
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer potential of nitric oxide (NO) in neuroblastoma treatment
Jenna L. Gordon a,
Kristin J. Hinsenb,
Melissa M. Reynolds*c,
Tyler A. Smithd,
Haley O. Tuckere and
Mark A. Brownf
a,
Kristin J. Hinsenb,
Melissa M. Reynolds*c,
Tyler A. Smithd,
Haley O. Tuckere and
Mark A. Brownf
aDepartment of Chemistry, Colorado State University, Fort Collins, CO 80521, USA. E-mail: jenna.short@colostate.edu
bDepartment of Biomedical Sciences, Colorado State University, Fort Collins, CO 80521, USA. E-mail: khinsen@rams.colostate.edu
cDepartment of Chemistry, School of Biomedical Engineering, Department of Chemical and Biological Engineering, Colorado State University, Campus Delivery 1872, Fort Collins, CO 80523, USA. E-mail: Melissa.reynolds@colostate.edu
dDepartment of Neuroscience, University of Texas Austin, 2500 Speedway, Austin, TX 78712, USA. E-mail: tylersmith128@utexas.edu
eDepartment of Molecular Genetics, Institute for Cellular and Molecular Biology, University of Texas Austin, Austin, TX 78712, USA. E-mail: haleytucker@austin.utexas.edu
fDepartment of Clinical Sciences, Colorado State University, Fort Collins, CO 80523, USA. E-mail: mark.brown@colostate.edu
First published on 1st March 2021
Abstract
The most common extracranial solid tumor in childhood, paediatric neuroblastoma, is frequently diagnosed at advanced stages and identified as high risk. High risk neuroblastoma is aggressive and unpredictable, resulting in poor prognosis and only ∼40% five-year survival rates. Herein, nitric oxide (NO) delivered via the S-nitrosothiol, S-nitrosoglutathione (GSNO), is explored as an anticancer therapeutic in various neuroblastoma lines. After 24 h of treatment with GSNO, cell viability assays, as assessed by resazurin and MTT ((3-4,5-dimethylthiazol-2-yl)-2,5-diphyltetrazolium bromide), consistently identified a moderate, ∼13–29%, decrease in metabolic activity, colony formation assays revealed notably significant reduction of clonogenic activity, and cytotoxicity assays revealed a visibly significant reduction of total number of cells and live cells as well as an increase in number of dead cells in treated cells versus untreated cells. Thrillingly, RNA-sequence analysis provided highly valuable information regarding the differentially expressed genes in treated samples versus control samples as well as insight into the mechanism of action of NO as an anticancer therapeutic. Favorably, the collective results from these analyses exhibited tumoricidal, non-tumour promoting, and discriminatory characteristics, illuminating the feasibility and significance of NO as a cytotoxic adjuvant in neuroblastoma treatment.
Introduction
Paediatric neuroblastoma is characterized by a majority (>60%) of initial diagnoses resulting in high risk categorization and recurrence.1,2 Although low to intermediate risk diagnoses have favourable five-year survival rates, >80%,3–6 high risk diagnoses reveal particularly poor prognosis, only ∼40% five-year survival rates.7,8 Largely, treatment options for high risk neuroblastoma include any variation of successive treatments including surgery, chemotherapy, radiation therapy, immunotherapy and bone marrow transplants.7,8 Despite the host of potential therapeutic possibilities, high risk and recurrent neuroblastoma continue to perplex doctors and anticancer researchers.As a result, recent work has focused on the development of various treatment options to combat the aggressive nature of high risk and recurrent neuroblastoma, such as enzyme and angiogenesis inhibition, targeted therapies, cytotoxic agents, and more.1 Furthermore, current research shows that bursts of elevated concentrations of nitric oxide (NO) effectively induce tumour-specific cytotoxicity in a range of malignancies, including ovarian, breast, prostate, and more.9–15 However, minimal focus has been placed on the use of NO as a treatment option for neuroblastoma.16,17 One major challenge in the use of NO as an anticancer therapeutic is the ability to control release kinetics and site-specific delivery. As such, various platforms for NO delivery have been explored, such as liposomes, diazeniumdiolates and S-nitrosothiols (RSNOs).11–13,18,19 Two major advantages to RSNOs are their natural occurrence as NO-donors in the human body and the ability to allow prolonged NO release.18–20 Yet the actual rate of NO release is dependent on the presence of light, metals, heat, and pH. In general, lower pH accelerates the rate of NO release from S-nitrosothiols.21–23 Theoretically, the decreased pH in tumour microenvironments will increase the rate of NO release,24 leading to higher concentration of NO near neoplastic cells than healthy cells. NO-delivery via S-nitrosothiols presents an impactful adjuvant to current neuroblastoma therapies.
Another perplexing aspect of the use of NO in anticancer treatment is the dual function of NO on malignancies.25–30 Explicitly, NO has been shown to induce both tumour-promoting, anti-apoptotic effects as well as tumoricidal, apoptotic effects.25–30 Even though these effects seem to be contradictory, current research indicates that two primary factors influence these outcomes, including concentration and exposure time.27–29 Generally, high levels of NO (micromolar concentrations) induce DNA-damage and therefore apoptotic effects while low levels of NO are linked to tumour progression and metastasis.28,29 Exposure time is not as straight forward, as increased exposure time can lead to increased cell death or tumour progression based on the NO concentration.27 Additional factors that impact the effect of NO on cancer include tumour type, location, microenvironment (including pH and composition), and heterogeneity.25,28
In a previous foundational study, we showed that micromolar concentrations of NO, delivered from 1 mM GSNO over 24 h, was moderately effective as a discriminatory therapeutic against murine N2a neuroblastoma cells.31 These studies revealed a consistent decrease in cell viability of N2a cells, ∼20–25%, assessed via multiple cellular viability assays as well as a complete cessation of colony formation capacity after therapeutic treatment. Healthy Human Dermal Fibroblast, adult (HDFa) cells were also exposed to 1 mM GNSO for 24 h and showed no decrease in cellular viability or colony formation capacity. Based on these initial results, complementary investigation of NO, delivered by GSNO, as an adjuvant anticancer agent against neuroblastoma was decidedly necessary.
Herein, anticancer applications of NO on murine (rat) and human neuroblastoma cell lines were expanded using S-nitrosoglutathione (GSNO) as a NO-donor. Prominently, various clinically relevant neuroblastoma cell lines, IMR-32 (human), SK-N-SH (human), and B104 (murine, rat), were exposed to 1 mM GSNO for 24 h and analysed via cellular viability, colony formation, cytotoxicity, and RNA-sequence analysis assays. The goal of this study was to feature NO as a selective agent in the impairment of neuroblastoma cellular viability, colony formation capacity, and initiate the investigation of its role in provocation of cell death and NO-mediated genetic alterations.
Experimental
Materials
Hydrochloric acid (HCl) and EPA vials were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Sodium nitrite (99.999% NaNO2) was purchased from Alfa Aesar (Ward Hill, MA, USA). Acetone (≥99.5%) was purchased from Sigma Aldrich (St. Louis, MO, USA). Dulbecco's Modified Eagle's Medium (DMEM), Eagle's Minimum Essential Medium (EMEM), and Penicillin–Streptomycin solution were purchased from Fisher Scientific (Hampton, NH, USA). EquaFETAL 100% Origin Bovine Serum was purchased from Atlas Biologicals (Fort Collins, CO, USA). Trypsin/EDTA solution was purchased from American Type Culture Collection (Manassas, VA, USA). Reduced glutathione (GSH; High Purity), CellTiter-Blue Cell Viability Assay (Resazurin), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphyltetrazolium bromide (MTT) were obtained from VWR International (Radnor, PA, USA). Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich. Calcein AM and propidium iodide were purchased from Invitrogen (Eugene, Oregon, USA). The neuroblastoma cell lines used include B104, IMR-32, and SK-N-SH.Synthesis of S-nitrosoglutathione (GSNO)
S-Nitrosoglutathione (GSNO) was synthesized through a previously developed synthesis. Succinctly, GSNO synthesis involved the addition of sodium nitrite to a solution of reduced glutathione (GSH) in Millipore water and 2 M hydrochloric acid. The GSNO mixture was constantly stirred in an ice bath for 40 min. The solution was then treated with acetone and allowed to continue reacting with constant stirring in an ice bath for 10 min (mixture turned red in colour). The red solution was filtered for 10 min with gravity filtration and then vacuum filtration for 3.5 h to isolate the GSNO precipitate. The final GSNO precipitate was washed successively with ice-water and acetone. The red filtrate solution was discarded as waste and the remaining solid pink powder (GSNO) was kept and analysed by UV-Vis spectrophotometry at 335 nm to confirm >95% purity.Cell culture
10% total volume foetal bovine serum and 1% total volume penicillin–streptomycin were added to DMEM/EMEM media to produce complete cell media (complete DMEM/EMEM). 1 mL (106 cells) were thawed for 1–2 min in a 37 °C water bath to prepare the stock culture. The thawed cells were then added to a 15 mL centrifuge tube containing 9 mL of complete media that had been warmed to 37 °C. Once centrifuged for 5 minutes at 4 °C, 2000 RPM, the supernatant was removed and discarded while the pellet was resuspended in 5 mL complete media. This was added to a T-25 cm2 flask containing 5 mL of complete media. The cell culture was incubated at 37 °C, 5% CO2 for at least 48 h before fresh complete media was appropriately provided every 24–72 h. The cell culture was counted and split based on both macroscopic observation and cell counting via a haemocytometer.Cell viability assays
Cells were plated in 96-well plates in 100 μL increments that contained between 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 cells per millilitre (mL). After 24 hours, the media was aspirated and discarded. The positive control samples (PC; ≥7 samples) received 100 μL of complete media, the functional control samples (GSH; ≥7 samples) received 100 μL of 1 mM GSH, and the test samples received 100 μL of 1 mM GSNO (sample; ≥7 samples). After another 24 h of incubation, the media was aspirated and replaced with 100 μL of fresh complete media in each well. The appropriate cell viability assay was then performed.
000–200000 cells per millilitre (mL). After 24 hours, the media was aspirated and discarded. The positive control samples (PC; ≥7 samples) received 100 μL of complete media, the functional control samples (GSH; ≥7 samples) received 100 μL of 1 mM GSH, and the test samples received 100 μL of 1 mM GSNO (sample; ≥7 samples). After another 24 h of incubation, the media was aspirated and replaced with 100 μL of fresh complete media in each well. The appropriate cell viability assay was then performed.
In the resazurin assay, cells were plated at 200000 cells per mL in a 96-well plate. Following the procedure above, 20 μL of pre-warmed resazurin stock solution was added to each well. The plate was incubated at 37 °C, 5% CO2 for 3 h before the absorbance was measured at 570 nm and 600 nm via a microplate reader.
In the MTT assay, cells were plated at 100000 cells per mL in a 96-well plate. Following the procedure above, 10 μL of pre-warmed 12 mM MTT stock solution was added to each well before the plate was placed back in the 37 °C, 5% CO2 incubator. After about 3 h of incubation, 50 μL DMSO was added to each well. The plate was again incubated for an additional 10 min before the absorbance was measured at 540 nm via a microplate reader.
A BioTek Synergy 2 Multi-Detection Microplate reader was used to detect absorbance measurements. The absorbance of each measured sample was compared to the calculated average and standard deviation of the PC cells. To determine the statistical difference of the data, ANOVA was performed.
Colony formation assays
Cells were plated in a 24-well plate in 1 mL increments of 100000 cells per mL and then placed in a 37 °C, 5% CO2 incubator. After 24 h of incubation, the media was aspirated and discarded. The positive control (PC; ≥4 samples) received 1 mL of fresh complete media, the functional control (GSH; ≥4 samples) received 1 mL of 1 mM GSH, and the remaining test samples received 1 mL of 1 mM GSNO (sample; ≥4 samples). After an additional 24 h of incubation, the media from each well was transferred to centrifuge tubes where the cells were collected via addition of trypsin and centrifugation. The cells were then re-plated in a new 24-well plate in 1 mL increments at 500 cells per mL and placed back into the incubator. The plates were checked every 24–72 h for three weeks using bright field microscopy to assess the formation of colonies—defined as masses of 50 or more cells.
LIVE/DEAD assays
Cells were plated in a 96-well plate in 100 μL increments of 100000 cells per mL and placed in a 37 °C, 5% CO2 incubator. After 24 h of incubation, the media was aspirated and discarded. The positive control (PC; ≥4 samples) received 100 μL of fresh complete media, the functional control (GSH; ≥4 samples) received 100 μL of 1 mM GSH, and the remaining test samples received 100 μL of 1 mM GSNO (sample; ≥4 samples). The plate was incubated for 24 h before the media was aspirated and discarded. 100 μL of 3 μM Calcein AM stock solution was added to each well before incubating for 30 min. The Calcein AM stock solution was aspirated and replaced with 100 μL of 5 μM PI stock solution and the plate was incubated for an additional 10 min. Fluorescence microscopy was used to capture images for qualitative comparison of the relative number of live (green) versus dead (red) cells in each sample.
RNA-sequence assay
Cells were plated in 125 cm2 flasks in 25 mL increments of 100000 cells per mL and placed in a 37 °C, 5% CO2 incubator. After 24 h of incubation, the media was aspirated and discarded. The positive control (PC; ≥4 samples) received 25 mL of fresh complete media, the functional control (GSH; ≥4 samples) received 25 mL of 1 mM GSH, and the remaining test samples received 25 mL of 1 mM GSNO (sample; ≥4 samples). All 12 flasks were incubated for 24 h before the media was aspirated and discarded. Cells were harvested from each flask in 15 mL centrifuge tubes, suspended in frozen 1 mL TriZol reagent, and frozen. The 12 frozen sample tubes were delivered to CSU MIP NGS Illumina Core facility (Microbiology, Pathology and Immunology Next Generation Sequence Facility) for RNA-sequence preparation and analysis. Libraries were prepared using NEBNext Ultra II Directional RNA kit using half reactions. Sequence analysis was performed on the NextSeq 500 using a High Output 75 cycle kit. Low-quality reads and PCR duplicates were filtered, resulting in ∼20 million reads/sample. Hisat2 Indexes were made using the Rattus norvegicus genome downloaded from http://uswest.ensembl.org/Rattus_norvegicus/Info/Index. Background noise was decreased using log fold shrinkage while preserving large differences.32 Further gene ontology analysis of biological processes was completed on the top 20 up- and downregulated transcripts via Enrichr (https://maayanlab.cloud/Enrichr/).
Data analysis and statistics
All statistical analysis was performed using one-way ANOVA with p < 0.01 to define statistically significant differences. Data points are represented by the mean ± standard deviation (SD).Results and discussion
Based on a previous study, 1 mM GSNO was applied in this work as a NO-donor and 1 mM GSH was used as a functional control31 (Fig. 1a and b). The results from the aforementioned study as well as analyses performed by other researchers, such as Kim et al. and Suchyta and Schoenfisch,11 informed the hypothesis that the micromolar concentrations of NO (0 h – ∼0.433 μmol to 24 h – ∼0.0650 μmol),31 delivered by 1 mM GSNO over 24 h, would proportionally decrease cellular viability and colony formation capacity while increasing cell death and genetic alterations in the neuroblastoma lines of interest, IMR-32, SK-N-SH, and B104. Excitingly, the results supported this hypothesis with remarkably distinct impacts on the reduction of colony formation capacity across all neuroblastomas analysed. Further, the results exhibited consistent, moderate reduction of cellular viability and obvious increase in cell death. These results provided impactful evidence of the potential of NO to act as an adjuvant anticancer therapeutic, increasing therapeutic impact while simultaneously decreasing harmful patient side effects. Markedly, valuable information about the process and occurrence of NO-mediated genetic alterations was acquired via RNA-sequence analysis, leading to a more in-depth understanding of the anticancer role of NO in neuroblastoma. Specifically, all 40 of the most differentially expressed genes are protein coding genes, involved in various molecular processes. After examining the top 20 upregulated transcripts in the GSNO group, it emerged that in neurons, NO is likely to cause ATP depletion, thereby inducing death via apoptosis.33 Additionally, several of these transcripts were discovered to be involved with oxidative stress and growth inhibition.34–41 Conclusively, the top 20 downregulated transcripts revealed that cell cycle arrest was a prominent effect of NO on these cells. | ||
| Fig. 1 Structures of S-nitrosoglutathione (a) and reduced glutathione (b). | ||
Cell viability assays
Initial assessment of the anticancer impact of NO on neuroblastomas of murine (rat) and human origin included the cellular viability assays resazurin and MTT. These assays were used synonymously to assess the ability of NO to decrease metabolic activity in vitro on three neuroblastoma cell lines, B104, SK-N-SH, and IMR-32. Notably, both of these assays consistently showed a statistically significant reduction of cellular viability, ∼13–29% in all three cell lines compared to control (untreated) cells and GSH-treated cells (Fig. 2a and b). Treated IMR-32 cells exhibited 80 ± 8% and 75 ± 3% viability, treated SK-N-SH cells revealed 87 ± 9% and 79 ± 2% viability, treated B104 cells displayed 83 ± 7% and 71 ± 4% viability via resazurin and MTT assays respectively. Control and GSH-treated cells were not statistically different in any case. Also, it is important to note the influential data collected in our previous study highlighting healthy HDFa cells were not impacted by identical NO treatment.31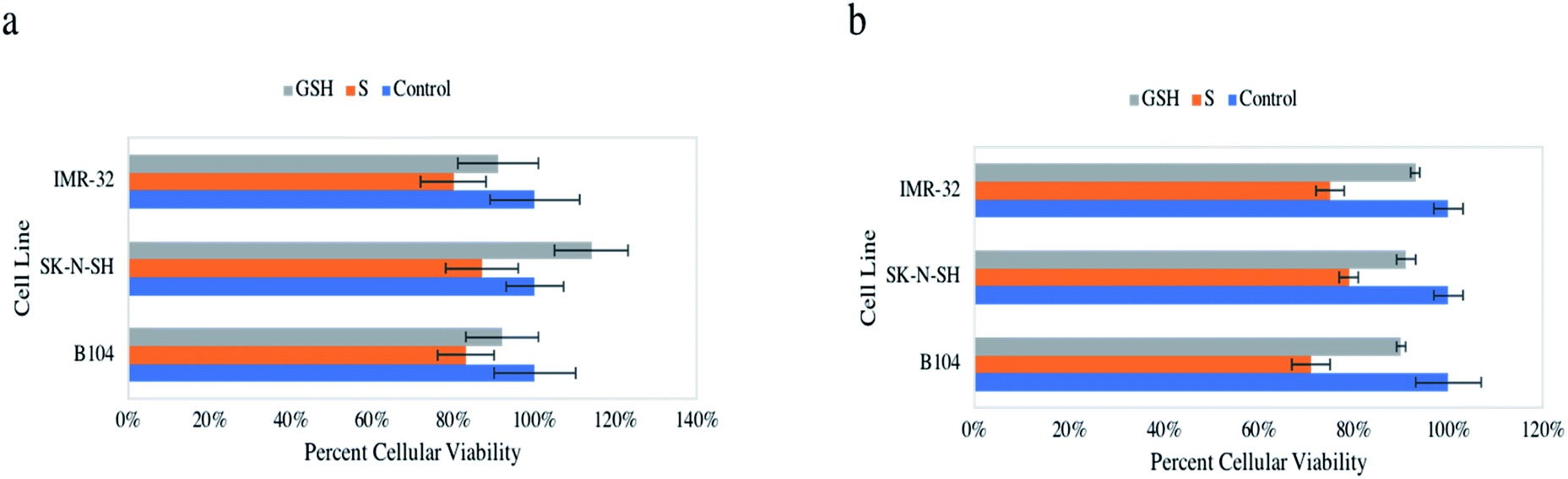 | ||
| Fig. 2 Percent cellular viability of IMR-32, SK-N-SH, and B104 neuroblastoma cells when untreated (blue), exposed to 1 mM GNSO for 24 h (orange), and 1 mM GSH for 24 h (grey), analysed with the resazurin assay (a) and the MTT assay (b). | ||
Colony formation
Consistent observation of a reduction in cellular viability across the neuroblastomas studied led to an interest in the impact of NO on colony formation capacity. Again, all three cell lines were exposed to 1 mM GSNO and GSH, which were both compared to an untreated control. Strikingly, treatment with NO yielded drastic, statistically significant reduction of colony formation capacity across all cell lines (Fig. 3). Again, our previous study remarkably showed no impact on clonogenic activity of identically treated healthy HDFa cells.31 This result is extraordinarily important, indicating a discriminatory effect of NO on neoplastic versus healthy cells.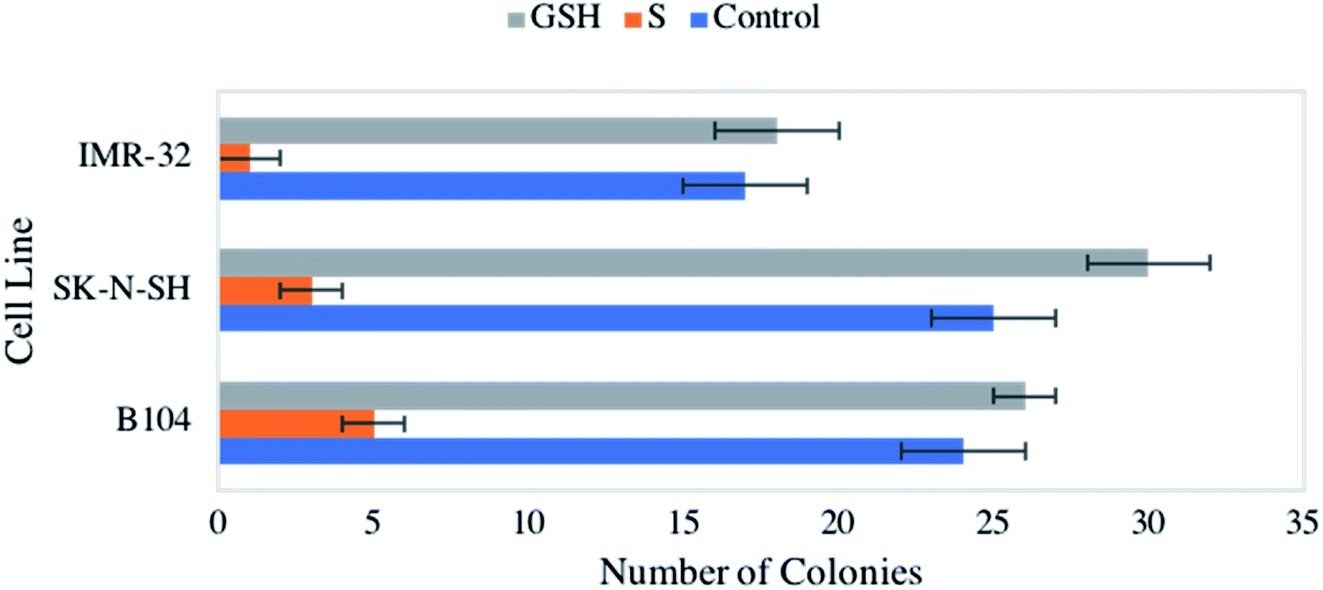 | ||
| Fig. 3 Clonogenic activity of IMR-32, SK-N-SH, and B104 neuroblastoma cells when untreated (blue), exposed to 1 mM GNSO for 24 h (orange), and 1 mM GSH for 24 h (grey), counted via brightfield microscopy. | ||
LIVE/DEAD cytotoxicity assays
Next, cytotoxicity analysis of NO-treated neuroblastomas was completed to qualitatively investigate the relative quantity of live and dead cells in untreated and treated (1 mM GSNO and GSH for 24 h) cells. Using PI/calcein AM staining and fluorescence microscopy, images of treated (GNSO and GSH) and untreated cells were captured. As shown in Fig. 4, it is blatantly clear that the number of live (green) cells is much higher in untreated samples than dead (red) cells. The opposite is true for the treated samples, which show a lower number of cells overall as well as a higher relative number of dead (red) cells than in their untreated counterparts. [The images of GSH-treated cells have not been included in this image to highlight the differences between the images of untreated and NO-treated cells.] It is important to address the obvious differences between cell lines. Since it was expected that NO would have different impacts on each cell line, it was important to analyse multiple neuroblastoma lines of various species. Explicitly, the B104 cells show an obvious presence of live and dead cells after treatment. This result clearly reflects the results seen via cell viability and clonogenic assays. However, treated SK-N-SH cells appear to show very few (almost indistinguishable) dead cells with a fair number of live cells still present. There are a couple of hypotheses to explain this: NO impacts the metabolic activity and colony formation capacity of SK-N-SH cells much more than the death of these cells (via necrosis or apoptosis) and the size of the conglomerations of live SK-N-SH cells is much larger than the size of the individual dead cells, making it difficult to capture both in the overlay (some of the live cells are overlapping the dead cells – can be seen when zoomed in). Finally, treated IMR-32 cells appear to show a much larger number of dead cells than live cells (while the cell viability is similar to that of the other two cell lines). Opposite to SK-N-SH cells, it is possible that NO causes more cell death (via apoptosis or necrosis) to IMR-32 cells than the other neuroblastoma cell lines investigated. In this case, the apparent lack of live cells can also be explained by size, as the size of the live IMR-32 cells is smaller than the size of the conglomerations of dead cells, making it difficult to capture the cells on the overlay (these cannot be seen as well due to the red colour concealing the green colour). | ||
| Fig. 4 LIVE/DEAD cytotoxicity fluorescence images for all three cell lines, untreated (control) and treated (1 mM GSNO for 24 h). These images serve as qualitative data to highlight the majority presence of live (green) cells in the control (untreated) samples versus the lack of overall cell count as well as presence of dead (red) cells in the treated samples. The white scale bar in each image represents 800 μM. | ||
RNA-sequence analysis assay
Ultimately, RNA-sequence analysis was performed on a single cell line, B104 to determine the differentially expressed genes after exposure to 1 mM GSNO for 24 h. All sample reads (∼20 million reads per sample) confirmed the genome to be Rattus norvegicus as expected (∼94%). After the initial analysis, successful log fold change (LFC) shrinkage was performed to decrease background noise while preserving large differences (Fig. 5a and b). LFC shrinkage data was used for further analyses. In the comparison of GSH versus control samples, 18% of genes were up-regulated (0 < LFC < 2) and 18% were down-regulated (0 > LFC > −2) out of 23078 genes with a nonzero total read count with a p-value < 0.1 (Fig. 5a). In the comparison of GSNO versus control samples, 26% of genes were up-regulated (0 < LFC < 2) and 24% were down-regulated (0 > LFC > −2) out of 23078 genes with a nonzero total read count with a p-value of <0.1 (Fig. 5b). This analysis powerfully evidenced that there were significantly more genes differentially expressed in the GSNO-treated samples compared to the GSH-treated samples. A heatmap showing the 30 most differentially expressed genes was generated to narrow down the results of this experiment and allow for interpretation (Fig. 6). Gene ontology (GO) analysis of biological processes was done only on the GSNO-treated samples due to a lack of transcripts available as input to generate reliable gene ontology annotations for the GSH-treated samples. The top 5 gene ontology biological processes for both up- and downregulated transcripts were specified in Fig. 7 and explored further to interpret the mechanism of NO-induced impact on B104 neuroblastoma. Imposingly, the biological processes implicated in the upregulated transcripts highlighted previously reported knowledge that NO regulates voltage-gated K+ channels as well as ATP-sensitive K+ channels in sensory neurons in a concentration-dependent manner. In ATP-sensitive K+ channels, NO has the opposite effect in which NO stimulates the channel. NO inhibits cellular respiration in astrocytes and contributes to resistance to NO-mediated cytotoxicity. However, neurons do not appear to utilize the same mechanism and are more likely to succumb to ATP depletion and die via apoptosis.33 Additionally, investigation of the top 20 upregulated transcripts in the GSNO-treated samples revealed several interesting transcripts were linked to oxidative stress, apoptosis, and growth inhibition: Fas cell surface death receptor (FAS), oxidative stress induced growth inhibitor 1 (OSGIN1 aka OKL38), heme oxygenase 1 (HMOX1), DNA damage inducible transcript 3 (DDIT3 aka CHOP/GADD153), ectodysplasin A2 receptor (EDA2R), tribbles pseudokinase 3 (TRIB3), tumour protein 53-induced nuclear protein 1 (TP53INP1), phorbol-12-myristrate-13-acetate-induced protein 1 (PMAIP1 aka NOXA) (Table 1).34–41 Specifically, NO and FAS are linked to DNA damage and p53 activation which induces adult motor neuron apoptosis;34 OSGIN1 was recognized as a cell growth inhibitor in response to oxidative stress as well as a chemotherapeutic sensor, in which it was upregulated in response to DNA damage and p53 activation and thereby induced apoptosis;35 neurons that overexpress HMOX1 resist oxidation-induced stress and apoptosis;36 DDIT3 induces cell cycle arrest and endoplasmic reticulum (ER)-mediated apoptosis in response to ER-related stress;37 EDAR2 stimulates apoptosis via trans-activation by p53;38 during ER-stress TRIB3 is involved in CHOP-dependent cell death by downregulating its own activation through repression of CHOP/ATF4 functions;39 TP53INP1 is a p53-target gene that is expressed in response to stress and is interrelated with anti-proliferative and pro-apoptotic characteristics;40 PMAIP1 upregulation is linked to downregulation of Usp9x (a deubiquitinase) and reduction of Mcl-1 expression, which fosters ubiquitination and degradation and leads to apoptosis of neoplastic cells.41 Finally, the biological processes indicated in the downregulated transcripts suggested that cell cycle arrest is a prominent effect of NO on these cells (Table 2).
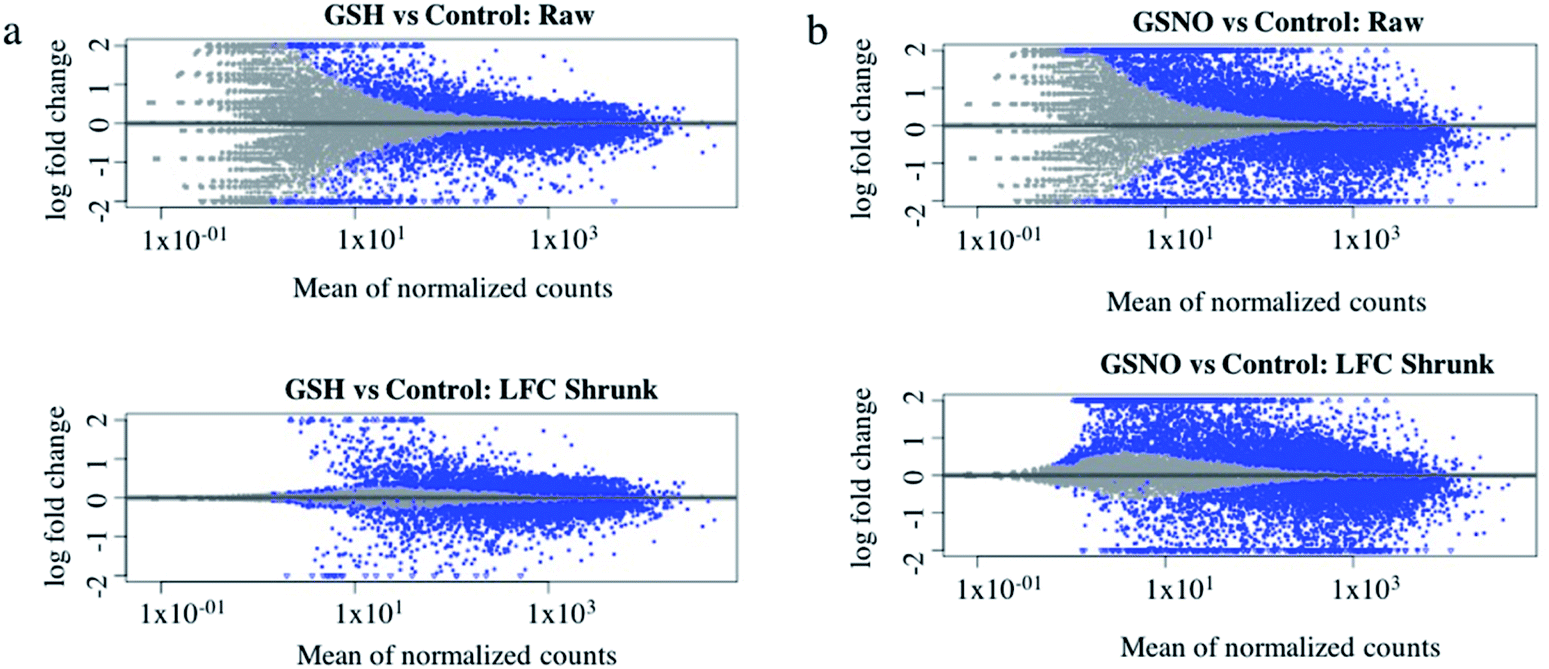 | ||
| Fig. 5 Differentially expressed genes in GSH (a) and GSNO (b) treated samples versus control. In GSH-treated samples, 18% of genes were up-regulated (0 < LFC < 2) and 18% of genes were down-regulated (0 > LFC > −2) out of 23078 total genes with a non-zero read count, p < 0.1. In GSNO-treated samples, 26% of genes were up-regulated (0 < LFC < 2) and 24% were down-regulated (0 > LFC > −2) out of 23078 genes with a nonzero total read count, p < 0.1 Log fold change (LFC) shrinkage (LFC Shrunk) was performed to minimize noise while preserving large differences. | ||
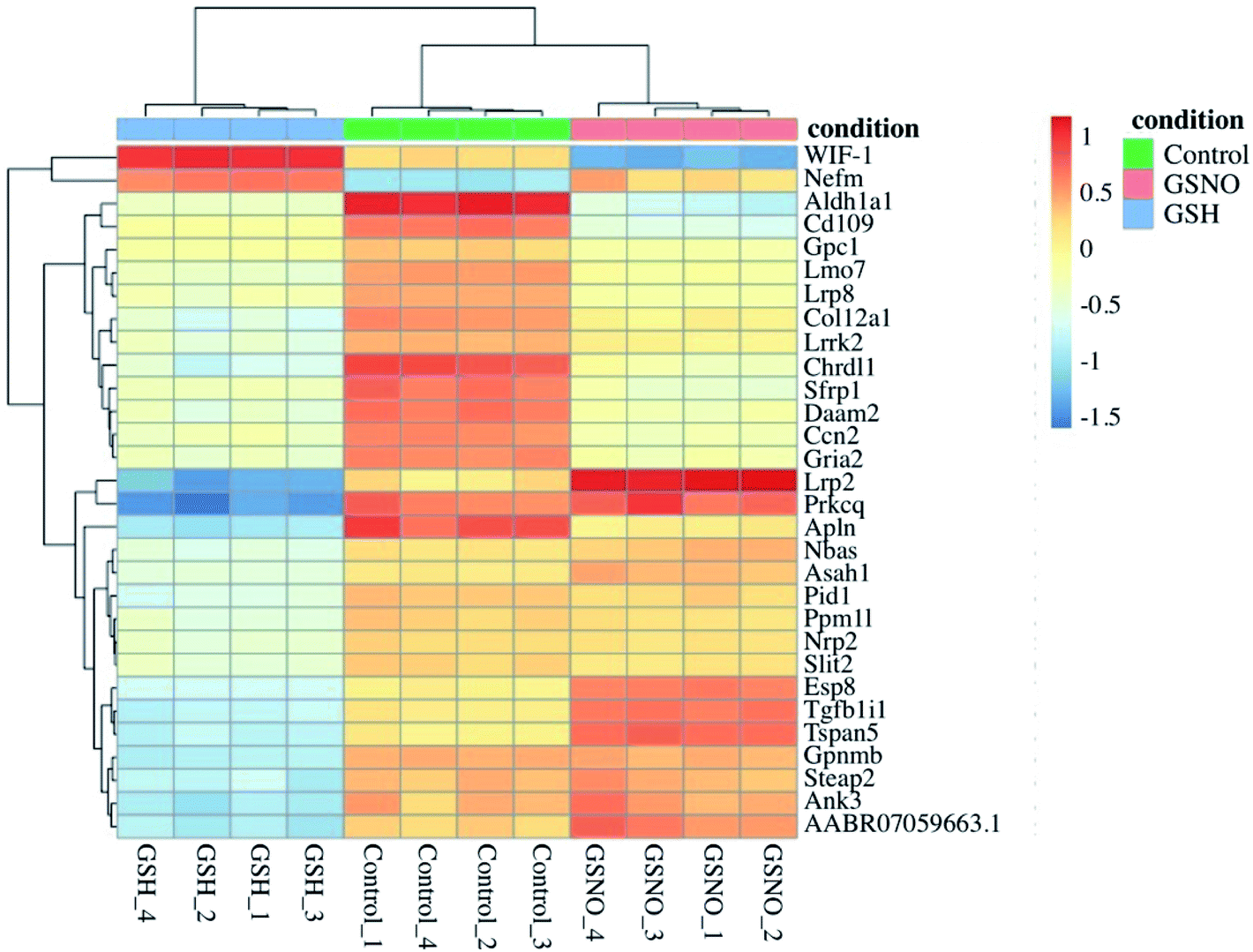 | ||
| Fig. 6 Heat map expressing the 30 most differentially expressed genes. This map displays the names of the differentially expressed genes as well as the extent to which each gene was differentially expressed in all samples. | ||
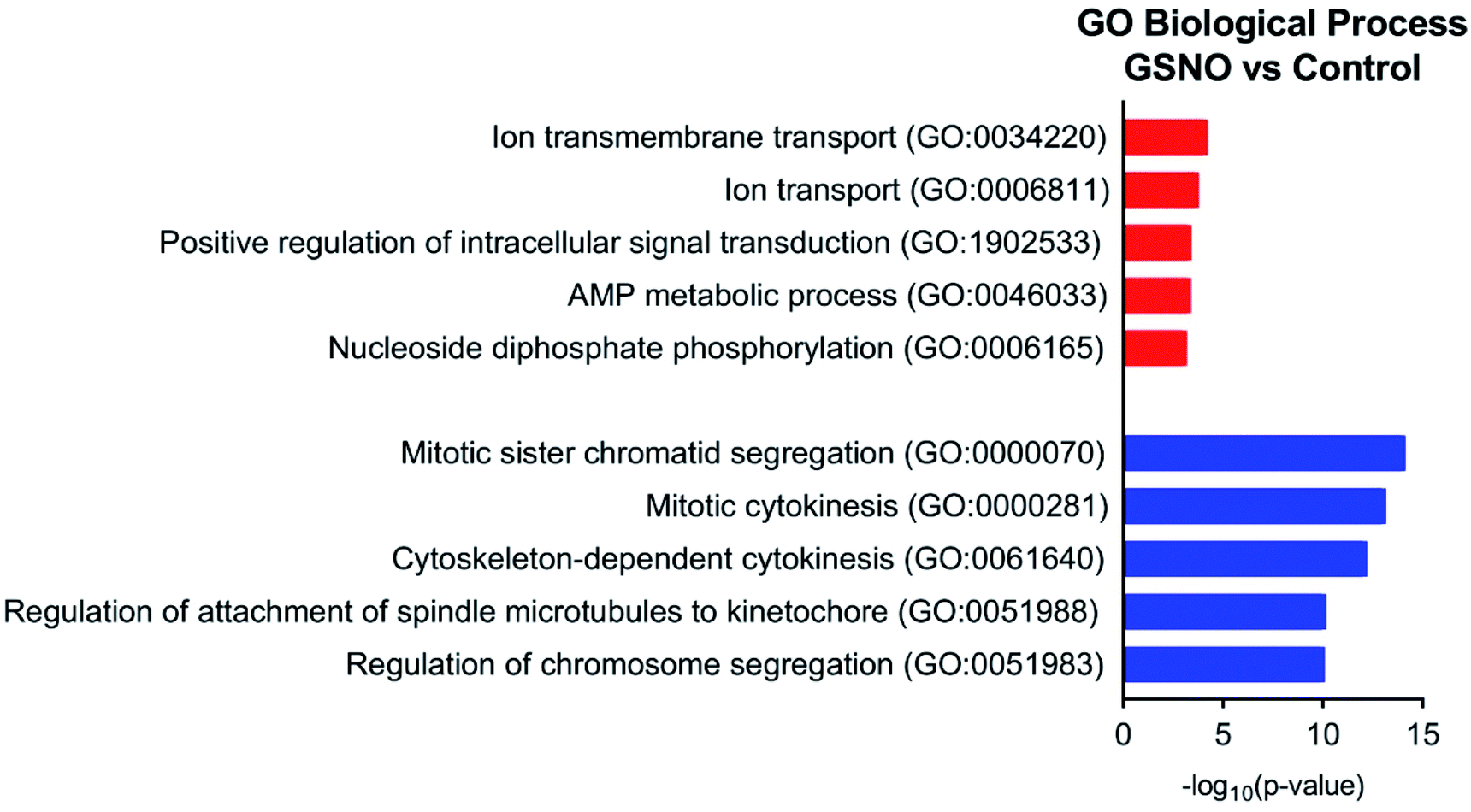 | ||
| Fig. 7 Top 5 gene ontology biological processes for up- (red) and downregulated (blue) transcripts in GSNO-treated samples versus control. | ||
| Ensembl ID | log2 fold change | padj | Name | Description |
|---|---|---|---|---|
| ENSRNOG00000014117 | 2.642690485 | 0 | Hmox1 | Heme oxygenase 1 [source: RGD symbol; Acc: 2806] |
| ENSRNOG00000018126 | 2.100712674 | 0 | Abca1 | ATP binding cassette subfamily A member 1 [source: RGD symbol; Acc: 631344] |
| ENSRNOG00000006789 | 2.108566582 | 3.22−262 | Ddit3 | DNA-damage inducible transcript 3 [source: RGD symbol; Acc: 62391] |
| ENSRNOG00000013484 | 2.934381196 | 6.59−212 | Gsta1 | Glutathione S-transferase alpha 1 [source: RGD symbol; Acc: 2753] |
| ENSRNOG00000037621 | 2.566554367 | 4.57−165 | Spata48 | Spermatogenesis associated 48 [source: RGD symbol; Acc: 1309870] |
| ENSRNOG00000054561 | 2.721372192 | 4.95−149 | Isg20 | Interferon stimulated exonuclease gene 20 [source: RGD symbol; Acc: 1306407] |
| ENSRNOG00000013018 | 2.455220167 | 5.22−139 | Eda2r | Ectodysplasin A2 receptor [source: RGD symbol; Acc: 1564025] |
| ENSRNOG00000047697 | 3.675674714 | 2.52−123 | Ggt1 | Gamma-glutamyltransferase 1 [source: RGD symbol; Acc: 2683] |
| ENSRNOG00000007964 | 2.145360583 | 3.79−107 | Tp53inp1 | Tumour protein p53 inducible nuclear protein 1 [source: RGD symbol; Acc: 631423] |
| ENSRNOG00000011316 | 3.689711899 | 3.87−106 | Fam167a | Family with sequence similarity 167, member A [source: RGD symbol; Acc: 1561302] |
| ENSRNOG00000007319 | 2.106674517 | 1.87−103 | Trib3 | Tribbles pseudokinase 3 [source: RGD symbol; Acc: 708432] |
| ENSRNOG00000027016 | 2.39177707 | 2.13−99 | Cobll1 | Cordon-bleu WH2 repeat protein-like 1 [source: RGD symbol; Acc: 1308954] |
| ENSRNOG00000018770 | 2.231130335 | 5.18−99 | Pmaip1 | Phorbol-12-myristate-13-acetate-induced protein 1 [source: RGD symbol; Acc: 1359266] |
| ENSRNOG00000014948 | 3.876937844 | 2.13−94 | Osgin1 | Oxidative stress induced growth inhibitor 1 [source: RGD symbol; Acc: 620679] |
| ENSRNOG00000003189 | 3.281285701 | 3.35−89 | Cited1 | Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 1 [source: RGD symbol; Acc: 620781] |
| ENSRNOG00000019142 | 2.052936986 | 3.16−86 | Fas | Fas cell surface death receptor [source: RGD symbol; Acc: 619831] |
| ENSRNOG00000012892 | 2.807251669 | 4.52−85 | Abca4 | ATP binding cassette subfamily A member 4 [source: RGD symbol; Acc: 1309445] |
| ENSRNOG00000036571 | 5.054889497 | 3.39−84 | Ces2c | Carboxylesterase 2C [source: RGD symbol; Acc: 621510] |
| ENSRNOG00000000245 | 2.020890852 | 1.18−69 | Slc16a6 | Solute carrier family 16, member 6 [source: RGD symbol; Acc: 735117] |
| ENSRNOG00000001527 | 2.735921532 | 1.52−64 | Cd80 | Cd80 molecule [source: RGD symbol; Acc: 2314] |
| Ensembl ID | log2 fold change | padj | Name | Description |
|---|---|---|---|---|
| ENSRNOG00000008040 | −3.993065851 | 0 | Pimreg | PICALM interacting mitotic regulator [source: RGD symbol; Acc: 1308747] |
| ENSRNOG00000032178 | −3.41779947 | 0 | Cenpa | Centromere protein A [source: RGD symbol; Acc: 1563607] |
| ENSRNOG00000037211 | −3.389787634 | 0 | Kif14 | Kinesin family member 14 [source: RGD symbol; Acc: 1310650] |
| ENSRNOG00000018815 | −3.271780618 | 0 | Plk1 | Polo-like kinase 1 [source: RGD symbol; Acc: 3352] |
| ENSRNOG00000027894 | −3.180043549 | 0 | Lqgap3 | IQ motif containing GTPase activating protein 3 [source: RGD symbol; Acc: 1305951] |
| ENSRNOG00000009946 | −3.114863936 | 0 | Ldlr | Low density lipoprotein receptor [source: RGD symbol; Acc: 2998] |
| ENSRNOG00000007906 | −3.011339242 | 0 | Bub1b | BUB1 mitotic checkpoint serine/threonine kinase B [source: RGD symbol; Acc: 619791] |
| ENSRNOG00000003388 | −2.946409613 | 0 | Cenpf | Centromere protein F [source: RGD symbol; Acc: 628667] |
| ENSRNOG00000008837 | −2.911178509 | 0 | Ass1 | Argininosuccinate synthase 1 [source: RGD symbol; Acc: 2163] |
| ENSRNOG00000058539 | −2.863922882 | 0 | Ccnb1 | Cyclin B1 [source: RGD symbol; Acc: 2291] |
| ENSRNOG00000028415 | −2.774143201 | 0 | Cdc20 | Cell division cycle 20 [source: RGD symbol; Acc: 620477] |
| ENSRNOG00000019100 | −2.748690893 | 0 | Kif2c | Kinesin family member 2C [source: RGD symbol; Acc: 620239] |
| ENSRNOG00000038035 | −2.742181645 | 0 | Kif4a | Kinesin family member 4A [source: RGD symbol; Acc: 620526] |
| ENSRNOG00000047314 | −2.731455572 | 0 | Tk1 | Thymidine kinase 1 [source: RGD symbol; Acc: 621014] |
| ENSRNOG00000011777 | −2.661567123 | 0 | Spag5 | Sperm associated antigen 5 [source: RGD symbol; Acc: 620152] |
| ENSRNOG00000053047 | −2.638046934 | 0 | Top2a | DNA topoisomerase II alpha [source: RGD symbol; Acc: 62048] |
| ENSRNOG00000017259 | −2.618329127 | 0 | Tacc3 | Transforming, acidic coiled-coil containing protein 3 [source: RGD symbol; Acc: 1302948] |
| ENSRNOG00000024428 | −2.555145555 | 0 | Kif20a | Kinesin family member 20A [source: RGD symbol; Acc: 1307695] |
| ENSRNOG00000004921 | −2.547345074 | 0 | Nusasp1 | Nucleolar and spindle associated protein 1 [source: RGD symbol; Acc: 1305764] |
| ENSRNOG00000000479 | −2.49309011 | 0 | Kifc1 | Kinesin family member C1 [source: RGD symbol; Acc: 1359118] |
Conclusions
Nitric oxide-based therapeutics offer exciting potential in anticancer applications due to accessibility, low-cost, and potential for tunability. Particularly, S-nitrosothiols (RSNOs) provide desirable characteristics for anticancer treatment, including endogenous production of many of these molecules and the prospective to influence timing and longevity of NO-release. Herein, analysis of NO, delivered by GSNO, as an anticancer agent was explored on human and an murine (rat) neuroblastoma cell lines (IMR-32, SK-N-SH, and B104) via cell viability, colony formation, cytotoxicity, and RNA-sequence analysis assays. Impactful results from two different cell viability assays, resazurin and MTT, consistently portrayed ∼13–29% decrease in viability of all three cell lines after 24 h of exposure to 1 mM GSNO. Remarkably, colony formation assays displayed a tremendously significant decrease in the ability of all three cell lines to form colonies after 24 h of exposure to an identical concentration of GSNO. Further, qualitative cytotoxicity assays performed on each cell line reinforced these findings, exhibiting a blatant decrease in the number of live cells as well as an increase in the number of dead cells after treatment with 1 mM GSNO. Finally, RNA-sequence analysis on B104 cells showcased a statistically significant increase in the number of up-regulated and down-regulated differentially expressed genes in the GSNO-treated samples as compared to the GSH-treated and control samples. Strikingly, the identity and biological processes of these genes provided valuable insight into the mechanism of action of NO on neuroblastoma cells, indicating its involvement in oxidative stress, apoptosis, growth inhibition, regulation of cell cycle progression, and mitosis. Inclusively, this data presents a convincing rationalization for use of NO, delivered via RSNOs, as anticancer adjuvants in treatment of neuroblastoma and incites further exploration of its conceivable application in other malignancies. Specifically, it would be highly informative to perform apoptosis and cell cycle analysis assays on these cell lines as well as RNA-sequence analysis assays on both SK-N-SH and IMR-32 cells to further elaborate on these results and their clinical translatability.Funding sources
This research was partially supported by Colorado State University Chemistry Department. H. O. T. received support from NIH Grant R01CA31534, Cancer Prevention Research Institute of Texas (CPRIT) Grants RP100612, RP120348; and the Marie Betzner Morrow Centennial Endowment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Marylee Layton with CSU MIP NGS Illumina Core facility for her knowledge and expertise regarding RNA-sequence preparation and analysis.References
- M. Hayat, Neuroblastoma, Springer, 2012 Search PubMed.
- J. Shohet and J. Foster, Br. Med. J., 2017, 357, j1863 CrossRef.
- J. G. Nuchtem, W. B. London, C. E. Barnewolt, A. Naranjo and P. W. McGrady, Ann. Surg., 2017, 256, 573–580 CrossRef.
- C. A. Perez, K. K. Matthay, J. B. Atkinson, R. C. Seeger, H. Shimada, G. M. Haase, D. O. Stram, R. B. Gerbing and J. N. Lukens, J. Clin. Oncol., 2000, 18, 18–26 CrossRef CAS.
- D. Pe, C. Le, C. Oscarlambret and H. Dieu, Br. J. Cancer, 2003, 89, 1605–1609 CrossRef.
- B. H. J. Nickerson, K. K. Matthay, R. C. Seeger, G. M. Brodeur, H. Shimada, C. Perez, J. B. Atkinson, M. Selch, R. B. Gerbing, D. O. Stram and J. Lukens, J. Clin. Oncol., 2000, 18, 477–486 CrossRef.
- K. K. Matthay, C. P. Reynolds, R. C. Seeger, H. Shimada, E. S. Adkins, D. Haas-Kogan, R. B. Gerbing, W. B. London and J. G. Villablanca, J. Clin. Oncol., 2009, 27, 1007–1013 CrossRef CAS.
- R. Ladenstein, U. Pötschgerulrike, D. Valteau-couanet, R. Luksch, V. Castel, S. Ash, G. Laureys, P. Brock, J. M. Michon, C. Owenscormac, T. Trahair, G. C. F. Chan, E. Ruud, H. Schroeder, M. Beck-popovic, G. Schreier, H. Loibner, P. Ambros, K. Holmes, M. R. Castellani, M. N. Gaze, A. Garaventa, A. D. J. Pearson and H. N. Lode, Cancers, 2020, 12, 1–19 CrossRef.
- S. Korde Choudhari, M. Chaudhary, S. Bagde, A. R. Gadbail and V. Joshi, World J. Surg. Oncol., 2013, 11, 1 CrossRef.
- E. V. Stevens, A. W. Carpenter, J. H. Shin, J. Liu, C. J. Der and M. H. Schoenfisch, Mol. Pharm., 2010, 7, 775–785 CrossRef CAS.
- D. J. Suchyta and M. H. Schoenfisch, RSC Adv., 2017, 7, 53236–53246 RSC.
- R. Dong, X. Wang, H. Wang, Z. Liu, J. Liu and J. E. Saavedra, Biomed. Pharmacother., 2017, 88, 367–373 CrossRef CAS.
- M. M. Reynolds, S. D. Witzeling, V. B. Damodaran, T. N. Medeiros, R. D. Knodle, M. A. Edwards, P. P. Lookian and M. A. Brown, Biochem. Biophys. Res. Commun., 2013, 431, 647–651 CrossRef CAS.
- A. Nortcliffe, A. G. Ekstrom, J. R. Black, J. A. Ross, F. K. Habib, N. P. Botting and D. O'Hagan, Bioorg. Med. Chem., 2014, 22, 756–761 CrossRef CAS.
- S. Y. Lee, Y. Rim, D. D. McPherson, S. L. Huang and H. Kim, Biomed. Mater. Eng., 2014, 24, 61–67 CAS.
- K. Oh-hashi, W. Maruyama, H. Yi, T. Takahashi, M. Naoi and K. Isobe, Biochem. Biophys. Res. Commun., 1999, 504–509 CrossRef CAS.
- B.-C. Kim, Y.-S. Kim, J.-W. Lee, J.-H. Seo, E.-S. Ji, H. Lee, Y.-I. Park and C.-J. Kim, Exp. Neurobiol., 2011, 20, 100 CrossRef.
- V. J. Findlay, D. M. Townsend, J. E. Saavedra, G. S. Buzard, M. L. Citro, L. K. Keefer, X. Ji and K. D. Tew, Mol. Pharmacol., 2004, 65, 1070–1079 CrossRef CAS.
- Y. Hou, J. Wang, P. R. Andreana, G. Cantauria, S. Tarasia, L. Sharp, P. G. Braunschweiger and P. G. Wang, Bioorg. Med. Chem. Lett., 1999, 9, 2255–2258 CrossRef CAS.
- E. Kogias, N. Osterberg, B. Baumer, N. Psarras, C. Koentges, A. Papazoglou, J. E. Saavedra, L. K. Keefer and A. Weyerbrock, Int. J. Cancer, 2012, 130, 1184–1194 CrossRef CAS.
- J. M. Joslin, B. H. Neufeld and M. M. Reynolds, RSC Adv., 2014, 4, 42039–42043 RSC.
- L. Grossi and P. Carlo, Chem.–Eur. J., 2002, 8, 380–387 CrossRef CAS.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 1 CrossRef.
- D. A. Wink, Y. Vodovotz, J. Laval, F. Laval, M. W. Dewhirst and J. B. Mitchell, Carcinogenesis, 1998, 19, 711–721 CrossRef CAS.
- B. J. Oleson and J. A. Corbett, Antioxid. Redox Signaling, 2018, 29, 1432–1445 CrossRef CAS.
- S. Pervin, R. Singh, S. Sen and G. Chaudhuri, Nitric Oxide (NO) and Cancer, 2010, pp. 39–57 Search PubMed.
- J. Hickok and D. Thomas, Curr. Pharm. Des., 2010, 16, 381–391 CrossRef CAS.
- Z. Huang, J. Fu and Y. Zhang, J. Med. Chem., 2017, 60, 7617–7635 CrossRef CAS.
- W. Badn and P. Siesjo, Curr. Pharm. Des., 2010, 16, 428–430 CrossRef CAS.
- J. L. Gordon, M. M. Reynolds and M. A. Brown, Vet. Sci., 2020, 7, 51 CrossRef.
- A. Zhu, J. G. Ibrahim and M. I. Love, Bioinformatics, 2019, 35, 2084–2092 CrossRef CAS.
- A. Almeida, S. Moncada and J. P. Bolaños, Nat. Cell Biol., 2004, 6, 45–51 CrossRef CAS.
- L. J. Martin, K. Chen and Z. Liu, J. Neurosci., 2005, 25, 6449–6459 CrossRef CAS.
- M. Liu, Y. Li, L. Chen, T. H. Man Chan, Y. Song, L. Fu, T. T. Zeng, Y. D. Dai, Y. H. Zhu, J. Chen, Y. F. Yuan and X. Y. Guan, Gastroenterology, 2014, 146, 1084–1096 CrossRef CAS.
- K. Chen, K. Gunter and M. D. Maines, J. Neurochem., 2000, 75, 304–313 CrossRef CAS.
- S. Oyadomari and M. Mori, Cell Death Differ., 2004, 11, 381–389 CrossRef CAS.
- R. Brosh, R. Sarig, E. B. Natan, A. Molchadsky, S. Madar, C. Bornstein, Y. Buganim, T. Shapira, N. Goldfinger, R. Paus and V. Rotter, FEBS Lett., 2010, 584, 2473–2477 CrossRef CAS.
- N. Ohoka, S. Yoshii, T. Hattori, K. Onozaki and H. Hayashi, EMBO J., 2005, 24, 1243–1255 CrossRef CAS.
- J. Shahbazi, R. Lock and T. Liu, Front. Genet., 2013, 4, 1–7 CAS.
- J. Yan, N. Zhong, G. Liu, K. Chen, X. Liu, L. Su and S. Singha, Cell Death Dis., 2014, 5, 1–7 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |