
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient photocatalytic degradation of dyes using photo-deposited Ag nanoparticles on ZnO structures: simple morphological control of ZnO
Hyeonhan Lim†
a,
Mohammad Yusuf† a,
Sehwan Songb,
Sungkyun Parkb and
Kang Hyun Park*a
a,
Sehwan Songb,
Sungkyun Parkb and
Kang Hyun Park*a
aDepartment of Chemistry, Chemistry Institute for Functional Materials, Pusan National University, Busan, 46241, Republic of Korea. E-mail: chemistry@pusan.ac.kr
bDepartment of Physics, Pusan National University, Busan, 46241, Republic of Korea
First published on 25th February 2021
Abstract
In this work, morphology-controlled ZnO structures were prepared via a hydrothermal method by simple adjustments in the NaOH concentration. The NaOH concentration variation from 0.2 to 1.2 M resulted in the formation of ZnO structures in shapes such as walnut, spherical flower, flower, rod, and urchin-like. The extent of OH− ions is the main factor influencing the growth of ZnO structures. Well-defined morphologies, good crystallinity, and optical properties were obtained for all ZnO structures. Among these ZnO structures, ZnOsf (spherical flower-like) structure showed a greater percentage of photodegradation of methyl orange and rhodamine B dyes. Surface plasmon resonance was achieved by modifying the surface of ZnO with Ag nanoparticles. ZnOsf was loaded with Ag nanoparticles by a facile photo-deposition method. Ag–ZnOsf showed superior photoactivity and recyclability for the degradation of methyl orange and rhodamine B. Therefore, modification of different ZnO structures can help realize potential catalysts for future environmental applications.
Introduction
Currently, excessive water contamination due to the discharge of organic dyes through industrial wastewater, primarily from the processing of products such as fabrics, pharmaceuticals, petrochemicals, foodstuffs, cosmetics, and leather, has become a severe problem given the importance of clean water in preserving life on Earth. The exposure of dyes in a small level of less than 1 mg L−1 can seriously affect the water quality of the aquatic environment. Methyl orange (MO), rhodamine B (RhB), and methylene blue (MB) are commonly used toxic, mutagenic, and non-biodegradable azo dyes hazardous to aquatic environments.1–4As an eco-friendly and sustainable treatment process, photocatalysis has been demonstrated to have considerable potential for the removal of dyes from wastewater.3,5,6 Furthermore, the development of photocatalysts is considered a promising prospect for future methods for generating renewable energy using abundantly-available and inexpensive natural sunlight radiation.
Photocatalysis is related to the reactions induced by ultraviolet-visible (UV-Vis) light incident on the surface of semiconductors. This reaction generates electrons and holes, which act as charge carriers. Semiconductor-based heterogeneous photocatalysts, such as TiO2, ZnS, and ZnO, have received significant interest for the renewable and sustainable harnessing of solar energy in order to solve energy issues and environmental problems.7–12 Among these photocatalysts, ZnO is an excellent semiconductor owing to its unique properties such as high photosensitivity, high electron mobility, and good physical and chemical stabilities.13,14 The synthesis and photochemical reactions of ZnO have been actively studied. However, the efficiency of ZnO is still limited due to the rapid recombination of electron–hole pairs,12,13 and the activity in the visible region is considerably lower than that in the ultraviolet region.15,16
In photocatalytic reactions, the morphology of the catalyst can dramatically affect the properties and performance of the catalyst. A considerable number of morphology-controlled TiO2 photocatalysts have been developed with various designs, including spheres, nanosheets, nanotubes, 3-D sea urchin-like, and 3-D hierarchically porous structures; these photocatalysts have been proposed and utilized in photocatalytic air purification. In addition, various morphologies of CuO obtained using the sonochemical method have been applied for studying the photocatalytic activity of MB. However, the control of the morphology was performed in the presence of different surfactants. Mesoporous Cu2O prepared through a morphology-control approach by the addition of reductants and surfactants was also studied in the photocatalytic degradation of MO.8,11,17–19 In addition, the presence of a base is one of the factors triggering the formation of the final structure. The diverse morphologies of ZnO were obtained by controlling different bases in the presence of surfactant or capping agent by various methods.20,21 For instance, ZnO nanorods and nanoflower morphologies were synthesized by the influence of ammonia. ZnO (flowerlike, spindle-like, sword-like, and umbrella-like) morphologies are formed by controlling NaOH and ethanolamine. ZnO (egg noodle and 2D plate-like) structures were obtained by sodium citrate and urea.22–24 NaOH is the most popular base since NaOH is easily ionized to Na+ and OH− in an aqueous solution and increases the solubility of Zn(OH)2 related to the formation [Zn(OH4)]2− complex leading to a high yield of ZnO product.21,25 After all, defining the correlation between the morphology and photocatalytic behavior of catalysts remains a challenge. The same variables mentioned above can be used to study the effect of morphology on the photocatalytic efficiency of ZnO structures.
Another method to improve photocatalytic performance is by loading the surface of the catalyst with noble metals such as Pd, Pt, Au, and Ag.26–31 Comparative experiments using Ag/TiO2, Au/TiO2, and Pt/TiO2 showed similar good photocatalytic activity between Ag/TiO2 and Pt/TiO2. However, Ag is more abundant and economical than Au and Pt. Therefore, using Ag would be more eco-friendly and easier to apply in the industry.32–38 The deposited Ag nanoparticles (Ag NPs) on the photocatalyst surface can effectively suppress the recombination of photogenerated electron–hole pairs due to the Schottky barrier formed at the interface between the metal and semiconductor. Moreover, surface plasmon resonance (SPR) of Ag NPs significantly improves the absorption of visible light.39–46 Thus, modifying ZnO with Ag can improve the photocatalytic activity of ZnO by altering its optical and electronic properties.
In this work, we present a simple method for the fabrication of ZnO via morphology control without the addition of a surfactant or capping agent. Furthermore, ZnO was loaded with Ag NPs through a convenient photo-deposition method under light irradiation. The photo-deposition reduction approach only involves irradiation with a light source, unlike most other techniques that require high temperatures, external redox agent, electrical potential, or multi-step operation.47,48 The composite Ag–ZnO structure was implemented as a photocatalyst for the degradation of various dyes such as MO and RhB.
Experimental
Chemicals
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99.0%), sodium hydroxide (NaOH, 99.0%), silver nitrate(ii) (AgNO3, 99.9%), MO, and RhB were purchased from Sigma-Aldrich. Ethanol (99.9%) and methanol were procured from Samchun Company.Synthesis of ZnO structures
Zn(NO3)2·6H2O (5 mmol) was dissolved in 20 mL of deionized water. Thereafter, 50 mL of NaOH solution (0.2, 0.6, 0.8, 0.9, or 1.2 M) was added to the Zn(NO3)2 solution. After stirring for 1 h (400 rpm, 25 °C), the mixture was transferred to a 120 mL Teflon-lined stainless-steel autoclave and heated at 100 °C for 12 h. The reaction mixture was naturally cooled, and the ZnO powder was washed with deionized water and ethanol. The product was then dried in an oven at 60 °C for 12 h.Synthesis of Ag–ZnOsf composite
Ag–ZnOsf composite was synthesized using the photoreduction method. First, 0.2 g of ZnO was dispersed in 35 mL of water and 20 mL of methanol solution. Thereafter, 5 mL of AgNO3 solution (7.65 mM) was added. The mixture was initially stirred with a magnetic bar (400 rpm, 25 °C, 30 min) to allow the absorption of Ag+ on the surface of ZnO. Then, the suspension was irradiated with a Xe lamp for 2 h. The Xe lamp was equipped with a liquid filter to prevent temperature rise by light. The experiments were conducted at pH = 7.8, 25 °C. The precipitates were centrifuged and washed with deionized water and ethanol. The product was then dried in an oven at 60 °C for 12 h.Photocatalytic activity
The photocatalytic activity of the as-synthesized catalyst was estimated based on the photocatalytic degradation of MO and RhB. First, 40 mg of the photocatalyst was dispersed in 80 mL of an aqueous organic dye solution (10 mg L−1). The solution was stirred in dark for 30 min to reach the adsorption–desorption equilibrium. The mixture was then exposed to UV-Vis irradiation using a Xe lamp. The degradation of the organic dye was monitored using UV-Vis spectroscopy after withdrawing and centrifuging the suspension. The pH of dyes was 8.8 for RhB and 8.7 for MO. The initial concentration of the dye used was maintained at 10 mg L−1 based on previous work, which indicated that ZnO catalysts were suitable at a relatively wide concentration of pollutants and the best efficiency was shown at 10 mg L−1 of MO concentration.49 The efficiency of MO and RhB degradation was determined by applying eqn (1).
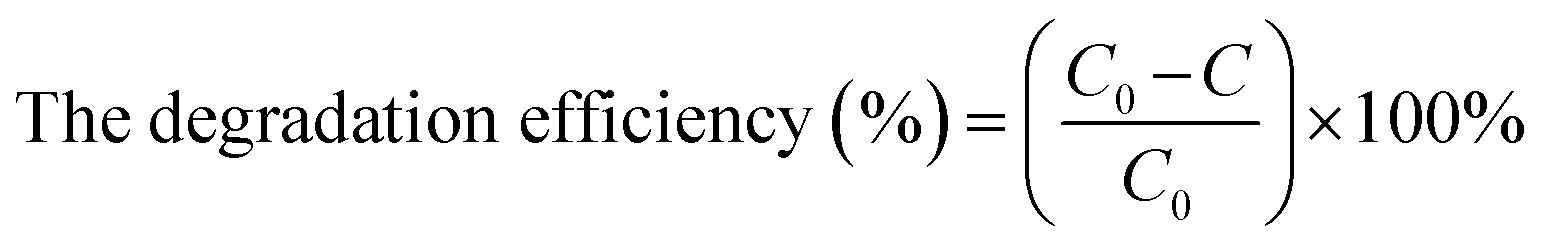 | (1) |
Material characterization
The morphologies of the samples were analyzed using high-resolution scanning electron microscopy (SEM) on a ZEISS SUPRA 40VP instrument (Carl ZEISS, Germany) and field-emission transmission electron microscopy (FE-TEM) on a TALOS F200X (200 keV) instrument (FEI, Netherlands). The TEM instrument was combined with energy-dispersive X-ray spectroscopy (EDX) elemental mapping analysis. Crystal structures in the synthesized samples were studied using X-ray diffraction (XRD) on an Xpert3 (Malvern Panalytical Ltd., Netherlands) diffractometer with Cu-Kα radiation at λ = 0.15406 nm. N2 sorption isotherms were constructed from the data acquired at −196 °C using a Micromeritics ASAP 2020 surface area analyzer (Micromeritics Instrument Corp., US). Before analysis, samples were degassed under N2 flow at 150 °C for 12 h. X-ray photoelectron spectroscopy (XPS) was utilized to investigate the surface chemical properties using an ESCALab250-AXIS SUPRA instrument (Kratos Analytical Ltd., UK). The optical properties were studied using UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) with V-770 (JASCO Corp., Japan) and photoluminescence (PL) spectroscopy with F-7000 spectrofluorometer (Hitachi, Japan). The photodegradation of MO and RhB was monitored using acquired UV-Vis absorption spectra UV-1800 instrument (Shimadzu, Japan).Results and discussion
Scheme 1 shows the method used for controlling the morphology of ZnO. ZnO catalysts with different shapes and sizes were prepared via a simple hydrothermal method, without the addition of any stabilizers, such as surfactants, polymers, and other functional molecules. Several parameters, including time, temperature, the concentration of precursors, the addition of surfactants, and solvents, can affect the morphology of materials. Here, the NaOH at the concentration varied from 0.2 to 1.2 M was only used to control the growth of ZnO particles. Ag–ZnOsf was synthesized using a photo-deposition process with AgNO3 solution as the source of Ag NPs (Scheme 2). The reduction of Ag was realized using the incident light from Xe lamp, which is a one-step process under mild conditions.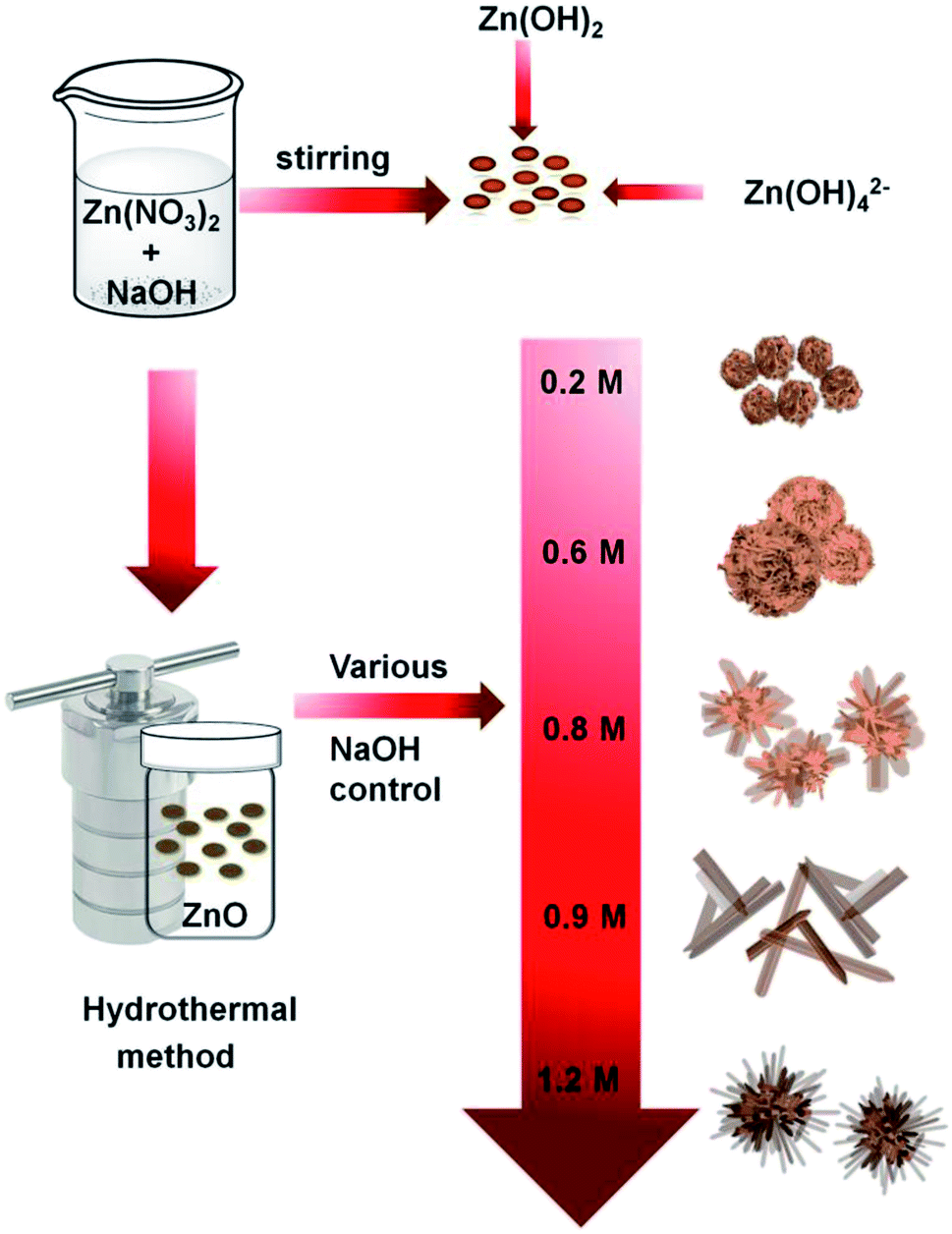 | ||
| Scheme 1 Synthetic procedure for various ZnO structures. | ||
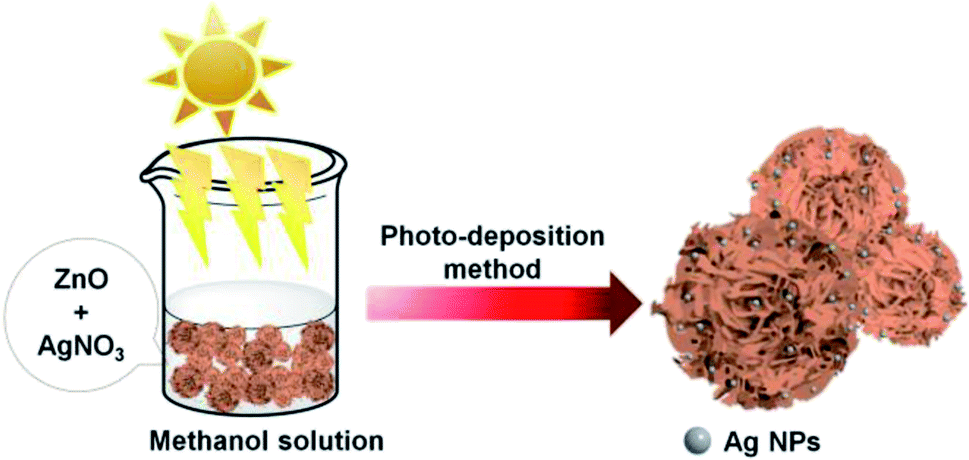 | ||
| Scheme 2 Synthesis of Ag–ZnO composite by photo-deposition method. | ||
The morphologies of the as-prepared ZnO samples were initially analyzed using SEM. Fig. 1 displays SEM images of ZnO with different shapes and sizes, formed by varying the NaOH concentration in the aqueous solution. As shown in Fig. 1(a) and (b), ZnO walnuts were formed when using 0.2 M NaOH. The diameter of these ZnO walnuts was approximately 360 nm. Fig. 1(c) and (d) shows the spherical flower-like ZnO structure produced with 0.6 M NaOH. The diameter of the spherical flower-like ZnO structure was approximately 3.1 μm. Furthermore, when the concentration of NaOH was increased to 0.8 M, the morphology of ZnO changed to a flower-like structure, with a diameter of approximately 4.6 μm, as shown in Fig. 1(e) and (f). Rod-like (380 nm) and urchin-like ZnO (4.8 μm) structures (Fig. 1(g) and (j)) were obtained with 0.9 M and 1.2 M NaOH, respectively.
 | ||
| Fig. 1 SEM images of various ZnO structures (a and b) walnut, (c and d) spherical flower, (e and f) flower, (g and h) rod, (i and j) urchin. | ||
The rate of the chemical reaction and the concentration of Zn2+ and OH− ions are the main factors influencing the growth of ZnO structures. The reactions for the synthesis of standard ZnO nanostructures can be described as follows:50–52
| Zn(NO3)2·6H2O + 2NaOH → Zn(OH)2 + 2NaNO3 + 6H2O | (2) |
| Zn(OH)2 + 2H2O → [Zn(OH)4]2− + 2H+ | (3) |
| [Zn(OH)4]2− → ZnO + H2O + 2OH− | (4) |
As sodium hydroxide is added to the zinc nitrate solution in the initial reaction, zinc hydroxide (Zn(OH)2), sodium nitrate (NaNO3), and water molecules are formed. The number of OH− ions in the solution (from NaOH) is a factor governing the length and width of the structures, as shown in Fig. 1. As molar concentration increases, the density of OH− ions increases, which leads to a significant growth rate and the generation of petal-like structures from walnut to spherical flower-like, flower-like, and rods. ZnO structure is a hexagonal unit cell consisting of polar (000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) low-index face with negative charge as oxygen-terminated and polar 0001 faces with positive charge as zinc-terminated sites, that stimulates a net dipole moment through the (0001) axis (c-axis).53,54 At low NaOH (0.2 M) concentration, ZnO walnuts were formed as the OH− ion concentration was too low to allow anisotropic growth. Therefore, ZnO nuclei-twining aggregate particles were formed since the [Zn(OH4)]2− was still inadequate and there was a lack of active sites. Increasing the amount of OH− ions leads to increasing [Zn(OH)4]2−. According to the reaction process (eqn (2)–(4)), ZnO nuclei were obtained by the dehydration of [Zn(OH)4]2− ions. During the crystal growth, the oriented attachment of nanoclusters was formed to minimize the total surface energy. Then, along with the aggregation of ZnO nanoclusters, the growth of nanosheets was generated, resulting in the formation of spherical flower-like morphology. The aggregated nanoclusters could rearrange themselves along to the c-axis due to ZnO's intrinsic anisotropic character and decrease the nanosheet concentration to form rod or flower-like morphology. Further increase of OH− ions, causes the [Zn(OH)4]2− ion to absorb by an electrostatic force in the positive polar plane of (0001), which enables the crystal to grow anisotropically in (0001) direction to obtain the rod morphology. However, with a further increase in NaOH concentration, the solution becomes saturated, leading to agglomeration, which indicates that rods begin to accumulate and form an urchin-like morphology.53,55,56
) low-index face with negative charge as oxygen-terminated and polar 0001 faces with positive charge as zinc-terminated sites, that stimulates a net dipole moment through the (0001) axis (c-axis).53,54 At low NaOH (0.2 M) concentration, ZnO walnuts were formed as the OH− ion concentration was too low to allow anisotropic growth. Therefore, ZnO nuclei-twining aggregate particles were formed since the [Zn(OH4)]2− was still inadequate and there was a lack of active sites. Increasing the amount of OH− ions leads to increasing [Zn(OH)4]2−. According to the reaction process (eqn (2)–(4)), ZnO nuclei were obtained by the dehydration of [Zn(OH)4]2− ions. During the crystal growth, the oriented attachment of nanoclusters was formed to minimize the total surface energy. Then, along with the aggregation of ZnO nanoclusters, the growth of nanosheets was generated, resulting in the formation of spherical flower-like morphology. The aggregated nanoclusters could rearrange themselves along to the c-axis due to ZnO's intrinsic anisotropic character and decrease the nanosheet concentration to form rod or flower-like morphology. Further increase of OH− ions, causes the [Zn(OH)4]2− ion to absorb by an electrostatic force in the positive polar plane of (0001), which enables the crystal to grow anisotropically in (0001) direction to obtain the rod morphology. However, with a further increase in NaOH concentration, the solution becomes saturated, leading to agglomeration, which indicates that rods begin to accumulate and form an urchin-like morphology.53,55,56
The crystalline structures of the synthesized materials were characterized using XRD. Fig. 2 shows the XRD patterns of the five types of ZnO morphologies (Fig. 2(a)) and Ag–ZnOsf (Fig. 2(b)) with a specific Ag particle. Diffraction peaks of ZnOsf were observed at 2θ = 32.1°, 34.8°, 36.6°, 47.8°, 56.9°, 63.1°, 68.2°, and 69.3°, which were attributed to the (100), (002), (101), (102), (110), (103), (112), and (201), crystalline planes, respectively, which corresponds to the hexagonal phase of ZnO (PDF# 36-1451). The XRD patterns of other ZnO materials are presented in the upper panel in Fig. 2, and the patterns were similar to that of ZnOsf, with slight differences in the peak intensity. Moreover, the diffraction peaks for Ag–ZnOsf at 2θ = 38.1°, 44.3°, and 64.5° were attributed to the (111), (200), and (220) crystalline planes, respectively, which correspond to the face-centered cubic (fcc) structure of Ag (PDF# 04-0783). Peaks corresponding to other impurities were not detected. The Ag crystallite size calculated from (111) faces by Scherrer equation was 14.51 nm. Furthermore, to collect additional information regarding the morphology, TEM studies were performed. Fig. 3 shows the structure of Ag NPs photo-deposited on ZnOsf, reveal that Ag was loaded on the surface of ZnO. EDX-mapping was also used to confirm the presence and distribution of Zn, O, and Ag (Fig. 3(c)–(f)). The Fourier-transform (FT) patterns of Ag–ZnOsf are shown in Fig. 3(g) and (h) (inset). The line spacing of approximately 0.242 nm, as shown in Fig. 3(g) inset, corresponds to the (101) lattice plane of ZnO, whereas the line spacing of approximately 0.240 nm, as shown in Fig. 3(h) inset, corresponds to the (111) lattice plane of the face-centered cubic Ag structure. These observations are in good agreement with the XRD pattern shown in Fig. 2(b).
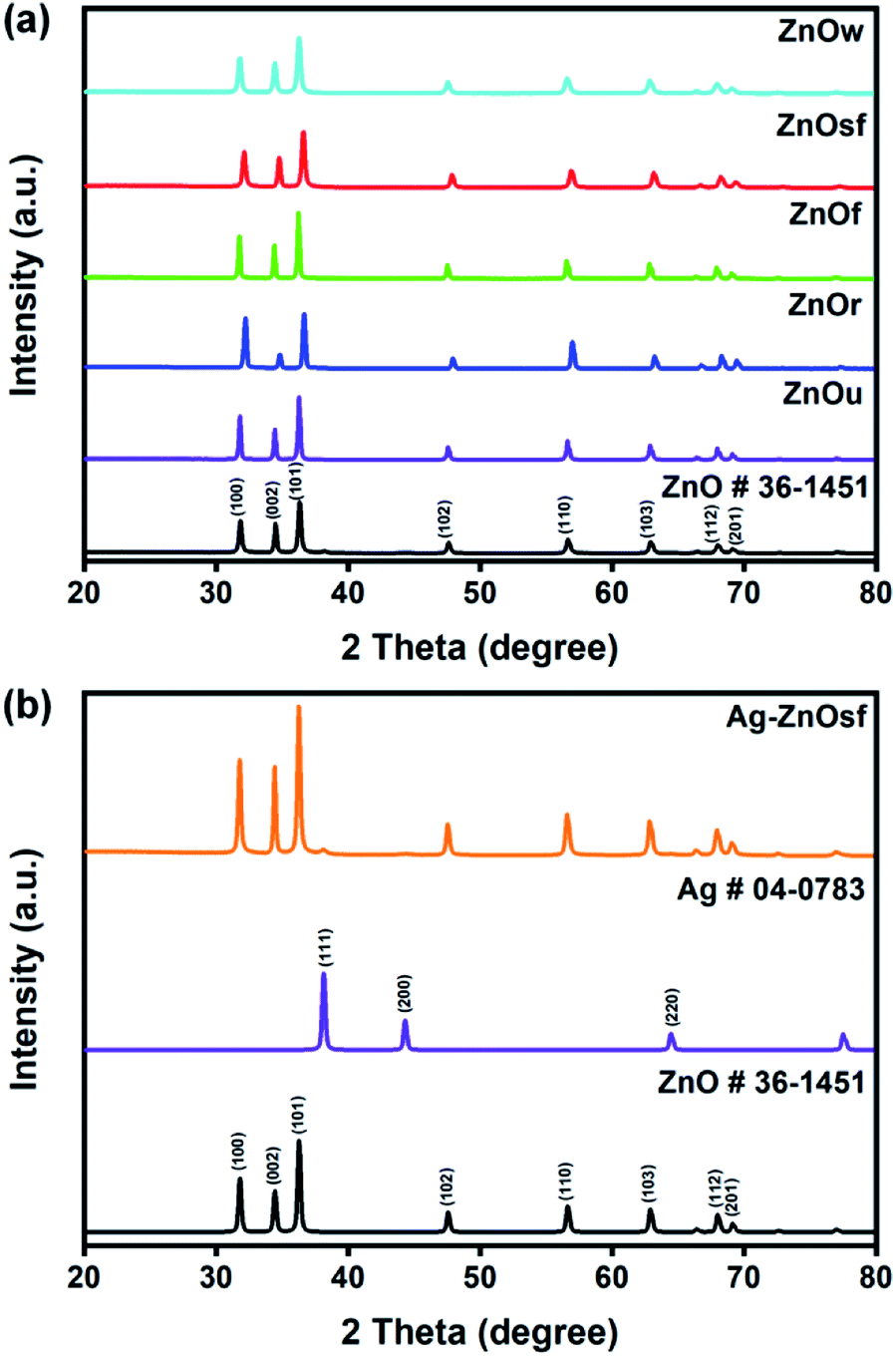 | ||
| Fig. 2 XRD patterns of (a) various ZnO structures and (b) Ag–ZnOsf composite. | ||
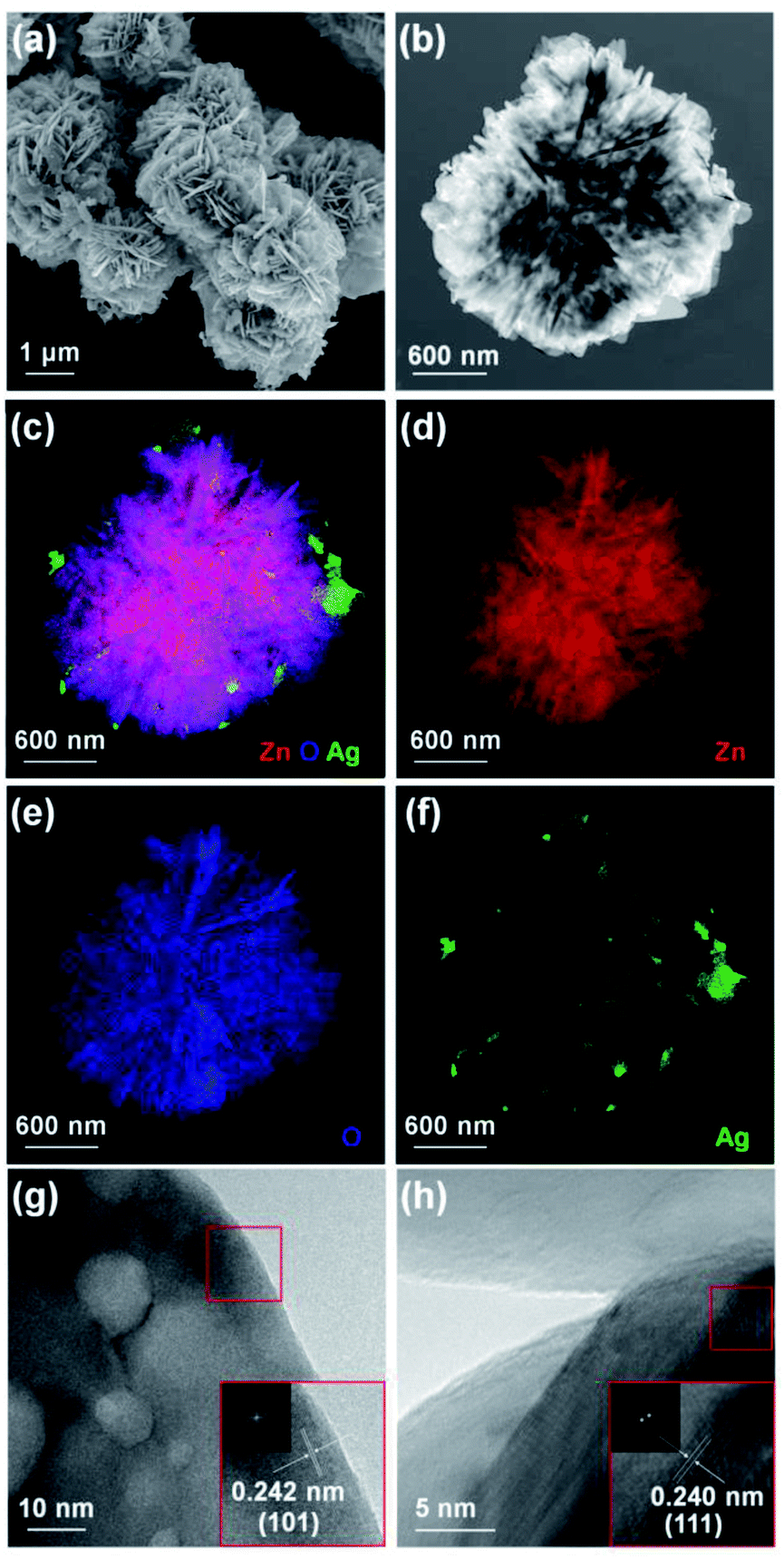 | ||
| Fig. 3 (a) SEM, (b) TEM, (c–f) EDX mapping, (g and h) HR-TEM images of Ag–ZnOsf composite. (Inset) FT patterns of ZnO and Ag. | ||
Furthermore, XPS analysis was performed to analyze the chemical composition of the Ag–ZnOsf surface. Fig. 4(a) presents the survey spectrum, showing signals of Ag, Zn, O, and C. The atomic ratio of Zn, O, and Ag were 58.43, 38.61 and 2.96%, respectively. The Zn 2p spectrum exhibits two main peaks of the Zn 2p1/2 and Zn 2p3/2 states (Fig. 4(b)). The binding energy (BE) peak of Zn 2p3/2 at 1020.85 eV is attributed to Zn–O bonds in ZnO. The O 1s spectrum (Fig. 4(c)) indicates peaks of Zn–O bonds (529.6 eV), and Zn–OH bonds (531.2 eV).57 The Ag 3d spectrum (Fig. 4(d)) shows a BE peak of Ag 3d5/2 at 366.92 eV, which is at a lower BE than that of the Ag metal (368.20 eV) and Ag2O (367.2 eV). This peak is attributed to the interaction between Ag NPs and ZnO.58 In addition, the XRD pattern only indicates the signals of Ag, which confirms the presence of metallic Ag.
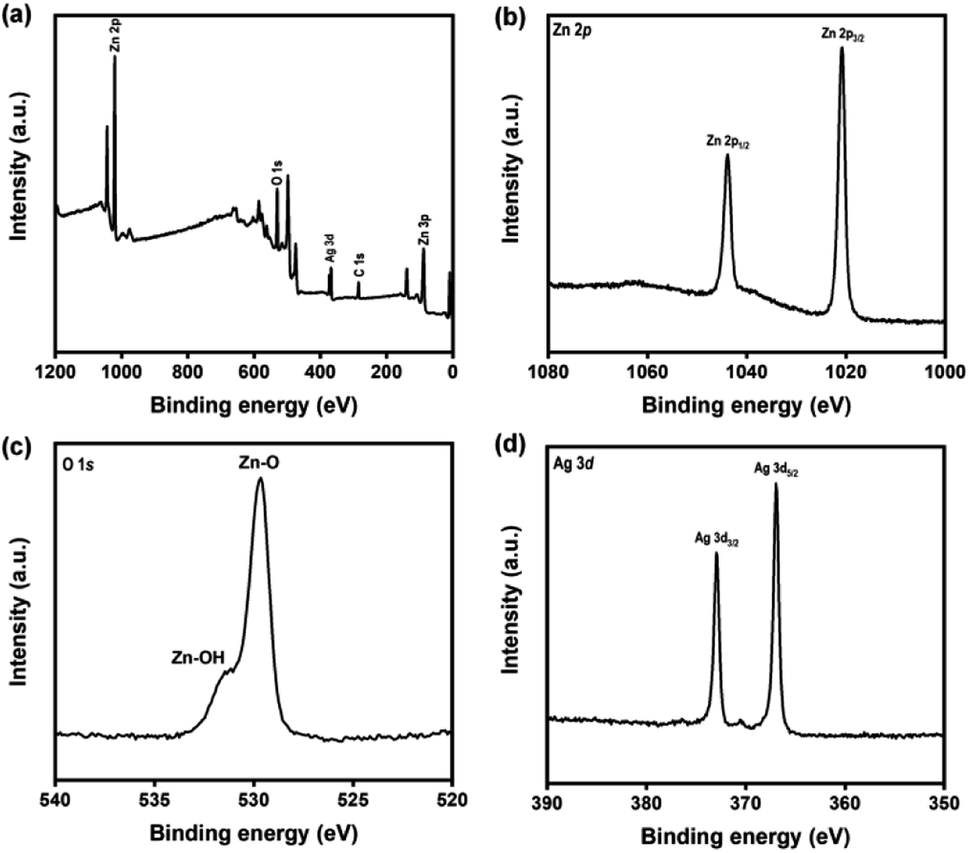 | ||
| Fig. 4 (a) XPS spectra of Ag–ZnOsf composite (a) survey, (b) Zn 2p lines, (c) O 1s lines and (d) Ag 2p lines. | ||
The surface structures were determined based on N2 sorption isotherms shown in Fig. 5(a). N2 adsorption/desorption isotherms of the as-prepared ZnO materials exhibited a typical type IV pattern for ZnOsf, ZnOw, ZnOr, ZnOf, indicative of mesoporous characteristics59 and typical type III pattern for ZnOu indicating a microporous material.59,60 The BET surface areas of various ZnO types were in the following order: ZnOsf (13.45 m2 g−1) > ZnOw (10.46 m2 g−1) > ZnOr (3.44 m2 g−1) > ZnOf (1.59 m2 g−1) > ZnOu (0.93 m2 g−1).
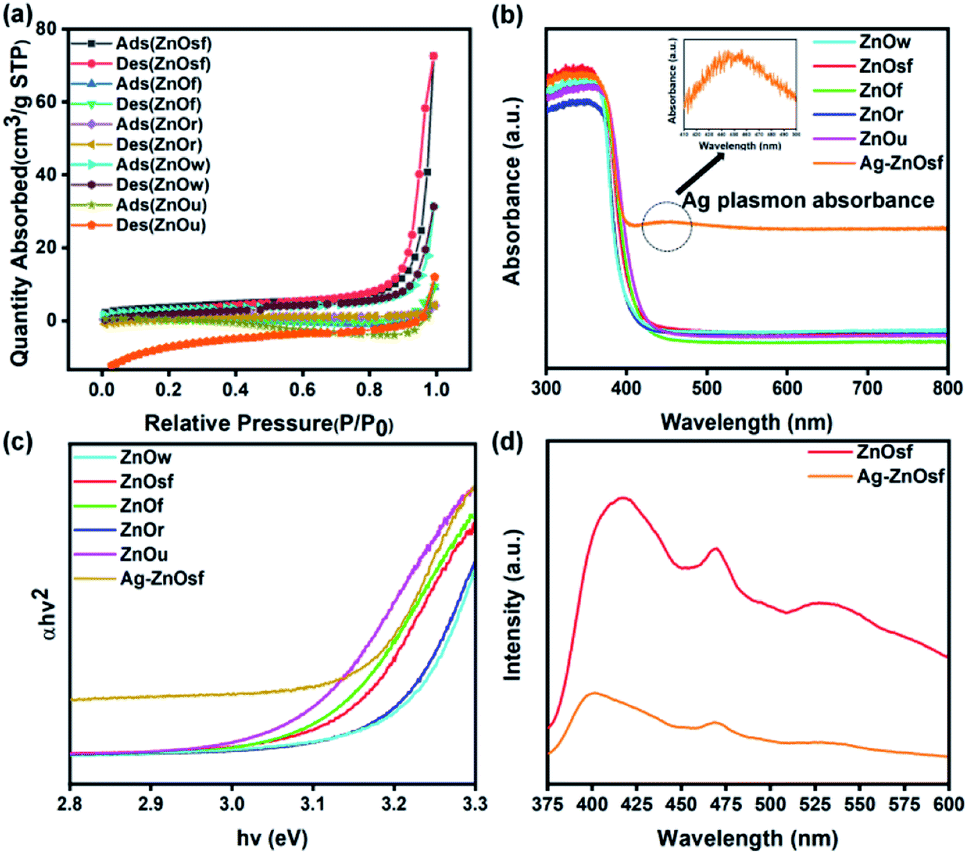 | ||
| Fig. 5 (a) N2 adsorption/desorption isotherms of various ZnO structures, (b) UV-Vis DRS of (c) the calculated band gap energy of various ZnO structures and Ag–ZnOsf (d) PL spectra for ZnOsf and Ag–ZnOsf composite. | ||
UV-Vis DRS was used to evaluate the optical properties and determine light absorption characteristics of various ZnO materials and after loading Ag NPs, as shown in Fig. 5(b). All the ZnO structures absorbed light in the ultraviolet to visible regions. Loading ZnOsf with Ag NPs gives an additional absorption peak in the visible region (410–550 nm), indicating the presence of Ag SPR. The calculated band-gap energies (Fig. 5(c)) of the as-prepared ZnO samples, i.e., ZnOw, ZnOsf, ZnOf, ZnOr, and ZnOu are 3.19, 3.09, 3.08, 3.17, and 3.05 eV, respectively. The measured band-gap of as-synthesized ZnO is less than that of the commercial ZnO (3.37 eV).61 The loading of Ag as SPR on ZnOsf shifted the band gap energy to low energy (3.07 eV) indicating the suppression of the recombination of photogenerated electron–hole pairs due to the improvement of the absorption of the visible light.39–41
PL spectra were also acquired to determine the charge recombination in Ag–ZnOsf relative to the ZnOsf. The PL spectra of ZnOsf and Ag–ZnOsf (Fig. 5(d)) were acquired at an excitation wavelength of 320 nm. The emission peaks were observed at 527, 470, and 417 nm. For Ag–ZnOsf, the intensity of the absorption peak declined significantly, indicating lower recombination of the electron–hole pairs, as compared to that in bare ZnOsf. Thus, the lifetime of the photogenerated charge carriers is improved with low photorecombination, which is expected to improve the photocatalytic activity of Ag–ZnOsf.
The photocatalytic activity of various ZnO structures was studied based on the degradation of MO and RhB, which were used as model dyes. The reaction under UV-Vis light irradiation using a Xe lamp at room temperature was investigated, and the decrease in the concentration of the dye was monitored using a UV-Vis spectrometer. The intensity of the maximum absorption peak of MO (464 nm) and RhB (554 nm) was monitored over time. Fig. 6(a) and (b) shows that the absorbance of MO and RhB decreased by approximately 99.7% and 94.2%, respectively, in the presence of ZnOsf after 50 min. The absorption without catalysts (Fig. 6(c) and (d)) was also explored to understand the role of ZnO during the photodegradation of the dyes.
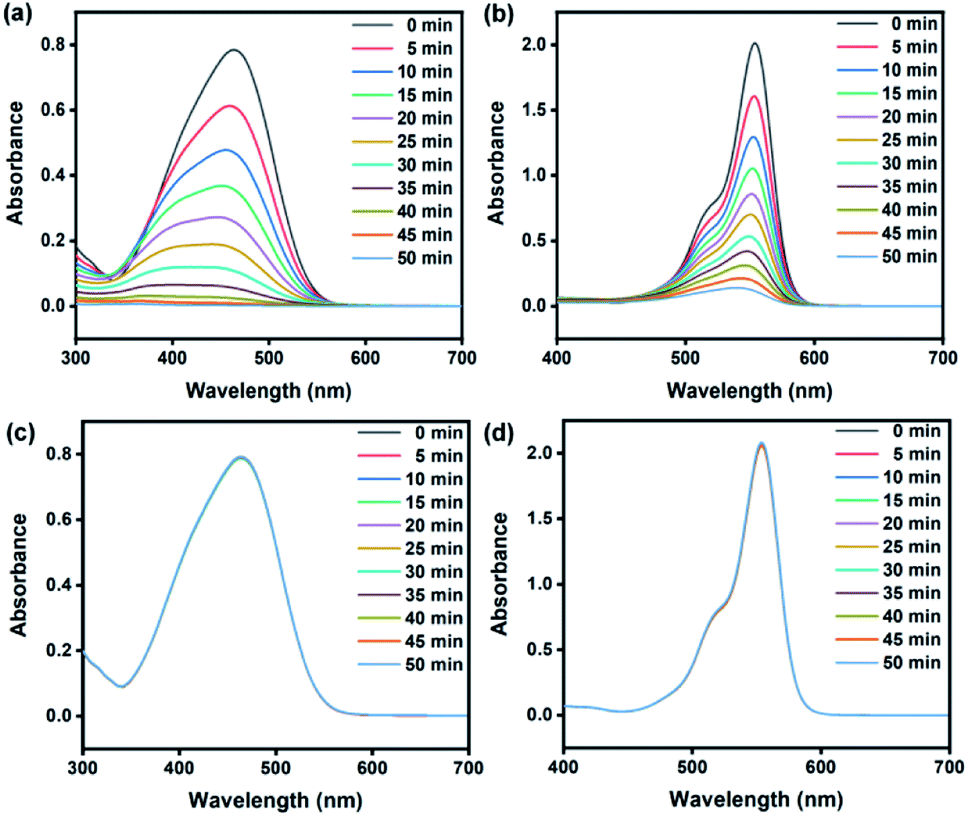 | ||
| Fig. 6 UV-Vis spectra degradation of (a) MO, (b) RhB using ZnOsf and (c and d) comparison without the catalyst. | ||
Moreover, to determine which ZnO structure afforded the optimal photocatalytic performance, C/C0 plots for the photodegradation of MO and RhB were constructed, as shown in Fig. 7(a) and (c). The percentage degradation afforded by ZnOsf, ZnOf, ZnOr, ZnOw, and ZnOu was estimated to be 99.7, 88.0, 80.8, 74.0, and 59.3% for MO and 94.2, 92.1, 90.8, 88.6, and 69.7% for RhB, respectively. The overall activity for MO and RhB degradation was in the following order: ZnOsf > ZnOf > ZnOr > ZnOw > ZnOu (Fig. 7). The kinetics of the photodegradation of MO and RhB can be defined by the following equation: ln(C0/C) = kt, where k is the first-order kinetics constant, and t is the duration of irradiation (Fig. 7(b) and (d)). The R2 value shows more than 0.9 for MO and RhB degradation for five types of the as-synthesized ZnO structures. The k values for ZnOsf (0.1104 min−1 (MO) and 0.0542 min−1 (RhB)) were the largest, as compared to those of the other structures, indicating that ZnOsf achieves better photodegradation performance. The higher photocatalytic activity of ZnOsf, as compared to those of ZnOf, ZnOr, ZnOw, and ZnOu, can be attributed to the higher surface area of ZnOsf. On the other hand, the surface morphology of the ZnOsf structure might also affect the catalytic activity because of the improved light absorption efficiency. Based on the structural tuning of MoS2 for the photodegradation of MB, it was found that MoS2 flower-like structures possess additional light paths that improve the light absorption efficiency, as compared to spherical and coil-like structures.62 The flower-like structure (ZnOsf and ZnOf) have a small band gap compared with the other structures indicating that they have more light absorption efficiency. However, even though ZnOu has a small band gap that is close to that of the flower-like structure, the photocatalytic activity is low due to the lower surface area. Besides, despite ZnOw having the second highest surface area, the photocatalytic activity is relatively low due to the highest bandgap energy. Therefore, the combination of surface area and light absorption efficiency produces the synergetic effect to impact the photodegradation performance.
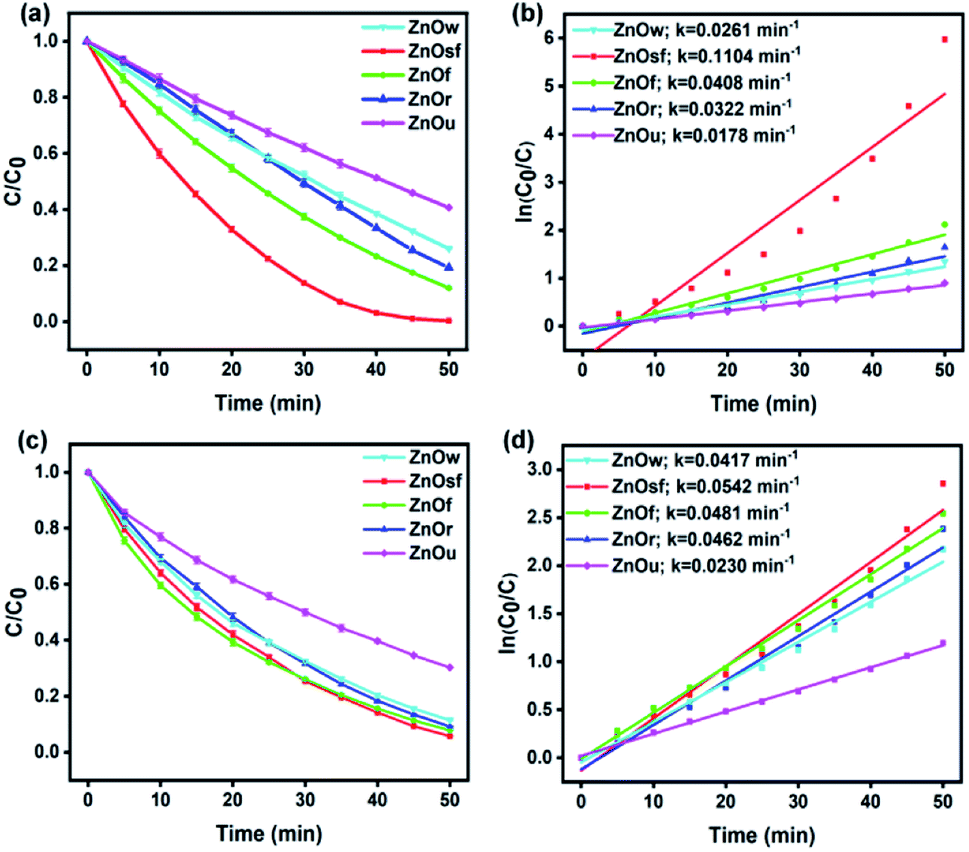 | ||
| Fig. 7 The degradation rate by time of various ZnO structure for (a and b) MO, (c and d) RhB. | ||
Furthermore, the SPR of Ag NPs loaded on ZnOf was also studied, indicating that the composite exhibited a significantly improved absorption of visible light. Fig. 8 shows the photocatalytic efficiency and kinetics of dye degradation by ZnOsf compared to those of Ag–ZnOsf. The Ag–ZnOsf composite degraded both MO and RhB within 35 min, with higher degradation percentages (MO: Ag–ZnOsf = 99.3% > ZnOsf = 93.0%; RhB: Ag–ZnOsf = 99.7% > ZnOf = 80.4%). The higher percentage degradation of the dyes is proportional to the k value.
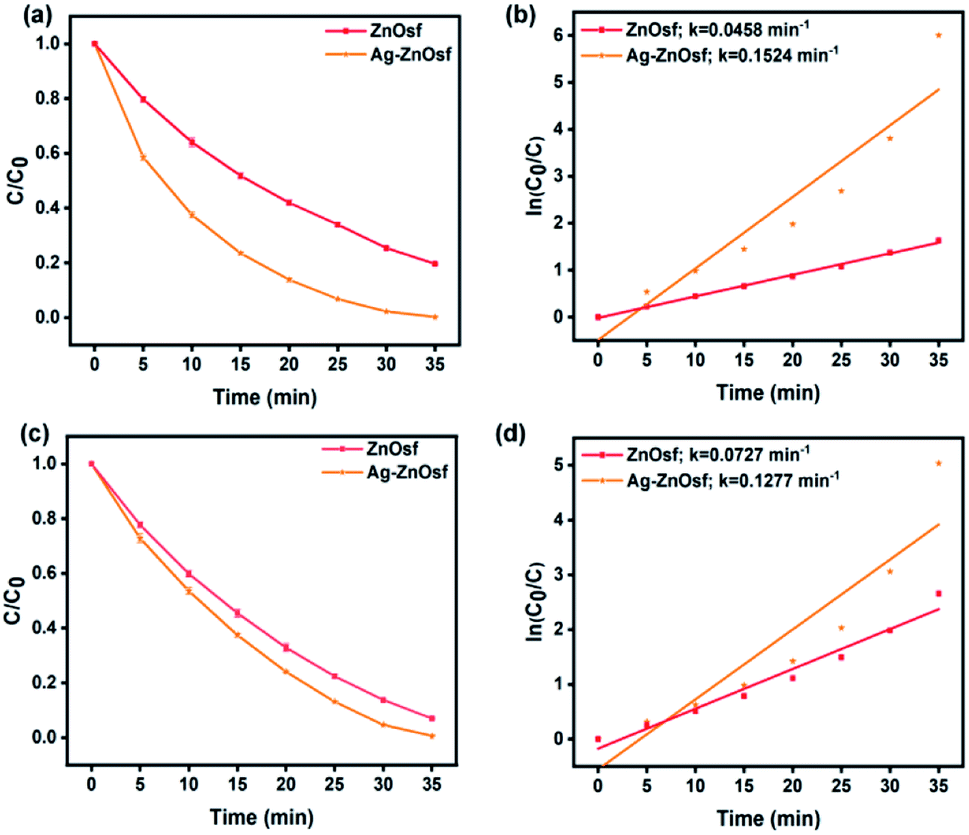 | ||
| Fig. 8 The degradation rate comparison between Ag–ZnOsf and ZnOsf for degradation of (a and b) MO and (c and d) RhB. | ||
The ideal photocatalytic performance of Ag–ZnOsf under incident UV-Vis light is due to the Schottky barrier between Ag and ZnO, which facilitates charge separation (Fig. 9). When Ag–ZnOsf undergoes induction under the incident light, electrons are excited from the valence band (VB) to the conduction band (CB). The excited electrons are quickly transferred to the Ag nanoparticles to prevent the recombination of electrons and holes; thus, the activity and lifetime of the catalyst increase. The electrons transferred to VB adsorb oxygen to form superoxide radical anions (O2˙−), which can react with H2O to supply hydroxyl radicals (˙OH). Simultaneously, VB, as a source of holes, interacts with H2O to produce ˙OH radicals. These generated O2˙− and ˙OH species attack the dyes (MO and RhB) and produce H2O and CO2 to complete the degradation process.8,63,64 The recyclability of the Ag–ZnOsf composite for the decomposition of dyes was determined based on the 10-cycle degradation of MO. The photocatalyst was used after separation from the medium and was washed with ethanol for each cycle. The photocatalytic efficiency of Ag–ZnOsf slightly decreased for the 5th run and continued to 80% after the 10th run (Fig. 10). The comparison for different catalysts based on ZnO and Ag using various methods for several dyes is displayed in Table 1. Ag–ZnOsf fabricated by a simple method gives a high degradation efficiency along with other catalysts. This suggests that the surface of the Ag–ZnO composite features numerous active sites, facilitating a dynamic catalytic reaction. Thus, the morphology-controlled ZnO and Ag–ZnO system can be used as potential photocatalysts for the degradation of other pollutants.
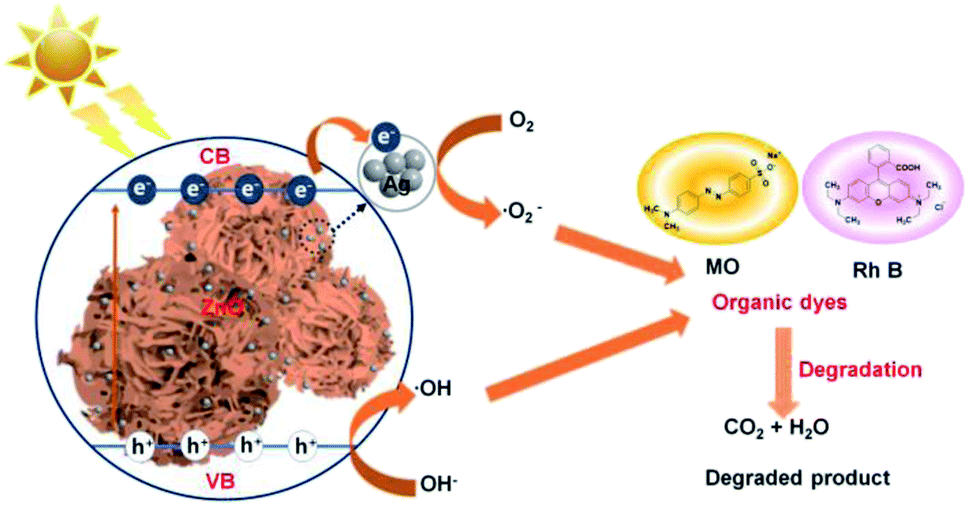 | ||
| Fig. 9 Probable mechanism for the reaction of Ag–ZnOsf composite for the degradation of dyes. | ||
| Photocatalyst | Synthesis method | Cat. (mg) | Dyes/conc. (mg L−1) | Irra. time (min) | Deg. rate (%) | Ref. |
|---|---|---|---|---|---|---|
| a Cat.: catalyst, Conc.: concentration, Irra.: irradiation, Deg.: degradation, MB: methylene blue, 4-NP: 4-nitrophenol, and CR: congo red. | ||||||
| Ag–ZnOsf | Hydrothermal, photodeposition | 40 | MO (10) | 35 | 99.7 | This work |
| RhB (10) | 99.3 | |||||
| Ag–ZnO flower like | Microwave | 150 | RhB (10) | 20 | 99 | 42 |
| 4-NP (20) | 80 | 95 | ||||
| Ag–ZnO heterostructure | Ball mill | 10 | MB (10) | 120 | 96 | 43 |
| Ag–ZnO 3D flower like | Precipitation (PVP template) | 40 | RhB (8) | 180 | 100 | 44 |
| Ag–TiO2 NPs | Sol–gel, vapor evaporation![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
500 | MO (0.5) | 80 | 99.3 | 45 |
| Ag–ZnO rod | Microwave, borohydride reduction | 50 | CR (16) | 60 | 81.6 | 46 |
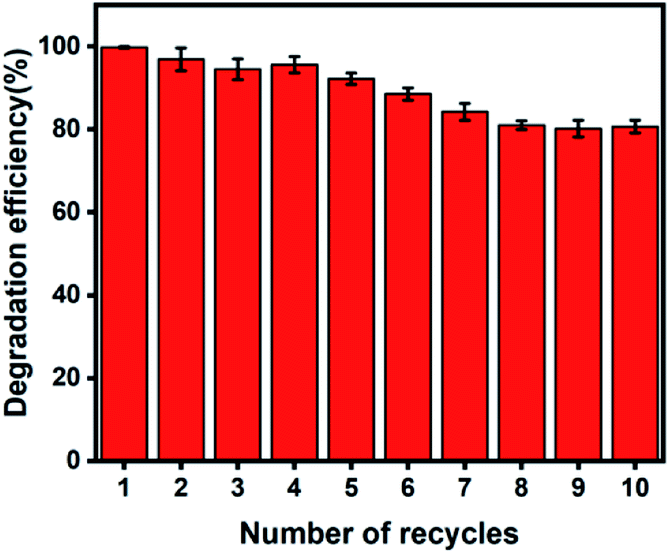 | ||
| Fig. 10 Recyclability test of Ag–ZnOsf composite with 10th cycles. | ||
Conclusions
In summary, five forms of ZnO (walnut, spherical flower-like, flower-like, rod-like, and urchin-like structures) were prepared by controlling the NaOH concentration during synthesis, without the addition of any surfactant or capping agent. Well-defined morphologies and good crystallinity were obtained for all ZnO structures. These five forms of ZnO possess good optical properties, with a small band gap. As-synthesized ZnOsf was more effective for photocatalytic degradation of MO and RhB than other ZnO structures. ZnOsf has a large surface area relative to the other structures. Ag NPs-loaded ZnOsf was successfully synthesized by the photo-deposition method. The as-prepared Ag–ZnOsf photocatalyst showed good photocatalytic activity and stability. The enhanced photocatalytic activity is attributed to the more rapid transport of surface electrons and the effective separation of electron–hole pairs due to the decoration by Ag NPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP) (NRF-2020R1I1A3067208, 2018R1D1A1B07045663, and NRF-2018H1A2A1062253) and PNU-RENovation (2019-2020) to H. L.References
- M. Ismail, K. Akhtar, M. I. Khan, T. Kamal, M. A. Khan, A. M. Asiri, J. Seo and S. B. Khan, Curr. Pharm. Des., 2019, 25, 3645–3663 CrossRef CAS.
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innov., 2019, 3, 275–290 CrossRef.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- S. Wijetunga, X. F. Li and C. Jian, J. Hazard. Mater., 2010, 177, 792–798 CrossRef CAS.
- M. M. Momeni, Appl. Phys. A: Mater. Sci. Process., 2015, 119, 1413–1422 CrossRef CAS.
- M. M. Mahlambi, C. J. Ngila and B. B. Mamba, J. Nanomater., 2015, 2015, 790173 Search PubMed.
- F. Zhang, X. Wang, H. Liu, C. Liu, Y. Wan, Y. Long and Z. Cai, Appl. Sci., 2019, 9(12), 2489 CrossRef CAS.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- S. Chakrabarti and B. K. Dutta, J. Hazard. Mater., 2004, 112, 269–278 CrossRef CAS.
- L. M. Torres-Martínez, I. Juárez-Ramírez and M. Z. Figueroa-Torres, in Nanomaterials for Environmental Protection, John Wiley & Sons, Inc, Hoboken, NJ, 2014, vol. 9781118496978, pp. 193–203 Search PubMed.
- F. Han, V. S. R. Kambala, M. Srinivasan, D. Rajarathnam and R. Naidu, Appl. Catal., A, 2009, 359, 25–40 CrossRef CAS.
- K. H. Kim and S. K. Ihm, J. Hazard. Mater., 2011, 186, 16–34 CrossRef CAS.
- A. Janotti and C. G. Van De Walle, Rep. Prog. Phys., 2009, 72, 126501 CrossRef.
- R. Kumar, G. Kumar and A. Umar, Nanosci. Nanotechnol. Lett., 2014, 6, 631–650 CrossRef CAS.
- H. Mou, C. Song, Y. Zhou, B. Zhang and D. Wang, Appl. Catal., B, 2018, 221, 565–573 CrossRef CAS.
- T. K. Le, T. M. T. Nguyen, H. T. P. Nguyen, T. K. L. Nguyen, T. Lund, H. K. H. Nguyen and T. K. X. Huynh, Arabian J. Chem., 2020, 13, 1032–1039 CrossRef CAS.
- F. Dufour, S. Pigeot-Remy, O. Durupthy, S. Cassaignon, V. Ruaux, S. Torelli, L. Mariey, F. Maugé and C. Chanéac, Appl. Catal., B, 2015, 174–175, 350–360 CrossRef CAS.
- X. Z. Chu, Z. P. Cheng, Y. J. Zhao, J. M. Xu, M. S. Li, L. Hu, S. Y. Zhou, F. Y. Wu and C. H. Lee, Ceram. Int., 2017, 43, 8222–8229 CrossRef CAS.
- N. F. Andrade Neto, P. M. Oliveira, R. M. Nascimento, C. A. Paskocimas, M. R. D. Bomio and F. V. Motta, Ceram. Int., 2019, 45, 651–658 CrossRef CAS.
- S. Baruah and J. Dutta, Sci. Technol. Adv. Mater., 2009, 10(1), 18 CrossRef.
- J. Wojnarowicz, T. Chudoba and W. Lojkowski, Nanomaterials, 2020, 10(6), 1086 CrossRef CAS.
- X. Wang, Q. Zhang, Q. Wan, G. Dai, C. Zhou and B. Zou, J. Phys. Chem. C, 2011, 115, 2769–2775 CrossRef CAS.
- V. Gerbreders, M. Krasovska, E. Sledevskis, A. Gerbreders, I. Mihailova, E. Tamanis and A. Ogurcovs, CrystEngComm, 2020, 22, 1346–1358 RSC.
- Z. B. Huang, X. P. Zou, G. Q. Yang, X. M. Lv, C. L. Wei, Z. Sun, H. Q. Zhou and B. L. Zhang, Adv. Mater. Res., 2014, 875–877, 1549–1553 Search PubMed.
- R. Wahab, A. Mishra, S. Il Yun, I. H. Hwang, J. Mussarat, A. A. Al-Khedhairy, Y. S. Kim and H. S. Shin, Biomass Bioenergy, 2012, 39, 227–236 CrossRef CAS.
- N. Güy, S. Çakar and M. Özacar, J. Colloid Interface Sci., 2016, 466, 128–137 CrossRef.
- J. Rodrigues, T. Hatami, J. M. Rosa, E. B. Tambourgi and L. H. I. Mei, Chem. Eng. Res. Des., 2020, 153, 294–305 CrossRef CAS.
- H. Liu, Y. Hu, Z. Zhang, X. Liu, H. Jia and B. Xu, Appl. Surf. Sci., 2015, 355, 644–652 CrossRef CAS.
- C. Jaramillo-Páez, J. A. Navío and M. C. Hidalgo, J. Photochem. Photobiol., A, 2018, 356, 112–122 CrossRef.
- M. Zayed, A. M. Ahmed and M. Shaban, Int. J. Hydrogen Energy, 2019, 44, 17630–17648 CrossRef CAS.
- S. Kuriakose, K. Sahu, S. A. Khan, A. Tripathi, D. K. Avasthi and S. Mohapatra, Opt. Mater., 2017, 64, 47–52 CrossRef CAS.
- R. Mohammadzadeh Kakhki, R. Tayebee and F. Ahsani, J. Mater. Sci.: Mater. Electron., 2017, 28, 5941–5952 CrossRef CAS.
- X. Liu, W. Li, N. Chen, X. Xing, C. Dong and Y. Wang, RSC Adv., 2015, 5, 34456–34465 RSC.
- B. Yu, Y. Zhou, P. Li, W. Tu, P. Li, L. Tang, J. Ye and Z. Zou, Nanoscale, 2016, 8, 11870–11874 RSC.
- M. Arab Chamjangali, G. Bagherian, A. Javid, S. Boroumand and N. Farzaneh, Spectrochim. Acta, Part A, 2015, 150, 230–237 CrossRef CAS.
- Y. Lin, C. Yang, S. Wu, X. Li, Y. Chen and W. L. Yang, Adv. Funct. Mater., 2020, 30, 2002918 CrossRef CAS.
- Y. Lin, H. Liu, C. Yang, X. Wu, C. Du, L. Jiang and Y. Zhong, Appl. Catal., B, 2020, 264, 118479 CrossRef.
- Y. Lin, S. Wu, X. Li, X. Wu, C. Yang, G. Zeng, Y. Peng, Q. Zhou and L. Lu, Appl. Catal., B, 2018, 227, 557–570 CrossRef CAS.
- A. Padmanaban, T. Dhanasekaran, S. Praveen Kumar, G. Gnanamoorthy, S. Munusamy, A. Stephen and V. Narayanan, Mechanics, Materials Science & Engineering, 2017, 9(1) DOI:10.2412/mmse.97.67.748.
- M. A. H. Devadi, M. Krishna, H. N. N. Murthy and B. S. Sathyanarayana, Procedia Mater. Sci., 2014, 5, 612–621 CrossRef CAS.
- R. M. Kulkarni, R. S. Malladi, M. S. Hanagadakar, M. R. Doddamani and U. K. Bhat, Desalin. Water Treat., 2016, 57, 16111–16118 CrossRef CAS.
- H. Liu, H. Liu, J. Yang, H. Zhai, X. Liu and H. Jia, Ceram. Int., 2019, 45, 20133–20140 CrossRef CAS.
- E. Mendoza-Mendoza, A. G. Nuñez-Briones, L. A. García-Cerda, R. D. Peralta-Rodríguez and A. J. Montes-Luna, Ceram. Int., 2018, 44, 6176–6180 CrossRef CAS.
- Y. Liang, N. Guo, L. Li, R. Li, G. Ji and S. Gan, New J. Chem., 2016, 40, 1587–1594 RSC.
- R. Saravanan, D. Manoj, J. Qin, M. Naushad, F. Gracia, A. F. Lee, M. M. Khan and M. A. Gracia-Pinilla, Process Saf. Environ. Prot., 2018, 120, 339–347 CrossRef CAS.
- N. Güy and M. Özacar, Int. J. Hydrogen Energy, 2016, 41, 20100–20112 CrossRef.
- Y. Lee, E. Kim, Y. Park, J. Kim, W. H. Ryu, J. Rho and K. Kim, J. Mater., 2018, 4, 83–94 Search PubMed.
- Y. H. Chiu, T. F. M. Chang, C. Y. Chen, M. Sone and Y. J. Hsu, Catalysts, 2019, 9(5), 430 CrossRef CAS.
- A. Zyoud, A. Zu’bi, M. H. S. Helal, D. H. Park, G. Campet and H. S. Hilal, J. Environ. Health Sci. Eng., 2015, 13, 1–10 CrossRef CAS.
- S. S. Alias, A. B. Ismail and A. A. Mohamad, J. Alloys Compd., 2010, 499, 231–237 CrossRef CAS.
- P. Chand, A. Gaur, A. Kumar and U. K. Gaur, Mater. Sci. Semicond. Process., 2015, 38, 72–80 CrossRef CAS.
- X. Zhao, S. Su, G. Wu, C. Li, Z. Qin, X. Lou and J. Zhou, Appl. Surf. Sci., 2017, 406, 254–264 CrossRef CAS.
- X. Zhao, M. Li and X. Lou, Adv. Powder Technol., 2014, 25, 372–378 CrossRef CAS.
- E. S. Jang, J. Korean Ceram. Soc., 2017, 54, 167–183 CrossRef CAS.
- F. Ahmed, N. Arshi, M. S. Anwar, R. Danish and B. H. Koo, RSC Adv., 2014, 4, 29249–29263 RSC.
- Y. Sun, L. Wang, X. Yu and K. Chen, CrystEngComm, 2012, 14, 3199–3204 RSC.
- R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef CAS.
- M. Li, W. Huang, W. Qian, B. Liu, H. Lin, W. Li, L. Wan and C. Dong, RSC Adv., 2017, 7, 46760–46766 RSC.
- Z. A. Alothman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- C. Tejada-Tovar, A. D. Gonzalez-Delgado and A. Villabona-Ortiz, Appl. Sci., 2019, 9, 4486 CrossRef CAS.
- N. Kamarulzaman, M. F. Kasim and R. Rusdi, Nanoscale Res. Lett., 2015, 10, 346 CrossRef.
- Y. Li, F. Xiang, W. Lou and X. Zhang, in IOP Conference Series: Earth and Environmental Science, Institute of Physics Publishing, 2019, vol. 300, p. 052021 Search PubMed.
- K. H. Leong, A. A. Aziz, L. C. Sim, P. Saravanan, M. Jang and D. Bahnemann, Beilstein J. Nanotechnol., 2018, 9, 628–648 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, RSC Adv., 2015, 5, 3306–3351 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |