
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances and prospects in the Zn-catalysed Mannich reaction
Salahudeen Shamna
a,
C. M. A. Afsina
a,
Rose Mary Philip
a and
Gopinathan Anilkumar
 *abc
*abc
aSchool of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills, Kottayam, Kerala 686560, India. E-mail: anilgi1@yahoo.com; anil@mgu.ac.in; Fax: +91-481-273-1036
bAdvanced Molecular Materials Research Centre (AMMRC), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India
cInstitute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India
First published on 1st March 2021
Abstract
Zn-catalysed reactions are ubiquitously important due to their inexpensive, generally less toxic and atom-economic nature. According to the modern criteria of sustainability, their use in a catalytic manner is a highly desirable goal, especially when using chiral ligands. Considering the relevance of well-established zinc-mediated C–C bond formation reactions, it is relatively surprising that the use of Zn as a catalyst is still underdeveloped, especially in comparison with other transition metals. The vast majority of natural molecules, including proteins, nucleic acids and most biologically active compounds, contain nitrogen. Consequently, developing new synthetic methods for the construction of nitrogenous molecules receives great attention from organic chemists. The Mannich reaction is a very basic and very useful platform for the development of several such nitrogen-containing molecules. In this review, we summarise the recent advancements in the Zn-catalysed Mannich reaction, covering the literature from 2011 to 2020.
 Salahudeen Shamna | Salahudeen Shamna was born in 1997 in Kerala, India. She received her B.Sc. degree (2017) from the Department of Chemistry, N. S. S College, Pandalam and her M.Sc. degree (2019) from the School of Chemical Sciences, Mahatma Gandhi University. Her research interests revolve around the development of new catalytic methodologies and employing these methodologies to make medicinally relevant compounds. |
 C. M. A. Afsina | C. M. A. Afsina was born in Kerala, India, in 1993. She obtained her B.Sc. degree from Kannur University (Government Brennen College, Thalassery) in 2014 and her M.Sc. degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam, Kerala, in 2016. She qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship and is currently pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Rose Mary Philip | Rose Mary Philip was born in Kottayam, Kerala, India. She obtained her dual degree of BSMS (Bachelor of Science and Master of Science) from the Indian Institute of Science Education and Research, Thiruvananthapuram in 2019. She qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship. Currently, she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and obtained his Ph.D in 1996 from Regional Research Laboratory (renamed the National Institute for Interdisciplinary Science and Technology (NIIST-CSIR)), Trivandrum, with Dr. Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently, he is a Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University, in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 120 papers in peer-reviewed journals, 7 patents, and 8 book chapters, and he has edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He received the Dr. S Vasudev Award from the Govt. of Kerala, India, for the best research (2016) and an Evonik research proposal competition award (second prize 2016). His h-index is 31. |
1. Introduction
The main motif of organic synthesis and medicinal chemistry is sustainability and efficiency in obtaining chemical compounds. In both cases, the structural complexity of molecules requires multistep synthetic routes, diversified substrates and reagents, aggressive reaction media and difficulties in the isolation and purification of products, leading to extremely laborious and costly experimental procedures.1 The science of organic synthesis is constantly enriched by the improvement of synthetic methodologies. The paradigms of organic synthesis have shifted from the traditional concept of using only chemical yield to define efficiency to one in which the economic and ecological values are also considered. Recently, multicomponent reactions (MCRs) have aroused considerable interest among synthetic chemists for the construction of complex molecules.2 Here, we disclose the multicomponent reactions representing a very interesting synthetic organic methodology performed either on a solid phase or in solution phase.3Multicomponent reactions are enormously useful for organic chemistry and are developing as a dominant method for the formation of new and complex molecular structures.4 In this context, multicomponent reactions represent a very interesting organic synthetic methodology due to the use of convergent one-pot reactions, atom economy, high-yielding steps, mild reaction conditions, accessible and cheap substrates, operational ease, and the ability to quickly build a library of structurally complex compounds. These factors are attributed to what chemists call the “ideal synthesis”.5,6 Multicomponent reactions can be classified in different ways depending on the nature of reagents, reaction conditions and mechanism of product formation. However, main classifications are (a) imine-based and (b) isocyanide-based MCRs. The application of MCRs includes the synthesis of bioactive compounds due to their diminished cost, quick reaction times, atom-economy, energy preservation, and the prevention of time-consuming product purification. The improvement of catalytic asymmetric reactions has been one of the main approaches for past two decades, and it remains a common chemical technique. From asymmetric multicomponent reactions (AMCRs), unimolecular or bimolecular syntheses can be achieved under catalytic conditions to afford products in high chemical yields with excellent enantioselectivities.7,8
A large variety of natural products and drugs possess optically active nitrogen-containing molecules such as proteins, nucleic acids and other biologically active compounds. They have attracted considerable attention from synthetic chemists and the pharmaceutical industry to stimulate the development of the asymmetric Mannich reaction, a useful transformation to access amine-containing building blocks.9
In 1912, Carl Mannich disclosed his studies on the three-component reaction between an enolizable C–H acidic substrate, an amine component and an aldehyde to form a β-amino carbonyl compound.10 The reaction generally involves a pre-equilibrium where the aldehyde and amine form an imine or iminium salt, which is subsequently attacked by the nucleophilic enol tautomer of the C–H acidic component.11 Reactions between aldimine and α-methylene carbonyls are also considered as Mannich reactions because these imines are formed between amines and aldehydes. Two key features make the Mannich reaction and its products very attractive: (a) the reaction tolerates a large variety of reactants12 and (b) the α-amino carbonyl products are valuable synthons for natural product synthesis and can be readily converted to derivatives that possess applications in the paint and polymer chemistry, medicinal chemistry and pharmaceutical industry.13
Recently, Mannich reactions have been catalysed by HCl,14 HBF4,15 InCl3,16 Y(OTf)3,17 Yb(PFO)3,18 Zn(BF4)2,19 Bi(OTf)3,20 PS-SO3H,21 a chiral Brønsted acid,22,23 phosphorodiamidic acid,24 iminodiacetic acid,25 heteropoly acid,26 dodecylbenzene sulfonic acid,27 nanoparticles,28,29 Troger's base,30 and enzymes.31 However, the major drawbacks of these catalyst systems include difficulty in catalyst extraction, catalyst recyclability issues, a less stereoselective reaction, and high temperature conditions; also, some catalysts are even corrosive, expensive, and volatile, and they often cause environmental problems. Recently, organometallic catalysts emerged as a reagent class representing a new methodology for green chemistry.32–34 Zinc is the most pervasive of all trace elements involved in human metabolism. More than 100 specific enzymes require zinc for their catalytic function. For example, in the catalytic centre of human carbonic anhydrase II, zinc(II) is coordinated by amino acids and water, as zinc-bound water and hydroxide/hydroxyl ions are excellent nucleophilic agents. The significance of Zn in catalysing life processes has opened a new era in the once-neglected field of Zn coordination chemistry. Zinc catalysis in different C–C bond forming reactions is also known.
In this review, we summarise the recent advancements in the Zn-catalysed Mannich reaction, covering the literature from 2011 to 2020. For better clarity and ease of understanding, the review is classified based on the type of Zn catalyst used in the Mannich reaction.
2. Zn-catalysed Mannich reactions
2.1 Zn-ProPhenol-catalysed reactions
Trost and co-workers disclosed the first enantio- and anti-diastereoselective Mannich reaction using α-fluoroketones for the synthesis of β-fluoroamine.35 They conducted Mannich reactions between fluoroindanone and 1.2 equivalents of Boc-protected aldimines in the presence of 20 mol% Et2Zn and 10 mol% (R,R)-ProPhenol in THF for 40 h at 60 °C to afford the products as diastereoisomers in 68–99% yields with excellent diastereoselectivity (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and enantioselectivity (up to 99%) (Scheme 1). Even the gram scale reactions provided excellent yields (up to 99%). Then, they generalised the reaction by using a variety of aromatic Boc-aldimines with electron-withdrawing and electron-donating groups that were tolerated in the ortho-, meta- and para-positions of the aromatic ring.
:1) and enantioselectivity (up to 99%) (Scheme 1). Even the gram scale reactions provided excellent yields (up to 99%). Then, they generalised the reaction by using a variety of aromatic Boc-aldimines with electron-withdrawing and electron-donating groups that were tolerated in the ortho-, meta- and para-positions of the aromatic ring.
 | ||
| Scheme 1 Synthesis of β-fluoroamine via an enantioselective Mannich reaction using a Zn-ProPhenol catalyst. | ||
Trost and co-workers described the first direct catalytic Mannich-type reaction between ynones and N-Boc imines, such as aromatic, aliphatic and α,β-unsaturated imines.36 Here, they took Boc-protected aldimine and 1.2 equivalents of silyl-protected ynone in the presence of 20 mol% Et2Zn catalyst and 10 mol% ligand in THF at 4 °C to produce the desired Mannich adduct as a single diastereomer in 75–99% yields and with enantioselectivity >90% and diastereoselectivity in the range of 9:1 to >20:1 (Scheme 2). Moreover, they expanded the substrate scope to various imines, such as non-aromatic imines, aromatic imines and α,β-unsaturated imines, under the optimized reaction conditions.
 | ||
| Scheme 2 Zn-catalysed Mannich reaction for the preparation of α-substituted β-amino ynones from ynones and N-Boc imines. | ||
In 2016, Trost et al. reported the first direct and scalable asymmetric Mannich reaction in the presence of α-branched ketone donors.37 They initiated their studies with 2-methyl indanone to explore the feasibility of transformation with Boc-protected aldimine in the presence of 5 mol% of (R,R-prophenol) and 10 mol% of Et2Zn in THF at 80 °C; they obtained the desired Mannich products in yields of 5–99% and with excellent diastereoselectivity (up to >20:1) and enantioselectivity (up to 99%) (Scheme 3). They investigated a variety of aromatic Boc-aldimines with electron-withdrawing and electron-donating substituents, heteroaromatic imines, vinyl imines, alkyl imines and Cbz-protected imines, which can be used as equally efficient partners in the Mannich reaction with highly functionalized aromatic ketones such as 5-membered aromatic ketones, tetralone, chromanone, and thiochromanone.
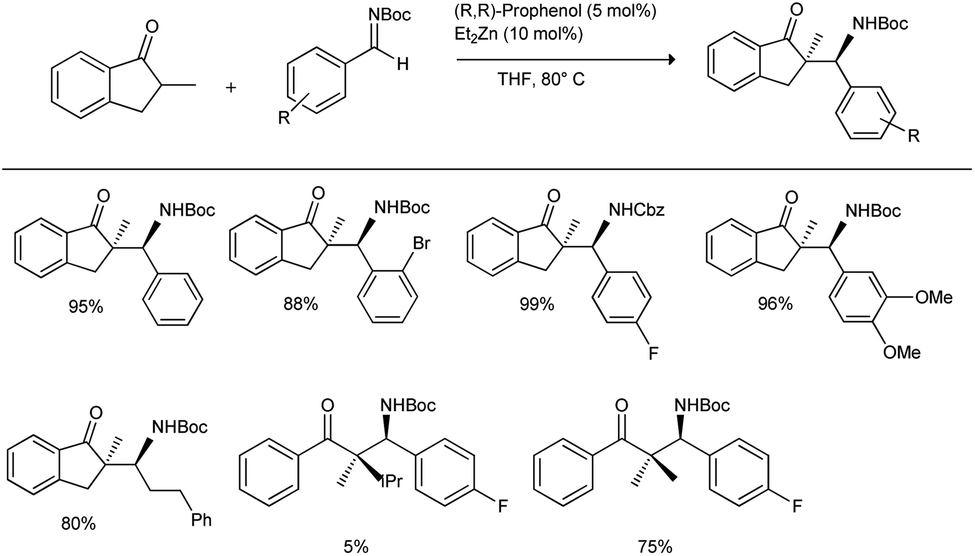 | ||
| Scheme 3 Zn-ProPhenol-catalysed direct catalytic asymmetric Mannich reactions for the construction of quaternary carbon stereocenters. | ||
The same group of researchers disclosed the first direct vinylogous Mannich reaction with easily deprotected N-Cbz imines and both α,β- and β,γ-butenolides bearing a variety of substituents with different substitution patterns (Scheme 4).38 Here, they treated 1 equiv. of α-angelica lactone, 1.2 equiv. of the imine, 10 mol% (R,R)-prophenol and 20 mol% Et2Zn at rt in THF for 8 h, which afforded the best results of 29–79% yield with excellent enantio- (97 to >99.5%) and diastereoselectivity (18:1) (Scheme 4). They further expanded the scope of the reaction by including a variety of imines and butenolides with phenyl and thienyl substitutions. They showed that α,β-butenolides are more easily synthesised and stored than the analogous β,γ-compounds due to conjugation with the carbonyl group in the former compounds.
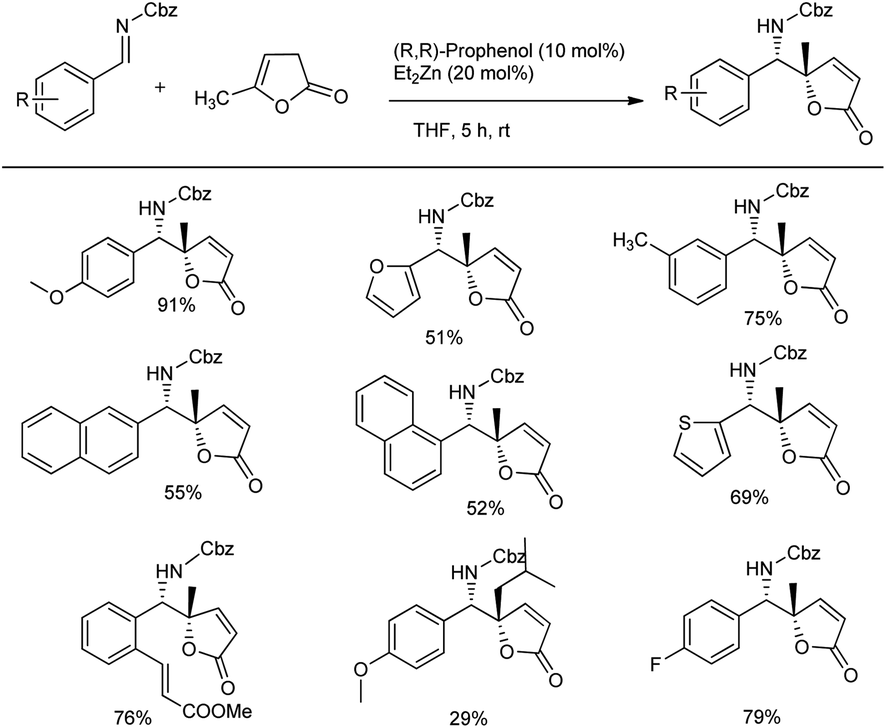 | ||
| Scheme 4 Preparation of tetrasubstituted vinylogous Mannich products from butenolides and imines using the Zn-ProPhenol-catalysed reaction. | ||
In this paper, Trost et al. unveiled Zn-ProPhenol-catalysed azaDarzens reaction for the access of chiral trisubstituted aziridines (Scheme 5).39 They started the studies by treating 0.20 mmol of chloroindanone with 0.22 mmol of imine in the presence of Zn-ProPhenol catalyst in 0.2 M Et2O at 40 °C for 1–16 h to afford the α-chloro-β-aminoketone in 65–99% yield and 87–99% ee and as a >20:1 mixture of diastereoisomers. They evaluated the generality of the reaction with a range of α-bromo and α-chloro-β-aminoketones and also carried out the one-pot synthesis. They also carried out the gram-scale reaction and obtained 95% yield of the product.
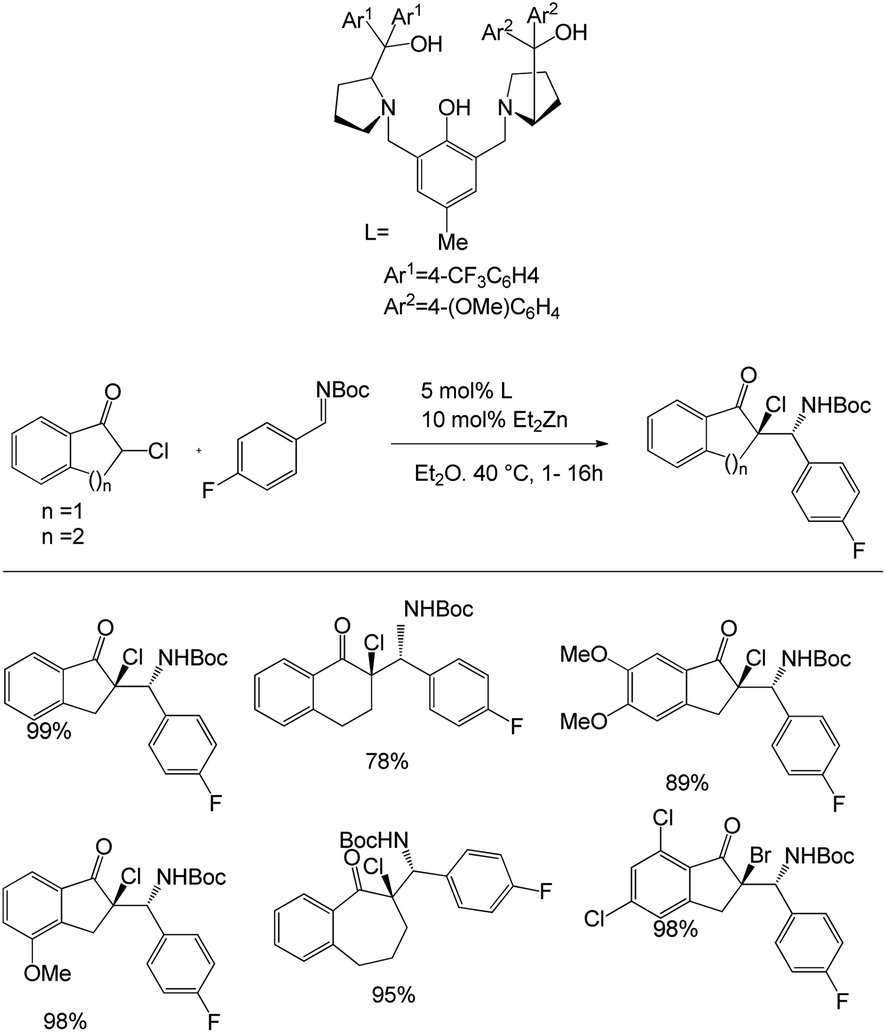 | ||
| Scheme 5 Synthesis of α-chloro-β-aminoketone from chloroindanone and imine. | ||
After the optimisation of α-chloro-β-aminoketones, the team successfully transformed the product into the corresponding aziridines (Scheme 6). There are two different protocols for the development of aziridine. Protocol A was applied to α-chloro-β-aminoketones derived from chloroindanones in Cs2CO3 in acetonitrile as the solvent, and the corresponding aziridines were obtained quantitatively. However, this protocol is limited to a few ketones, namely the substrates derived from chlorotetralone, chloro-furanocyclohexanone or chlorochromanones. To overcome this circumstance, they introduced another protocol (protocol B) using NaH in THF. Both protocols positively responded to the substrates and offered yields of up to 99%.
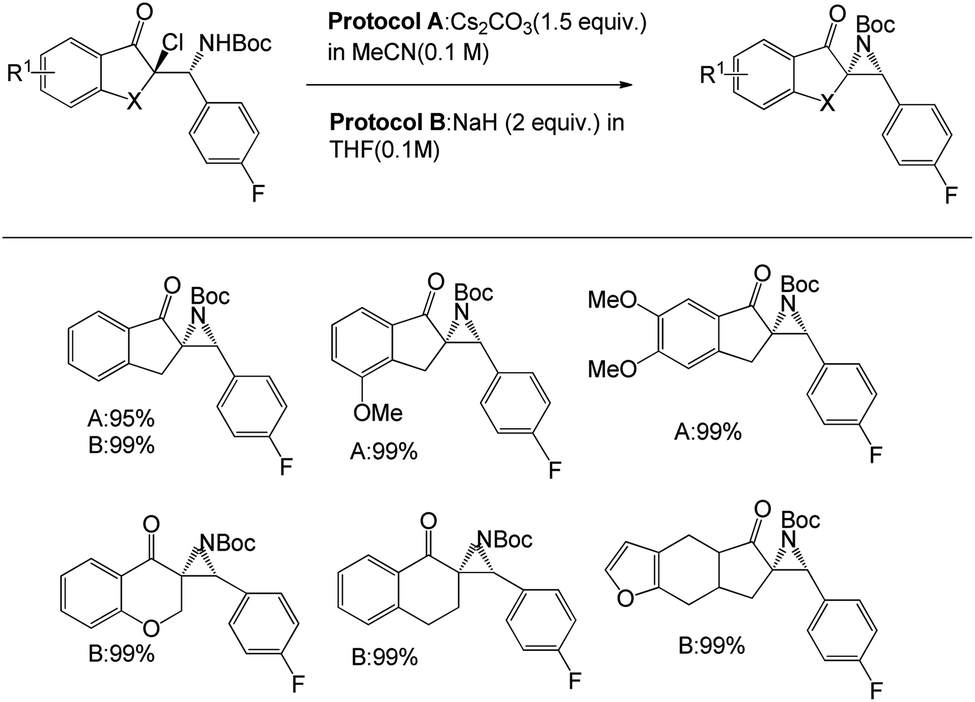 | ||
| Scheme 6 Synthesis of aziridines from β-fluoroamines. | ||
Trost et al. disclosed the enantioselective and stereodivergent synthesis of 1,3-aminoalcohols using a bimetallic Zn-ProPhenol catalyst (Scheme 7).40 They unveiled the feasibility of the proposed Mannich reaction by treating 0.24 mmol isobutyraldehyde with 0.26 mmol imine Boc-imine in the presence of Zn-ProPhenol catalyst in toluene at 4–23 °C for 14–24 h, which afforded the Mannich adduct with excellent yields of 56–99% and 99% selectivity. The optimised reaction conditions were used to study the scope of the Mannich reaction with a range of imines, such as aliphatic, aromatic, heteroaromatic and vinyl imines, as well as with a variety of aldehydes, which positively responded to the reaction conditions.
 | ||
| Scheme 7 Enantioselective and stereodivergent synthesis of 1,3-aminoalcohols. | ||
Trost and co-workers unveiled a method for the synthesis of two contiguous tetrasubstituted stereogenic centres containing vinylogous products by a Zn-ProPhenol-catalysed asymmetric Mannich reaction (Scheme 8).41 They started their studies using 1 equiv. of ketamine and treated it with 1.2 equiv. of α-angelica lactone in the presence of Zn catalyst at rt in 0.3 M THF for 40 h, which afforded the desired products in a range of yields of 35–86% and with an enantiomeric excess of up to 99%. With the optimised conditions, they expanded the scope of the reaction using various fluorinated ketimines with β,γ-butenolides, affording good yields.
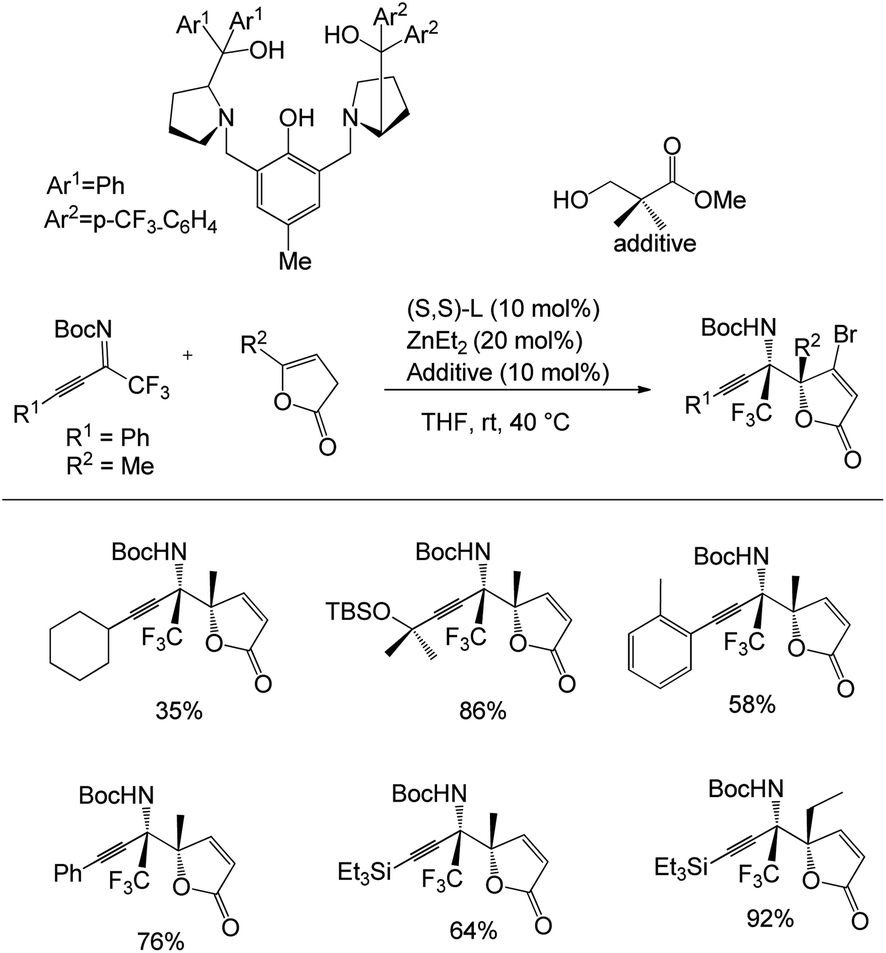 | ||
| Scheme 8 Mannich reaction between butenolides and polyfluorinated alkynyl ketimines. | ||
In continuation of their interest in the field, in 2019, Trost and co-workers introduced a Mannich reaction with N-Cbz imine and aldehydes to construct an o-bromo motif in the presence of Zn-ProPhenol catalyst (Scheme 9).42 To improve the diastereoselectivity, different ligands and reaction temperatures were evaluated. Unfortunately, even with 10 mol% catalyst loading, no reaction was observed at elevated temperatures. Interestingly, lowering the temperature from room temperature to 4 °C drastically increased the diastereomeric ratio (dr) to 12:1, and PhMe was found to be the best solvent to afford the products in better yields of 68–82%.
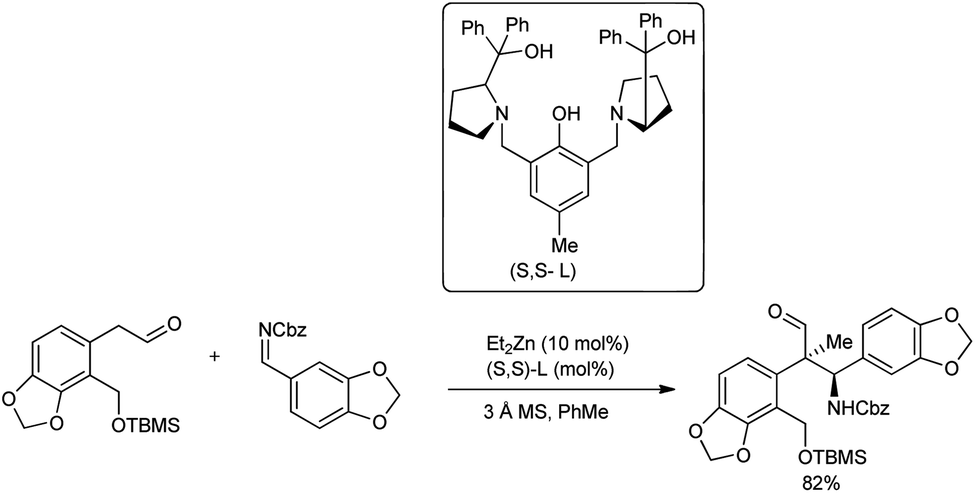 | ||
| Scheme 9 Enantioselective synthesis of isoquinoline alkaloids by a Zn-ProPhenol-catalysed asymmetric Mannich reaction. | ||
In the same year, they proposed a method to access chiral pyrrolidines wherein the stereoselective Mannich reaction forms the key step. They conducted the synthesis of β-amino ynones in enantioenriched form via Mannich reactions between aldimines and gem dimethyl ynone with 10 mol% Zn-ProPhenol catalyst in THF at room temperature for 12 h to obtain 46–87% yields with good E/Z selectivity and enantioselectivities (70–99% ee) (Scheme 10).43 Sequential cycloisomerization of neutral and electron-rich aromatic rings produced comparable results in the cyclization step under the standard conditions; however, aliphatic and vinyl imines were exceptions. They also carried out the reaction in gram scale with high yields. They proposed a reaction mechanism, which is shown below (Scheme 11).
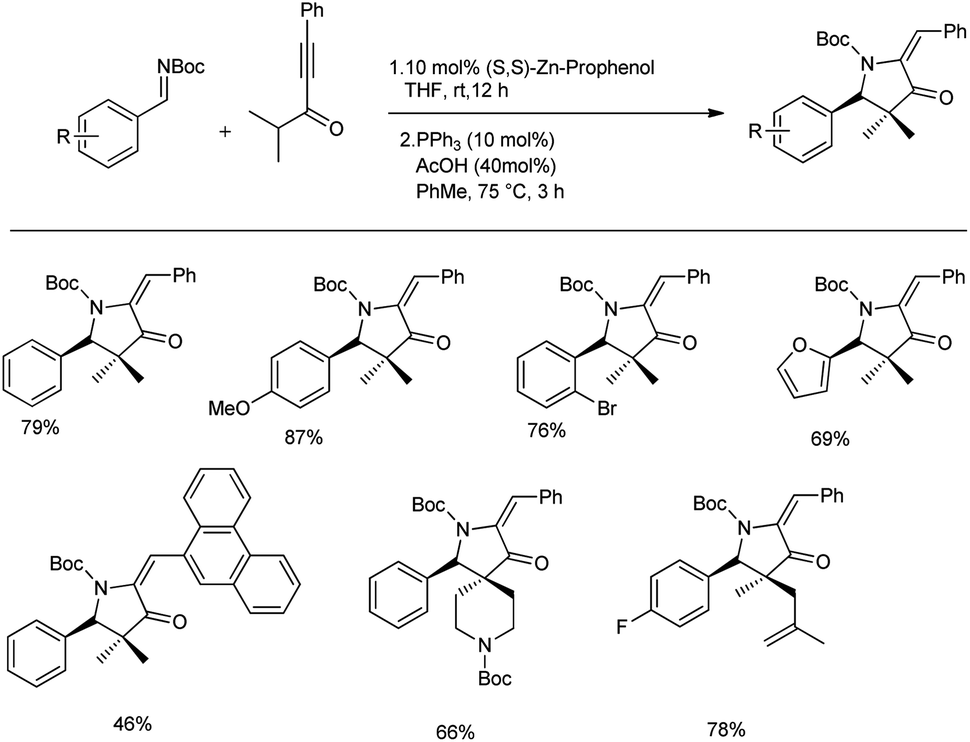 | ||
| Scheme 10 Synthesis of chiral pyrrolidones via a Zn-ProPhenol-catalysed Mannich reaction. | ||
 | ||
| Scheme 11 Proposed mechanism for the synthesis of pyrrolidones via the Zn-ProPhenol-catalysed Mannich reaction. [Reproduced with permission from ref. 43]. | ||
Trost's group also studied the Mannich reaction between 1.2 equiv. of enone as the nucleophile and N-Cbz aldimine as the electrophilic partner in the presence of 10 mol% Zn-(R,R)-L1 in THF at 60 °C for 16 h to afford the desired Mannich adduct with better yield (84–95%) and good enantioselectivity (up to 99%) (Scheme 12).44 With the optimized conditions in hand, imines with electron-withdrawing and donating groups on the aromatic ring and heteroaromatic imines reacted well with exocyclic and endocyclic enones to deliver highly functionalized chiral β-amino ketones. Gram scale reactions also afforded 78–95% yields.
 | ||
| Scheme 12 Mannich reactions between N-carbamoyl imines and α-branched ketones via the Zn-ProPhenol-catalysed Mannich reaction. | ||
Recently, they reported an efficient, scalable protocol to access enantioenriched β-amino ketones bearing a tetrasubstituted α-CF3 and α-SCF3 stereogenic center.45 They used 1.0 equivalents of trifluoromethyl indanone, 1.2 equiv. of imine, 10 mol% of (S,S)-extra pull ProPhenol, and 20 mol% of Et2Zn in hexane and PhMe at 4 °C for 16 h in the presence of molecular sieves to afford the desired Mannich adducts with 71–98% yields and excellent diastereoselectivity (dr > 20:1) (Scheme 13). The Mannich reaction of CF3-substituted ketones with aromatic imines bearing substitutions at the para, meta, and ortho positions afforded excellent yields and selectivities. Moreover, the reaction was found to be compatible with the alkyl-substituted imines; however, the substitutions on the indanone scaffold showed little influence on the reactivity. Also, Zn-ProPhenol-catalysed Mannich reactions of aromatic imines with α-SCF3 ketones proceeded well under the same conditions, wherein an imine substituted with a conjugated alkene and acyclic α-SCF3 ketone afforded the desired adduct. Furthermore, Boc-protected imines gave improved selectivities compared to Cbz-protected imines.
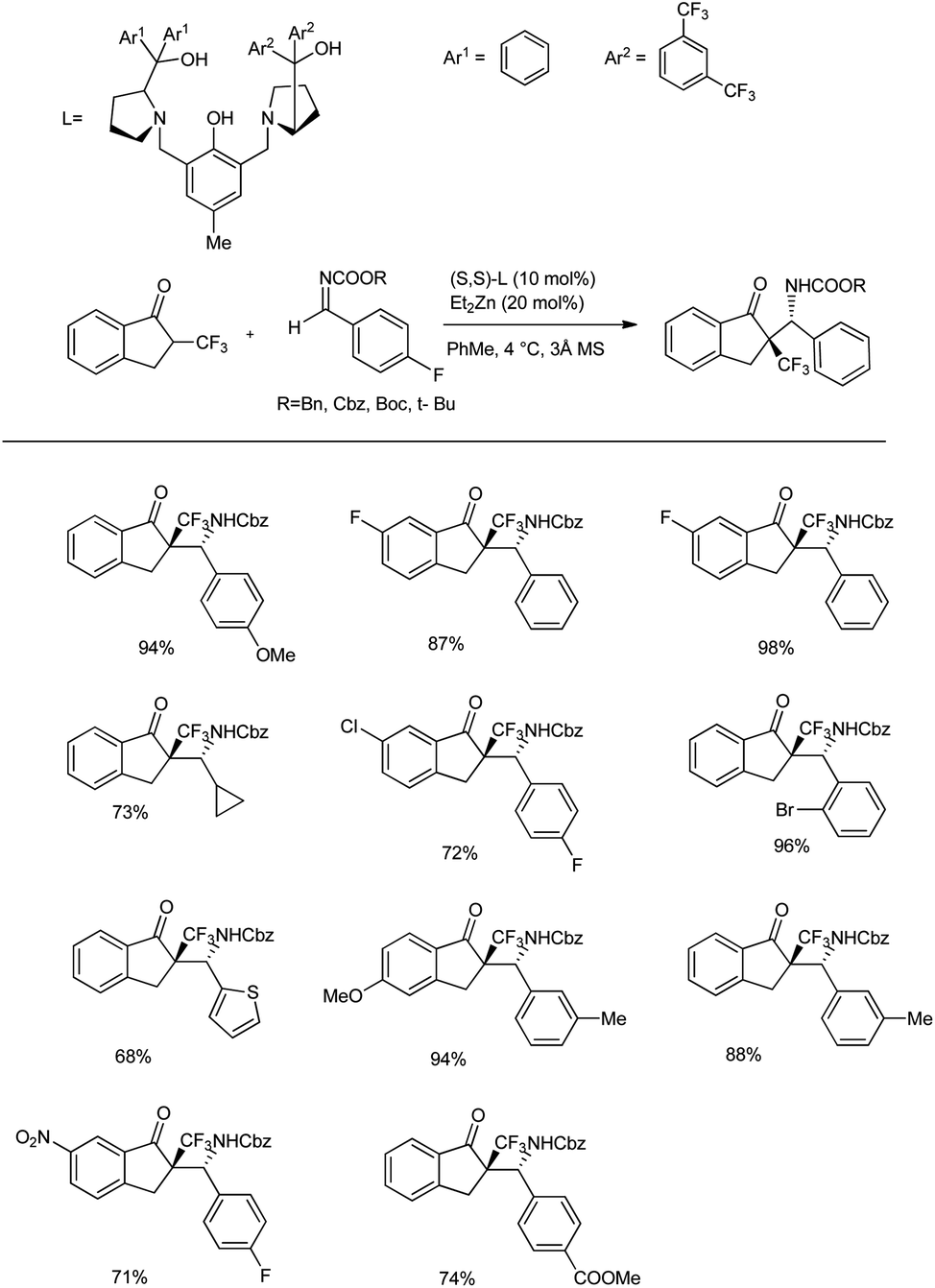 | ||
| Scheme 13 Mannich addition of Zn-ProPhenol-catalysed α-trifluoromethyl- and α-trifluoromethylthio-substituted ketones. | ||
A double Mannich reaction was established by Trost and co-workers where the 1,3-diamines generated upon consecutive Mannich reactions were achieved in 61–96% yields and excellent enantioselectivity (up to 99.5%) and diastereoselectivity (up to >20:1) when they were treated with 2.2 equiv. of the imine with 1.0 equiv. of ynone in the presence of 10 mol% Et2Zn, 5 mol% (S,S)-ProPhenol in THF at room temperature (Scheme 14).46 Under these optimized conditions, the asymmetric double Mannich addition was investigated with diverse imines and a variety of ynones (Scheme 14).
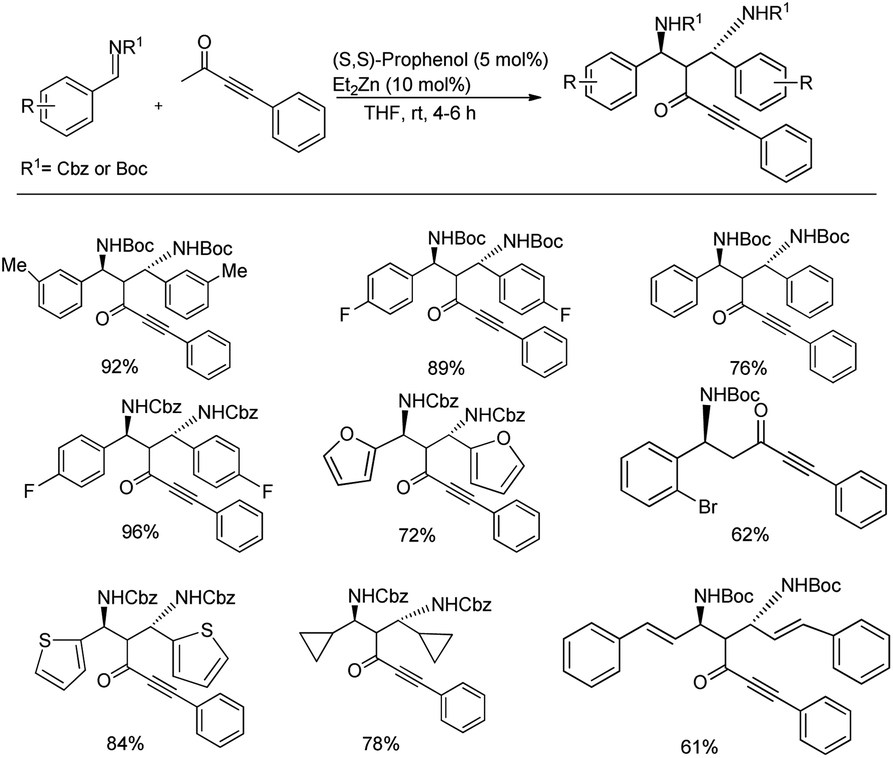 | ||
| Scheme 14 Zn-ProPhenol-catalysed double Mannich reaction between ketones and imines for the preparation of 1,3-diamines. | ||
Very recently, Trost and co-workers investigated the usage of acyclic vinyl α-fluoroketone in the Mannich reaction with 2 equivalents of Boc-protected imine in the presence of 10 mol% Zn/ProPhenol catalyst derived from L2 in a 1:2 ratio of THF and diethyl ether for 16–18 h, which led to the anticipated Mannich adduct in 72–91% yield and with diastereoselectivity (16:1 dr) (Scheme 15).47 Substrates with different substitutions, such as longer alkyl chains, sterically demanding branched alkyl groups, vinyl groups, aromatic rings and heteroaromatic rings, tolerated the reaction well.
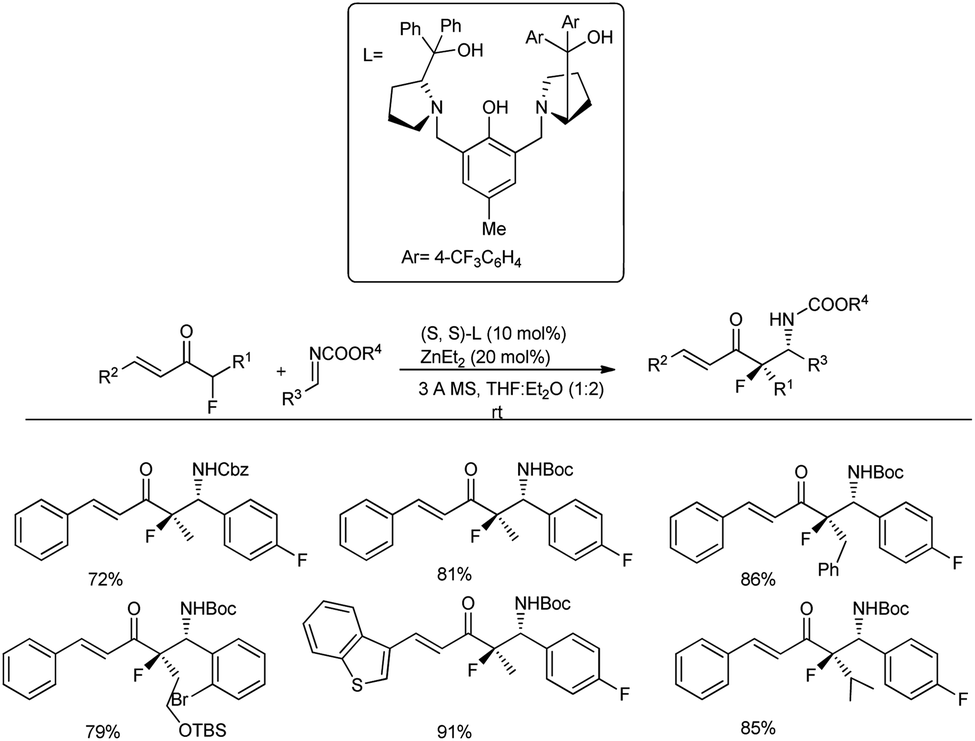 | ||
| Scheme 15 Preparation of acyclic chiral β-fluoro amines by a direct Zn-ProPhenol-catalysed Mannich reaction. | ||
Trost et al. described the synthesis of various chiral aziridines by a Zn-ProPhenol-catalysed Mannich reaction of cyclic 2H-azirines with alkynyl ketones. They treated 0.1 mmol of cyclobutyl-substituted alkynyl ketone with 1.5 equivalents of 2H-azirine in the presence of Zn-ProPhenol catalyst at rt in 0.33 M THF for 12 h to synthesise chiral aziridines with a maximum yield of 91% (Scheme 16).48 The N–H bond acetylation of the formed aziridine was carried out using 2 equivalents of acetic anhydride and double the amount of triethyl amine in DCM for 2 h at rt to produce the desired product with overall 91% yield and 98% ee. Moreover, the optimised conditions were used to evaluate the scope of various alkynyl ketones with electron-neutral, electron-withdrawing and electron-donating groups for the synthesis of aziridines.
 | ||
| Scheme 16 The enantioselective Mannich reaction of cyclic 2H-azirines with alkynyl ketones. | ||
They also proposed a mechanism for the Mannich reaction (Scheme 17). It is initiated by the formation of dinuclear Zn–ProPhenol complex. The zinc enolate A is formed by coordination, followed by deprotonation of the alkynyl ketone by the catalyst. Then, the Lewis acidic site of A coordinates to the 2H-azirine to deliver the complex B. The formation of complex C occurs by the reface attack of the alkynyl ketone to 2H-azirine. Afterwards, the protonation and decomplexation of C with the alkynyl ketone provide the Mannich aziridine adduct and regenerate the zinc enolate A.
 | ||
| Scheme 17 Proposed mechanism for the reaction between cyclic 2H-azirines and alkynyl ketones. [Reproduced with permission from ref. 48]. | ||
2.2 ZnO nanoparticle-catalysed reaction
In 2011, MaGee et al. reported a rapid and efficient method for the one-pot Mannich reaction of aldehydes, ketones and amines in the presence of ZnO-nanoparticles.49 Under the optimized procedure, the reaction was carried out with 4-chlorobenzaldehyde, aniline and cyclohexanone using 0.5 mmol ZnO nanoparticles as the catalyst in water at 60 °C to afford the products in high yields (Scheme 18). This procedure was applied to a wide variety of aromatic aldehydes and amines bearing electron-withdrawing and electron-donating groups to offer facile access to β-amino ketones in excellent yields (74–93%) and with anti-selectivity.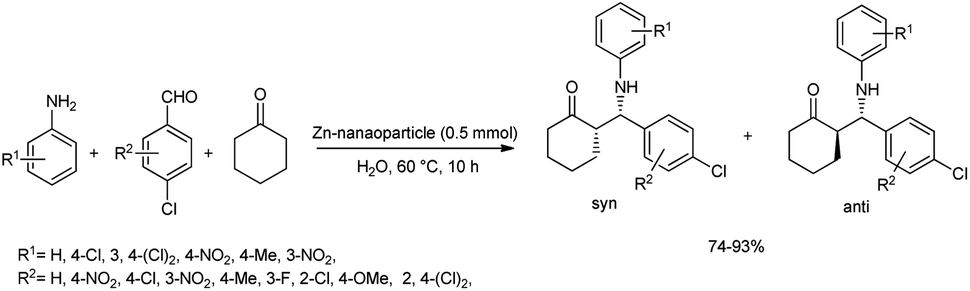 | ||
| Scheme 18 ZnO-nanoparticle-mediated Mannich reaction for the synthesis of β-amino carbonyl compounds. | ||
Sharghi et al. developed a rapid eco-friendly method for the synthesis of lariat ethers in which a ZnO-catalysed and solvent-free Mannich reaction forms a vital step. The lariat ether was prepared from anthraquinone aza crown ether, which was prepared in two steps.50 In the initial step, 1,8-dihydroxyanthraquinone was reacted with ethyl 2-bromoacetate in the presence of a K2CO3 as the base and dry acetone as solvent to produce di-ester anthraquinone in high yields. In the second step, di-ester anthraquinone was reacted with ethylene triamine in ethanol to afford the desired anthraquinone aza crown ether. The synthesised anthraquinone aza crown ether was treated with eugenol and paraformaldehyde in the presence of ZnO nanoparticles at 100 °C for the generation of novel anthraquinone lariat ethers in solvent-free conditions via the three-component Mannich reaction (Scheme 19).
 | ||
| Scheme 19 Mannich reaction for the preparation of new lariat ethers based on anthraquinone using ZnO nanoparticles. | ||
2.3 Lewis acid-promoted Zn catalysed reaction
A zinc-catalysed Mannich reaction of the imine with silyl enol ether was disclosed by Vazdar and co-workers.51 They chose 1-methoxy-1-trimethylsilyloxy-2,2-dimethylethene as the nucleophilic partner with azetidin-2-one-tethered imines in the presence of catalytic and equimolar amounts of zinc(II) iodide (20 mol%) in toluene at −20 °C to deliver the product in good chemical yield (Scheme 20). They also studied the effect of imines with phenyl, ferrocenyl, alkyl and aryl substituents on the diastereomeric ratio of the products obtained.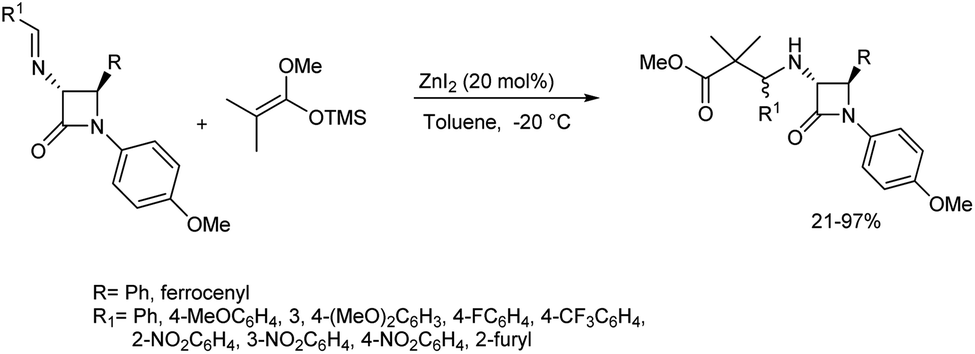 | ||
| Scheme 20 Mannich reaction between β-lactam-tethered aldimines and 1-methoxy-1-trimethylsilyloxy-2,2-dimethylethene. | ||
Dong et al. devised a method for the simple and efficient synthesis of β-amino carbonyl compounds through one-pot Mannich reactions of aldehydes, ketones and aromatic amines using zinc nitrate as the catalyst in anhydrous ethanol for a 6 h reaction time (Scheme 21).52 Given its excellent catalytic activity and low cost, the tartaric acid–zinc nitrate system was found to be the best catalytic system for this Mannich reaction. Although excellent yields of the products were obtained with aromatic aldehydes with electron-withdrawing and electron-donating substituents, aliphatic aldehydes did not afford the desired products.
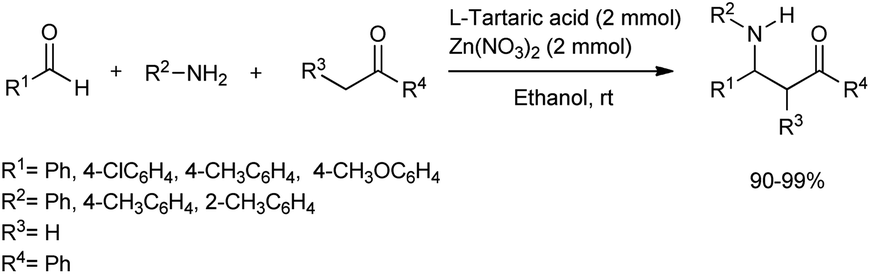 | ||
| Scheme 21 Mannich reaction catalysed via tartaric acid and zinc nitrate. | ||
2.4 Heteronuclear Zn-catalysed reaction
Khalifeh et al. reported the synthesis of a novel series of heterogeneous catalytic complexes for the preparation of propargyl amines under mild conditions without any co-catalyst, activator or base.53 The condensation reaction of aryl acetylene, morpholine and different aldehydes using 2 mol% of Zn complex ([Zn(II)BHPPDAH]) as the catalyst at 70 °C for 2 h in water provided good yields of the products (Scheme 22). Importantly, when amines and terminal aliphatic alkynes were reacted with formalin in the presence of this catalytic system, propargyl amines were obtained in good to excellent yields. This Mannich reaction when carried out on a gram scale afforded 70–95% yields in 2 h.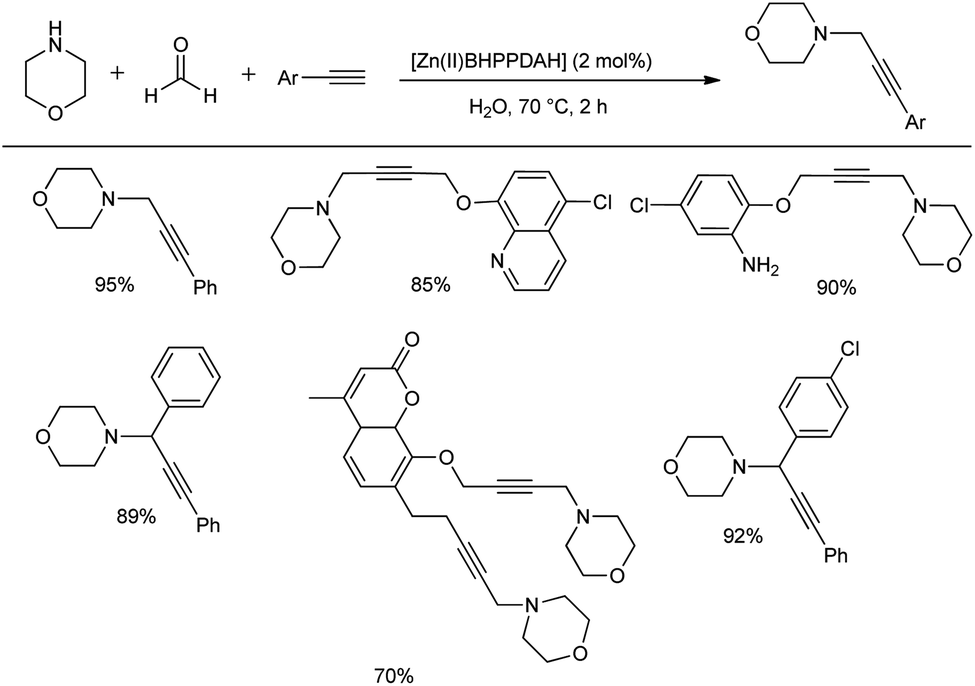 | ||
| Scheme 22 Synthesis of propargyl amines using Zn(II)BHPPDAH catalyst. | ||
Kumar and co-workers pioneered the introduction of the Petasis borono-Mannich (PBR) reaction. It is a multicomponent Mannich-type condensation that involves heteronuclear Zn2Ln2 coordination clusters (CCs) as catalysts with amines, aldehydes and boronic acid as reacting substrates.54 Here, they performed the reaction between salicylic aldehyde, indoline and phenylboronic acid in the presence of 1.0 mol% heteronuclear Zn/Ln coordination clusters at room temperature in DME, and the optimized reaction afforded 84–98% yields (Scheme 23). To illustrate the applicability of the given reaction, different secondary amines, aldehydes and boronic acids were employed in this MCR using Zn/Dy cluster as the best catalyst among other lanthanide clusters.
 | ||
| Scheme 23 Zn-mediated Petasis borono-Mannich (RBR) reaction. | ||
2.5 Zn acetate/Zn-acetic acid-mediated reaction
In 2011, Kidwai et al. developed a protocol for the synthesis of β-amino carbonyl compounds from substituted anilines, benzaldehydes, and cyclohexanone.55 The reaction is carried out at room temperature in acetic acid and water as a solvent using Zn[(L)proline]2 as the catalyst for 12 h to afford a 90% product yield (Scheme 24).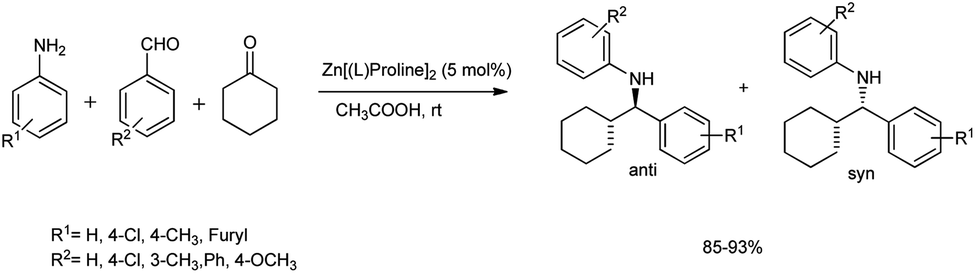 | ||
| Scheme 24 Mannich reaction for the synthesis of β-amino carbonyls using bis[(L)prolinato-N,O]Zn in acetic acid–water. | ||
A plausible mechanism for the first stereoselective three-component Zn[(L)proline]2-catalysed Mannich reaction is shown in Scheme 25. The proposed mechanism shows the preparation of the imine from the aldehyde and amine at room temperature in the presence of two drops of acetic acid in water with cyclohexanone in the presence of Zn[(L)proline]2.
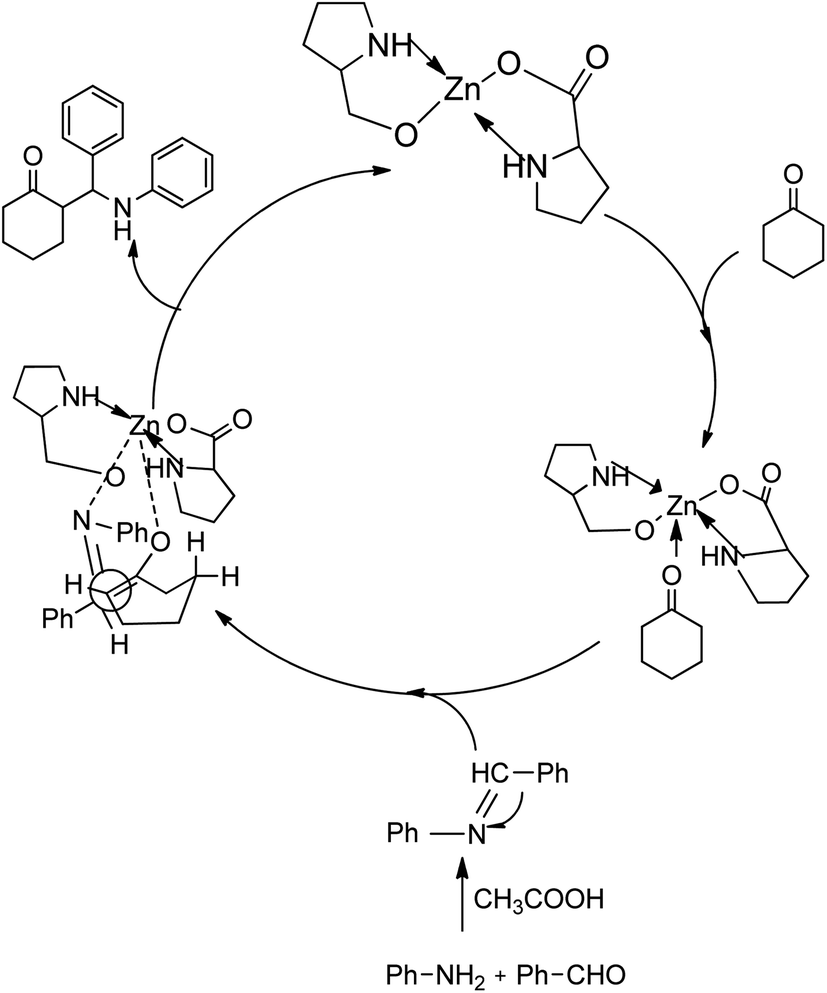 | ||
| Scheme 25 Mechanism for the synthesis of β-amino carbonyls. [Reproduced with permission from ref. 55]. | ||
Vedamurthy and co-workers established a protocol for the synthesis of phenol-functionalized nylon 6 with enhanced dielectric and hydrophobic properties through a zinc acetate-catalysed Mannich reaction.56 The reaction procedure must be fixed to obtain the desired product; otherwise, it can lead to a mixture of polymer products containing more of the phenolic resin as a gel instead of the expected phenol-functionalized nylon 6 polymers. Initially, nylon 6 was dissolved in formic acid, followed by the addition of formaldehyde. Then, the mixture was stirred vigorously at room temperature for 2 h, which led to the formation of hydroxymethyl amide-6. Subjecting hydroxymethyl amide to chloroform, phenol and Zn(II) acetate at room temperature for 3 days provided a slightly turbid solution, and the final product was obtained after washing with hot water to remove any unreacted phenol, formaldehyde or zinc acetate (Scheme 26).
 | ||
| Scheme 26 Mannich reaction for the synthesis of phenol-functionalized nylon 6 polymers using a zinc acetate catalyst. | ||
2.6 Miscellaneous
An asymmetric Mannich reaction of 5H-oxazol-4-ones was accomplished by Zhao and co-workers.57 They carried out the reaction between the N-Dpp (Dpp = diphenylphosphinoyl)imine and 5H-oxazol-4-one catalysed by L1/Et2Zn (10 mol%) in the presence of dialkyl phosphoramidates and THF at 0 °C (Scheme 27). The reaction resulted in structurally important α-alkyl norstatine derivatives in good yields and excellent diastereoselectivities. They also attempted a series of reactions with different substrates employing the developed procedure; further, the reaction was performed on the gram-scale.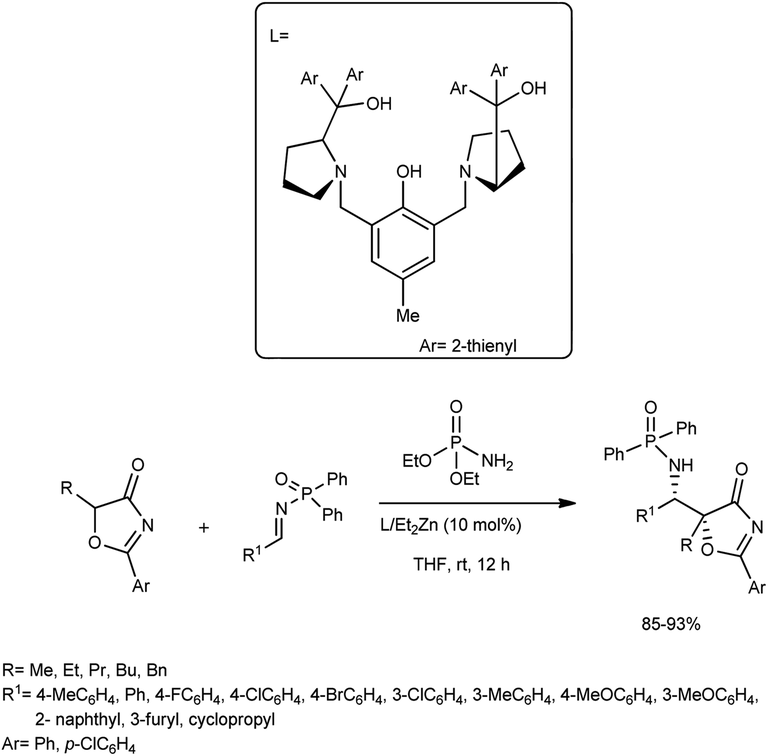 | ||
| Scheme 27 Catalytic asymmetric Mannich reactions for the synthesis of α-alkyl norstatine derivatives. | ||
Zhao et al. proposed a method for the synthesis of vicinal tetrasubstituted stereocenters from silyl ketene imines with isatin-derived ketimines.58 They took 0.10 mmol isatin-derived N-Boc ketimine with 0.20 mmol silyl ketene imines in 2.5 mol% Zn(OTf)2/L-PrPh at −45 °C for 12 h in 0.5 mL THF as a solvent to produce the desired β-amino nitriles with high yields (above 90%) and good enantioselectivity (up to 99%) and diastereoselectivity in >19:1. They also evaluated the scope of the reaction with various isatin-derived ketimines and multiple kinds of silyl ketene imines (Scheme 28).
 | ||
| Scheme 28 Synthesis of vicinal tetrasubstituted stereocenters using silyl ketene imines with isatin-derived ketimines. | ||
Yuan et al. proposed the Mannich reaction of difluoroenoxysilanes with hydrazone using imidazoline-anchored phosphine ligand–zinc(II) complexes. While conducting the reaction, they employed 10 mol% of Lewis acid Zn(NTf2)2 together with an imidazoline–phosphine ligand as the catalytic combination in a mixture of THF and MeOH at 5 °C for 24 h to ensure effective Mannich reactions of the substrates used (Scheme 29).59 They found that the reaction proceeded smoothly to afford the desired product in good chemical yield and enantiomeric excess.
 | ||
| Scheme 29 Mannich-type reaction using imidazoline-anchored phosphine ligand Zn(NTf2)2 complexes. | ||
A strategy for the synthesis of pyrrolidin-2-ones by a conjugate addition/nitro-Mannich/lactamization sequence was developed by Anderson and co-workers (Scheme 30).60 Here, they conducted the conjugate addition of a diorgano zinc reagent to nitroacrylate, followed by the trapping of the resultant nitronate anion with the imine; this resulted in a β-nitroamine intermediate which cyclized in situ to afford the desired pyrrolidin-2-ones (Scheme 30). With the optimal conditions in hand, other N-protecting groups on the imine derived from benzaldehyde also enabled the reaction to provide access to many densely functionalized pyrrolidines.
 | ||
| Scheme 30 Mannich reaction for the synthesis of densely functionalized pyrrolidin-2-ones. | ||
Gall et al. suggested an unusual method for the zinc and cobalt-catalysed synthesis of α,β-disubstituted N-sulfonyl β-amino esters.61 They treated N-benzylidene-4-methylbenzenesulfonamide in acetonitrile with 4.5 equiv. of acrylate and 2.4 equiv. of bromobenzene in the presence of a reducing metal such as Zn and cobalt bromide to attain the Mannich adduct in 30 minutes (Scheme 31). The experiment was performed with different aromatic bromides containing either electron-withdrawing or electron-donating groups to furnish good yields of the products. A plausible reaction mechanism is depicted in Scheme 32. The reaction starts with a zinc-mediated redox process from the aryl bromide for the formation of organocobalt. The acrylate and organometallic intermediate undergo conjugate addition to produce a zinc enolate that undergoes a Mannich-type addition to the activated imine to yield the final product.
 | ||
| Scheme 31 Zinc-mediated Mannich reaction for the preparation of α,β-disubstituted N-sulfonyl β-amino esters. | ||
 | ||
| Scheme 32 Possible mechanism of the Mannich reaction. [Reproduced with permission from ref. 61]. | ||
Nakamura et al. developed an enantioselective vinylogous Mannich reaction of trimethyl siloxy furans with ketimines using N-picolinoyl-9-amino-9-deoxy-epi-cinchonine and Zn(OTf)2 catalyst.62 While conducting the reaction, γ-butenolide and ketimines were treated with 10 mol% of Zn(OTf)2 as a Lewis acid, Et3N as the base and a chiral ligand derived from cinchona alkaloids at 0 °C in THF for 24–72 h to yield the desired products with high enantioselectivity (Scheme 33). Under the optimized conditions, they screened a broad range of ketimines, including heteroaryl imines, to obtain products in high yields and excellent diastereo- and enantioselectivities. From the proposed mechanism, we can conclude that the Zn(OTf)2 reacts with the ligand to produce the complex A. Coordination of the γ-proton with Zn(II) and successful deprotonation of γ-proton with Et3N produce complex B containing the dienolate. On the Zn(II) cation, the ligand exchange reaction of triflate for ketimine affords complex C, and further reaction with dienolate affords complex D (Scheme 34).
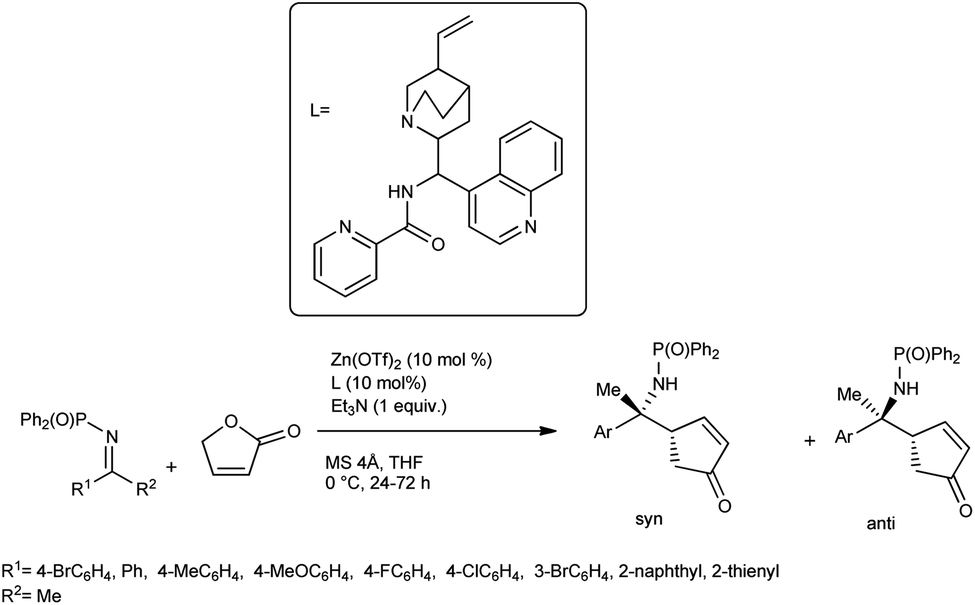 | ||
| Scheme 33 Cinchona alkaloid amide/zinc(II)-catalysed Mannich reaction of ketimines with γ-butenolide. | ||
 | ||
| Scheme 34 Plausible reaction mechanism for the asymmetric direct vinylogous Mannich reaction of ketamine with γ-butenolide. [Reproduced with permission from ref. 62]. | ||
Chrzanowski and co-workers developed a protocol in which enantiopure heteroatom-containing chiral ligands were used in the formation of asymmetric C–C bonds.63 Here, they chose 1-hydroxypropan-2-one, 4-nitrobenzaldehyde and 4-methoxyaniline as the substrates under optimized reaction conditions of 15 mol% Zn(OTf)2, L1, L2, L3 and L4 as the ligands, acetic acid, and DMO in the presence of ultrasound to afford excellent yields of the products (Scheme 35).
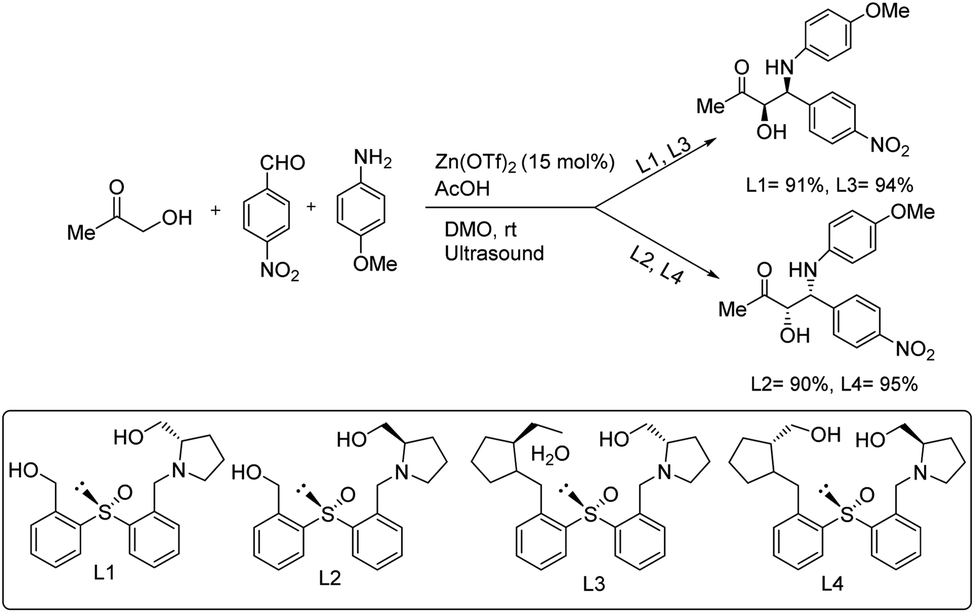 | ||
| Scheme 35 Asymmetric C–C bond formation via a Zn-catalysed Mannich reaction. | ||
Saikia et al. demonstrated a method to synthesise three multifaceted acidic 1,3-disulfoimidazolium chlorometallate systems, such as [Dsim]2[ZnCl4], in the solid state from the reaction between 1,3-disulfoimidazolium chlorides ([Dsim]Cl) with ZnCl2 in different molar ratios (1:1 and 2:1) at 60 °C for 2 h followed by an acid-catalysed multicomponent Mannich-type reaction between acetophenone, aromatic aldehydes and substituted anilines in ethanol at room temperature to produce β-amino carbonyl compounds (Scheme 36).64
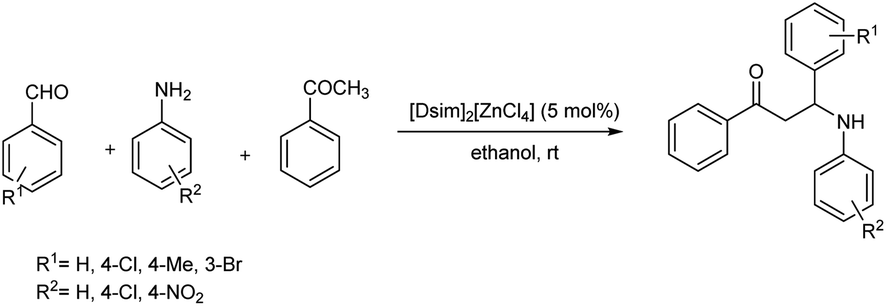 | ||
| Scheme 36 Synthesis of β-amino carbonyl compounds using [Dsim]2[ZnCl4]. | ||
The Presset research group developed a new method for the synthesis of indolines by a Zn-mediated Mannich reaction.65 Indolines are important structural parts in numerous naturally occurring alkaloids, such as strychnine or oleracein A–D, and in other bioactive compounds, such as pentopril. They carried out an organometallic Mannich reaction between 2-bromobenzyl bromide, aniline and 4-tolualdehyde in the presence of 2 equiv. of zinc dust in acetonitrile at room temperature to furnish the desired ortho-halogenated β-arylethylamine products in 36–88% yields (Scheme 37). Unfortunately, no reaction was observed when primary amides, ketones, pivaldehyde or methyl pyruvate were used as carbonyl compounds.
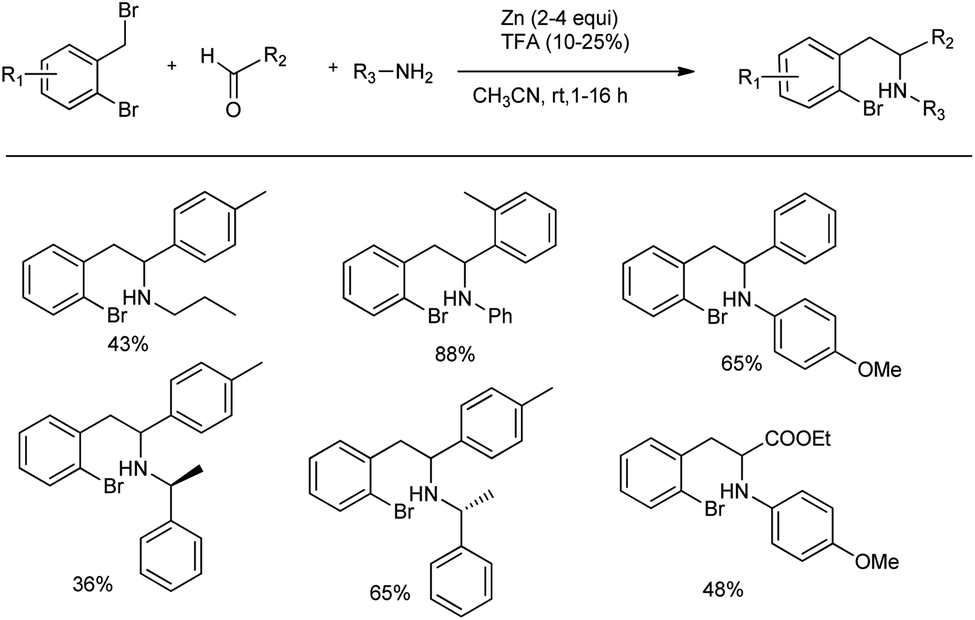 | ||
| Scheme 37 Zn-mediated Mannich reaction for the synthesis of indolines. | ||
3. Conclusions
The present review summarizes the developments in zinc-catalysed Mannich reactions. The Mannich reaction is a classical method for the preparation of β-amino ketones and aldehydes. Additionally, three-component Mannich reactions are important in organic synthesis for preparing essential synthetic intermediates due to the possibility of the introduction of several elements to a molecule in a single step.Zinc-catalysed Mannich reactions using Zn-ProPhenol complexes were developed majorly by Trost's group and afforded a large class of functionalized β-amino carbonyl compounds. Their work based on ynone acceptors tolerates a variety of functional groups on the donors and acceptors, allowing rapid construction of chiral and heavily functionalized α-substituted β-amino ynones.
Most of the protocols exhibit high chemo-, diastereo-, and enantioselectivities and high functional group tolerance. More importantly, the reaction offers rapid access to a broad spectrum of amine-containing skeletal types, including pyrrolidin-2-ones, propargyl amines and indoline derivatives. This is suggestive of the application of the products in medicinal chemistry and the preparation of pharmaceutical analogues and complex natural products. Moreover, zinc-catalysed reactions have gained much attention because of their low-cost, less toxic and environmentally friendly nature. Considering the broad scope and functional group compatibility of the present methodology along with the environmentally benign catalytic systems, we anticipate that the process can be expanded to other acceptor and donor systems for the Mannich reaction in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CMA and RMP thank the Council of scientific and Industrial Research (CSIR), New Delhi for the award of junior research fellowships. GA thanks the Kerala State Council for Science Technology and Environment (KSCSTE) Trivandrum, India for a research grant (Order No. 341/2013/KSCSTE dated 15.03.2013).References
- S. Kobayashi, R. Matusubara, Y. Nakamura, H. Kitagawa and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 2507 CrossRef CAS.
- C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51 CrossRef CAS.
- D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602 CrossRef CAS.
- R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123 CrossRef CAS.
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3169 CrossRef.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS.
- W. A. Nugent, T. V. RajanBabu and M. J. Burk, Science, 1993, 259, 479 CrossRef CAS.
- M. Ikunaka, Chem.–Eur. J., 2003, 9, 378 CrossRef CAS.
- M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044 CrossRef.
- C. Mannich and W. Krosche, Arch. Pharm., 1912, 250, 647 CrossRef CAS.
- (a) D. C. Cole, Tetrahedron, 1994, 50, 9517 CrossRef CAS; (b) M. J. Brown, Heterocycles, 1989, 29, 2225 CrossRef CAS.
- R. J. Robinson, J. Chem. Soc., Trans., 1917, 111, 762 RSC.
- S. Saito, K. Hatanaka and H. Yamamoto, Tetrahedron, 2001, 57, 875 CrossRef CAS.
- T. Akiyama, K. Matsuda and K. Fuchibe, Synlett, 2005, 322 CrossRef CAS.
- T. Akiyama, J. Takaya and H. Kagoshima, Synlett, 1999, 1024 Search PubMed.
- T. P. Loh, S. B. K. W. Liung, K. L. Tan and L. L. Wei, Tetrahedron, 2000, 56, 3227 CrossRef CAS.
- C. X. Zhang, J. C. Dong, T. M. Cheng and R. T. Li, Tetrahedron Lett., 2001, 42, 461 CrossRef CAS.
- L. M. Wang, J. W. Han, J. Sheng, H. Tian and Z. Y. Fan, Catal. Commun., 2005, 6, 201–204 CrossRef CAS.
- B. C. Ranu, S. Samanta and S. K. Guchhait, Tetrahedron Lett., 2002, 58, 983–988 CrossRef CAS.
- T. Ollevier and E. Nadeau, J. Org. Chem., 2004, 69, 9292 CrossRef CAS.
- S. Iimura, D. Nobutou, K. Manabe and S. Kobayashi, Chem. Commun., 2003, 1644 RSC.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS.
- A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 3175 CrossRef CAS.
- M. Terada, K. Sorimachi and D. Uraguchi, Synlett, 2006, 133 CrossRef CAS.
- M. Urbaniak and W. Iwanek, Tetrahedron, 2006, 62, 1508 CrossRef CAS.
- N. Azizi, L. Torkiyan and M. R. Saidi, Org. Lett., 2006, 8, 2079 CrossRef CAS.
- A. S. Gonzalez, R. G. Arrayas and J. C. Carretero, Org. Lett., 2006, 8, 2977 CrossRef CAS.
- K. Manabe and S. Kobayashi, Org. Lett., 1999, 1, 1965 CrossRef CAS.
- M. Kidwai, N. K. Mishra, V. Bansal, A. Kumar and S. Mozumdar, Tetrahedron Lett., 2009, 50, 1355 CrossRef CAS.
- P. D. Sawant, J. Josena, V. V. Balasubramanian, A. Katsuhiko, S. Pavuluri, V. Sivan, H. Shivappa and B. V. Ajayan, Chem.–Eur. J., 2008, 14, 3200 CrossRef.
- H. Wu, X. Chen, W. L. Ye, H. Xin, H. Xu, C. Yue, L. Pang, R. Maa and D. Shi, Tetrahedron Lett., 2009, 50, 1062 CrossRef CAS.
- K. Li, T. He, C. Li, X. W. Feng, N. Wang and X. Q. Yu, Green Chem., 2009, 11, 777–779 RSC.
- M. F. Pilz, C. Limberg, B. B. Lazarov, K. C. Hultzsch and B. Ziemer, Organometallics, 2007, 3668 CrossRef CAS.
- S. Doherty, P. Goodrich, C. Hardacre, H. K. Luo, M. Nieuwenhuyzen and R. K. Rath, Organometallics, 2005, 24, 5945 CrossRef CAS.
- B. M. Trost, T. Saget, A. Lerchen and C. J. Hung, Angew. Chem., Int. Ed., 2016, 55, 781 CrossRef CAS.
- B. M. Trost and C. J. Hung, J. Am. Chem. Soc., 2015, 15940 CrossRef CAS.
- B. M. Trost, T. Saget and C. J. Hung, J. Am. Chem. Soc., 2016, 138, 3659 CrossRef CAS.
- B. M. Trost, E. Gnanamani, J. S. Tracy and C. A. Kalnmals, J. Am. Chem. Soc., 2017, 139, 18198 CrossRef CAS.
- B. M. Trost, T. Saget and C. J. Hung, Angew. Chem., Int. Ed., 2017, 56, 2440 CrossRef CAS.
- B. M. Trost, C. J. Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523 CrossRef CAS.
- B. M. Trost, C. J. Hung and M. I. Scharf, Angew. Chem., Int. Ed., 2018, 57, 11408 CrossRef CAS.
- B. M. Trost, C. J. Hung and Z. Jiao, J. Am. Chem. Soc., 2019, 141, 16085 CrossRef CAS.
- B. M. Trost, E. Gnanamani, C. J. Hung and C. A. Kalnmals, Org. Lett., 2019, 21(6), 1890 CrossRef CAS.
- B. M. Trost, C. J. Hung and E. Gnanamani, ACS Catal., 2019, 9, 1549 CrossRef CAS.
- B. M. Trost, C. J. Hung, G. Mata, Y. Liu, Y. Lu and E. Gnanamani, Org. Lett., 2020, 2437 CrossRef CAS.
- B. M. Trost and E. Gnanamani, Org. Lett., 2020, 22, 1675 CrossRef CAS.
- B. M. Trost, J. S. Tracy, T. Yusoontorn and C. J. Hung, Angew. Chem., Int. Ed., 2020, 59, 2370 CrossRef CAS.
- B. M. Trost and C. Zhu, Org. Lett., 2020, 22, 9683 CrossRef CAS.
- D. I. MaGee, M. Dbiri, P. Salehi and L. Torkianb, ARKIVOC, 2011, 11, 156 Search PubMed.
- H. Sharghi, A. Khoshnood, M. M. Doroodmand and R. Khalifeh, J. Heterocycl. Chem., 2016, 53, 164 CrossRef CAS.
- K. Vazdar, D. Margetić and I. Habuš, Heterocycles, 2011, 83, 63 CrossRef CAS.
- H. Dong, Q. Liua, Y. Tiana and Y. Qiaob, J. Chem. Res., 2018, 42, 463 CrossRef CAS.
- R. Khalifeh, H. Sharghi and Z. Rashidi, Heteroat. Chem., 2013, 24, 372 CrossRef CAS.
- P. Kumar, K. Griffiths, S. Lymperopoulou and G. E. Kostakis, RSC Adv., 2016, 6, 79180–79184 RSC.
- M. Kidwai, A. Jain, R. Poddar and S. Bhardwaj, Appl. Organomet. Chem., 2011, 25, 335 CrossRef CAS.
- T. Vedamurthy and M. Murugesa, Mater. Chem. Phys., 2018, 216, 51 CrossRef.
- D. Zhao, L. Wang, D. Yang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 1 CrossRef.
- J. N. Zhao, B. Fang, W. W. Luo, X. Y. Hao, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 241 CrossRef CAS.
- Z. Yuan, L. Mei, Y. Wei, M. Shi, P. V. Kattamuri, P. McDowell and G. Li, Org. Biomol. Chem., 2012, 10, 2509 RSC.
- J. C. Anderson, L. R. Horsfall, A. S. Kalogirou, M. R. Mills, G. J. Stepney and G. J. Tizzard, J. Org. Chem., 2012, 77, 6186 CrossRef CAS.
- E. l. Gall, S. Sengmany, I. Samb, S. Benakrour, C. Colin, A. Pignon and E. Léonel, Org. Biomol. Chem., 2014, 12, 3423 RSC.
- S. Nakamura, R. Yamaj and M. Hayashi, Chem.–Eur. J., 2015, 21, 1 CrossRef.
- J. Chrzanowski, M. Rachwalski, A. M. Pieczonka, S. Lesniak, J. Drabowicza and P. Basinski, Tetrahedron, 2016, 72, 2649 CrossRef CAS.
- S. G. Saikia, A. K. Dutta, P. Sarmah and R. Borah, J. Mol. Catal. A: Chem., 2016, 416, 63 CrossRef CAS.
- M. Presset, A. Pignon, J. Paul, E. L. Gall, E. Leonel and T. Martens, J. Org. Chem., 2017, 82, 3302 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |