carraro, A. Sartorel, G. Scorrano, C. Maccato, M. H. Dickman, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2008, 47, 1–6 CrossRef.
- M. Stuckart and K. Y. Monakhov, Chem. Sci., 2019, 10, 4364–4376 RSC.
- (a) G. Zhang, J. Zhang, Y. Wang, Y. Wu, Q. Li, Y. Liang, W. Qi, H. Rao, R. Su and Z. He, J. Colloid Interface Sci., 2020, 578, 218–228 CrossRef CAS; (b) J. Li, X. Li, F. Jiang, X. Li, X. Xie, L. Wu, L. Wang, M. Lee and W. Li, Chem.–Eur. J., 2017, 23, 13510–13517 CrossRef CAS; (c) G. Wang, W. Guan, B. Li and L. Wu, Curr. Opin. Colloid Interface Sci., 2018, 35, 91–103 CrossRef CAS; (d) P. Gao, Y. Wu and L. Wu, Soft Matter, 2016, 12, 8464–8479 RSC.
- (a) J. Li, X. Li, J. Xu, Y. Wang, L. Wu, Y. Wang, L. Wang, M. Lee and W. Li, Chem.–Eur. J., 2016, 22, 15751–15759 CrossRef CAS; (b) J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2592–2595 CrossRef CAS.
- J. Luo, B. Zhang, C. Yvon, M. Hutin, S. Gerislioglu, C. Wesdemiotis, L. Cronin and T. Liu, Eur. J. Inorg. Chem., 2019, 380–386 CrossRef CAS.
- M. B. Čolović, M. Lacković, J. Lalatović, A. S. Mougharbel, U. Kortz and D. Z. Krstić, Curr. Med. Chem., 2020, 27, 1–18 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10218k |
| This journal is © The Royal Society of Chemistry 2021 |