
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination versus hydrogen bonds in the structures of different tris(pyridin-2-yl)phosphoric triamide derivatives†
Marjan Sebghatia,
Atekeh Tarahhomi *a,
Marjan Sadat Bozorgvara,
Dan G. Dumitrescub and
Arie van der Leec
*a,
Marjan Sadat Bozorgvara,
Dan G. Dumitrescub and
Arie van der Leec
aDepartment of Chemistry, Semnan University, Semnan 35351-19111, Iran. E-mail: tarahhomi.at@semnan.ac.ir; Fax: +98 23 33654110; Tel: +98 23 31533199
bXRD2 beamline, Elettra – Sincrotrone Trieste S.C.p.A., 34149 Basovizza, Trieste, Italy
cInstitut Européen des Membranes, IEM, UMR-5635, Université de Montpellier, ENSCM, CNRS, Place Eugéne Bataillon, 34095, Montpellier cedex 5, France
First published on 22nd February 2021
Abstract
The crystalline forms of tris(pyridin-2-yl)phosphoric triamide, OP[NH-2Py]3 were investigated using single crystal X-ray diffraction, both as a metal complex [Co{(O)P[NH-2Py]2[NH-2PyH]}2]Cl3, compound 1, and as the purely organic pseudopolymorphs, the pure/anhydrate OP[NH-2Py]3, 2, monohydrate 2OP[NH-2Py]3·H2O, 3 and solvate 2OP[NH-2Py]3·2DMF·H2O, 4, respectively. An improved model of the metal organic framework (MOF) structure of Cu(II)O6 {[Cu(OCHO)3][(CH3)2NH2]}n, 5 is also reported. Compound 1 is the first example of a discrete chelate phosphoric triamide (PT) complex with an [N]3P(O)-based backbone which in the PT compound acts as a flexible tridentate ligand. The structures 1, 3 and 4 crystallize in the monoclinic space group P21/n, while the anhydrous form 2 crystallizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) trigonal space group. 5 which was obtained as a byproduct of the synthesis process of coordination compounds of OP[NH-2Py]3, crystallizes in the monoclinic space group I2/c. Hydrogen bonding pattern analysis for the different solid state forms of OP[NH-2Py]3 (2–4) shows a 2D sheet of the N–H⋯N linked molecules for 2, and a 1D chain and a ten-membered ring motif, both formed via the hydrogen bonds N–H⋯O, N–H⋯N and O–H⋯O, for 3 and 4, respectively. Of note is the unusual absence of the expected hydrogen bond interaction N–H⋯O
trigonal space group. 5 which was obtained as a byproduct of the synthesis process of coordination compounds of OP[NH-2Py]3, crystallizes in the monoclinic space group I2/c. Hydrogen bonding pattern analysis for the different solid state forms of OP[NH-2Py]3 (2–4) shows a 2D sheet of the N–H⋯N linked molecules for 2, and a 1D chain and a ten-membered ring motif, both formed via the hydrogen bonds N–H⋯O, N–H⋯N and O–H⋯O, for 3 and 4, respectively. Of note is the unusual absence of the expected hydrogen bond interaction N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P of PT compounds and also a lack of any significant π interactions in structure 2. The role of the solvent/hydrate and substituent (aminopyridine) in the formation of the expected hydrogen bond interactions in the studied solid state forms is also investigated. For these structures, crystal packing analysis using 3D Hirshfeld surface (HS), 2D fingerprint plot (FP) and enrichment ratio (E) calculations confirm a competition between the pyridinyl nitrogen and phosphoryl oxygen atoms as H-bond acceptors to construct the N–H⋯N or N–H⋯O contacts. Moreover, the significant differences between the anhydrous form of OP[NH-2Py]3, 2, and the monohydrate and solvate forms, 3 and 4, can be correlated to the pseudo-infinite (in 2) versus finite values (in 3 and 4) for the upper values of de and di on the related full FPs.
P of PT compounds and also a lack of any significant π interactions in structure 2. The role of the solvent/hydrate and substituent (aminopyridine) in the formation of the expected hydrogen bond interactions in the studied solid state forms is also investigated. For these structures, crystal packing analysis using 3D Hirshfeld surface (HS), 2D fingerprint plot (FP) and enrichment ratio (E) calculations confirm a competition between the pyridinyl nitrogen and phosphoryl oxygen atoms as H-bond acceptors to construct the N–H⋯N or N–H⋯O contacts. Moreover, the significant differences between the anhydrous form of OP[NH-2Py]3, 2, and the monohydrate and solvate forms, 3 and 4, can be correlated to the pseudo-infinite (in 2) versus finite values (in 3 and 4) for the upper values of de and di on the related full FPs.
Introduction
The design of crystalline solids and their molecular architectures with a focus on emerging technological applications as one of the main goals in crystal engineering has attracted a growing interest in the recent years. Designing and modelling such supramolecular architectures require a good understanding and control of various intermolecular interactions.1–3 A common route to direct the formation of a crystalline structure is the formation of metal–ligand coordination bonds, and many examples from various fields are produced by these interactions.4,5 Structural variation can also be achieved using purely organic compounds, which can have different solid forms such as polymorphs, hydrates and solvates.6,7 The study of the patterns and types of intermolecular interactions in a crystal structures are paramount for understanding and predicting the specific arrangement of the molecules in space, and, by extension, various properties such as stability, solubility and structure.8,9Phosphoramide compounds as a class of amino-functionalized P(V) derivatives containing phosphorous imido bound (OP–N) have recently received considerable attention, as they can belong to an ideal class of certain flexible (half-rigid) ligands in coordination chemistry. Such ligands having both N- and O-donor functionalities can be provide multiple metal binding sites offering an attractive pathway in crystal engineering to construct novel various coordination assemblies such as molecular cages, metal–organic frameworks (MOFs) and porous coordination polymers.10–12 Applications of phosphoramide-based compounds include their use as Lewis base catalysts when in chiral form, as flame-retardants, as urease inhibitors, or as dyes for adsorption of light in dye sensitized solar cells.13–16
Among phosphoramide ligands, pyridyl-functionalized amino ligands with the general formula OP[NH-Py]3 and their phosphate and phosphonium salts have been shown great potential to generate interesting examples of multi-metallic architectures.10,11,17–20 Such produced compounds can be used as suitable materials with potential technological applications such as gas adsorption17 and fluorescence.18 Based on a search on the Cambridge Structural Database (CSD, version 5.40, updated to February 2019 (ref. 21)), for OP[NH-Py]3 ligands with all three substitutions 2-, 3- and 4-pyridinyl (OP[NH-2/3/4Py]3), there are some reported metal–ligand coordination compounds, but for OP[NH-2Py]3 only a few complexes of silver salts have been recently published (with the CSD refcodes VEFVAF, VEFVEJ, VEFVEJ01 and VEFVIN).18 As such, the X-ray structures of these ligands OP[NH-2/3/4Py]3 have been reported (OLOCEY for OP[NH-3Py]3 and PIHROP for OP[NH-4Py]3) which in the case of OP[NH-2Py]3, both the non-solvated (LAFNAI22 and LAFNAI01 (ref. 12)) and solvate (VEFTUX23) crystals. However, an in-depth analysis of the structures using Hirshfeld surface analyses and theoretical approaches has not been performed so far.
In view of the above interesting examples of ligand and metal–ligand assemblies, we employ in this study the amino–P(V) ligand containing 2-pyridyl substituent in order to synthesize new coordination compounds based on tris(pyridin-2-yl)phosphoric triamide. We report the crystal structure of the first discrete chelate complex ([Co{(O)P[NH-2Py]2[NH-2PyH]}2]Cl3 (compound 1) with a tridentate [N]3P(O)-based ligand derived from the OP[NH-2Py]3. The structural diversity of new anhydrate (a pseudo-polymorph of LAFNAI01,12 OP[NH-2Py]3 (2)), monohydrate (2OP[NH-2Py]3·H2O (3)) and solvate (2OP[NH-2Py]3·2DMF·H2O (4)) crystals of pyridyl-functionalized amino P(V) ligands are also analyzed and compared to each other. Moreover, the crystal structure of an improved model of a very old reported metal organic framework (MOF) of Cu(II)O6 ({[Cu(OCHO)3][(CH3)2NH2]}n (compound 5)) in the year 1973 with CSD refcode MACUFR24 obtained during the synthesis process of complexes of OP[NH-2Py]3 is presented. The pattern and type of intermolecular interactions in the quoted forms of selected phosphoric triamide (PT) ligand OP[NH-2Py]3 were investigated by 3D Hirshfeld surfaces, 2D fingerprint plots, enrichment ratios (E) and interaction energies frameworks. Our objective is to provide suitable insights into the structural chemistry of these tris(pyridin-2-yl)phosphoric triamide-based compounds and their impact on the crystal packing structure.
Experimental
Materials and measurements
All materials and solvents were purchased from commercial sources (Merck or Aldrich) and used as received. A Bruker ALPHA FT-IR spectrometer was used to record Infrared (IR) spectra using a KBr disk.Syntheses and crystallization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) in acetonitrile (15 ml) slowly was added and the mixture was refluxed for three days. Tiny blue single crystals were obtained from the reaction solution by slow evaporation at room temperature. IR Data (KBr,
:1 molar ratio) in acetonitrile (15 ml) slowly was added and the mixture was refluxed for three days. Tiny blue single crystals were obtained from the reaction solution by slow evaporation at room temperature. IR Data (KBr, ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) , cm−1): 3177, 3159, 1664, 1610, 1541, 1477, 1420, 1381, 1325, 1277, 1165, 1041, 1003, 928, 872, 768, 719, 548, 501.:1:1) at room temperature. IR Data (KBr, , cm−1): 3124, 3082, 3074, 3051, 3040, 3026, 2878, 1664, 1599, 1576, 1487, 1462, 1445, 1406, 1294, 1271, 1205, 1153, 1097, 1053, 968, 953, 824, 770, 735, 646, 600, 525, 509, 484.:1) (for 4) under reflux, followed by slow evaporation of the related solutions at room temperature. IR Data (KBr, , cm−1) for 3: 3385, 3144, 1630, 1609, 1595, 1483, 1458, 1452, 1419, 1400, 1331, 1277, 1227, 1165, 1040, 1003, 926, 771, 652, 586, 546, 503; for 4: 3439, 3111, 3088, 3055, 2974, 2939, 2881, 2854, 2816, 2777, 2733, 2698, 1663, 1599, 1581, 1487, 1448, 1414, 1387, 1294, 1265, 1244, 1215, 1203, 1153, 1103, 1051, 997, 957, 876, 773, 735, 727, 636, 609, 523, 503, 478, 409., cm−1): 3425, 3059, 2775, 2496, 1668, 1578, 1576, 1472, 1385, 1375, 1342, 1250, 1165, 1024, 889, 825, 804, 771, 625, 553, 517.
, cm−1): 3177, 3159, 1664, 1610, 1541, 1477, 1420, 1381, 1325, 1277, 1165, 1041, 1003, 928, 872, 768, 719, 548, 501.:1:1) at room temperature. IR Data (KBr, , cm−1): 3124, 3082, 3074, 3051, 3040, 3026, 2878, 1664, 1599, 1576, 1487, 1462, 1445, 1406, 1294, 1271, 1205, 1153, 1097, 1053, 968, 953, 824, 770, 735, 646, 600, 525, 509, 484.:1) (for 4) under reflux, followed by slow evaporation of the related solutions at room temperature. IR Data (KBr, , cm−1) for 3: 3385, 3144, 1630, 1609, 1595, 1483, 1458, 1452, 1419, 1400, 1331, 1277, 1227, 1165, 1040, 1003, 926, 771, 652, 586, 546, 503; for 4: 3439, 3111, 3088, 3055, 2974, 2939, 2881, 2854, 2816, 2777, 2733, 2698, 1663, 1599, 1581, 1487, 1448, 1414, 1387, 1294, 1265, 1244, 1215, 1203, 1153, 1103, 1051, 997, 957, 876, 773, 735, 727, 636, 609, 523, 503, 478, 409., cm−1): 3425, 3059, 2775, 2496, 1668, 1578, 1576, 1472, 1385, 1375, 1342, 1250, 1165, 1024, 889, 825, 804, 771, 625, 553, 517.Crystal structure determinations
All single crystal X-ray diffraction experiments were performed at the XRD2 structural biology beamline, Sincrotrone Elettra Trieste SCpA. The light source used by the beamline is a superconducting wiggler, and a λ of 0.7000 Å was selected using a cryogenically cooled dual crystal Si monochromator. The diffraction setup consisted of an Arinax MD2S high throughput diffractometer and Pilatus 6M detector. The beam was defined using a 100 μm aperture and further cleaned using a 200 μm capillary, while sample cooling was performed using an open flow nitrogen cryostat at 100 K. Details of crystal data and structure refinement are provided in Tables 1 and 2.| Compound | 1 | 2 | 3 |
|---|---|---|---|
| CCDC number | 1956238 | 1960317 | 1956236 |
| Chemical formula | 2(C15H15·5Co0·5N6OP)·1.5(Cl2) | C15H15N6OP | 2(C15H16N6OP)·0.398H2O |
| Mr (gr/mol) | 818.89 | 326.3 | 659.76 |
| Temperature (K) | 100 | 100 | 100 |
| Crystal system, space group | Monoclinic, P21/n | Trigonal, R |
Monoclinic, P21/n |
| a, b, c (Å) | 8.5130 (17), 15.816 (3), 26.981 (5) | 10.0040 (14), 28.420 (6) | 10.568 (2), 13.475 (3), 21.853 (4) |
| β (°) | 93.06 (3) | — | 92.35 (3) |
| V (Å3) | 3627.6 (13) | 2463.1 (8) | 3109.3 (11) |
| Z | 4 | 6 | 4 |
| Radiation type | Synchrotron, λ = 0.700 Å | Synchrotron, λ = 0.700 Å | Synchrotron, λ = 0.700 Å |
| μ (mm−1) | 0.79 | 0.18 | 0.19 |
| F(000) | 1676 | 1020 | 1376 |
| Crystal size (mm) | 0.11 × 0.10 × 0.04 | 0.12 × 0.05 × 0.03 | 0.09 × 0.05 × 0.02 |
| Crystal color/habit | Colorless/Plate | Colorless/Block | Colorless/Block |
| Diffractometer | Huber 4-circles Kappa Goniometer, Pilatus 2M detector | Huber 4-circles Kappa Goniometer, Pilatus 2M detector | Huber 4-circles Kappa Goniometer, Pilatus 2M detector |
| Theta range for data collection (°) | 1.5 to 27.7 | 2.9 to 28.6 | 2.7 to 28.1 |
| Absorption correction | Multi-scan/XDS | Multi-scan/XDS | Multi-scan/XDS |
| No. of measured, independent and observed [I > 2.0σ(I)] reflections | 51573, 8709, 7105 |
11798, 1339, 1335 |
44499, 7572, 7353 |
| Rint | 0.023 | 0.038 | 0.025 |
| (sinθ/λ)max (Å−1) |
0.663 | 0.663 | 0.663 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.068, 0.234, 1.07 | 0.048, 0.122, 1.18 | 0.037, 0.099, 1.06 |
| No. of reflections | 8709 | 1339 | 7572 |
| No. of parameters | 492 | 91 | 453 |
| No. of restraints | 4 | 0 | 0 |
| H-atom treatment | H-atom parameters constrained | All H-atom parameters refined | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 1.99, −0.95 | 0.51, −0.82 | 0.49, −0.47 |
| Compound | 4 | 5 |
|---|---|---|
| CCDC number | 1956239 | 1956237 |
| Chemical formula | 2(C15H15N6OP)·2(C3H7NO)·H2O | C3H3CuO6·C2H8N |
| Mr (gr mol−1) | 816.81 | 244.69 |
| Temperature (K) | 100 | 100 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, I2/a |
| a, b, c (Å) | 10.625 (2), 18.707 (4), 20.479 (4) | 11.5393 (16), 8.6391 (6), 8.8358 (9) |
| β (°) | 98.25 (3) | 95.746 (11) |
| V (Å3) | 4028.3 (14) | 876.41 (16) |
| Z | 4 | 4 |
| Radiation type | Synchrotron, λ = 0.700 Å | Synchrotron, λ = 0.700 Å |
| μ (mm−1) | 0.16 | 2.39 |
| F(000) | 1720 | 500 |
| Crystal size (mm) | 0.08 × 0.02 × 0.01 | 0.11 × 0.09 × 0.08 |
| Crystal color/habit | Colorless/Needle | Colorless/prism |
| Diffractometer | Huber 4-circles Kappa Goniometer, Pilatus 2M detector | Huber 4-circles Kappa Goniometer, Pilatus 2M detector |
| Theta range for data collection (°) | 2.6 to 28.6 | 2.7 to 31.2 |
| Absorption correction | Multi-scan/XDS | Multi-scan/CrysAlis PRO |
| No. of measured, independent and observed [I > 2.0σ(I)] reflections | 57512, 9811, 9586 |
3048, 1239, 951 |
| Rint | 0.032 | 0.047 |
| (sinθ/λ)max (Å−1) |
0.663 | 0.752 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.037, 0.098, 1.07 | 0.073, 0.282, 1.17 |
| No. of reflections | 9811 | 1239 |
| No. of parameters | 551 | 63 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.31, −0.47 | 1.37, −1.03 |
For each of the samples a minimum of four crystals were tested in order to ensure data quality and check the sample purity. A standard data collection was used, consisting of a 360° ϕ-scan. Frame integration and scale and absorption corrections were done with XDS25 and space group assignment was provided by Pointless from the CCP4 software suite,26 as implemented in the XDS4Elettra interface. The structures were solved using ShelxT and refined using ShelxL as implemented in the OLEX2 package.27 The H atoms were either located using the difference maps or added geometrically in the case of those attached to carbon atoms after which the positions were refined with riding constraints.28
ORTEP and molecular packing diagrams are produced using by the programs PLATON29 and Mercury.30 Selected bond lengths and angles are listed in Tables S1 and S2.†
Hirshfeld surface analysis
Three dimensional (3D) Hirshfeld surfaces (HSs) were generated by using the Crystallographic Information File (CIF) as the input file for the software package Crystal Explorer 17.5.31 In this case, the bond lengths of C–H, N–H and O–H are automatically set to standard neutron values (1.083 Å, 1.009 Å and 0.983 Å, respectively) during the calculations. For the 3D colored Hirshfeld map, the distance dnorm is employed which is a normalized function of two distances de and di, the distances from the surface to the exterior (for former) or interior (for later) nearest atom to the surface, and the van der Waals (vdW) radius of the appropriate atom as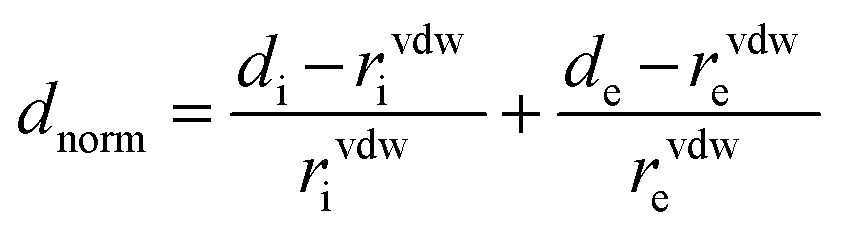 . Thus, a suitable red-white-blue color scheme is produced which highlights contacts shorter than the sum of the vdW radii (by color red), around the vdW separation (white) and the longer than vdW radii (blue).32 Another important feature of HS is the shape index which is employed to recognize the particular packing modes such as π-effects displayed as a red π-hole (C–H⋯π) or red-blue triangles (π⋯π) on the surface.33 Moreover, information collected from the HS analysis can be summarized as a fingerprint plot (FP) designed on a 2D grid formed by (de, di) pairs which each point on this plot illustrates a hit formed by a (de, di) pair.34 Finally, the enrichment ratio (EXX/XY) is derived from the HS analysis which represents the propensity of pairs of elements (X, X) or (X, Y) in X⋯Y interactions.35 This ratio is defined as
. Thus, a suitable red-white-blue color scheme is produced which highlights contacts shorter than the sum of the vdW radii (by color red), around the vdW separation (white) and the longer than vdW radii (blue).32 Another important feature of HS is the shape index which is employed to recognize the particular packing modes such as π-effects displayed as a red π-hole (C–H⋯π) or red-blue triangles (π⋯π) on the surface.33 Moreover, information collected from the HS analysis can be summarized as a fingerprint plot (FP) designed on a 2D grid formed by (de, di) pairs which each point on this plot illustrates a hit formed by a (de, di) pair.34 Finally, the enrichment ratio (EXX/XY) is derived from the HS analysis which represents the propensity of pairs of elements (X, X) or (X, Y) in X⋯Y interactions.35 This ratio is defined as 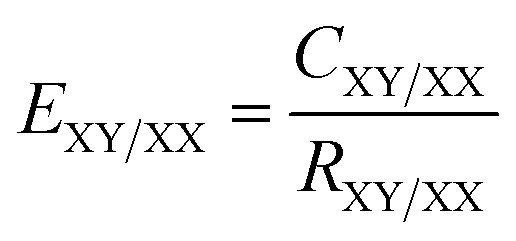 , where CXY/XX is the ratio between the proportion of actual contacts and RXY/XX is the theoretical proportion of random contacts calculated by the summation (SX) of proportions of all contacts containing chemical type X on the HS. The high and low propensity to form contacts in crystals are parameterized as an enrichment ratio which is larger (E > 1) or lower (E < 1) than unity, respectively, and are alternatively called to be ‘favored’ for the former and ‘dis-favored’ for the latter.
, where CXY/XX is the ratio between the proportion of actual contacts and RXY/XX is the theoretical proportion of random contacts calculated by the summation (SX) of proportions of all contacts containing chemical type X on the HS. The high and low propensity to form contacts in crystals are parameterized as an enrichment ratio which is larger (E > 1) or lower (E < 1) than unity, respectively, and are alternatively called to be ‘favored’ for the former and ‘dis-favored’ for the latter.
Results and discussion
X-ray crystallography investigations
The reaction between the PT ligand OP[NH-2Py]3 and CoCl2·6H2O in the solution of methanol/acetonitrile leads to the preparation of complex 1 (Fig. 1) crystallized in the monoclinic space group P21/n. The asymmetric unit contains 2 × 0.5 {Co(II){(O)P[NH-2Py]2[NH-2PyH]]2} cations and 3 × 1 Cl anions (highly disordered). The crystal structure is composed of alternating sheets of complex molecules and of disordered chlorine anions. The chlorine atoms are disordered over several positions, in spite of being stabilized through H-bonds by NH groups. Interestingly, in the structure there are three chlorine atoms for every Co, indicating an extra positive charge in the complex molecule. An analysis of the H-bond network indicates that two pyridine moieties are also partially protonated stabilizing the anionic chlorine sheets through N–H⋯Cl hydrogen bonds. These hydrogen bonds (Table 3) lead to the formation of a two-dimensional supramolecular parallel to the (110) plane (Fig. 2).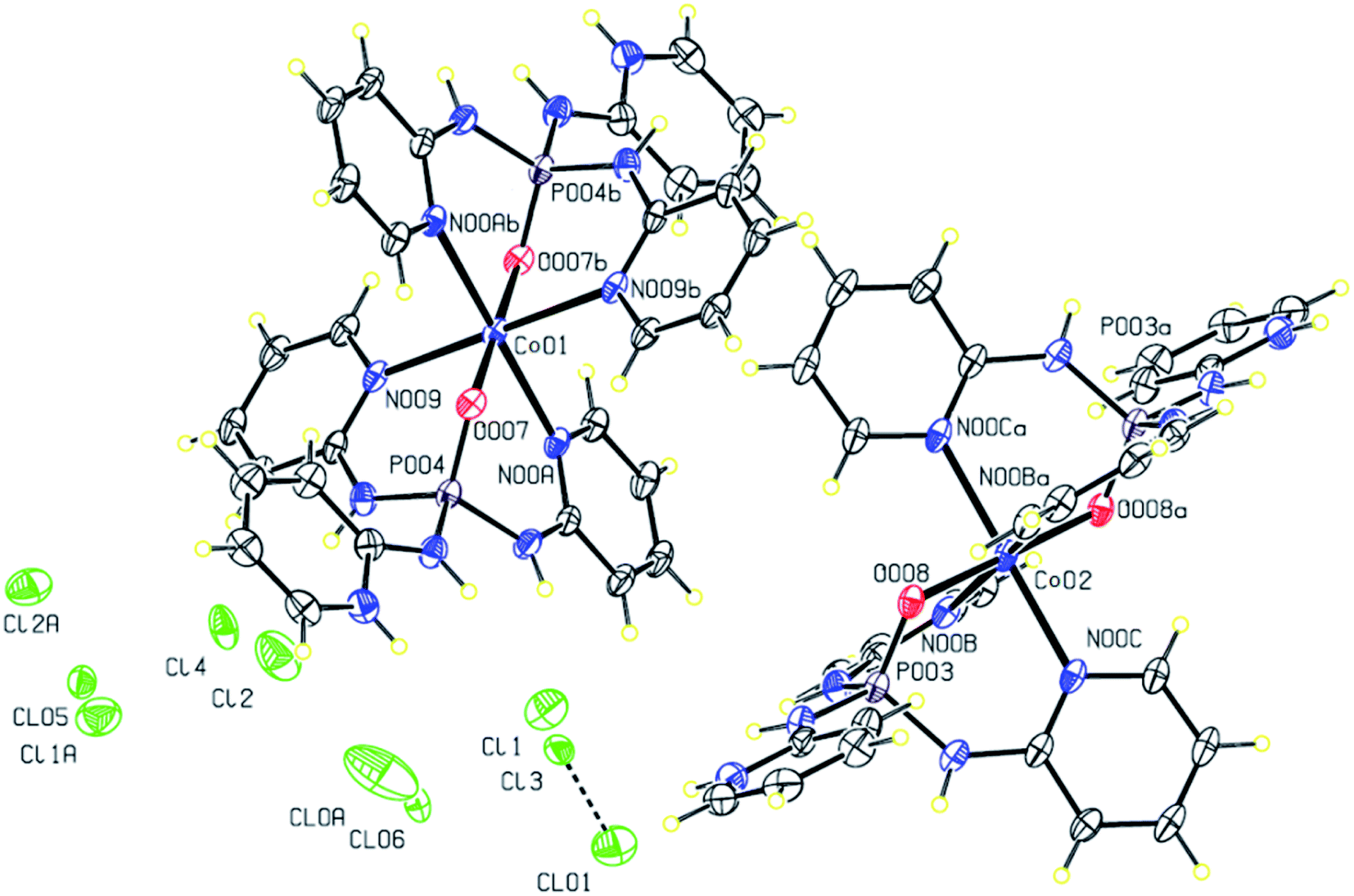 | ||
| Fig. 1 Displacement ellipsoid plot (30% probability level) and the atom numbering (for Cl, P, Co and the coordinated atoms) scheme for the structure 1. H atoms are drawn as circles of arbitrary radii. | ||
| 1 | ||||
|---|---|---|---|---|
| D–H⋯A | D–H | H⋯A | D⋯A | ∠D–H⋯A |
a Symmetry transformations used to generate equivalent atoms for 1: (i) 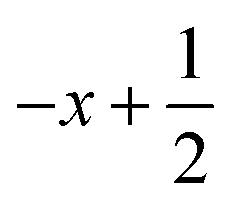 , , 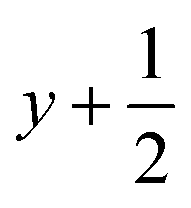 , , 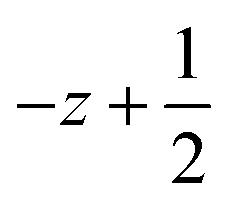 ; (ii) ; (ii) 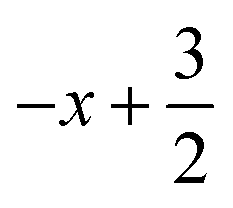 , , 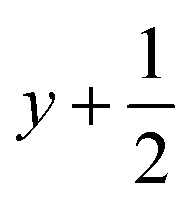 , , 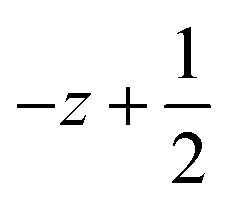 ; (iii) ; (iii) 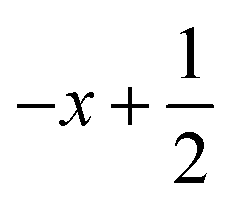 , , 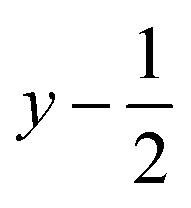 , , 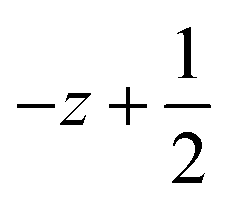 ; (iv) x − 1, y + 1, z; (v) ; (iv) x − 1, y + 1, z; (v) 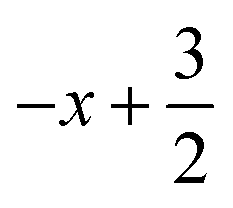 , ,  , , 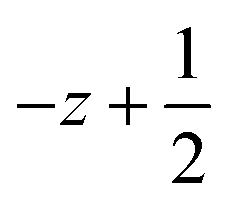 ; *for π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. ; *for π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. |
||||
| N00I–H00I⋯Cl1Ai | 0.860 | 2.20 | 3.03 (1) | 163.0 |
| N00D–H00D⋯Cl1Ai | 0.860 | 2.32 | 3.12 (1) | 154.7 |
| N00H–H00H⋯Cl0Ai | 0.860 | 2.210 | 3.053 (9) | 166.7 |
| N00H–H00H⋯Cl06i | 0.860 | 2.205 | 3.057 (3) | 170.7 |
| N00J–H00J⋯Cl1 | 0.860 | 2.654 | 3.161 (4) | 119.0 |
| N00J–H00J⋯Cl01 | 0.860 | 2.114 | 2.948 (4) | 163.3 |
| N00J–H00J⋯Cl3 | 0.860 | 2.295 | 3.037 (4) | 144.5 |
| N00E–H00E⋯Cl1 | 0.860 | 2.443 | 3.194 (4) | 146.2 |
| N00G–H00G⋯Cl1 | 0.860 | 2.085 | 2.912 (4) | 161.1 |
| N00K–H00K⋯Cl1Aii | 0.860 | 2.59 | 3.22 (1) | 131.2 |
| N00K–H00K⋯Cl2Aii | 0.860 | 2.30 | 3.11 (1) | 156.1 |
| N00K–H00K⋯Cl05ii | 0.860 | 2.346 | 3.153 (2) | 156.4 |
| N00F–H00F⋯Cl2 | 0.860 | 2.036 | 2.880 (4) | 166.7 |
| N00F–H00F⋯Cl4 | 0.860 | 2.216 | 3.057 (5) | 165.8 |
| C00L–H00L⋯Cl01iii | 0.950 | 2.793 | 3.482 (5) | 130.2 |
| C00N–H00N⋯Cl05iv | 0.950 | 2.80 | 3.52 (1) | 133.3 |
| C01D–H01D⋯Cl07v | 0.950 | 2.498 | 3.357 (5) | 150.2 |
| C01D–H01D⋯Cl08v | 0.950 | 2.78 | 3.49 (1) | 131.9 |
| C01F–H01F⋯Cl10ii | 0.950 | 2.566 | 3.38 (1) | 144.0 |
| C01F⋯C01Dv | — | — | 3.253 (6) | — |
| C01E–H01E⋯π* | — | 2.892 | — | 140.87 |
| C01A–H01A⋯π* | — | 2.929 | — | 125.2 |
| C015–H015⋯π* | — | 3.184 | — | 131.80 |
| C015–H015⋯π* | — | 3.224 | — | 138.1 |
| C016–H016⋯π* | — | 3.215 | — | 130.59 |
| C016–H016⋯π* | — | 3.267 | — | 121.61 |
| π⋯π* | — | — | 3.585 | — |
| π⋯π* | — | — | 3.619 | — |
 | ||
| Fig. 2 View of two-dimensional supramolecular structure of 1 constructed through the N–H⋯Cl (black dashed lines, Table 3) interactions parallel to the ab plane. Different colors have been shown for symmetrically independent molecules. | ||
The Co(II) cation is found in a hexa-coordinate environment Co(N)4(O)2 adopting a distorted octahedral geometry, and is bonded to pyridine nitrogens and phosphoryl oxygens from two different PT ligands (Fig. 1). Indeed, in this structure, each PT molecule acts as a tridentate N,N,O-donor ligand providing three sites around the central cation Co(II) leading to the formation of the first discrete chelate PT complex with two tridentate [N]3P(O)-based ligands. It should be noted that in each PT ligand the remaining pyridine nitrogen atom which is not coordinated to the metalic centre is protonated as NH+. The Co–N distances range from 2.210 (3) to 2.246 (2) Å, and those of type Co–O are 2.011 (2) and 2.013 (2) Å. The N–Co–N angles change from 87.24 (9) to 92.76 (9)° for cis and are about 180.0° for trans angles. The cis N–Co–O angles are different from 88.47 (9) to 91.53 (9)° and a value of about 180° is found for trans O–Co–O angles. These values for angles around the central cation Co(II) confirm a disordered geometry in both symmetrically independent cationic complexes. Moreover, the oxygen atoms of the PO groups of two PT ligands are in trans-position with respect to each other and their bond lengths (1.501 (2) and 1.498(2) Å) are slightly longer than those in the free ligand (1.4783 (14) Å).
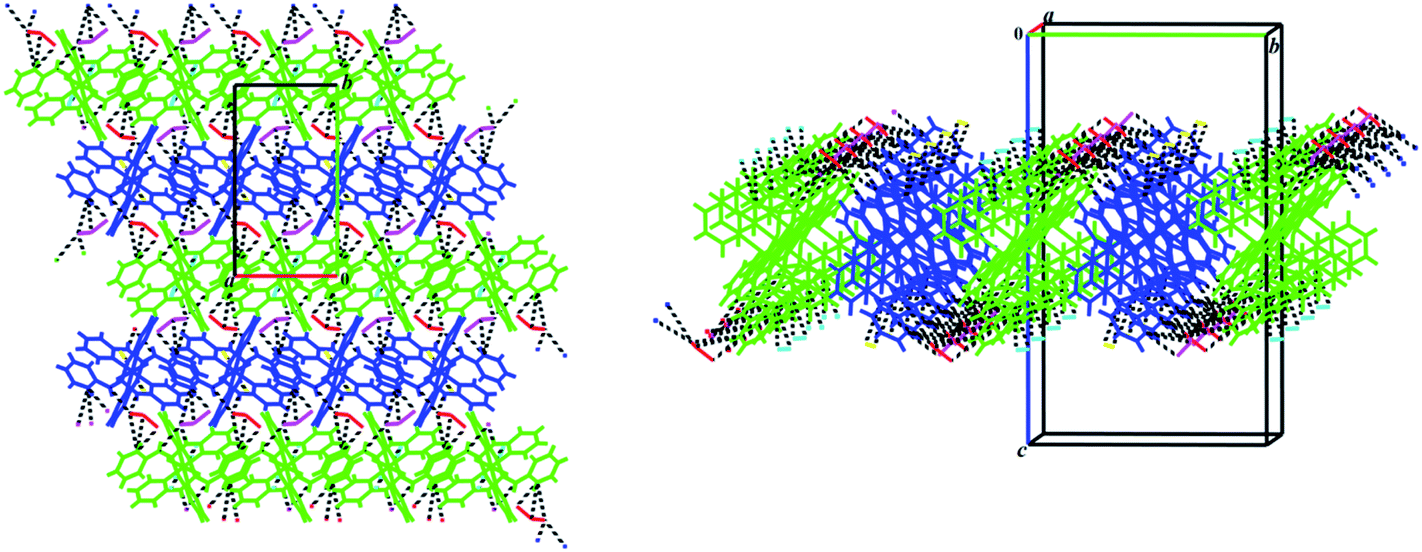
The above-mentioned 2D network of 1 constructed by the N–H⋯Cl hydrogen bond interactions (Table 3) becomes more stable via the participation of pyridine rings in some π-effect interactions (C–H⋯π and π⋯π, Table 3, Fig. 3). Furthermore, intermolecular interactions of the C–H⋯Cl and C⋯C types (Table 3) link these 2D layers into a 3D extended network (Fig. 3).
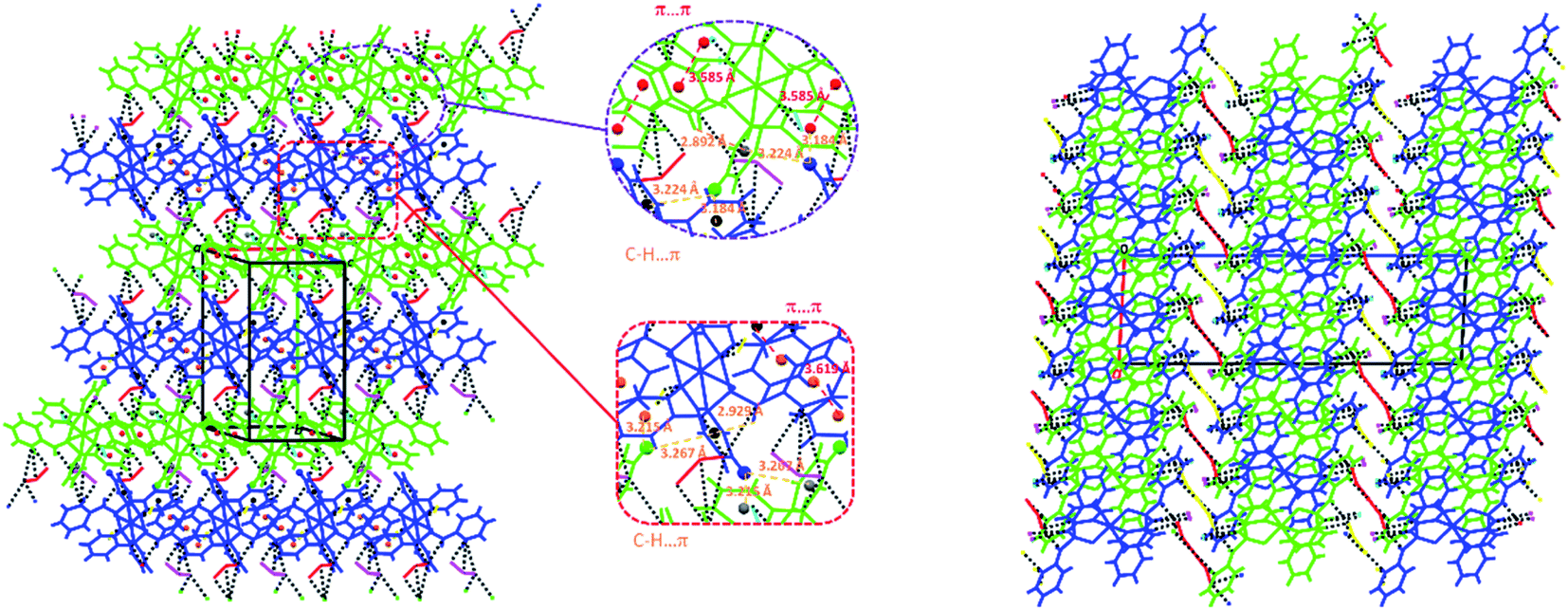 | ||
| Fig. 3 Left: presentation of π-effect (C–H⋯π and π⋯π, Table 3) interactions involved in the N–H⋯Cl (black dashed lines) 2D network of 1; right: a view along b axis of the 3D network formed by linking the N–H⋯Cl 2D layers (three layers are seen in figure) via the C–H⋯Cl and C⋯C interactions. Symmetrically-independent molecules are shown by different colors (dark blue and green for cationic complexes and red, yellow, light blue and pink for chlorine anions). | ||
The molecular structures of 2–4 are displayed in Fig. 4 and 5. The structures display the anhydrate, monohydrate and dimethylformamide/water (DMF/H2O) solvate crystals of tris(pyridin-2-yl)phosphoric triamide ligand, respectively, crystallized in the trigonal space group R for 2 and the monoclinic space group P21/n for 3 and 4. The structure 2 is a pseudo-polymorph of the previously reported structure OP[NH-2Py]3 (CSD refcode LAFNAI01)12 crystallized in the rhombohedral space group R. The asymmetric unit of 2 is composed of one third of the molecule with the PO bond aligned along the 3-fold axis, while for 3 it consists of two PT molecules (P001 and P002) and one water molecule. In the case of 4, two molecules P001 and P002, two dimethylformamide molecules and one water molecule make up the asymmetric unit.
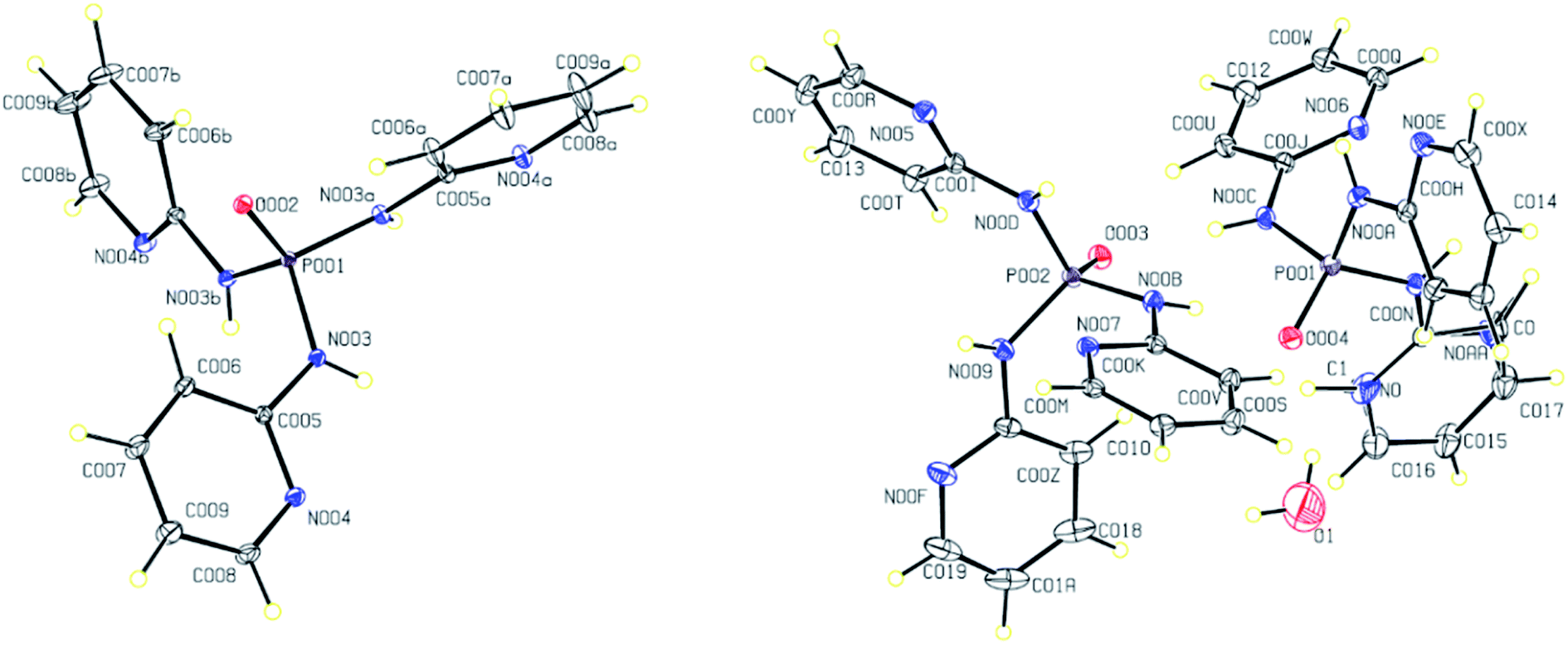 | ||
| Fig. 4 Displacement ellipsoid plots (30% probability level) and the atom numbering schemes for the structures 2 (left) and 3 (right) in the asymmetric unit. H atoms are drawn as circles of arbitrary radii. | ||
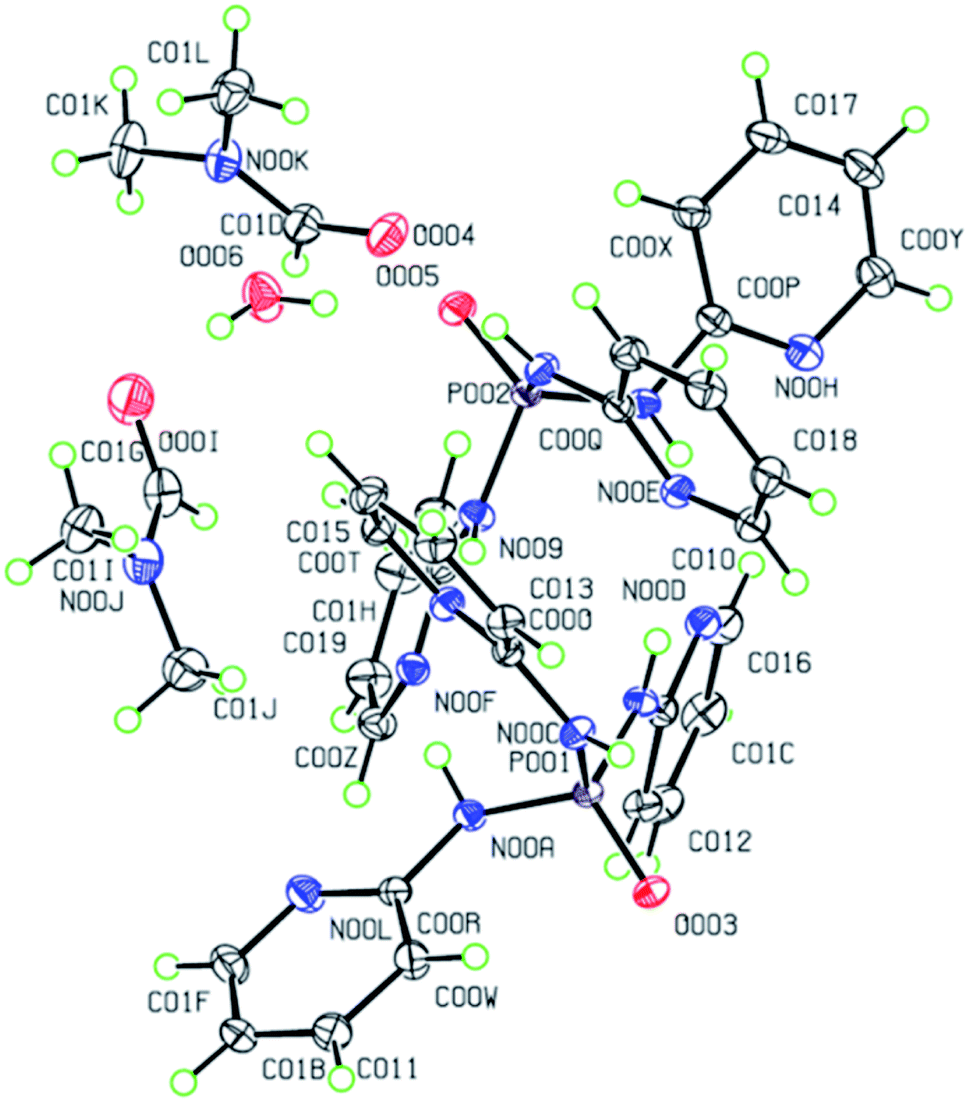 | ||
| Fig. 5 Displacement ellipsoid plot (50% probability level) and the atom numbering scheme for the structure 4 in the asymmetric unit. H atoms are drawn as circles of arbitrary radii. | ||
The P atom is in all structures in a distorted tetrahedral environment made up by a terminal oxygen and three nitrogen atoms of amide substitutes with standard bond angles of O–P–N and N–P–N for the phosphoramides36 (Tables S1 and S2†). The PO and P–N bond lengths and C–N–P angles (Tables S1 and S2†) are similar to those in previous reported compounds.40
In the crystal structure of 2 the molecules are packed in a 2D plane arrangement along the ab plane by the N–H⋯N classical hydrogen bond interactions (Table 4) generating the hexagonal unit composed of six molecules with the ring cavity in the middle (Fig. 6). In this packing, the phosphoryl groups do not take part in any classical H-bonds, although they are known to act as H-bond acceptor sites in similar phosphoramide compounds.38 The ab planes hydrogen bond together via the (C–H…)3OP hydrogen bond interactions forming a 3D network (Table 4, Fig. 6).
| D–H⋯A | D–H | H⋯A | D⋯A | ∠D–H⋯A |
|---|---|---|---|---|
a Symmetry transformations used to generate equivalent atoms for 2: (i) 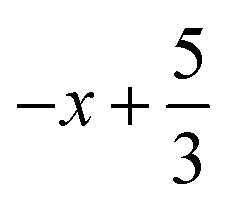 , , 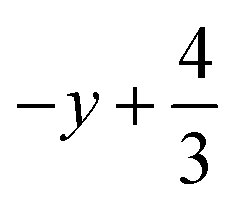 , , 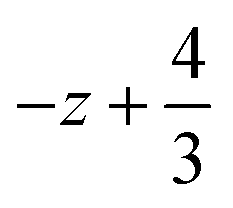 ; (ii) ; (ii) 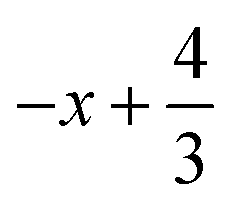 , , 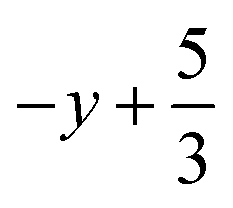 , , 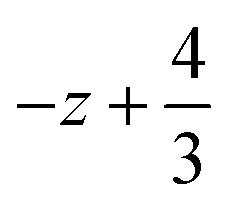 ; for 3: (i) ; for 3: (i) 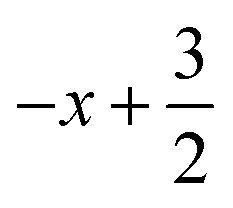 , , 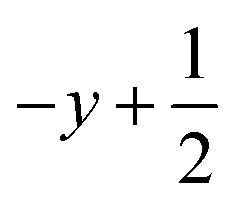 , , 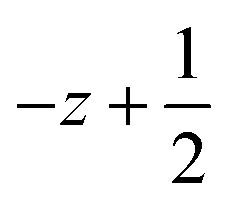 ; (ii) ; (ii) 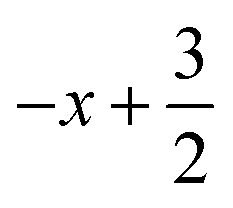 , , 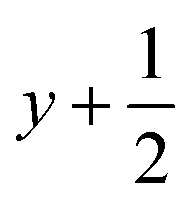 , , 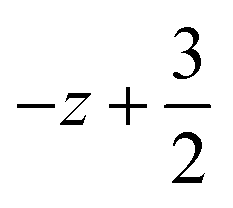 ; (iii) −x, −y +1, −z +1; *For π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. ; (iii) −x, −y +1, −z +1; *For π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. |
||||
| 2 | ||||
| N003–H003⋯N004i | 0.83 (2) | 2.18 (2) | 3.007 (2) | 176 (2) |
| C007–H007⋯O002ii | 1.00 (2) | 2.41 (2) | 3.388 | 165 |
| 3 | ||||
|
||||
| O1–H1B⋯O004 | 0.850 | 1.9771 | 2.773 (7) | 155.4 |
| N00B–H00B⋯O004 | 0.84 (2) | 2.00 (2) | 2.832 (1) | 173 (2) |
| N00C–H00C⋯O003 | 0.85 (2) | 2.09 (2) | 2.932 (1) | 169 (2) |
| N00D–H00D⋯N006i | 0.88 (2) | 2.17 (2) | 3.043 (1) | 173 (2) |
| N008–H008⋯N005i | 0.88 (2) | 2.10 (2) | 2.953 (1) | 162 (1) |
| N00A–H00A⋯N007i | 0.87 (2) | 2.15 (2) | 2.995 (1) | 163 (2) |
| N009–H009⋯N00Ei | 0.82 (2) | 2.13 (2) | 2.938 (1) | 173 (2) |
| C00U–H00U⋯N00Fii | 0.950 | 2.527 | 3.318 (2) | 140.72 |
| C012–H012⋯C00Mii | 0.950 | 2.822 | 3.542 (2) | 133.38 |
| C014–H014⋯O1iii | 0.950 | 2.532 | 3.352 (8) | 144.5 |
| π⋯π* | — | — | 3.678 | — |
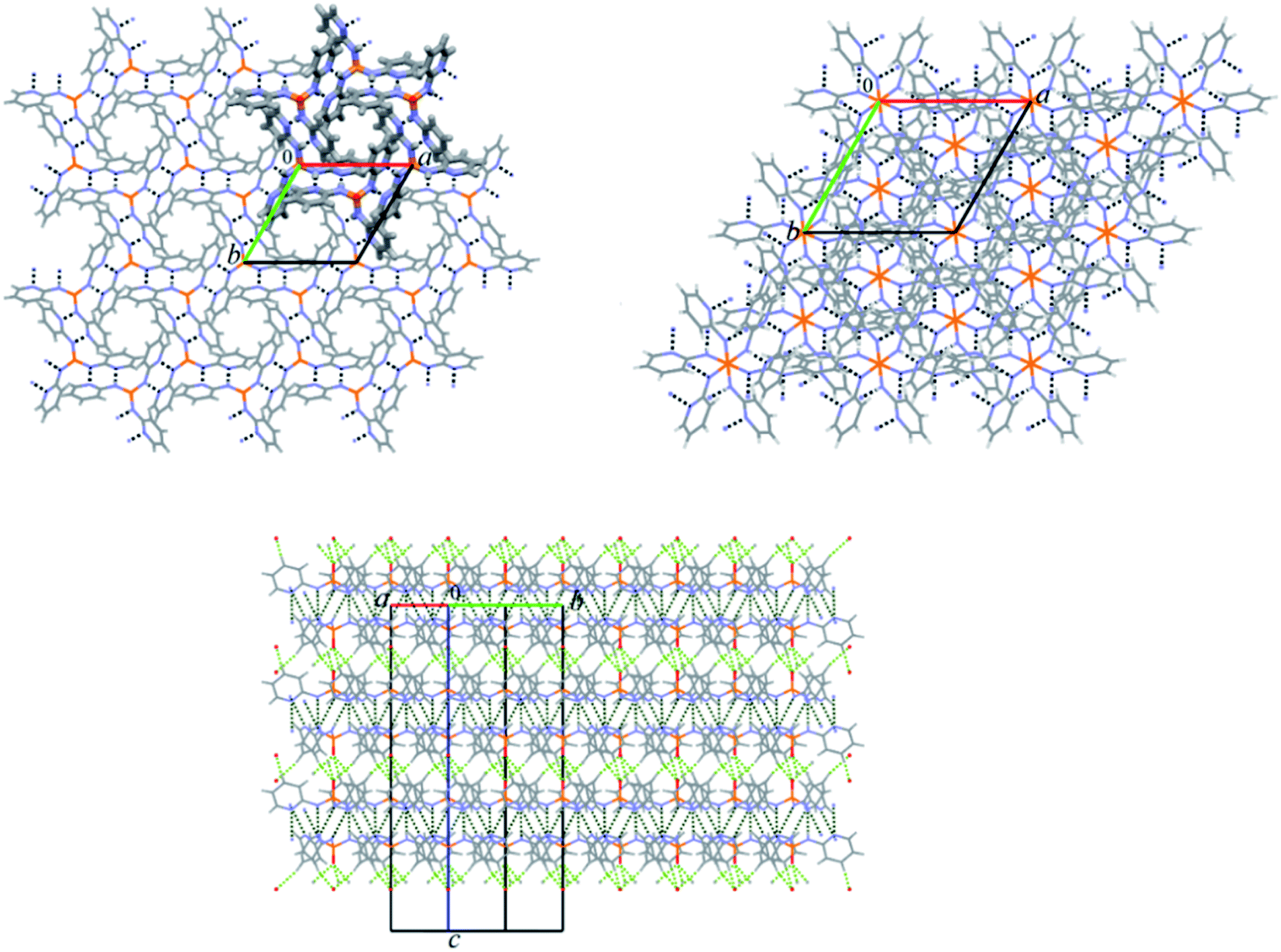 | ||
| Fig. 6 Up: Views of partial packing diagrams of compound 2 viewed along the c axis are displayed; left up: a 2D plane arrangement is built by N–H⋯N hydrogen bond interactions (black dash lines, Table 4) along the ab plane generating the hexagonal unit bolded by capped stick style with the ring cavity in the middle; right up: the 2 × 2 × 2 unit cell packing diagram of 2 showing the 2D planes taken together represent the non-interacting phosphoryl groups occur within the ring cavities; down: a 3D network is formed by connecting the 2D hydrogen bonded planes via (C–H…)3OP hydrogen bond interactions (green dash lines, Table 4). | ||
In the crystal structure of monohydrated crystals 3, the one-dimensional (1D) chains along the b axis formed via the N–H⋯O, N–H⋯N and O–H⋯O (Table 4) hydrogen bond interactions connected via π⋯π stacking interaction to build a two-dimensional (2D) molecular assembly parallel to the (101) plane (Fig. 7). Furthermore, the crystal structure is stabilized by more weak hydrogen bonds such as C–H⋯N, C–H⋯C and C–H⋯O (Table 4) forming a 3D network.
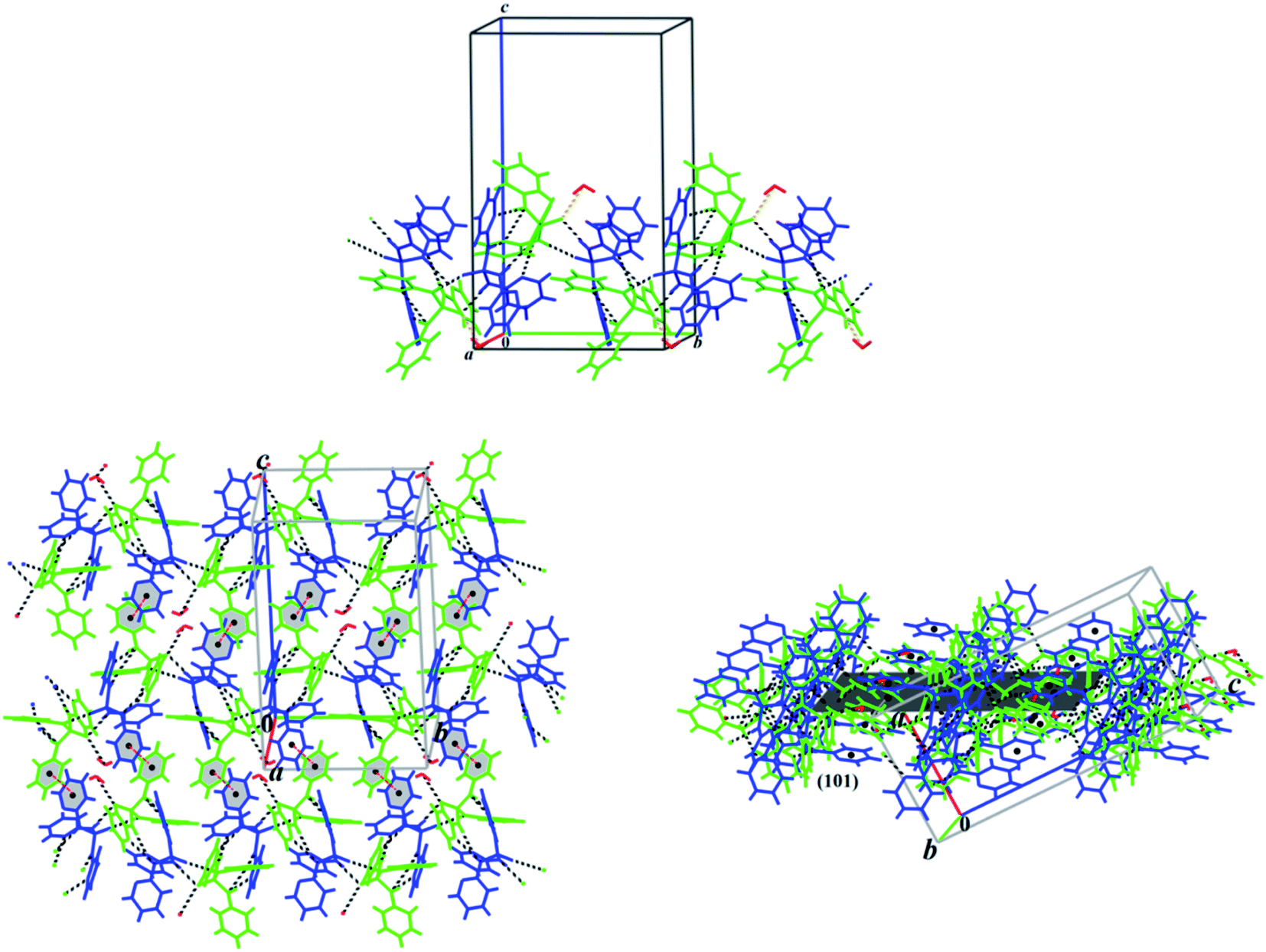 | ||
| Fig. 7 Up: partial view of one-dimensional (1D) chain formed via N–H⋯O and N–H⋯N (black dash lines), and O–H⋯O (orange dash lines) hydrogen bond interactions along the b axis in the crystal structure of 3. Down: the 1D chains are connected via π⋯π stacking interaction (red dash lines) building a two-dimensional (2D) molecular assembly parallel to the (101) plane showed in two views. The symmetrically independent molecules are specified by different colors. | ||
In the solvate structure 4, the adjacent molecules are linked via N–H⋯O, N–H⋯N and O–H⋯O classical hydrogen bonds (Table 5, Fig. 8) forming a hydrogen-bonded ten-membered motif constructed from five symmetrically-independent molecules (two PT molecules P001 and P002, one water molecule and two DMF molecules). In this arrangement, a PT molecule (P001) is connected to same molecule (P001) via the N–H⋯OP hydrogen bonds, while it is connected to other PT molecule (P002) by the N–H⋯N hydrogen bonds. Then, the N–H⋯O and O–H⋯O hydrogen bond interactions interlink one of PT molecules (P002) to water and DMF molecules. By considering additional interactions especially π-effects, further stabilization of the structure 4 is achieved. For example, the arrangement of molecules can be regarded as a two-dimensional network parallel to plane (−101) by the quoted classical hydrogen bonds, the non-classical hydrogen bond C–H⋯O, and the π-stacking interactions C–H⋯π and π⋯π (Table 5, Fig. 9). In this 2D aggregation, the mentioned hydrogen-bonded ten-membered motifs are linked together via C–H⋯O and C–H⋯π interactions. Some π⋯π interactions are also found between aromatic rings in ten-membered motifs.
| D–H⋯A | D–H | H⋯A | D⋯A | ∠D–H⋯A |
|---|---|---|---|---|
a Symmetry transformations used to generate equivalent atoms for 4: (i) −x + 1, −y +2, −z +1; (ii) 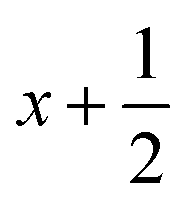 , , 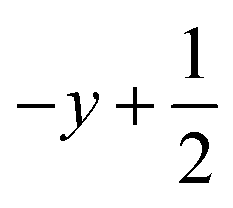 , , 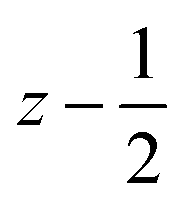 . *For π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. . *For π⋯π interactions d(D⋯A) = Cg⋯Cg and for C–H⋯π interactions d(H⋯A) = H⋯Cg, where Cg = centroid of the aromatic pyridine rings. |
||||
| 4 | ||||
| N00C–H00C⋯O003i | 0.860 | 1.9687 | 2.789 (1) | 159.14 |
| N00G–H00G⋯O004 | 0.860 | 1.915 | 2.775 (2) | 178.10 |
| O006–H00B⋯O005 | 0.850 | 1.9330 | 2.779 (1) | 173.59 |
| O006–H00D⋯O00I | 0.850 | 1.927 | 2.776 (2) | 176.20 |
| N00A–H00A⋯N00F | 0.860 | 2.329 | 2.901 (1) | 124.25 |
| N009–H009⋯N00B | 0.860 | 2.137 | 2.979 (1) | 165.99 |
| N008–H008⋯N00E | 0.860 | 2.188 | 3.042 (1) | 171.91 |
| N007–H007⋯N00D | 0.860 | 2.309 | 2.885 (2) | 124.58 |
| C01B–H01B⋯O006i | 0.930 | 2.561 | 3.413 (2) | 152.45 |
| C012–H012⋯C01Iii | 0.950 | 2.704 | 3.330 (2) | 123.95 |
| C01L–H01Q⋯π* | — | 2.999 | — | 126.02 |
| C00Y–H00Y⋯π* | — | 3.047 | — | 147.64 |
| C01L–H01P⋯π* | — | 3.260 | — | 110.84 |
| π⋯π* | — | — | 3.607 | — |
| π⋯π* | — | — | 3.668 | — |
| 5 | ||||
|
||||
| N005–H00A⋯O003 | 0.890 | 1.938 | 2.818 | 170.0 |
| C006–H006⋯O002 | 0.930 | 2.542 | 3.040 (5) | 113.9 |
| C006–H006⋯O004 | 0.930 | 2.903 | 3.411 (6) | 115.7 |
| C007–H00E⋯C006 | 0.960 | 2.753 | 3.650 (7) | 155.7 |
| C007⋯C006 | — | — | 3.338 (8) | — |
| C007⋯C008 | — | — | 3.433 | — |
| O003⋯O004 | — | — | 3.128 | — |
| Cu01⋯O002 | — | — | 2.004 (4) | — |
| Cu01⋯O004 | — | — | 1.994 (3) | — |
| Cu01⋯O003 | — | — | 2.451 | — |
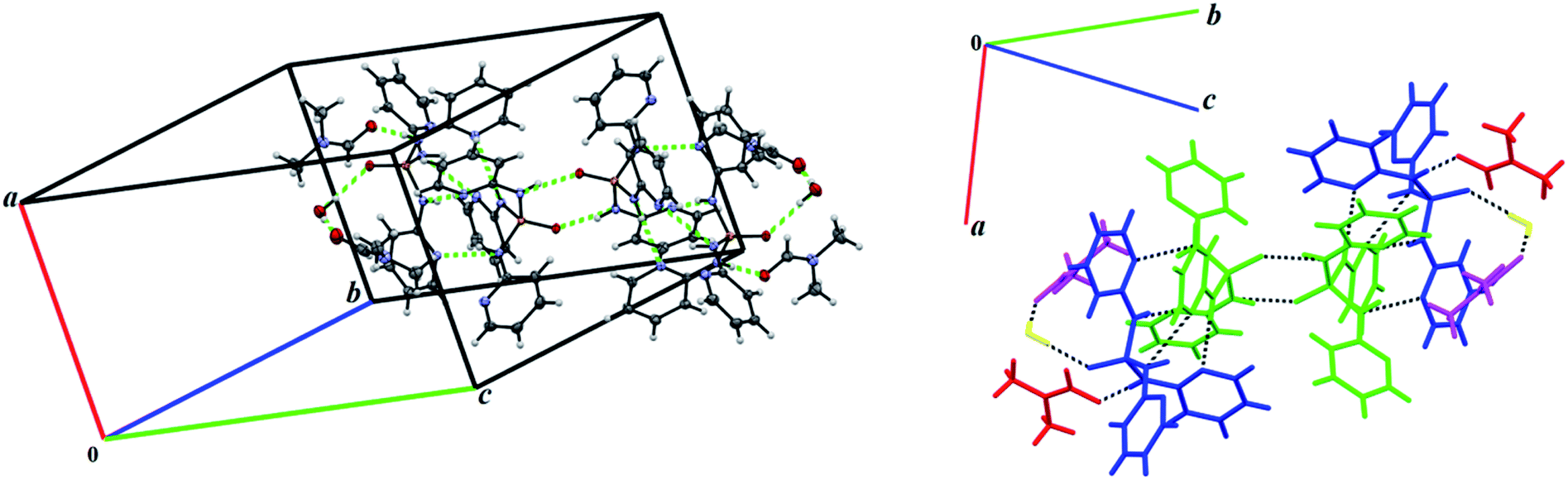 | ||
| Fig. 8 Views of hydrogen-bonded ten-membered motif constructed from five symmetrically independent molecules in the structure 4. Left: adjacent molecules are linked via the N–H⋯O, N–H⋯N and O–H⋯O hydrogen bonds (green dashed lines, Table 5); Right: for a better vision, different colors are used for symmetrically independent molecules PT (blue and green), DMF (red and purple) and water (yellow) linked by the mentioned different hydrogen bonds (black dashed lines). | ||
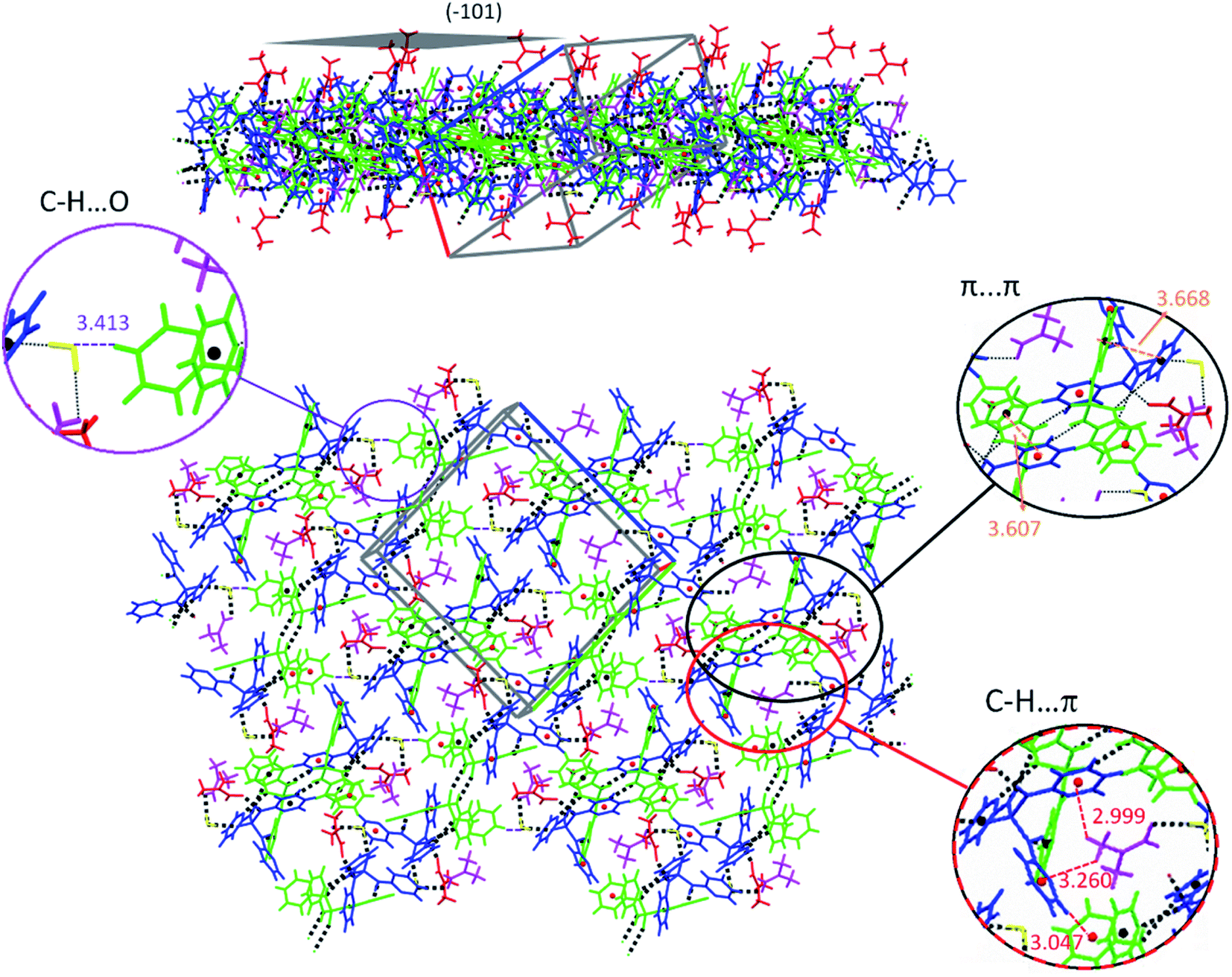 | ||
| Fig. 9 Views of 2D molecular network formed by classical (N–H⋯O, N–H⋯N and O–H⋯O), non-classical (C–H⋯O) and π-effect (C–H⋯π and π⋯π) interactions parallel to the (−101) plane in the structure 4. Symmetrically independent molecules are shown by different colors. | ||
As is apparent from the structural description above, the inclusion of solvent or water molecules results in the formation of typical hydrogen bond interaction N–H⋯OP in PT compounds. Such H-bond patterns can be found in the crystal packing feature of monohydrate and solvate crystals of OP[NH-2Py]3 (structures 3 and 4), while it is absent in anhydrate/pure form 2. Moreover, monohydrate and solvate forms crystallize in the common crystal system/space group of monoclinic/P21/n but the anhydrous analogue shows a less common crystal system/space group of trigonal/R. This is unusual as a CSD search for PT structures with a backbone OP[N]3 manifests that for some of such structures both pure (anhydrate) and solvate crystals show usually the same crystal system/space group, for example, OP[NMe2]3 (POTJAJ and POTJAJ01) and its co-crystals and solvates. As pyridine and its derivatives can force structures to pack in different modes and also, to crystallize in different crystal systems, it transforms the PT molecule OP[NH-2Py]3 into a flexible tridentate ligand constructing achelate complex (structure 1). Indeed, this substituent provides, besides the lone pairs on the oxygen atom of the phosphoryl group PO, other suitable sites as both H-bond acceptors and metal binding sites, i.e. the lone pair on the nitrogen atom of the pyridine ring. This leads to an N,N,O-donor ligand in the structure 1 and a competition to form the N–H⋯N normal hydrogen bond interaction versus N–H⋯OP in the different solid forms of OP[NH-2Py]3 in the structures 2–4. Whereas, in the pure crystal of 2, only the N–H⋯N hydrogen bond interaction is preferred, in contrast both the N–H⋯N and N–H⋯OP interactions are present in the crystal packing structures of the monohydrate and solvate forms of 3 and 4.
The Cu(II) complex 5 obtained from the unexpected reaction of formate anions results from the hydrolysis of the DMF solvent. This structure is an improved model of the previously published complex {[Cu(OCHO)3][(CH3)2NH2]}n (CSD refcode MACUFR24) that crystallizes in the monoclinic space group I2/c. A view of the structure of 5 is shown in Fig. 10.
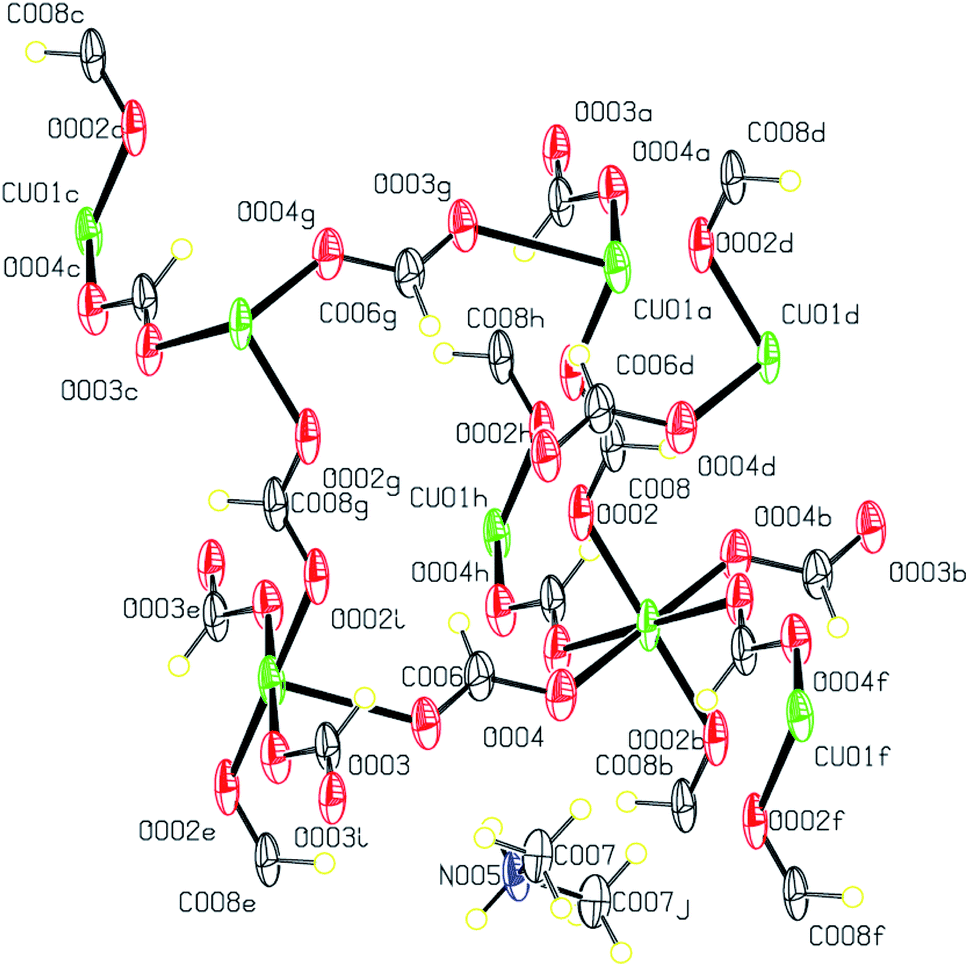 | ||
| Fig. 10 Displacement ellipsoid plot (50% probability level) and the atom numbering scheme for the structure 5. H atoms are drawn as circles of arbitrary radii. | ||
It is noted that complex 5 displays uniaxial negative thermal expansion along one of its orthogonal axes. The structure in this study was determined at 100 K, while the previous reported structure, MACUFR, was measured at room temperature. Fig. 11 gives the percentual expansions along three orthogonal axes in the structure: the calculated thermal expansion coefficients are 62.65 (for X1), 13.50 (X2), 44.40 (X2) and −5.27 MK−1 for the volume.
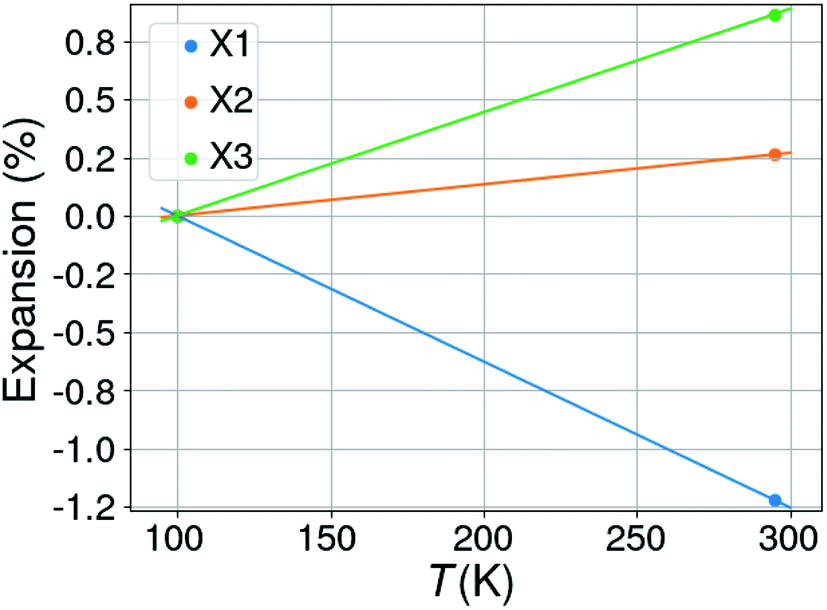 | ||
| Fig. 11 Percentual thermal expansion in the structure of 5; X1, X2 and X3 are the three orthogonal axes in the structure (of which one is aligned with the crystallographic b-axis). | ||
The asymmetric unit of 5 is composed of one-half of a Cu(II) cation, an anionic ligand (OCHO)− and an anionic ligand (CHO)−0.5, complemented by one-half of [NH2(CH3)2]+ counter-ion. The Cu(II) atom is hexa-coordinated by six anionic formate ligands, adopting a distorted octahedral geometry. This distorted structure has different Cu–O distances (Table S2†) with the average value of 2.150 (3) Å and the bond angles around Cu(II) are 88.38 (13)° and 91.62 (13)° for cis O–Cu–O angles and 180.0° for trans O–Cu–O angles. The anionic formate groups acts in a bidentate bridge between two neighbouring atoms Cu(II) bridged by μ2-formate linkers. In this way, the connection of these six-coordinated Cu(II) by μ2-formate linkers constructs a three-dimensional metal–organic framework (MOF). Dimethylammonium cations provide charge balance and are located in the quoted interstices of this Cu(II) MOF, tethered by the N–H⋯O hydrogen bond interactions (Table 5). It is should be noted that such anionic 3D network of coordinate formate anions captured by the metal ions are common, but dimethylammonium cations captured in crystals are very infrequent.39
A view of 2 × 2 × 2 unit cell packing diagram is shown in Fig. 12. As it is clearly seen, this 3D network illustrates a scarce porous framework for formate coordination polymer structures.
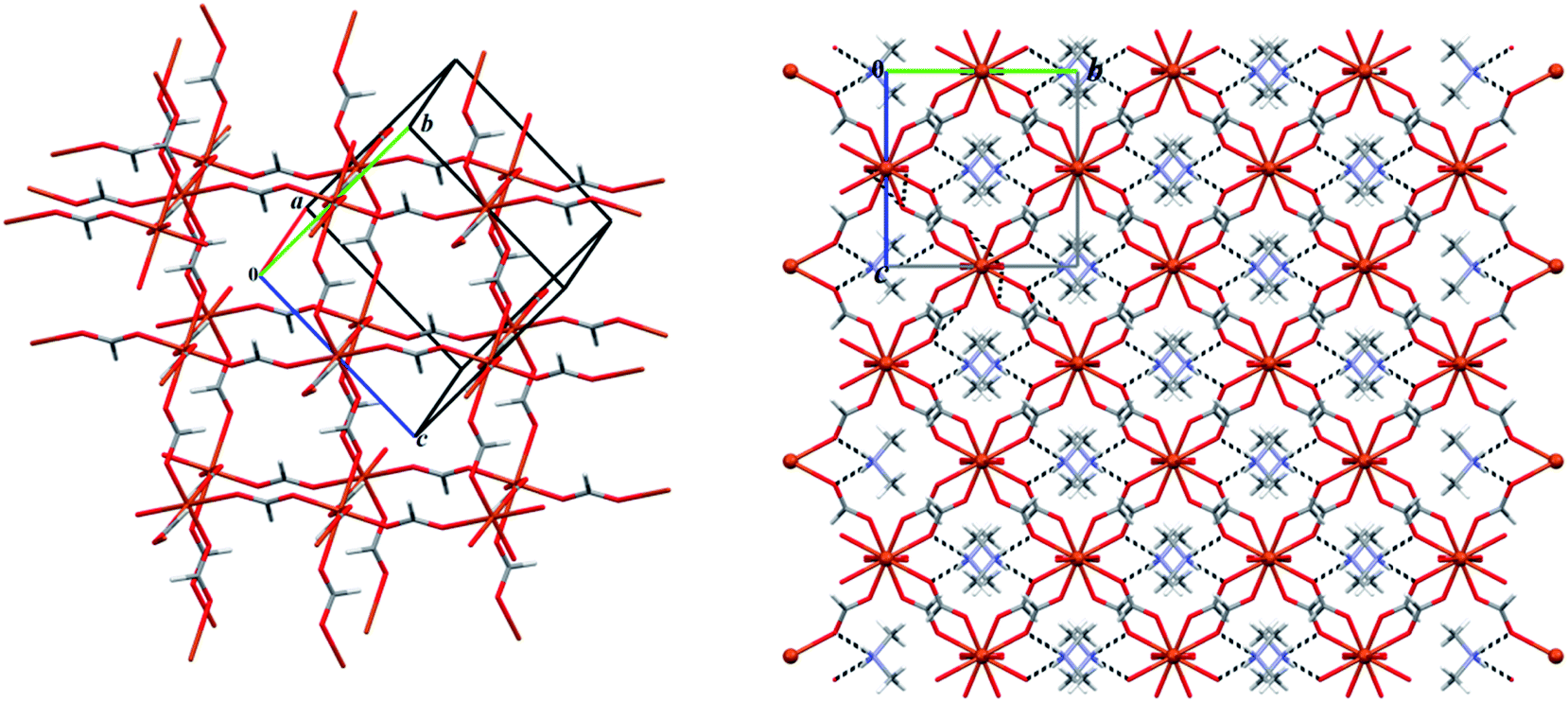 | ||
| Fig. 12 Left: a partial view of the anionic network formed in the structure 5 exhibiting the void spaces as cubic cavities; right: the 2 × 2 × 2 unit cell packing diagram of 5 showing dimethylammonium cations occur within the cubic cavities by the N–H⋯O (black dashed lines, Table 5) hydrogen bond interactions. | ||
Hirshfeld surface (HS) analysis and enrichment ratio (EXY)
For this section, all discussed intermolecular interactions are summarized in Tables 6 (for 1 and 2), 7 (for 3 and 4) and S3† (for 5). The relative contributions of the close contacts to the corresponding HS areas and the enrichment ratios (EXY) are listed in Tables 5 (for 1 and 2), 6 (for 3 and 4) and S3† (for 5). The Hirshfeld surfaces mapped over the dnorm property are shown in Fig. 13 (for 1), 16 (for 2 and molecules P001 of 3 and 4), S1† (molecules P002 of 3 and 4) and 19 (for 5). The full 2D fingerprint plots (FPs) are presented in Fig. 15 (for 1), 17 (2–4) and 19 (5). Fig. S2–S6† are also exhibited the 2D decomposed FPs of the compounds 2–4. Shape index surfaces plotted on some of the studied structures are displayed in Fig. 14 (molecule Co01 of 1), 18 (molecules P001 of 3 and 4) and S7† (molecules P002 of 3 and 4). It is should be noted that for 5, HS has been generated on the [Cu(OCHO)4]2− unit of this structure. Moreover, in the following discussion the label of the metal or phosphorous atoms are used to introduce the symmetrically independent molecules of a structure.| Contacts | Co1 of 1 | Co2 of 1 | 2 | SX | H | C | N | O | Cl |
|---|---|---|---|---|---|---|---|---|---|
| H⋯H | 45.7 | 43.5 | 44.2 | Co1 of 1 | 68.90 | 19.35 | 4.15 | 0.65 | 6.85 |
| H⋯C/C⋯H | 24.9 | 26.8 | 19.3 | Co2 of 1 | 68.10 | 19.45 | 4.25 | 0.60 | 6.90 |
| H⋯N/N⋯H | 6.5 | 7.4 | 21.5 | 2 | 66.75 | 19.65 | 10.75 | 2.15 | — |
| H⋯O/O⋯H | 1.3 | 1.2 | 4.3 | ||||||
| H⋯Cl | 13.7 | 13.8 | — | ||||||
| C⋯C | 6.0 | 5.5 | 10.0 | ||||||
| C⋯N/N⋯C | 1.8 | 1.1 | — |
| Atoms | H | C | O | N | Cl | H | C | O | N | Cl | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Co1 of 1 | RXX/RXY | EXX/EXY | Co1 of 1 | ||||||||||
| H | 47.47 | 26.66 | 5.72 | 0.89 | 9.44 | H | 0.96 | 0.93 | 1.14 | 1.46 | 1.45 | ||
| C | 26.66 | 3.74 | — | 1.61 | — | C | 0.93 | 1.60 | — | 1.12 | — | ||
| N | 5.72 | 1.61 | — | — | — | N | 1.14 | 1.12 | — | — | — | ||
| O | 0.89 | — | — | — | — | O | 1.46 | — | — | — | — | ||
| Cl | 9.44 | — | — | — | — | Cl | 1.45 | — | — | — | — | ||
| Co2 of 1 | RXX/RXY | EXX/EXY | Co2 of 1 | ||||||||||
|
|||||||||||||
| H | 46.38 | 26.49 | 5.79 | 0.82 | 9.40 | H | 0.94 | 1.01 | 1.28 | 1.46 | 1.47 | ||
| C | 26.49 | 3.78 | — | 1.65 | C | 1.01 | 1.45 | — | 0.67 | ||||
| N | 5.79 | 1.65 | — | — | N | 1.28 | 0.67 | — | — | ||||
| O | 0.82 | — | — | — | O | 1.46 | — | — | — | ||||
| Cl | 9.40 | Cl | 1.47 | ||||||||||
| Atoms | H | C | O | N | H | C | O | N | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | RXX/RXY | EXX/EXY | 2 | ||||||||
| H | 44.55 | 26.23 | 14.35 | 2.87 | H | 0.99 | 0.73 | 1.50 | 1.50 | ||
| C | 26.23 | 3.86 | — | — | C | 0.73 | 2.59 | — | — | ||
| N | 14.35 | — | — | — | N | 1.50 | — | — | — | ||
| O | 2.87 | — | — | — | O | 1.50 | — | — | — |
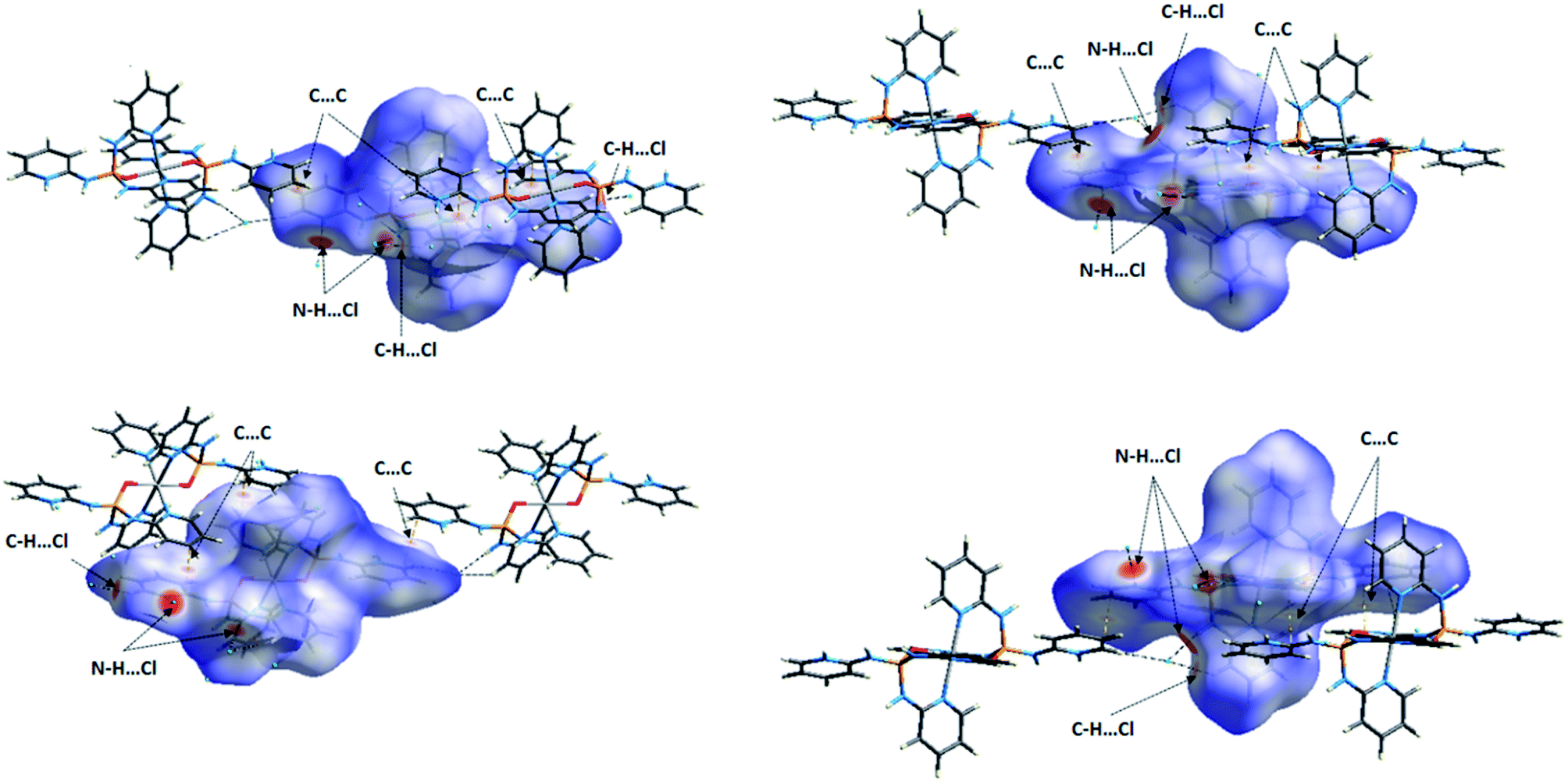 | ||
| Fig. 13 Views of the dnorm Hirshfeld surfaces plotted on the molecules Co01 (right) and Co02 (left) of 1 in two orientations. The close contacts N–H⋯Cl, C–H⋯Cl and C⋯C between the selected molecule inside the surface and its neighboring molecules are introduced in figure. For these close contacts refer to Table 3. | ||
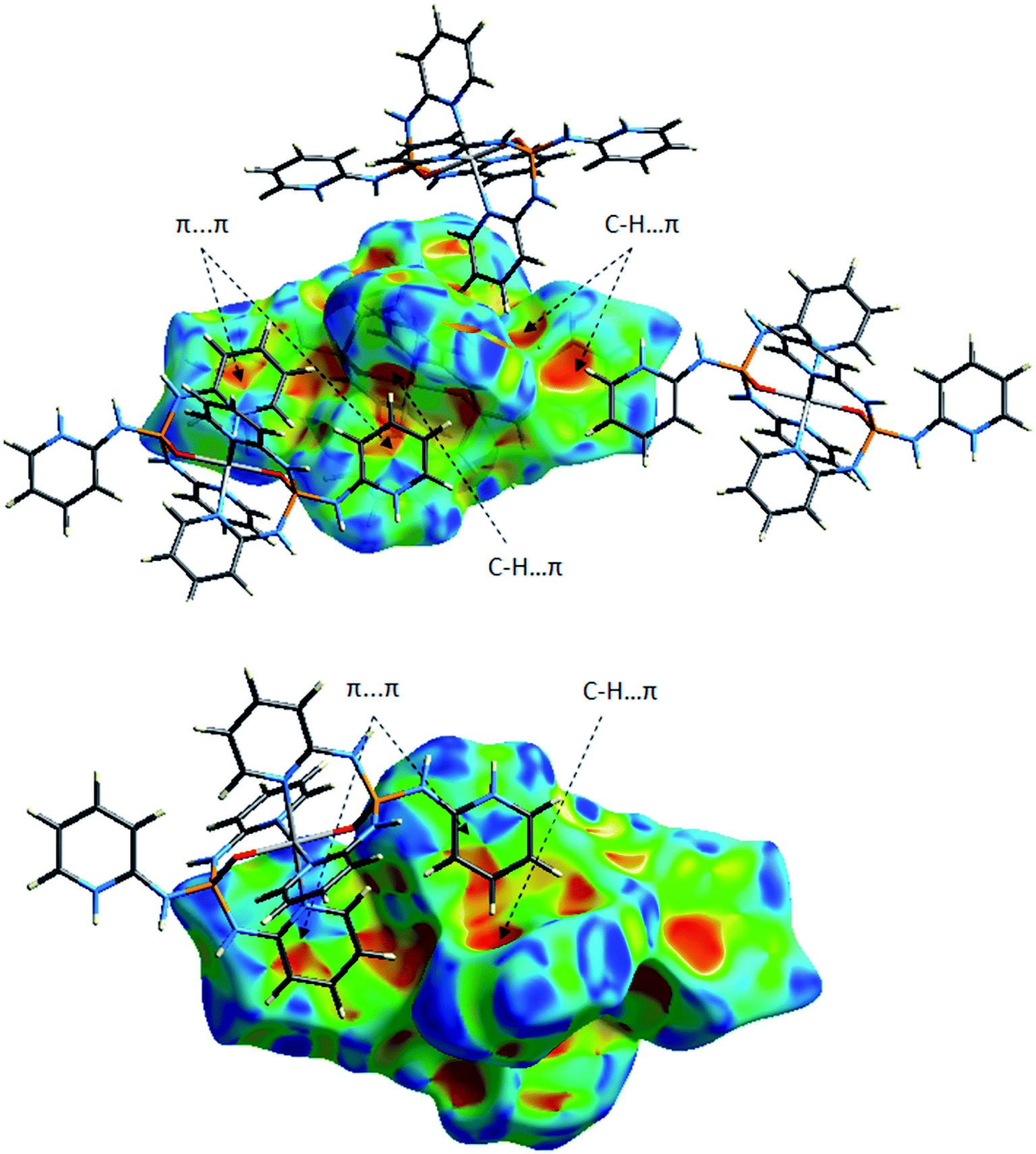 | ||
| Fig. 14 Two views of the Hirshfeld surface mapped with the shape index for the molecule Co01 of 1 illustrating some π-effect interactions. | ||
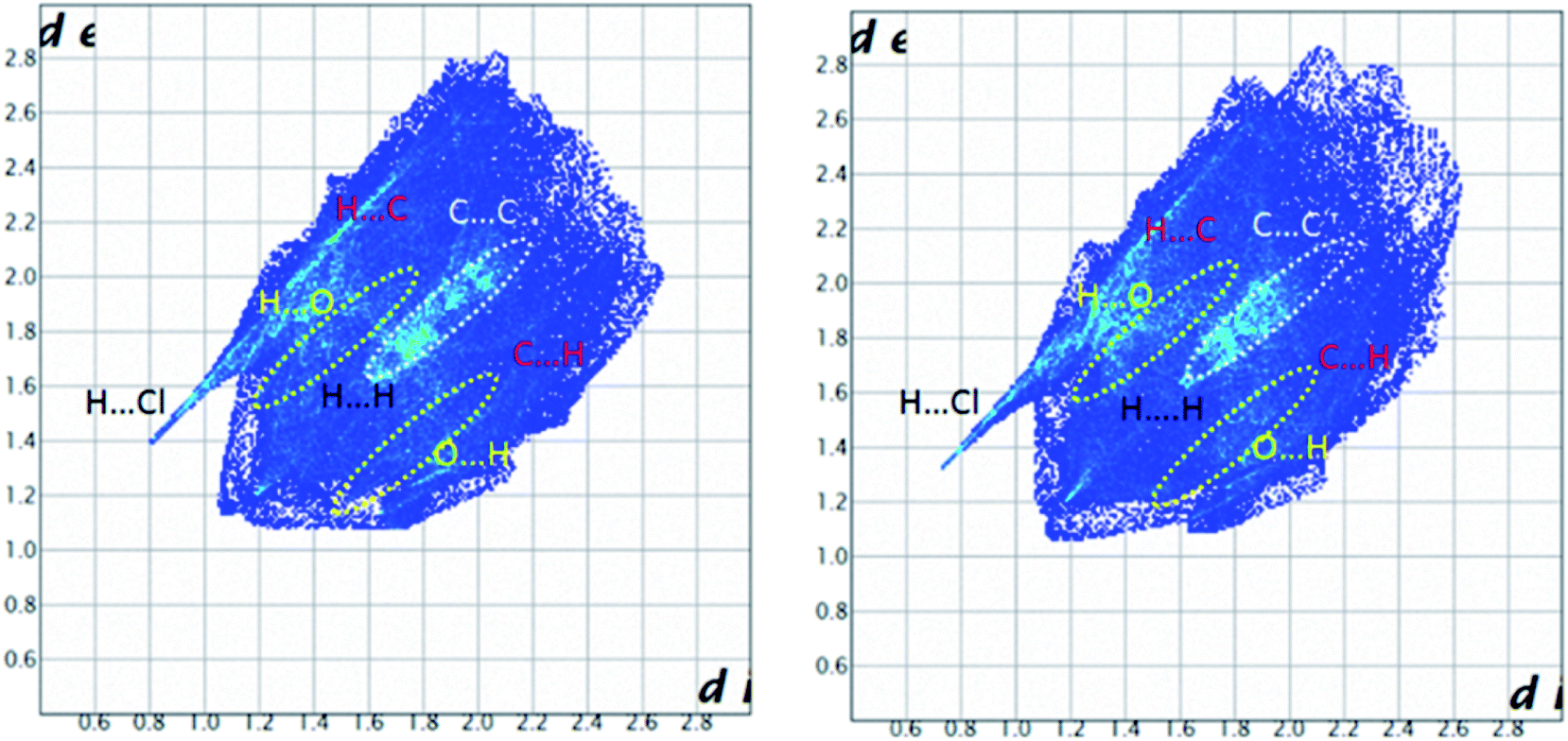 | ||
| Fig. 15 Full 2D fingerprint plots highlighting the main close contacts for the molecules Co01 (right) and Co02 (left) of 1 are displayed. The decomposed FPs of various intermolecular contacts are represented at ESI.† | ||
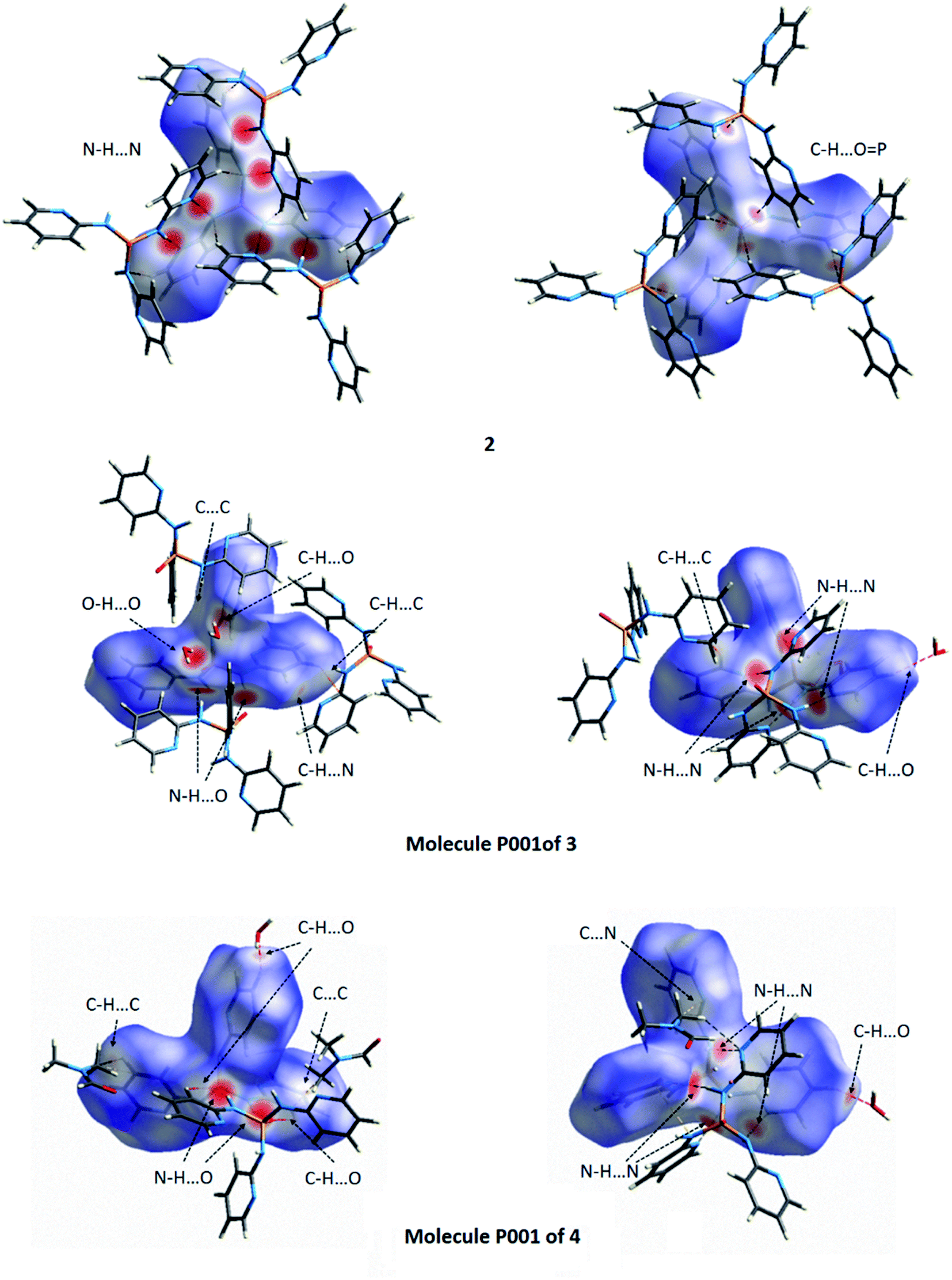 | ||
| Fig. 16 Views of the dnorm Hirshfeld surfaces plotted on 2 and the molecules P001 of 3 and 4 in two orientations. The various close contacts N–H⋯N, N–H⋯O, C–H⋯O, C–H⋯C, C–H⋯N, O–H⋯O, C⋯C and C⋯N between the selected molecule inside the surface and its neighboring molecules are introduced in figure. For these close contacts refer to Tables 4 and 5. | ||
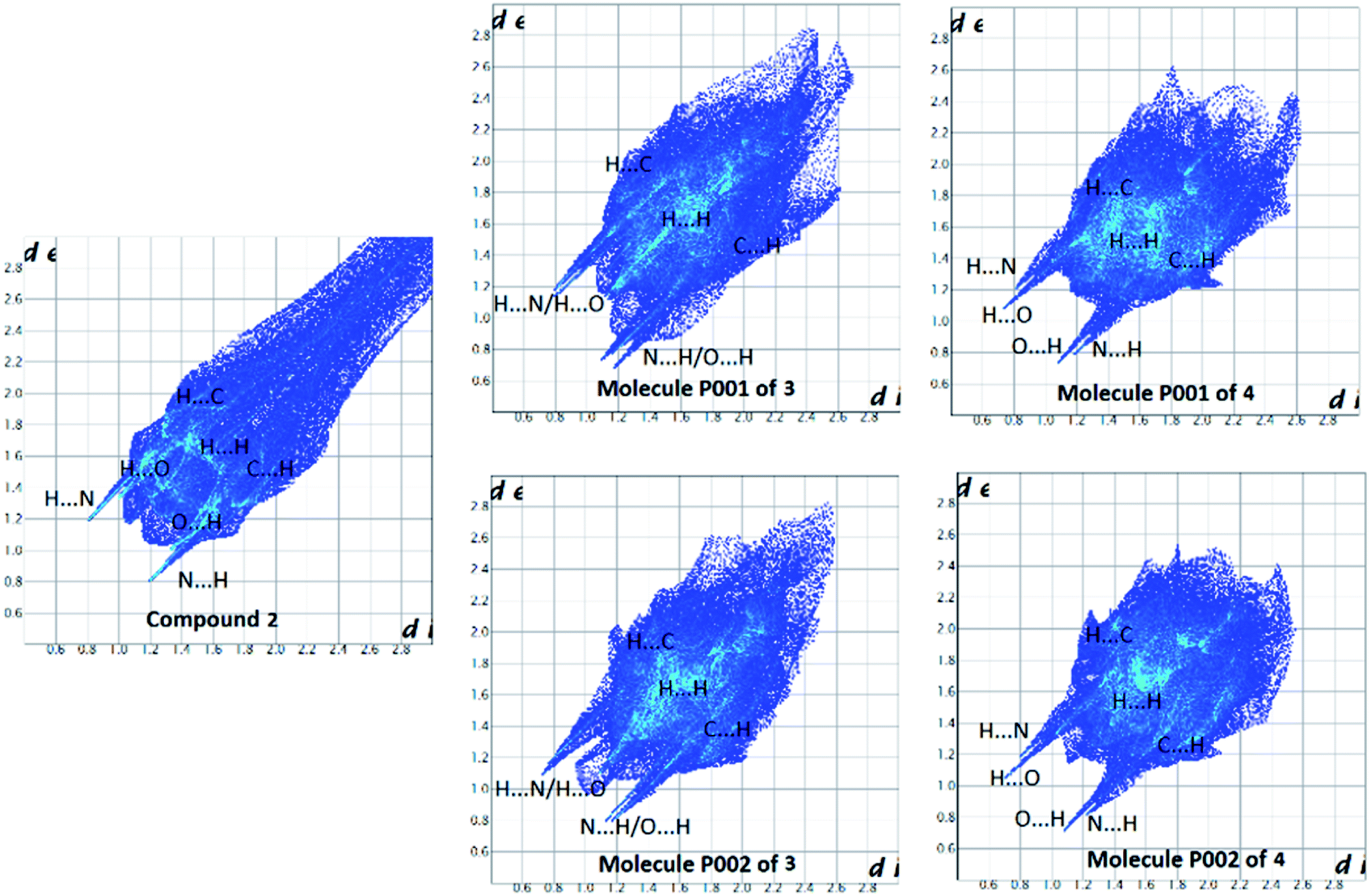 | ||
| Fig. 17 Full 2D fingerprint plots highlighting the main close contacts for compounds 2–4 are represented. For decomposed FPs of various intermolecular contacts refer to ESI.† | ||
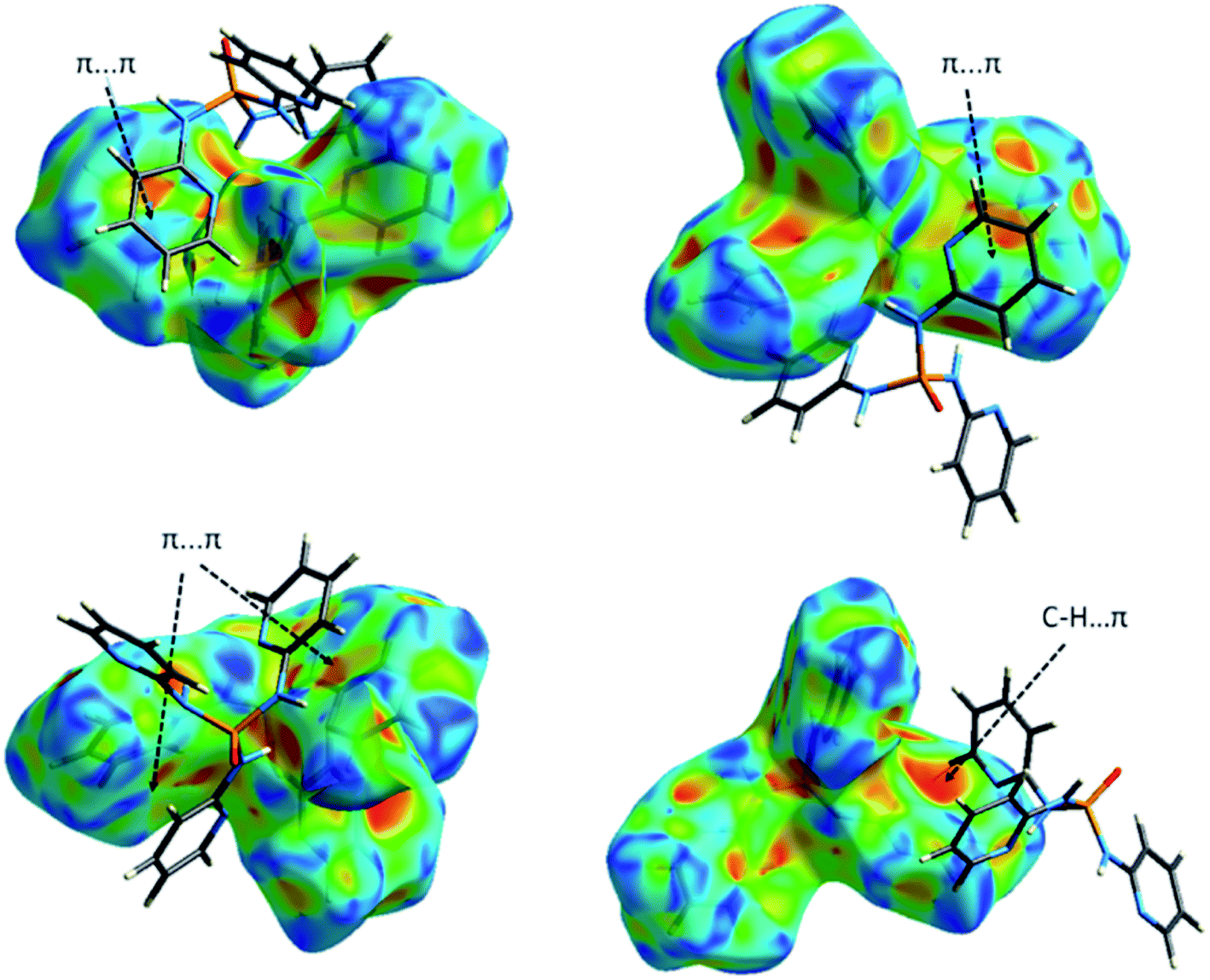 | ||
| Fig. 18 Views of the Hirshfeld surface mapped with the shape index for the molecules P001 of 3 (up) and 4 (down) illustrating some π-effect interactions. | ||
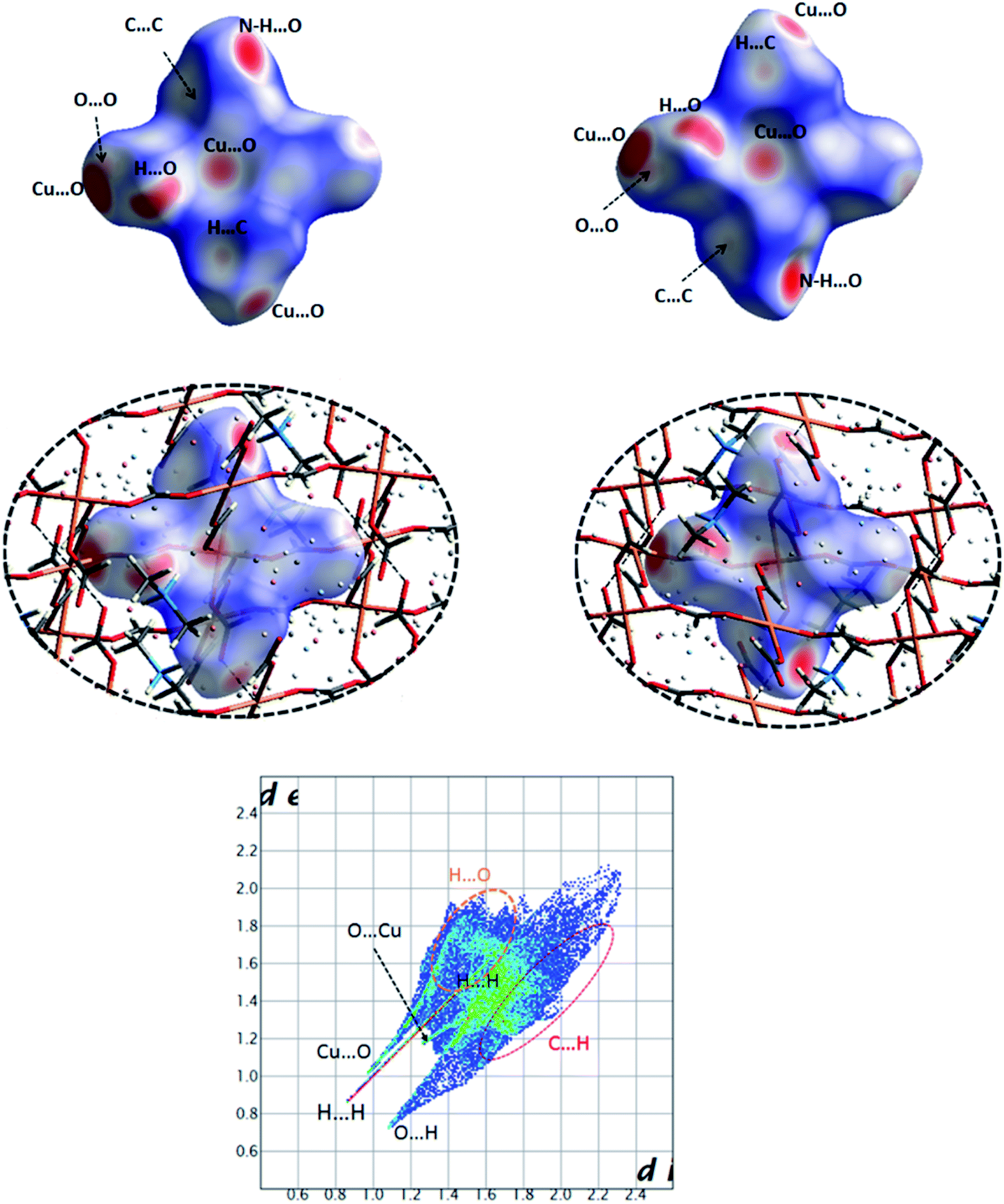 | ||
| Fig. 19 Views of the Hirshfeld surface mapped over dnorm for the compound 5 in two orientations illustrating various contacts Cu⋯O, N–H⋯O, H⋯O (C–H⋯O), C⋯C, O⋯O and H⋯C (top) with surrounding molecules in the crystal structure (middle). Full 2D fingerprint plot highlighting the main close contacts for 5 has been shown in down. | ||
Coordinated PO ligand in Co(II) complex
The obvious red areas on the Hirshfeld surfaces (Fig. 13) of the molecules Co01 and Co02 of 1 depict H⋯Cl contacts are associated to the N–H⋯Cl and C–H⋯Cl hydrogen bonds. However, those related to the C–H⋯Cl interactions are smaller and more diffuse. Other pale red spots on the surfaces correspond to C⋯C contacts characteristic of π-stacking interactions. Such interactions which represent π⋯π stacking arrangement of pyridine rings of PT ligands are simply recognized by pattern of alternated red and blue triangles on the shape index surfaces displayed in Fig. 14. Other π-effect interactions of type C–H⋯π are also observed on the shape index surfaces as red π-holes.From the FPs of the molecules Co01 and Co02 of 1 in Fig. 15, the major finding refers to the H⋯Cl contacts represented as a narrow spike with the minimum values of di + de ≈ 2.2 Å (for Co01) and 2.1 Å (for Co02) which are less than 2× the van der Waals radius of hydrogen and chlorine atoms (≈2.85 Å). This outcome confirms the results of Hirshfeld surfaces above-mentioned to accept the H⋯Cl contacts as the main short contacts in this structure. However, this type of contacts, i.e. H⋯Cl, only comprise about 14% of the total surface areas as it is presented in Table 6. The pale blue/green region on the diagonal at around 1.6 Å < de, di < 2.1 Å depicts the C⋯C contacts comprising about 6% of the surface (Table 6) characteristic of π⋯π stacking interactions. Other important result from FPs is corresponded to the H⋯H and H⋯C/C⋯H contacts which have the major contributions (Table 6) due to the relative high proportion of carbon and hydrogen atoms in this structure. Moreover, in this coordinated compound, the H⋯N/N⋯H contacts associated to the N–H⋯N hydrogen bonds comprise a little portion of the HS (around 7%), where the N–H⋯N hydrogen bonds are of important interactions in the crystal structure of free PT ligand OP[NH-2Py]3 as it is discussed in the following sections.
For this structure, the most favored intermolecular interactions quantified by considering the enrichment ratios (Table 6) are attributed to the C⋯C contacts for the molecule Co01 with E = 1.60 and the C⋯C, H⋯Cl and H⋯O/O⋯H contacts with almost the same values of E (≈1.45) for the molecule Co02. However, for both molecules Co01 and Co02, these interactions do not comprise the significant contributions of the corresponding surface areas. In both molecules, the H⋯H contacts with the high percentage contributions are introduced as disfavored with E < 1, where this result interestingly is converged with the favored ones with the low contributions. For the molecule Co01, other disfavored contacts are of type H⋯C/C⋯H and those of type H⋯Cl, H⋯O/O⋯H, H⋯N/N⋯H and C⋯N/N⋯C are favored. However, for the molecule Co02, the H⋯C/C⋯H contacts show an enrichment ratio around unity and the C⋯N/N⋯C contacts with E = 0.67 are recognized as disfavored.
Comparison of packing modes in anhydrate, monohydrate and DMF/H2O solvate crystals of PT ligand OP[NH-2Py]3
As seen from Fig. 16, deep red areas are observed for the dnorm HS of the anhydrate structure 2 nearby the nitrogen atom of pyridine ring and the hydrogen atom of NH unit, both of the 2-aminopyridine substituent group, result from the N–H⋯N hydrogen bond interactions. Some little brighter red spots on the surface are a manifestation of the C–H⋯OP hydrogen bond interactions. In the dnorm HS of structure 3 (Fig. 16 and S1†), the vivid red regions arise from the N–H⋯N, N–H⋯OP and O–H⋯OP hydrogen bond interactions. For the molecule P001 of 4 (Fig. 16), there are the red and bright red areas on the surface which exhibit the N–H⋯OP and N–H⋯N hydrogen bond interactions, respectively. However, for the molecule P002 of 4 (Fig. S1†), the obvious red regions on the dnorm HS are related to the N–H⋯O (between the NH unit of 2-aminopyridine substituent group as donor and the oxygen atom of the carbonyl group of DMF as acceptor) and O–H⋯O (between the OH unit of water molecule as donor and the oxygen atom of the phosphoryl group as acceptor) hydrogen bond interactions. The N–H⋯N hydrogen bond interactions are also visible on the surface as the lighter red regions. Some small red spots on the surfaces correspond to the C–H⋯O (for 3 and 4), C–H⋯N (for 3) and C–H⋯C (for 4) interactions. Some white areas on the dnorm HS of 3 and the molecule P001 of 4 are related to the C–H⋯C (for 3), C⋯C (for 3 and 4) and C⋯N (for 4) interactions.
Collected information from FPs (Fig. 17, S2–S6† and Tables 6 and 7) testify the above-mentioned results. Thus, in the structure 2, the H⋯N/N⋯H contacts are represented by a pair of sharp spikes with the minimum values of di + de = 2.0 Å and a pair of very small spikes are seen for the H⋯O/O⋯H with di + de ≥ 2.3 Å (Fig. S2†). In contrast, for 3 and 4, both the H⋯N/N⋯H and H⋯O/O⋯H contacts are displayed as sharp spikes with minimum values of di + de ≈ 2.0 Å for the H⋯N/N⋯H and ≈ 1.9 Å or 1.8 Å for the H⋯O/O⋯H. Moreover, the contribution of H⋯N/N⋯H contacts is greater in the case of 2 compared to 3 and 4 and the H⋯O/O⋯H contacts conversely cover much less contribution than H⋯N/N⋯H in 2. This is supplementary evidence that support the higher importance of N–H⋯N hydrogen bond interactions than N–H⋯O in structure 2. Another important finding is related to C⋯N/N⋯C contacts, where the reduction of contribution of H⋯N/N⋯H contacts in the structures 3 and 4 is compensated by the development of C⋯N/N⋯C contacts versus the absence of such contacts in 2. Furthermore, the C⋯C contacts are concentrated for 3 (with 1.6 Å < di, de < 2.8 Å) and 4 (1.7 Å < di, de < 2.4 Å) providing some strong π⋯π interactions, whereas, they are much expanded in 2 (2.2 Å < di, de < 3.0 Å). The shape index surfaces of 3 and 4 shown in Fig. 18 and S7† confirm the presence of such π-effect interactions (C–H⋯π and π⋯π) in these structures. The shape index surface of 2 is in contrast almost empty of any important π-effect interactions. The X-ray diffraction data of 2 support this result, as the packing structure of 2 discussed in the section “X-ray crystallography investigations” do not show important π-effect interactions. Eventually, a visual inspection of FPs is a proper tool to unfold the important role of weak interactions (with high distances de and di) in the crystal packing, where the upper de and di values on the full FPs are more expanded in the case of 2 compared to those in 3 and 4. Indeed, it is seems that the Full FP of 2 shows the pseudo-infinite values for the upper values of de and di.
| Contacts | P001 of 3 | P002 of 3 | P001 of 4 | P002 of 4 | SX | H | C | N | O |
|---|---|---|---|---|---|---|---|---|---|
| H⋯C/C⋯H | 18.0 | 18.1 | 18.1 | 21.8 | P001 of 3 | 69.10 | 17.05 | 9.70 | 3.95 |
| H⋯N/N⋯H | 16.5 | 15.5 | 12.3 | 13.8 | P002 of 3 | 68.65 | 17.25 | 9.05 | 4.55 |
| H⋯O/O⋯H | 7.9 | 9.1 | 11.0 | 10.7 | P001 of 4 | 72.50 | 14.05 | 7.35 | 5.50 |
| C⋯C | 6.6 | 6.9 | 3.8 | 3.7 | P002 of 4 | 70.35 | 15.75 | 8.05 | 5.35 |
| C⋯N | 2.9 | 2.6 | 2.4 | 2.3 |
| Atoms | H | C | N | O | H | C | N | O | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P001 of 3 | RXX/RXY | EXX/EXY | P001 of 3 | ||||||||
| H | 47.75 | 23.56 | 13.40 | 5.46 | H | 1.00 | 0.76 | 1.23 | 1.47 | ||
| C | 23.56 | 2.91 | 3.31 | — | C | 0.76 | 2.27 | 0.88 | — | ||
| N | 13.40 | 3.31 | — | — | N | 1.23 | 0.88 | — | — | ||
| O | 5.46 | — | — | — | O | 1.47 | — | — | — | ||
| P002 of 3 | RXX/RXY | EXX/EXY | P002 of 3 | ||||||||
|
|||||||||||
| H | 47.13 | 23.68 | 12.43 | 6.25 | H | 1.00 | 0.76 | 1.25 | 1.46 | ||
| C | 23.68 | 2.97 | 3.12 | — | C | 0.76 | 2.32 | 0.83 | — | ||
| N | 12.43 | 3.12 | — | — | N | 1.25 | 0.83 | — | — | ||
| O | 6.25 | — | — | — | O | 1.46 | — | — | — | ||
| P001 of 4 | RXX/RXY | EXX/EXY | P001 of 4 | ||||||||
|
|||||||||||
| H | 52.56 | 20.37 | 10.66 | 7.97 | H | 0.99 | 0.89 | 1.15 | 1.38 | ||
| C | 20.37 | 1.97 | 2.07 | — | C | 0.89 | 1.93 | 1.16 | — | ||
| N | 10.66 | 2.07 | — | — | N | 1.15 | 1.16 | — | — | ||
| O | 7.97 | — | — | — | O | 1.38 | — | — | — | ||
| P002 of 4 | RXX/RXY | EXX/EXY | P002 of 4 | ||||||||
|
|||||||||||
| H | 49.49 | 22.16 | 11.33 | 7.53 | H | 0.95 | 0.98 | 1.22 | 1.42 | ||
| C | 22.16 | 2.48 | 2.53 | — | C | 0.98 | 1.49 | 0.91 | — | ||
| N | 11.33 | 2.53 | — | — | N | 1.22 | 0.91 | — | — | ||
| O | 7.53 | — | — | — | O | 1.42 | — | — | — | ||
The enrichment ratio listed in Tables 6 and 7 mainly disclose the similarities of the structures 2–4. For all three structures, the H⋯N/N⋯H and H⋯O/O⋯H contacts with E > 1 (associated to the favored hydrogen bonds N–H⋯N and N–H⋯O in such compounds) and those of type H⋯H and H⋯C/C⋯H with E < 1 are enriched (accepted as favored) and non-enriched (disfavored), respectively. However, for both molecules P001 and P002 of 3, the enrichment ratio for H⋯H is exactly equal to unity. The C⋯C contacts display the largest enrichment ratios among the enriched contacts for all three structures which is an expected result in such aromatic compounds due to π⋯π stacking interactions in their crystal packing. However, for 2, these C⋯C contacts cover only weak interactions as they are expanded to high values of di, de in the C⋯C FP (Fig. S2,† 2.2 Å < di, de < 3.0 Å) as was discussed before.
An interesting outcome from HS analysis is the relatively significant difference between the two molecules P001 and P002 of 4. The X-ray diffraction analysis reveals an unusual H-bonded pattern, i.e. a ten-membered motif constructed from five symmetrically independent molecules for this structure which is uncommon among its similar analogous phosphoric triamide structures.37 Indeed, the different environments of two phosphoric triamide molecules P001 and P002 in the crystal structure of 4 are perfectly illustrated by 3D HSs and 2D FPs. The main red regions are related to the N–H⋯OP and N–H⋯N hydrogen bond interactions for P001, whereas these regions correspond to the N–H⋯OC (carbonyl group of DMF), O–H⋯OP (OH group of water) and N–H⋯N hydrogen bonds for P002. In the FPs, the contributions of the H⋯H and H⋯C/C⋯H contacts have the most different behavior, with a difference of 4.6% for the former contact and 3.7% for the latter, in the two quoted molecules. Moreover, the enriched contacts with E > 1 are of the H⋯N/N⋯H, H⋯O/O⋯H and C⋯C type for both molecules P001 and P002, but the C⋯N/N⋯C contact has a different behavior, in the sense that this contact is enriched for P001 and non-enriched for P002.
Based on the results from the HS analysis, it can be concluded that the anhydrate crystal of 2 features only the pyridine nitrogen atom as a potential H-bond acceptor, whereas for the monohydrate and DMF/H2O solvate crystals of 3 and 4, respectively, the phosphoryl oxygen atom competes to be a good H-bond acceptor beside the pyridine nitrogen atom for 3 or to be a better H-bond acceptor than the pyridine nitrogen atom for 4. Nevertheless, based on our previous studies,38 the N–H⋯O interactions contribute the most to the packing stability in phosphoric triamides but the O atom of the PO group is the most significant H-atom acceptor. On the other hand the solvent (water or non-water) plays a crucial role in the crystal packing modes of structures and the absence or presence can change the space group symmetry from the trigonal space group R (for 2) to monoclinic P21/n (for 3 and 4). It is also worthwhile to note that the R structure for 2 with an overall C3 symmetry along the axis of the PO bond and the arrangement of molecules as the hexagonal units with the ring cavity in the middle can influence the absence of the phosphoryl group in the classical interactions. As it can be reasoned, the quoted results are in a same trend with the X-ray diffraction data and confirm those.
Finally, a comparison of HS results for the coordinated PT ligand OP[NH-2Py]3 in the structure 1 and non-coordinated ones in the structures 2–4 (free ligands) illustrates that the most important differences are related to the increment of H⋯C/C⋯H contacts and the decrement of H⋯N/N⋯H and H⋯O/O⋯H in the coordinated ligand compared to free ligands. Although, in 1 the decrement of H⋯N/N⋯H and H⋯O/O⋯H contacts is recovered by H⋯Cl contacts. There are not considerable differences for the enrichment ratio (Tables 6 and 7).
Polymeric Cu(II) complex of O-donor formate ligand
As observed in Fig. 19, the deep red circular regions on the HS of 5 are related to the Cu⋯O close contact with the smallest bond length (Table S2†) and the N–H⋯O hydrogen bond interaction. The Cu⋯O contacts with the larger bond lengths are visible as the pale red regions on the surface, where such Cu⋯O contacts are responsible for the polymeric structure of 5. The mentioned N–H⋯O hydrogen bond corresponds to an interaction between the oxygen atom of the μ2-formate linker within the surface and the N–H unit of dimethylammonium cation outside of this surface. The dimethylammonium cations captured in the cubic cavities of anionic network of 5 are connected to the polymeric structure by this hydrogen bond interaction. Other visible pale red regions on the surface are indicative of H⋯O interactions related to C–H⋯O hydrogen bonds between the O atoms and CH groups of μ2-formate linkers (Table 5). The small diminutive red spots near the carbon atom of formate linkers represent its participation as an acceptor in a weak C–H⋯C hydrogen bond (Table 5) with the CH group of dimethylammonium cation outside of the surface as a donor. Additional very small faint red spots on the white regions of the surface originate from weak O⋯O interactions, between the oxygen atoms of μ2-formate linkers, and C⋯C interactions between the carbon atoms of μ2-formate linker within the surface and of dimethylammonium cation outside of the surface.A visual view of fingerprint plot (FP) of 5 in Fig. 19 reveals that the main contacts appeared as three sharp spikes are related to Cu⋯O, H⋯H and O⋯H representing short contacts with the minimum values of ≈2.0 (for Cu⋯O), 1.7 (H⋯H), 1.8 Å (O⋯H) for di + de. As one can be seen, these values are less than 2 × the van der Waals radius of related atoms (≈1.9 Å for Cu⋯O, 2.2 Å for H⋯H and 2.6 Å for O⋯H). The complementary pairs of these contacts, i.e. O⋯Cu and H⋯O contacts are presented as a very short spike (marked in Fig. 19) for the former and a more extended area for the latter (highlighted in Fig. 19) in the top-left part of the plot. Among these contacts, the H⋯O/O⋯H contacts associated to C–H⋯O hydrogen bonds have the major contribution (53.1%), where the main portion is found for the O⋯H contacts (50.8%). The occurrence frequency of the H⋯H contacts increases in the region of 0.8 Å < de, di < 1.5 Å visualized by a color change from blue/green to red, while for other contacts a medium occurrence frequency revealed by green color is found. Moreover, the C⋯H contacts are points in the right regions of the plot relative to its diagonal (de < di) associated to C–H⋯C contacts.
Finally, a survey of the resulted enrichment ratios in Table S3† shows that the most enriched contacts with E > 2 are of the Cu⋯O type, as is expected in a polymeric structure with a main CuO6 segment. However, these contacts only cover about 11% of the total surface. Other enriched contacts with E > 1 are indicative of H⋯C and H⋯O recognized as favoured contacts. The H⋯H and O⋯O contacts are disfavored (E < 1), while the surface contacts for H and O atoms (SH and SO) are significant (Table S3†).
For a comparison, 3D HSs, 2D FPs (Fig. S8†) and E ratios (Table S3†) are also calculated for the reported structure MACUFR. The most important differences between 5 and MACUFR are found in the H⋯O/O⋯H contacts, where these contacts are displayed as the red spots on the HS of 5, while there are only very small pale red spots for these contacts on the HS of MACUFR. Another considerable difference is related to the percentage of contributions of H⋯H contacts which are decreasing from 23.7% in 5 to 19.3% in MACUFR. Moreover, the sharp long spike related to the shortest H⋯H contacts (with di + de ≈ 1.7 Å) observed in the FP of 5 is converted to a very short spike on the FP of MACUFR with the minimum values of di + de ≈ 2.4 Å. This value is larger than twice the van der Waals radius of hydrogen atoms. These results can be attributed to the different H-atom treatment in the refinement of these structures which affect the contacts involving hydrogen atoms. For the E ratios of 5 and MACUFR (Table S3†), the considerable difference is not found.
Conclusion
The crystal structures of four different solid state forms of OP[NH-2Py]3 as coordinated, pure, monohydrate and solvate crystals (1–4) along with an improved model of a MOF of Cu(II)O6 (5) are reported. A structural study using X-ray diffraction and Hirshfeld surface analyses features interesting findings which are presumably due to the presence of pyridine rings in the studied PT compounds. These are as follows:(1) The OP[NH-2Py]3 provides three metal binding sites of type N,N,O in its coordinated form in the structure 1, and such a design of an [N]3P(O)-based ligand in discrete PT complexes has not been observed up to now.
(2) The monohydrate and solvate forms of OP[NH-2Py]3 (3 and 4) crystallize in the common monoclinic space group P21/n, whereas the pure form (2) shows a less common trigonal space group R.
(3) In the crystal structure of 2, the molecular arrangement as the hexagonal 2D sheet with the PO units rendered in above and below this sheet leads to remain the phosphoryl group as non-interacting in any typical hydrogen bond interactions, where the absence of the PT characteristic hydrogen bond N–H⋯O is evident in this structure, but instead N–H⋯N hydrogen bonds are observed.
(4) In all three structures of 2–4, the N–H⋯N hydrogen bond interaction is the characteristic feature of the H-bonding network, where the red areas on the HSs of these structures are observed nearby the pyridyl nitrogen atom as a potential H-bond acceptor. However, the PT typical hydrogen bonds of the N–H⋯OP type are found in the structures of 3 and 4 confirmed by the assignment of the vivid red areas on the HSs of 3 and 4 to the phosphoryl oxygen atom beside the pyridyl nitrogen as H-bond acceptors.
(5) The presence of aromatic pyridine rings in the structure of 2, does not lead to considerable π-effect interactions in the crystal packing. However, the pseudo-infinite values for the upper values of de and di on the C⋯C FP of 2 confirm the presence of very weak π⋯π interactions in this crystal structure.
(6) An improved model of Cu(II) MOF constructed using the formate ligand (5) is structurally investigated and compared to the previously reported structure MACUFR by HS analysis. The considerable differences between two structures corresponds to the contacts involving hydrogen atoms (H⋯O/O⋯H and H⋯H) which are likely attributed to the different H-atom treatment in the refinement of these structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of this investigation by Semnan University is gratefully acknowledged.References
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS.
- R. Pepinsky, Phys. Rev., 1955, 100, 971 CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797 CrossRef CAS.
- D. E. Braun, T. Gelbrich, V. Kahlenberg and U. J. Griesser, CrystEngComm, 2015, 17, 2504 RSC.
- C. E. S. Bernardes and M. E. M. da Piedade, Cryst. Growth Des., 2012, 12, 2932 CrossRef CAS.
- L. Vella-Zarb, D. Braga, A. Guy Orpend and U. Baisch, CrystEngComm, 2014, 16, 8147 RSC.
- A. T. Hulme, A. Johnston, A. J. Florence, P. Fernandes, K. Shankland, C. T. Bedford, G. W. A. Welch, G. Sadiq, D. A. Haynes, W. D. S. Motherwell, D. A. Tocher and S. L. Price, J. Am. Chem. Soc., 2007, 129, 3649 CrossRef CAS.
- N. Li, F. Jiang, L. Chen, X. Li, Q. Chen and M. Hong, Chem. Commun., 2011, 47, 2327 RSC.
- A. K. Gupta, A. K. Srivastava, I. K. Mahawar and R. Boomishankar, Cryst. Growth Des., 2014, 14, 1701 CrossRef CAS.
- A. K. Gupta, F. A. S. Chipem and R. Boomishankar, Dalton Trans., 2012, 41, 1848 RSC.
- S. E. Denmark, S. B. D. Winter, X. Su and K.-T. Wong, J. Am. Chem. Soc., 1996, 118, 7404 CrossRef CAS.
- C. Dong, L. Sun, X. Ma, Z. Lu, P. He and P. Zhu, Polymers, 2019, 11, 1829 CrossRef CAS.
- M. J. Domínguez, C. Sanmartín, M. Font, J. A. Palop, S. San Francisco, O. Urrutia, F. Houdusse and J. M. García-Mina, J. Agric. Food Chem., 2008, 56, 3721 CrossRef.
- Z. Shariatinia and R. S. Tuba, J. Sol. Energy Eng., 2015, 137, 011006 CrossRef.
- A. K. Gupta, S. S. Nagarkar and R. Boomishankar, Dalton Trans., 2013, 42, 10964 RSC.
- A. K. Gupta, A. Steiner and R. Boomishankar, Dalton Trans., 2012, 41, 9753 RSC.
- Y.-Q. Gong, J.-W. Gao and T.-Q. Mi, Chin. J. Struct. Chem., 2016, 35, 747 CAS.
- M. Yu, L. Chen, F. Jiang, K. Zhou, C. Liu, C. Sun, X. Li, Y. Yang and M. Hong, Chem. Mater., 2017, 29, 8093 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS.
- K. Gholivand, M. D. Alavi and M. Pourayoubi, Z. Kristallogr.–New Cryst. Struct., 2004, 219, 124 CAS.
- A. Kumar Srivastava, P. Divya, B. Praveenkumar and R. Boomishankar, Chem. Mater., 2015, 27, 5222 CrossRef.
- E. Sletten and L. H. Jensen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1752 CrossRef CAS.
- W. Kabsch, XDS. Acta Crystallogr., Sect. D: Struct. Biol., 2010, 66, 125 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer 17.5, University of Western Australia, Australia, 2017 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2004, 60, 627 CrossRef.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119 CrossRef CAS.
- M. Sebghati, A. Tarahhomi and A. Kozakiewicz, ChemistrySelec, 2020, 5, 185 CrossRef CAS.
- A. Tarahhomi and A. van der Lee, Monatsh. Chem., 2018, 149, 1759 CrossRef CAS.
- A. Tarahhomi, A. van der Lee and B. Ośmiałowski, Polyhedron, 2019, 158, 215 CrossRef CAS.
- Y. Wang, R. Cao, W. Bi, X. Li, D. Yuan and D. Sun, Microporous Mesoporous Mater., 2006, 91, 215 CrossRef CAS.
- M. Pourayoubi, M. Toghraee, J. Zhu, M. Dušek, P. J. Bereciartua and V. Eigner, CrystEngComm, 2014, 16, 10870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1956236–1956239 and 1960317 for the five reported structures (1–5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10539b |
| This journal is © The Royal Society of Chemistry 2021 |