
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidation of boron-doped nickel–iron layered double hydroxide for facile charge transfer in oxygen evolution electrocatalysts†
In-Kyoung Ahna,
So-Yeon Leea,
Hyoung Gyun Kima,
Gi-Baek Leea,
Ji-Hoon Leeb,
Miyoung Kima and
Young-Chang Joo *acd
*acd
aDepartment of Materials Science & Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: ycjoo@snu.ac.kr
bMaterials Center for Energy Convergence, Surface Technology Division, Korea Institute of Materials Science (KIMS), Changwon, Gyeongnam 51508, Republic of Korea
cResearch Institute of Advanced Materials (RIAM), Seoul National University, Seoul 08826, Republic of Korea
dAdvanced Institute of Convergence Technology, 145 Gwanggyo-ro, Yeongtong-gu, Suwon 16229, Republic of Korea
First published on 19th February 2021
Abstract
The oxygen evolution reaction (OER) is the key reaction in water splitting systems, but compared with the hydrogen evolution reaction (HER), the OER exhibits slow reaction kinetics. In this work, boron doping into nickel–iron layered double hydroxide (NiFe LDH) was evaluated for the enhancement of OER electrocatalytic activity. To fabricate boron-doped NiFe LDH (B:NiFe LDH), gaseous boronization, a gas–solid reaction between boron gas and NiFe LDH, was conducted at a relatively low temperature. Subsequently, catalyst activation was performed through electrochemical oxidation for maximization of boron doping and improved OER performance. As a result, it was possible to obtain a remarkably reduced overpotential of 229 mV at 10 mA cm−2 compared to that of pristine NiFe LDH (315 mV) due to the effect of facile charge-transfer resistance by boron doping and improved active sites by electrochemical oxidation.
Introduction
Recently, the demand for innovative energy production methods, such as ecofriendly and clean energy sources, has rapidly increased due to the depletion of fossil fuels and the increase of environmental pollution caused by the use of fossil fuels.1–7 Among many candidates, hydrogen production through electrochemical water splitting, which is called green hydrogen production, or production through electrochemical CO2 reduction are not only promising solutions to solve this problem but also can be conducted in combination with solar cells, increasing production sustainability.2,8–10 The oxygen evolution reaction (OER) is essential to energy production reactions based on electrochemistry.11–13 The OER plays the role of counterpart for each reaction occurring in electrochemical energy production, and its efficiency changes depending on the performance of the electrocatalyst.14–17 In particular, water splitting through the use of a hydrogen evolution reaction (HER) electrode and an OER electrode generally shows a very large difference in performance between the two electrodes.5,18–20 Generally, OER exhibits sluggish kinetics and should be applied with a large potential in water splitting because four electrons are involved in bond formation and breakage, whereas the HER is relatively simple. Thus, improving the electrocatalytic performance of the OER would effectively improve water splitting performance.13,21 Although OER electrocatalysts for use under alkaline conditions commonly contain noble metals, such as Ir and Ru, noble metals present disadvantages such as supply shortages and exhaustion, high cost, and limitations in applications requiring mass production.12,16,22 For this reason, electrocatalysts based on earth-abundant elements, such as transition metal oxides, layered double hydroxides (LDHs), sulfides, single atom catalysts (SACs), two-dimensional materials, etc.,3,10,13,16,23–27,63–66 have been extensively studied to replace precious metals. Among such electrocatalyst candidates, LDHs show outstanding electrocatalytic activity and electrochemical stability under OER conditions. Nevertheless, LDHs also need to enhance the OER catalytic activity to increase the possibility of water splitting commercialization. LDHs should be modified to overcome their relatively poor intrinsic conductivity and should be supplemented with more active sites for improved water oxidation efficiency.To increase the OER performance of LDHs in recent studies, the active sites of LDHs have been improved to affect partial charge transfer through LDHs combined with various transition metals, such as Ni, Fe, Mn, V, and Cr.25,28–32 Another strategy is heterogeneous element doping in LDHs to adjust their electronic structure for improved conductivity or charge-transfer resistance and improve the active sites of electrocatalysts.27,33,34 Furthermore, activation of transition metal compounds under OER conditions or during the conversion of transition metal compounds to oxyhydroxides, so-called precatalysts, has been reported.35 This activation step increases the proportion of oxyhydroxides in LDHs, which has a positive effect on OER performance.36,37
Herein, an efficient OER electrocatalyst with a hierarchically designed structure to facilitate charge transfer was suggested using gas–solid boronization and electrochemical oxidation. First, nickel–iron LDH (NiFe LDH) was synthesized using a simple hydrothermal method. Second, boron-doped NiFe LDH (B:NiFe LDH) was fabricated through a boronization step, which was conducted through annealing of the NiFe LDH and boron source. Boronization is a simple method that could be easily applied in mass production processes. Boronization was performed simply through a boron source with a relatively low boiling point. Next, synthesized B:NiFe LDH was electrochemically oxidized for activation. It is well known that transition metal-based electrocatalysts are irreversibly converted to oxyhydroxides through electrochemical oxidation under anodic conditions.35,38 In this work, B:NiFe LDH exhibited low charge-transfer resistance, which was reflected as a better catalytic performance than that of pristine NiFe LDH. Furthermore, electrochemical oxidation of B:NiFe LDH confirmed that oxyhydroxides were formed on the surface, which showed better OER electrocatalytic performance than that of directly synthesized NiFe LDH due to the facile charge transfer structure and improved active sites of B:NiFe LDH. The designed electrocatalyst achieved an overpotential of 229 mV at 10 mA cm−2, which was approximately 140 mV lower than that of Ir under the same conditions.
Experimental
Preparation of galox-B:NiFe LDH electrocatalyst
NiFe LDH was used as a starting material before boronization and was synthesized through a hydrothermal method. First, each precursor was dissolved in DI water (50 mL) and stirred for 30 min. The molarity of nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Sigma-Aldrich), iron sulfate heptahydrate (FeSO4·7H2O, Sigma-Aldrich), ammonium fluoride (NH4F, Sigma-Aldrich), and urea (NH2CONH2, Sigma-Aldrich) was 50 mM, 9 mM, 250 mM, and 625 mM, respectively. The prepared solution with Ni foam (1 cm × 5 cm) was sealed in a Teflon-lined stainless steel autoclave. Then, the autoclave was heated at 120 °C for 18 hours in a dry oven. Second, B:NiFe LDH was synthesized through thermal treatment. Three pieces of NiFe LDH and 3 g of boric acid (H3BO3, Sigma-Aldrich) were loaded into a tube furnace. Then, the furnace was thermally treated under 30 sccm argon gas flow at 350 °C for 3 hours at a ramp rate of 5 °C min−1. Third, electrochemical oxidation of B:NiFe LDH was conducted through a conventional three-electrode cell using a potentiostat (ZIVE MP2A, Wonatech, Korea) in a 1.0 M KOH electrolyte (Sigma-Aldrich). The working, counter, and reference electrodes were the B:NiFe LDH electrode, Pt foil electrode, and standard calomel electrode (SCE), respectively. Electrochemical oxidation was conducted using the galvanostatic method at 0.1 mA cm−2 for 12 hours and started at the stabilized open circuit voltage of the working electrode.Material characterization
To analyze the phase of the synthesized electrocatalyst samples, XRD (New D8 Advances, Bruker) was performed. Microstructural analysis of the synthesized electrodes was conducted using TEM (JEM-2100F, JEOL Ltd.) and FE-SEM (SUPRA 55VP, Carl Zeiss). Additionally, to observe the surface area of the electrodes, a BET analyzer with N2 adsorption/desorption at 77.3 K (ASAP2420, Micromeritics Instruments) was used. Chemical spectral characterization was conducted via Raman spectroscopy (LabRAM HR Evolution, HORIBA) and FT-IR (Nicolet 6700, Thermo Scientific). To verify the ratio of boron in B:NiFe LDH, ICP-AES was conducted through Ar plasma (OPTIMA 8300, Perkin-Elmer, USA).Electrochemical characterization
The polarization curves of each electrode were obtained under the same configuration as the electrochemical oxidation step. The potential range of the polarization curves was 1.1 to 1.8 V (vs. RHE) with a scan rate of 1 mV s−1. ECSA acquisition was conducted through cyclic voltammetry (CV). The CV potential range was 0.18 to 0.28 V (vs. RHE) at scan rates of 5, 10, 25, 50, 100, 200, 400, 600, 800, and 1000 mV s−1. The linear slope and ECSA were calculated from CV data by plotting Ja–Jc at 0.23 V (vs. RHE) depending on the scan rate. EIS was performed within a frequency range from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz to 0.01 Hz at a constant voltage of 350 mV (vs. RHE). The evolved gas was detected through gas chromatography (Micro GC Fusion gas analyzer, INFICON) during the stability test at a constant current density of 10 mA cm−2.
000 Hz to 0.01 Hz at a constant voltage of 350 mV (vs. RHE). The evolved gas was detected through gas chromatography (Micro GC Fusion gas analyzer, INFICON) during the stability test at a constant current density of 10 mA cm−2.
Results and discussion
Boronization and electrochemical oxidation of electrocatalyst
Fig. 1 shows the overall fabrication scheme and microstructural characterization of the synthesized electrocatalyst. Fig. 1(a) displays a schematic illustration of the synthesis of galvanostatic oxidized B:NiFe LDH (galox-B:NiFe LDH) to achieve high OER performance. The synthesis process could be divided into two steps: boronization and electrochemical oxidation. In boronization, NiFe LDH on hydrothermally synthesized nickel foam (Ni:Fe, 4:1 atomic ratio) was thermally annealed with boric acid as the boron source in a furnace at 350 °C, which is higher than the boiling point of boric acid, for 3 hours. Next, galox-B:NiFe LDH was synthesized by the galvanostatic method at a constant current density of 0.1 mA cm−2 for 12 hours using a general electrochemical analyzer. The potential profile of galvanostatic oxidation is depicted in Fig. S1.†
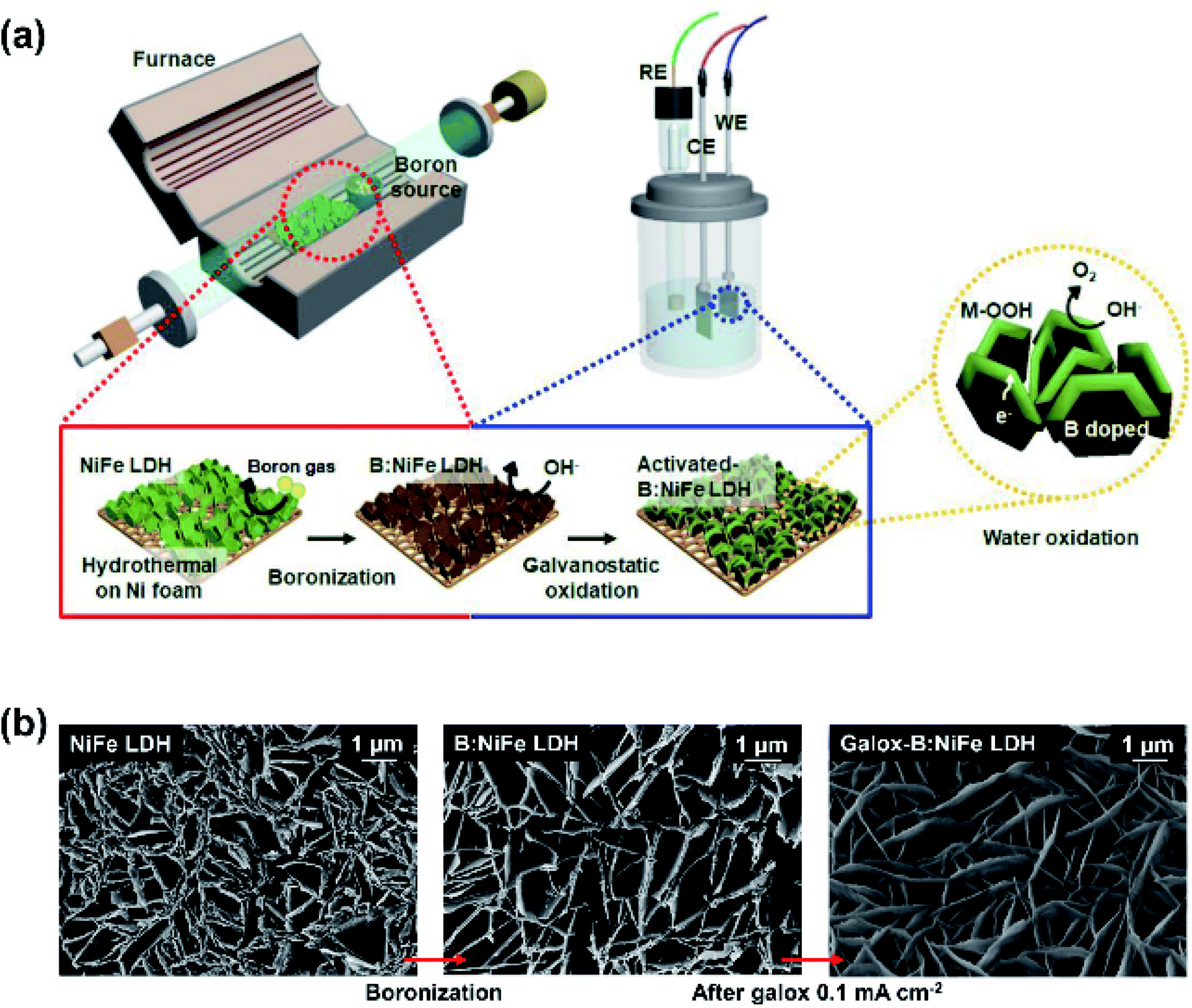 | ||
| Fig. 1 Summary of the fabrication scheme and structural and morphological characterization of galvanostatic oxidized nickel–iron layered double hydroxide (NiFe LDH) with boron doping. (a) Schematic illustration of the synthesis of galvanostatic oxidized boron-doped NiFe LDH (galox-B:NiFe LDH) through gas–solid boronization in a furnace and electrochemical oxidation. (b) Field-emission scanning electron microscopy (FE-SEM) images of the NiFe LDH-based electrocatalyst under each condition. | ||
The morphology of the as-prepared NiFe LDH-based electrocatalysts is shown in Fig. 1(b). The FE-SEM images show that nanoplates formed in all samples. Even after boronization and galvanostatic oxidation were conducted, the nanoplate structure did not collapse or undergo delamination. However, it was observed that the thickness of the nanoplates was relatively thin after heat treatment during boronization.
Energy dispersive X-ray spectroscopy (EDS) analysis of the surface verified successful boron doping, as shown in Fig. S2.† The intensity of the boron peak was notably higher in the EDS spectrum of B:NiFe LDH than in that of pristine NiFe LDH (Fig. S2(a)†). Furthermore, EDS mapping confirmed that the chemical composition of B:NiFe LDH was uniformly distributed over the entire surface of the Ni foam (Fig. S2(b–f)†).
Characterizations of galox-B:NiFe LDH
Fig. 2 shows the results of microstructural characterization of the NiFe LDH-based electrocatalysts through transmission electron microscopy (TEM) analysis. To further verify the successful completion of boron doping and electrochemical oxidation of LDH, as shown in Fig. 2(a and b), TEM was performed, and selected area electron diffraction (SAED) patterns and EDS maps were obtained. As observed in the previous SEM results, there was no significant change in morphology after boronization and electrochemical oxidation were performed (TEM image in Fig. 2(a)). SAED was conducted after boronization and electrochemical oxidation to determine the diffraction patterns that corresponded to each d-spacing. First, the SAED pattern of pristine NiFe LDH contained the (101), (110), (202), (212), (033), and (220) planes of NiFe LDH. Similar planes were observed in the SAED patter of B:NiFe LDH. In both cases, hexagonal symmetry was observed. Furthermore, after an OER sweep, B:NiFe LDH and galox-B:NiFe LDH showed similar SAED patterns. As shown in Fig. 2(a), that the lattice of LDH was unaffected by boronization. Additionally, when an OER sweep and electrochemical oxidation of B:NiFe LDH were conducted, it was confirmed that the oxyhydroxide form was obtained. The d-spacing results of high-resolution TEM and the corresponding fast Fourier transformation (FFT) of each NiFe LDH-based electrocatalyst are shown in Fig. S3.† Furthermore, the STEM images (Fig. S4†) and corresponding EDS mapping (Fig. 2(b)) showed that Ni, Fe, B, and O in B:NiFe LDH were uniformly distributed. The inductively couple plasma atomic emission spectrometer (ICP-AES) was conducted to quantitatively verify the ratio of boron (Table S1†).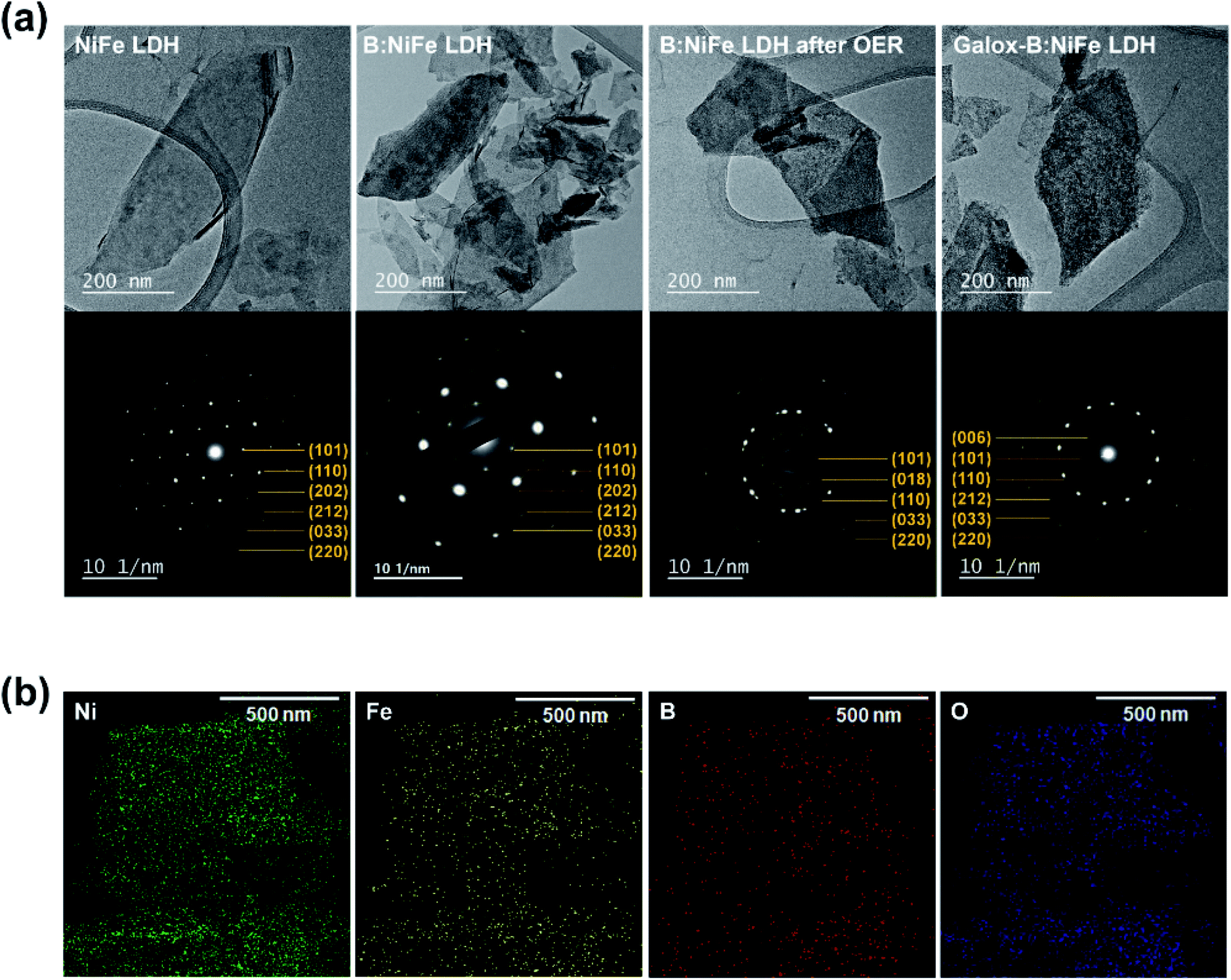 | ||
| Fig. 2 Further morphological characterization of B:NiFe LDH before and after galvanostatic oxidation. (a) Transmission Electron Microscopy (TEM) analysis (top of each row) and selected area electron diffraction (SAED) pattern (bottom of each row) of the NiFe LDH-based electrocatalyst under each condition. (b) EDS mapping of TEM images of the B:NiFe LDH chemical composition. | ||
The unchanged SAED results and the distribution of boron observed through EDS indicated that boron was successfully-doped into NiFe LDH. Therefore, a structure capable of facile charge transfer was obtained by forming activated oxyhydroxide on the B:NiFe LDH surface through boronization and galvanostatic oxidation.
In Fig. 3, the structural and chemical characteristics of galox-B:NiFe LDH were compared with those of the electrocatalyst before boronization and electrochemical oxidation by X-ray diffraction (XRD), Raman spectroscopy, and Fourier transformation infrared (FT-IR) spectroscopy.
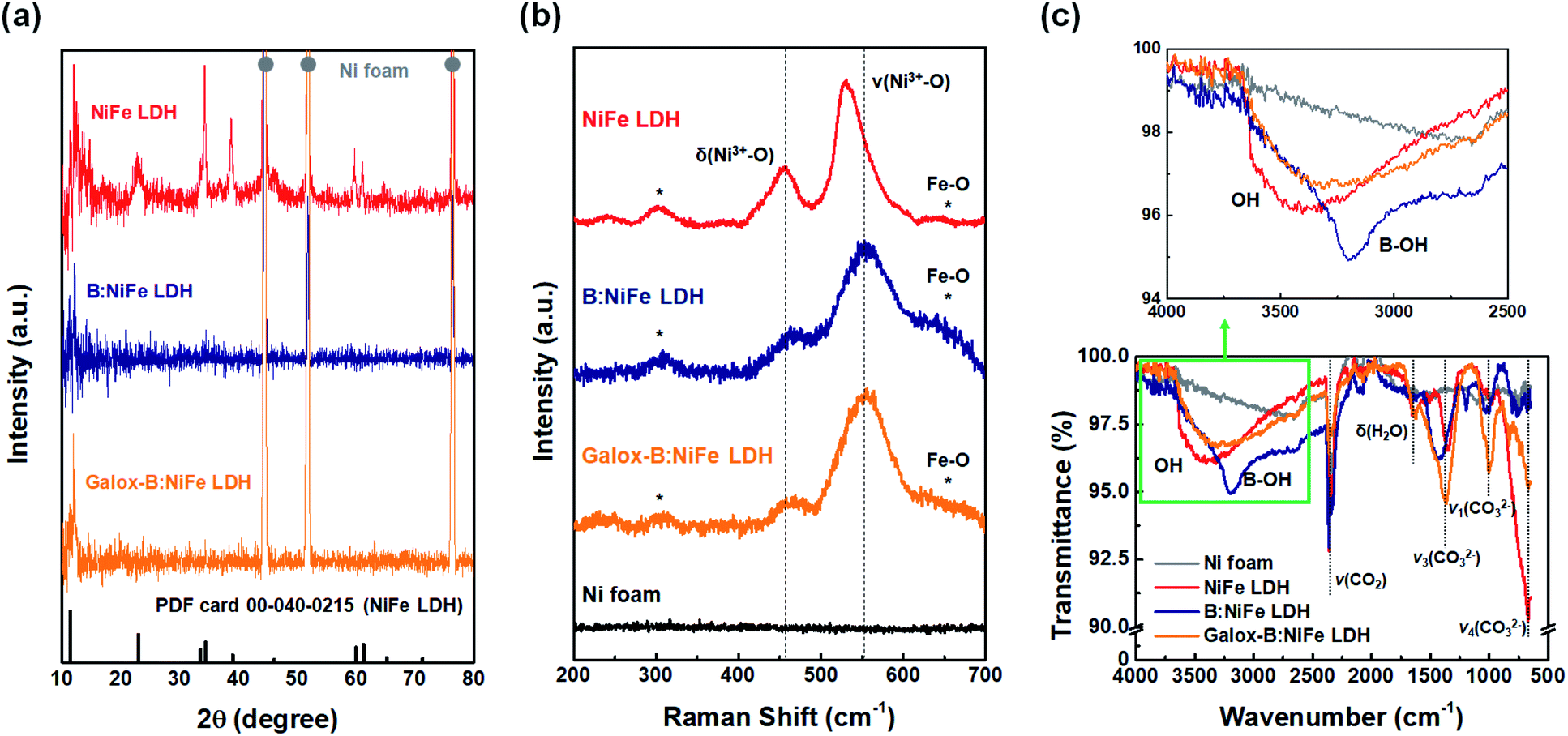 | ||
| Fig. 3 Structural characterization and spectral analysis to confirm successful gaseous boronization and electrochemical oxidation. (a) X-ray diffraction (XRD) analysis for verification of electrochemical oxidation and boronization. (b) Raman spectral analysis of the NiFe LDH-based electrocatalysts. (c) Fourier transformation infrared (FT-IR) spectral analysis to confirm boron doping of NiFe LDH. | ||
Fig. 3(a) depicts the phase analysis of the prepared NiFe LDH-based electrocatalysts at different stages throughout the synthesis process. The red, blue, and orange lines represent the electrocatalyst after hydrothermal NiFe LDH synthesis, boronization, and electrochemical oxidation, respectively. The XRD pattern of NiFe LDH was in agreement with the reference NiFe LDH pattern (PDF 00-040-0215).34 In the case of the B:NiFe LDH and galox-B:NiFe LDH, although all peaks except for the main peak were difficult to distinguish, the main peak of LDH at approximately 11° was clear.34,39,40
Fig. 3(b) shows the Raman spectra of the electrocatalysts before and after boronization and electrochemical oxidation. In these spectra, the pristine Ni foam, which was not functionalized at all, did not show any peaks. The peaks of the NiFe LDH electrode spectrum (red line) appeared at 300, 457, 531, and 650 cm−1 and correspond to the vibrations of the NiFe–O bond.2 Additionally, B:NiFe LDH and galox-B:NiFe LDH had similar Raman spectra to that of pristine NiFe LDH. peaks detected at 477 and 556 cm−1 corresponded to typical δ(Ni3+–O) and ν(Ni3+–O) vibrations of γ-NiOOH.15,38,41 As demonstrated by the vibrational peaks in the spectrum of the crystalline LDH form, the Raman results confirmed that the NiFe–O bonds remained after gaseous boronization and electrochemical oxidation.
Further investigation through FT-IR spectroscopy exhibited a change in the chemical bonding state after boron doping of the NiFe LDH-based electrocatalyst (Fig. 3(c)). In the full FT-IR spectra (bottom of Fig. 3(c)), several peaks were commonly detected below approximately 2500 cm−1 in all NiFe LDH-based electrocatalysts. The spectral band at 1620 cm−1 corresponds to the vibration of water molecules.42 Between 1400 and 600 cm−1, the vibration peaks of CO32− appearing at 1360, 1000, and 680 cm−1 and correspond to asymmetrical bending vibration (ν3), symmetrical stretching vibration (ν1), and asymmetrical bending vibration (ν4), respectively.14,39 Additionally, the peaks corresponding to the bridge vibration mode of H2O–CO32− and CO2 appear at approximately 2700 and 2360 cm−1, respectively, in Ni foam- and the NiFe LDH-based electrocatalysts.28,43 Moreover, the broad absorption band between 3500 and 3000 cm−1 was different depending on the electrocatalyst (top of Fig. 3(c)). This spectral band corresponds to the O–H stretching vibrations of the hydroxyl groups in the hydrotalcite layers and in interlayer water molecules.2,44 While no peak in the region was observed in the Ni foam spectra, a broad peak was detected in the NiFe LDH-based electrocatalyst spectra. In particular, an additional peak was detected at 3201 cm−1 in the spectra of B:NiFe LDH, corresponding to the B–OH stretching mode.45 Thus, it could be suggested that a heterogeneous atom, boron, had been successfully doped into NiFe LDH, as confirmed by the XRD, Raman, and FT-IR results.
Water oxidation performance of galox-B:NiFe LDH
The electrochemical performance of the NiFe LDH-based electrocatalysts was investigated under a 1.0 M KOH alkaline electrolyte. As shown in Fig. 4, to examine the oxygen evolution electrocatalytic activity of galox-B:NiFe LDH, linear sweep voltammetry (LSV) was conducted for each electrode. The OER polarization curves were obtained with a conventional three-electrode system in 1.0 M KOH. The potential range of polarization measurements for the OER was from 1.1 to 1.8 V (vs. a reversible hydrogen electrode (RHE)) at a scan rate of 1.0 mV s−1. First, the OER performance of the NiFe LDH-based electrocatalysts was measured as the overpotential at a current density of 10 mA cm−2, which is generally used as a reference to evaluate electrocatalysts.13,46 Fig. 4(a) displays the polarization curves of the OER for each electrode. The galox-B:NiFe LDH electrocatalyst required an overpotential of 229 mV to reach a current density of 10 mA cm−2, which is the lowest overpotential among this work. However, the overpotentials of the Ni foam, Ir foil, NiFe LDH, and B:NiFe LDH were 465, 371, 315, and 281 mV, respectively, under the same conditions. In particular, it was confirmed that the overpotential was further improved after boronization and galvanostatic oxidation of NiFe LDH and B:NiFe LDH, respectively. Additionally, to use an OER electrocatalyst for solar-driven hydrogen production, an overpotential of at least 250 mV is required.47 Thus, the current density required to achieve an overpotential of 250 mV for galox-B:NiFe LDH was 15.0 mA cm−2, while that for the Ni foam, Ir foil, NiFe LDH, and B:NiFe LDH was 0.1, 0.1, 1.9, and 4.6 mA cm−2, respectively.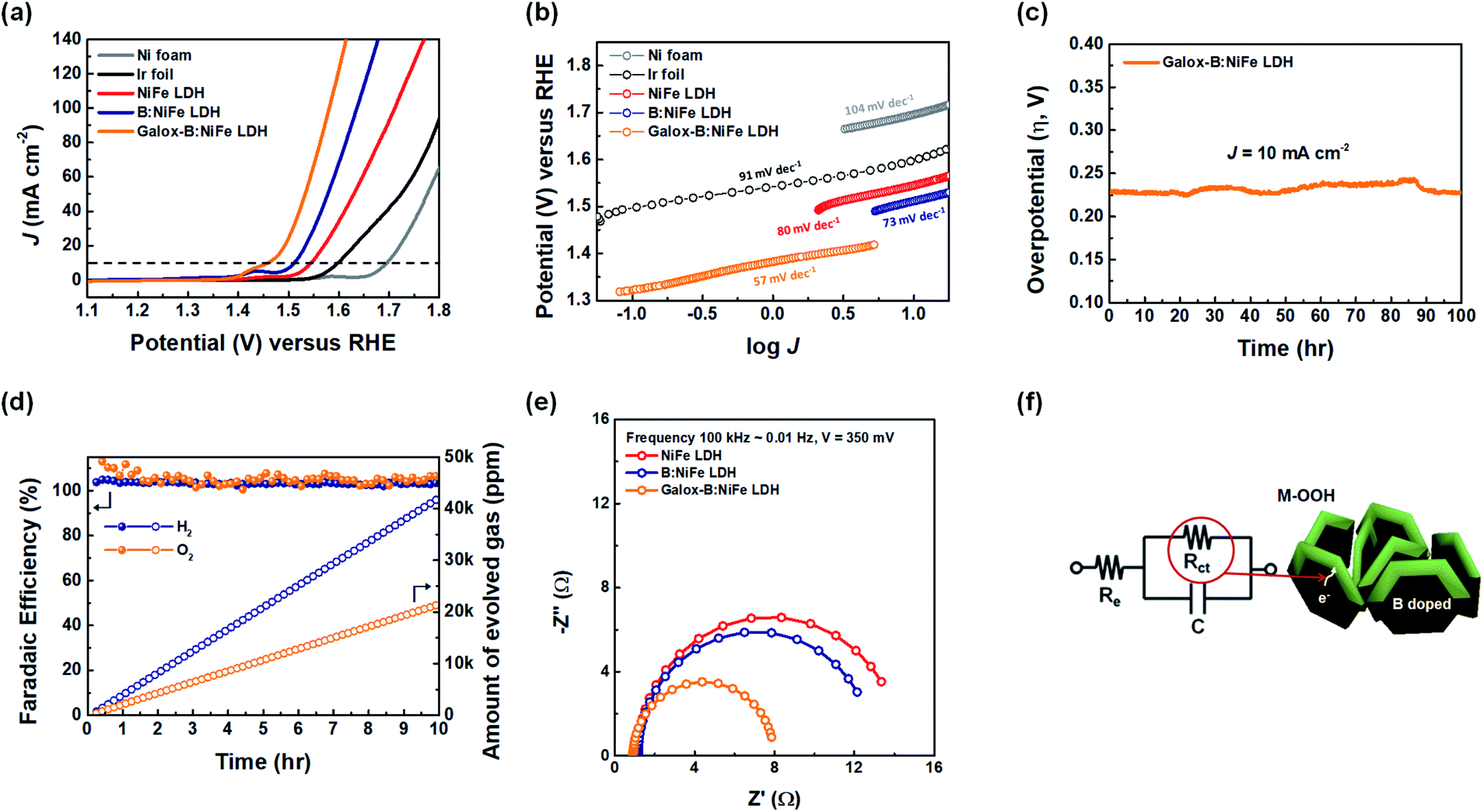 | ||
| Fig. 4 Electrochemical characterization of the NiFe LDH-based electrocatalyst electrodes for water oxidation in 1.0 M KOH. (a) OER polarization curves after a linear sweep with the different electrodes at a scan rate of 1.0 mV s−1. (b) Calculation of Tafel slopes corresponding to the OER polarization curves. (c) Chronopotentiometric stability test of galox-B:NiFe LDH at 10 mA cm−2. (d) Faradaic efficiency and amount of O2 and H2 evolved during water splitting at 10 mA cm−2. (e) Nyquist plots at 350 mV (vs. RHE) for different electrodes. (f) Equivalent circuit model and schematic diagram of the possibility of charge transfer in B:NiFe LDH. | ||
As shown in Fig. 4(b), the Tafel plot was displayed to analyze the electrocatalyst reaction kinetics, which were plotted based on the potential against log(J), and the Tafel slope was calculated. The galox-B:NiFe LDH electrocatalyst showed a low Tafel slope of 57 mV dec−1, while that for the Ni foam, Ir foil, NiFe LDH, and B:NiFe LDH was 104, 91, 80, and 73 mV dec−1, respectively. In this respect, the reaction kinetics of the galox-B:NiFe LDH were the fastest among those of the measured electrocatalysts, which may mean improved charge transfer through B:NiFe LDH.
To verify the stability of the galox-B:NiFe LDH electrocatalyst, a long-term durability test was conducted through the chronopotentiometric method (Fig. 4(c)). At a constant current density of 10 mA cm−2, the galox-B:NiFe LDH electrocatalyst delivered a stable overpotential of approximately 230 mV over 100 hours. Similarly, the overpotentials of NiFe LDH and B:NiFe LDH were also maintained at 320 and 280 mV, respectively (Fig. S5(a)†).
Fig. 4(d) shows the faradaic efficiency and amount of evolved gas (O2, H2) during the stability test at a constant current density of 10 mA cm−2 for galox-B:NiFe LDH. The faradaic efficiency was calculated using the number of electrons required to evolve 1 mol gas, and the number of moles were obtained using the amount of evolved gas. The number of electrons is 2 for H2 and 4 for O2. The amount of evolved gas was measured using gas chromatography, and the image of equipment of gas chromatography is depicted in Fig. S6.† As shown in Fig. 4(d), it was confirmed that the faradaic efficiency (left Y-axis) was maintained at almost 100% for both the working and counter electrodes at a constant current density of 10 mA cm−2. Additionally, the ratio of gas generated during the stability test was maintained at 1:2 (O2:H2).
To further clarify the effect of the boron-doped NiFe LDH on facile charge transfer during the OER step, an analysis through electrochemical impedance spectroscopy (EIS) was conducted in the frequency range from 100000 to 0.01 Hz at a constant overpotential of 350 mV. Fig. 4(e) and (f) display the Nyquist plots of each electrocatalyst and an equivalent-circuit model (EIS results for different potential conditions: Fig. S5(b and c)).† The resistances of the circuit consist of electrolyte resistance (Re) and charge-transfer resistance (Rct) in Fig. 4(f). As shown in Fig. 4(e), the Rct of the galox-B:NiFe LDH electrocatalyst was the lowest. This result means that facile charge transfer is possible as a result of forming activated oxyhydroxide on the B:NiFe LDH electrode through galvanostatic oxidation.
It has been previously reported that the valence state of transition metal element in LDH affects OER performance.52–57 As reported by Dionigi et al., the synergetic effects through the O-bridged Fe-M (Ni or Co) reaction center formed between the electronic structure of the Fe site and the adjacent M site stabilize the OER intermediates which are disadvantageous on Ni centers or pure Fe sites.58 In this work, the reaction center was adjusted by adding Fe element. Furthermore, the electronic structure of NiFe LDH was modified through boron doping to facilitate charge transfer. The boron doping would induce amorphization of LDH which creates oxygen vacancy. The formed vacancy effects on the improvement of charge transfer capability.59–62 Additionally, it has been previously reported that when the B:NiFe structure is activated under anodic conditions, it undergoes a structural change reminiscent of the deformation of the NiBi thin film prepared by electrodeposition.48 In this step, Jahn–Teller-distorted Ni(III) centers are activated to bis-oxo/bridged Ni centers that are organized into sheets of edge-sharing octahedral NiO6, which is a well-known possible active site for the OER.40,49,50 Considering these results and those of previous reports, it can be seen that catalytic activity is improved due to B doping.
Change of surface state caused by boron doping and oxidation
In Fig. 5, the characterization of surface composition, valance state, and specific surface area was performed to confirm the improved OER performance. X-ray photoelectron spectroscopy (XPS) was conducted to investigate the effect of B doping on the chemical composition and change in valance state due to the formation of oxyhydroxide caused by activation through electrochemical oxidation (Fig. 5(a–c)). Fig. 5(a) shows the O 1s XPS spectra for NiFe LDH after different steps in the synthesis process. It could be confirmed that the O 1s spectrum of B:NiFe LDH shifted toward a higher energy than that of the NiFe LDH O 1s spectrum due to O–B bonding after boron doping.51 On the other hand, it appeared that the O 1s spectrum of galox-B:NiFe LDH shifted toward lower energy due to the formation of oxyhydroxide. Furthermore, lattice oxygen (O2−) and absorbed molecular water also affected this shift.47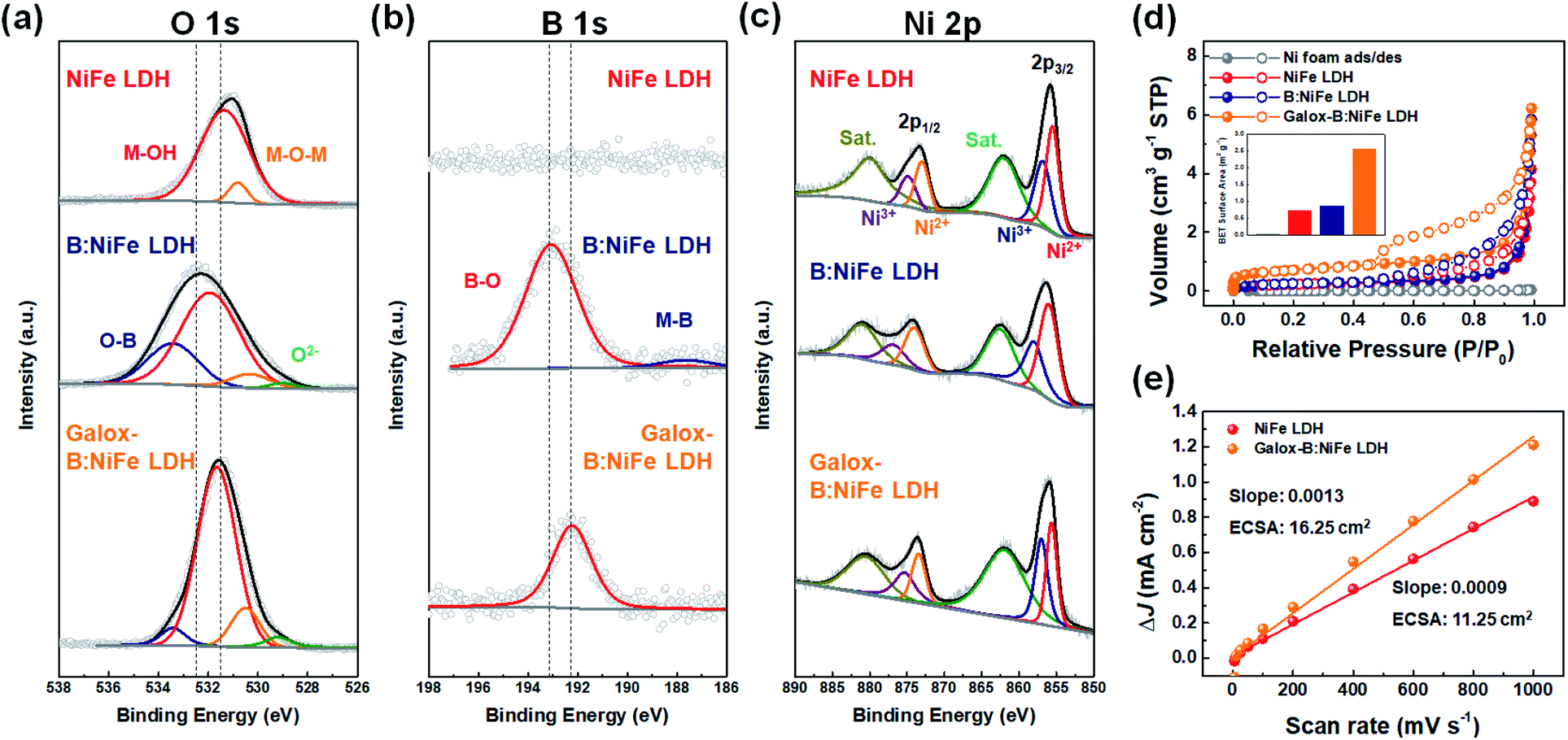 | ||
| Fig. 5 Chemical composition and specific surface area characterization for verification of enhanced catalytic activity. (a–c) X-ray photoelectron spectroscopy (XPS) analysis for observation of chemical composition. (a) O 1s, (b) B 1s, and (c) Ni 2p XPS spectra of NiFe LDH-based electrocatalysts under each condition. (d) Brunauer–Emmett–Teller (BET) analysis of an isothermal plot with N2 adsorption/desorption (inset: comparison of BET surface area under each condition). (e) Electrochemical surface area (ECSA) of the NiFe LDH base electrocatalyst. The linear slope is related to the double layer capacitance. | ||
In Fig. 5(b), the B 1s XPS spectrum for NiFe LDH was empty since it did not react with boric acid. However, the B 1s spectra for B:NiFe LDH and galox-B:NiFe LDH demonstrated the formation of a boron peak. In particular, the B 1s spectrum for B:NiFe LDH showed peaks at 188 eV and 193 eV, which correspond to M–B and surface oxidized B (B–O), respectively.46 The M–B peak (188 eV) decreased when exposed to air and the electrolyte during electrochemical oxidation, as observed in the B 1s spectrum of galox-B:NiFe LDH.36 As observed in the O 1s spectra, the B–O peak of galox-NiFe LDH shifted toward a lower energy than that of B:NiFe LDH.
In Fig. 5(c), the Ni 2p spectra exhibited peaks corresponding to Ni2+ (855.5 (2p3/2) and 873.0 eV (2p1/2)) and Ni3+ (856.8 (2p3/2) and 874.8 eV (2p1/2)). The intensity of the Ni3+ peaks of galox-B:NiFe LDH was higher than that of the corresponding peaks of NiFe LDH and B:NiFe LDH due to the formation of oxyhydroxide through electrochemical oxidation.2 Similar to the Ni 2p trends, the intensity of the Fe3+ peaks in the Fe 2p spectrum of galox-B:NiFe LDH also increased compared to that of the Fe3+ peaks in the Fe 2p spectrum of pristine NiFe LDH and B:NiFe LDH (Fig. S7†).
To further characterize the physical surface area of NiFe LDH, B:NiFe LDH, and galox-B:NiFe LDH, Brunauer–Emmett–Teller (BET) analysis was conducted (Fig. 5(d)), where the BET specific surface area (inset of Fig. 5(d)) and pore distribution (Fig. S8†) were obtained. In the case of galox-B:NiFe LDH, the BET specific surface area in Fig. 5(d) was 2.54 m2 g−1, and that of the Ni foam, NiFe LDH, and B:NiFe LDH was 0.02, 0.72, and 0.85 m2 g−1, respectively. These BET analysis results exhibited a similar tendency to the electrochemical surface areas (ECSAs) in Fig. 5(e). Compared to pristine NiFe LDH, the galox-B:NiFe LDH electrocatalyst showed a relatively large ECSA. As observed via characterization of the specific surface area, galox-B:NiFe LDH had relatively improved active sites, which had a positive effect on OER performance.
After carrying out the OER durability test, post-analysis was performed (Fig. S10†). As shown in Fig. S10(a),† the XRD pattern of galox-B:NiFe LDH hardly changed after OER. Additionally, although O2 gas was vigorously emitted during the OER, galox-B:NiFe LDH was well retained on the Ni foam in the SEM images (Fig. S10(b)).† In particular, it was confirmed that no microstructure collapse was observed even under high current density conditions. In both the XRD and SEM results, the pattern and morphology of KOH was detected since it was the electrolyte implemented during the OER. According to the O 1s of XPS result of galox-B:NiFe LDH after durability test, it is similar to result of NiFe LDH before boron doping. Furthermore, the Ni and Fe 2p spectrum results show that the intensity of Ni3+ and Fe3+ increased even more after the durability test compared with galox-B:NiFe LDH. According to the post-analysis, it can be confirmed that the structural, phase change did not occur, and the ratio of oxyhydroxide increases after the stability test.2
Conclusions
In this work, we demonstrated that an electrocatalyst developed with a low-cost, earth-abundant transition metal was a highly efficient water splitting anode. NiFe LDH was successfully boron-doped through simple gaseous boronization using a boron source at a relatively low temperature, although boron doping is not easily achieved due to the high boiling point and melting point of boron. Boron doping was effective at enhancing the OER electrocatalytic activity, reaching values of 281 mV at 10 mA cm−2, of B:NiFe LDH electrodes. Furthermore, the effect of boron doping was maximized by activation through electrochemical oxidation, and the galox-B:NiFe LDH electrocatalyst reached an overpotential of 229 mV at 10 mA cm−2. The morphological and chemical spectral characterization of the synthesized NiFe LDH-based electrocatalysts confirmed successful boron doping of NiFe LDH and activation through electrochemical oxidation. Through potentiostat EIS investigation of each electrocatalyst electrode, it was confirmed that boron doping kinetically improved the electrocatalyst due to a low charge-transfer resistance. The relatively simple boron doping method presented in this study suggest the possibility of mass LDH catalyst production based on the enhanced OER performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2017M3D1A1040688). This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2019R1A2C2090859). The SEM and TEM analysis was supported by the Research Institute of Advanced Materials (RIAM) at Seoul National University.Notes and references
- I. K. Ahn, Y. J. Lee, S. Na, S. Y. Lee, D. H. Nam, J. H. Lee and Y. C. Joo, ACS Appl. Mater. Interfaces, 2018, 10, 2242–2248 CrossRef CAS.
- H. Park, I. J. Park, M. G. Lee, K. C. Kwon, S. P. Hong, D. H. Kim, S. A. Lee, T. H. Lee, C. Kim, C. W. Moon, D. Y. Son, G. H. Jung, H. S. Yang, J. R. Lee, J. Lee, N. G. Park, S. Y. Kim, J. Y. Kim and H. W. Jang, ACS Appl. Mater. Interfaces, 2019, 11, 33835–33843 CrossRef CAS.
- S. Y. Lim, S. Park, S. W. Im, H. Ha, H. Seo and K. T. Nam, ACS Catal., 2019, 10, 235–244 CrossRef.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- R. D. Smith, M. S. Prevot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS.
- H. Liang, F. Meng, M. Caban-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS.
- J. Park, S. Choi, A. Oh, H. Jin, J. Joo, H. Baik and K. Lee, Nanoscale Horiz., 2019, 4, 727–734 RSC.
- J.-C. Lee, J.-Y. Kim, W.-H. Joo, D. Hong, S.-H. Oh, B. Kim, G.-D. Lee, M. Kim, J. Oh and Y.-C. Joo, J. Mater. Chem. A, 2020, 8, 11632–11641 RSC.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- M. Lee, H.-S. Oh, M. K. Cho, J.-P. Ahn, Y. J. Hwang and B. K. Min, Appl. Catal., B, 2018, 233, 130–135 CrossRef CAS.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- I. K. Ahn, W. Joo, J. H. Lee, H. G. Kim, S. Y. Lee, Y. Jung, J. Y. Kim, G. B. Lee, M. Kim and Y. C. Joo, Sci. Rep., 2019, 9, 19539 CrossRef CAS.
- L. Dang, H. Liang, J. Zhuo, B. K. Lamb, H. Sheng, Y. Yang and S. Jin, Chem. Mater., 2018, 30, 4321–4330 CrossRef CAS.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS.
- J. Hao, W. Yang, J. Hou, B. Mao, Z. Huang and W. Shi, J. Mater. Chem. A, 2017, 5, 17811–17816 RSC.
- X. Ma, W. Zhang, Y. Deng, C. Zhong, W. Hu and X. Han, Nanoscale, 2018, 10, 4816–4824 RSC.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS.
- X. Zheng, X. Han, Y. Zhang, J. Wang, C. Zhong, Y. Deng and W. Hu, Nanoscale, 2019, 11, 5646–5654 RSC.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- Y. Wang, D. Yan, S. El Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef.
- Z. Lu, L. Qian, Y. Tian, Y. Li, X. Sun and X. Duan, Chem. Commun., 2016, 52, 908–911 RSC.
- S. Park, Y. H. Lee, S. Choi, H. M. Seo, M. Y. Lee, M. Balamurugan and K. T. Nam, Energy Environ. Sci., 2020, 13(8), 2310–2340 RSC.
- W. Xu, J. Chen, M. Yu, Y. Zeng, Y. Long, X. Lu and Y. Tong, J. Mater. Chem. A, 2016, 4, 10779–10785 RSC.
- X. Xue, F. Yu, B. Peng, G. Wang, Y. Lv, L. Chen, Y. Yao, B. Dai, Y. Shi and X. Guo, Sustainable Energy Fuels, 2019, 3, 237–244 RSC.
- Y. Yang, L. Dang, M. J. Shearer, H. Sheng, W. Li, J. Chen, P. Xiao, Y. Zhang, R. J. Hamers and S. Jin, Adv. Energy Mater., 2018, 8, 1703189 CrossRef.
- X. Long, S. Xiao, Z. Wang, X. Zheng and S. Yang, Chem. Commun., 2015, 51, 1120–1123 RSC.
- H. Liu, Y. Wang, X. Lu, Y. Hu, G. Zhu, R. Chen, L. Ma, H. Zhu, Z. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 35, 350–357 CrossRef CAS.
- K. N. Dinh, P. Zheng, Z. Dai, Y. Zhang, R. Dangol, Y. Zheng, B. Li, Y. Zong and Q. Yan, Small, 2018, 14, 1703257 CrossRef.
- Z. Tan, P. Liu, H. Zhang, Y. Wang, M. Al-Mamun, H. G. Yang, D. Wang, Z. Tang and H. Zhao, Chem. Commun., 2015, 51, 5695–5697 RSC.
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC.
- W. Li, D. Xiong, X. Gao and L. Liu, Chem. Commun., 2019, 55, 8744–8763 RSC.
- J. Masa, I. Sinev, H. Mistry, E. Ventosa, M. de la Mata, J. Arbiol, M. Muhler, B. Roldan Cuenya and W. Schuhmann, Adv. Energy Mater., 2017, 7, 1700381 CrossRef.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734–1743 RSC.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS.
- J. Li, H. Chen, Y. Liu, R. Gao and X. Zou, J. Mater. Chem. A, 2019, 7, 5288–5294 RSC.
- M. Asrofi, H. Abral, Y. K. Putra, S. M. Sapuan and H. J. Kim, Int. J. Biol. Macromol., 2018, 108, 167–176 CrossRef CAS.
- N. L. Drenchev, K. K. Chakarova, O. V. Lagunov, M. Y. Mihaylov, E. Z. Ivanova, I. Strauss and K. I. Hadjiivanov, J. Visualized Exp., 2020, 156, e60285 Search PubMed.
- M. N. Iliev, D. Mazumdar, J. X. Ma, A. Gupta, F. Rigato and J. Fontcuberta, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 014108 CrossRef.
- J. Romanos, M. Beckner, D. Stalla, A. Tekeei, G. Suppes, S. Jalisatgi, M. Lee, F. Hawthorne, J. D. Robertson, L. Firlej, B. Kuchta, C. Wexler, P. Yu and P. Pfeifer, Carbon, 2013, 54, 208–214 CrossRef CAS.
- H. Han, Y. R. Hong, J. Woo, S. Mhin, K. M. Kim, J. Kwon, H. Choi, Y. C. Chung and T. Song, Adv. Energy Mater., 2019, 9, 1803799 CrossRef.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS.
- X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef CAS.
- Y. Yang, L. Zhuang, R. Lin, M. Li, X. Xu, T. E. Rufford and Z. Zhu, J. Power Sources, 2017, 349, 68–74 CrossRef CAS.
- B. J. Matsoso, K. Ranganathan, B. K. Mutuma, T. Lerotholi, G. Jones and N. J. Coville, New J. Chem., 2017, 41, 9497–9504 RSC.
- L. Lv, Z. Yang, K. Chen, C. Wang and Y. Xiong, Adv. Energy Mater., 2019, 9, 1803358 CrossRef.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS.
- B. S. Brunschwig, M. H. Chou, C. Creutz, P. Ghosh and N. Sutin, J. Am. Chem. Soc., 1983, 105, 4832–4833 CrossRef CAS.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. G. de Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS.
- M. Gorlin, P. Chernev, J. Ferreira de Araujo, T. Reier, S. r. Dresp, B. Paul, R. Krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef.
- Z. Cai, D. Zhou, M. Wang, S. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li and W. Liu, Angew. Chem., 2018, 130, 9536–9540 CrossRef.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS.
- H. Yang, Z. Chen, P. Guo, B. Fei and R. Wu, Appl. Catal., B, 2020, 261, 118240 CrossRef CAS.
- X. Lu, H. Xue, H. Gong, M. Bai, D. Tang, R. Ma and T. Sasaki, Nano-Micro Lett., 2020, 12, 86 CrossRef CAS.
- X. H. Lu, G. M. Wang, S. L. Xie, J. Y. Shi, W. Li, Y. X. Tong and Y. Li, Chem. Commun., 2012, 48, 7717–7719 RSC.
- X. H. Lu, G. M. Wang, T. Zhai, M. H. Yu, J. Y. Gan, Y. X. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS.
- L. Bai, Z. Duan, X. Wen and J. Guan, J. Catal., 2019, 378, 353–362 CrossRef CAS.
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- S. Li, J. Sun and J. Guan, Chin. J. Catal., 2021, 42, 511–556 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10169a |
| This journal is © The Royal Society of Chemistry 2021 |