
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel two-dimensional metal–organic framework as a recyclable heterogeneous catalyst for the dehydrogenative oxidation of alcohol and the N-arylation of azole compounds†
Chengxin Liua,
Jin Cui*b,
Yufang Wangc and
Mingjie Zhang *a
*a
aDepartment of Chemistry, School of Sciences, Tianjin University, Tianjin 30035, P. R. China
bNational Foodstuff Inspection Center, Tianjin Product Quality Inspection Technology Research Institute, Tianjin 300384, P. R. China
cScientific Research Department, Shijiazhuang University of Applied Technology, Shijiazhuang 050081, P. R. China
First published on 22nd March 2021
Abstract
A novel metal–organic framework (MOF) with two-dimensional (2D) crystal structure was developed using Cu(NO3)2·3H2O and 2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarboxylic acid. Further, its structure was characterized using infrared spectroscopy, thermogravimetry, X-ray diffraction, and X-ray crystallography. The activated Cu-MOF was used to catalyze the dehydrogenative oxidation of alcohol and N-arylation of azole compounds. Furthermore, it could be easily recovered and reused.
Organic synthesis comprises reaction steps that involve different types of catalysts; the usage of transition metals as catalysts occupies an important position.1 For instance, the dehydrogenative oxidation of alcohols catalyzed by a transition metal is an important development with respect to the industrial production of many fine and bulk chemicals including agrochemicals, fragrance materials, dyestuffs, insecticides, cosmetics, flame-retardant materials, vitamins, and drugs.2–4 In another example, one of the most important synthetic methodologies for N-aryl compounds involves the transition metals used to catalyze the C–N cross-coupling reactions, such as Goldberg-type reactions5–7 and Ullmann-type reactions.8–11 The development of recyclable Cu catalysts, which is of considerable significance, is of particular interest.12 The metal–organic frameworks (MOFs) comprising metal ions and organic ligands are widely used as heterogeneous catalysts because of their excellent stability, functional tunability, and unique porosity.13–17 Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2 and MIL-101, reported by Long18 and S. Kaskel19 in 2008, respectively, have exposed metal coordination sites. They have been identified as size-selective Lewis acid catalysts. In 2011, MIL-47 and MOF48 were reported as heterogeneous recyclable catalysts for converting methane to acetic acid, relying on metal sites within the secondary building units (SBUs).20 Carboxylic acid derivatives are the ligands that are traditionally used to construct MOFs because they can be easily synthesized and modified.21–23 For example, MOF-199, which constructed from H3BTC, can be used as an excellent heterogeneous recyclable catalyst upon the loss of coordinated solvates.24,25 4,4′-Biphenyldicarboxylic acid and its derivatives are one kind of the earliest carboxylic acid ligands used to construct MOFs.26–30 In 2012, Janiak reported a Cu-MOF with a dinuclear Cu-paddle wheel SBU constructed from 2,2′,6,6′-tetramethyl-4,4-biphenyldicarboxylic acid and Cu(NO3)2·H2O.31 The twist between the two benzene rings and the benzene ring with the plane of the four carbons of the SBU are both close to 90°, which leads to its three-dimensional (3D) interpenetrating structure. A similar crystal structure is maintained after the removal of the coordination solvents. However, in 3D MOFs, ligands coordinate with the metal ions in all directions, which restricts the interactions between metal ions and substrate molecules or other compounds in the reaction. It is difficult for the molecules to concentrate near active sites once they enter the pores in the MOF,32–34 resulting in reduced performance. It is necessary to develop a 2D Cu-MOF with catalytic activity.35 There are only two reports about 2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarboxylic acid, both of which regarded this compound as an intermediate and not the final product and used large amounts of bromine during synthesis.36,37 The work presented herein constructed a new type of 2D Cu-MOF using 2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarboxylic acid, wherein a recyclable heterogeneous catalyst was used. The results of this study demonstrate the efficacy of an alternative process for the future synthesis and catalysis of organic, which is less costly and toxic to the environment.
Using 2,5-dimethoxybenzoic acid as the raw material, the ligand (2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarboxylic acid) was synthesized and heated with Cu(NO3)2·3H2O, methanol, and N,N-diethylformamide (DEF) to obtain the green octahedral crystals of the Cu-MOF (for images of Cu-MOF, see Fig. 2(d)). The single-crystal X-ray diffraction analysis revealed that the Cu-MOF crystallized in the orthorhombic system with the Cmca space group and exhibited a 2D dinuclear Cu paddlewheel crystal structure in the secondary building unit (SBU). In this SBU, Cu(II) formed Cu–Cu bonds between Cu atoms, four Cu–O bonds with COO−, and Cu–O bonds with methanols, producing a six-coordinate structure (Fig. 1(a)).
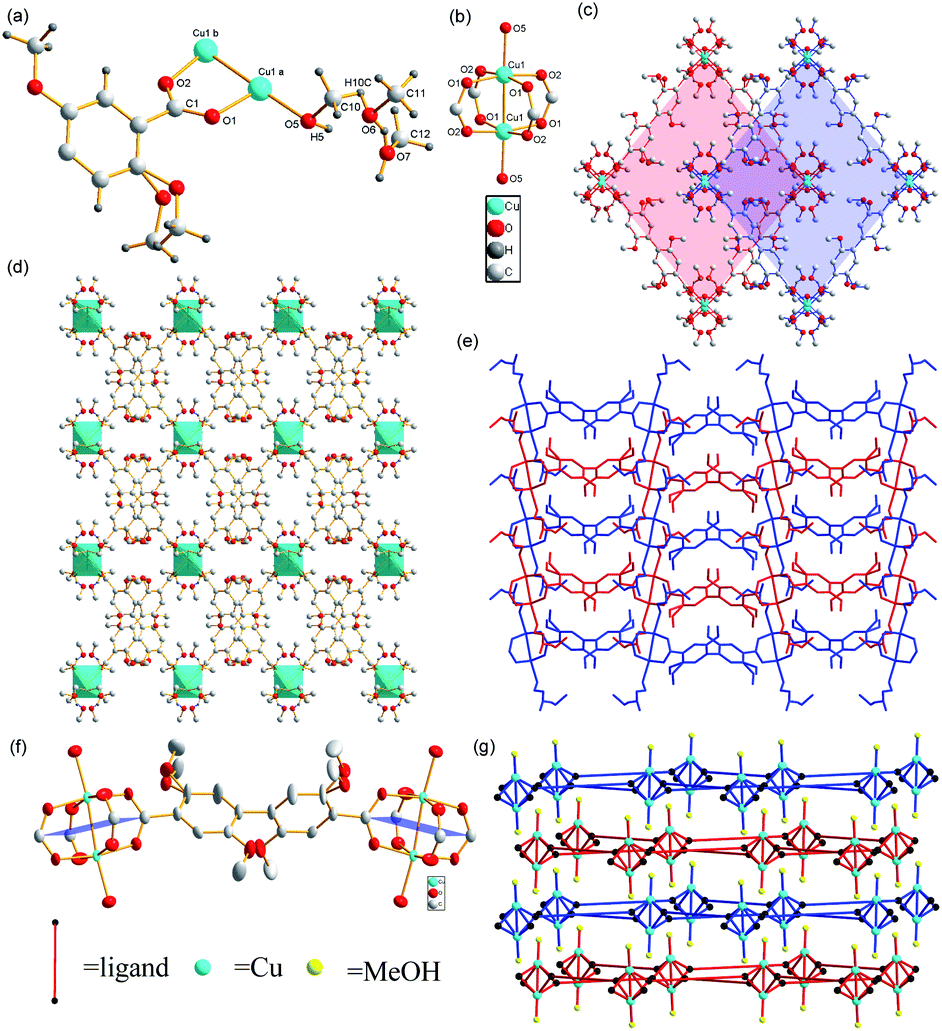 | ||
| Fig. 1 (a) Unit-cell crystal structure of the Cu-MOF. Here, a = x, y, z and b = x, −y, −z. (b) Secondary building unit (SBU) of the Cu-MOF. (c) Crystal structure of a Cu-MOF viewed along the [010] direction. Red and blue planes represent adjacent layers. (d) Crystal structure of a Cu-MOF viewed along the [010] direction. (e) Crystal structure of a Cu-MOF viewed along the [100] direction. Red and blue represent neighboring layers. (f) Two paddlewheel SBUs linked by 2,2′,5,5′-tetramethyl-4,4′-biphenyldicarboxylate to illustrate the twist. The single O atom in the apical position on Cu belongs to a MeOH molecule. (g) 2D structure schematic representation of Cu-MOF. | ||
2,2′,5,5′-tetramethoxy-4,4-biphenyldicarboxylic acid and 2,2′,6,6′-tetramethyl-4,4-biphenyldicarboxylic acid exhibit similar structures; the twists between the two benzene rings are both close to 90°.31 One difference is that the twist between the benzene ring and the plane of the four carbons of SBU in 2,2′,5,5′-tetramethoxy-4,4-biphenyldicarboxylic acid is close to 45° instead of 90° in 2,2′,6,6′-tetramethyl-4,4-biphenyldicarboxylic acid. Therefore, the torsion angle between the plane of the four carbons of two adjacent SBUs is closer to 180° instead of 90°, resulting in the 2D split-layer crystal structure of Cu-MOF (Fig. 1(f)). Layers are closely packed to form 3D supramolecular framework. Although the twisted chain comprising solvents penetrates into the adjacent layer, there is no strong interaction between them (Fig. 1(e)). Therefore, the unactivated Cu-MOF has not a 3D but a 2D crystal structure. This feature becomes more obvious after simplifying the ligand and solvent as shown in Fig. 1(g). A schematic of the simplified node is shown in Fig. S1.† Unlike the carboxylic acid ligand, methanol, which is not deprotonated, coordinated with Cu(II) to form a Cu–O bond that coordinated with the lone pair of O electrons and the empty orbital in Cu. The valence of Cu remained +2, but the bond length of Cu1–O5 (2.116 (7) Å) was longer than those of Cu1–O1 (1.961 (5) Å) and Cu1–O2 (1.966 (5) Å). This explains why Cu1–O5 is prone to fracture when heated. The bond lengths of C10–H10 (0.980 (2) Å) and O10–H10 (0.9892 (167) Å) are almost identical, indicating that the fracturing temperatures of these bonds during heating may also be identical. The methanol molecules in Cu-MOF connect to each other via hydrogen bonds and coordinate with MOFs through the Cu1–O5 bond. Therefore, we propose that the methanol molecules were lost during heating. This hypothesis was consistent with the thermogravimetric data.
The crystal structure of Cu-MOF can be clearly observed along the [010] direction (Fig. 1(c)). The ligands at right angles to each other extend from the SBUs to form a layer, which is parallel to the adjacent layers but staggered. The SBUs and all the methanols in any layer occur at the extension of the center of the parallelogram hole into the adjacent layers because of the large quantity and steric hindrance of methoxy groups. The observations in the [100] direction (Fig. 1(e)) reveal that the methanol molecules in one layer are connected via hydrogen bonds into twisted chains located in a position subparallel to adjacent layers. Because of the unique structure of this Cu-MOF, we infer that the substrates connect via Cu(II) and exhibit a five-coordinate structure because of the loss of solvent during the catalytic reaction. The substrates diffuse between layers and concentrate around the center of the parallelogram holes because of the steric hindrance of the methoxy groups on ligands. This location is close to the halfway point between two binuclear SBUs in adjacent layers, enabling substrates to interact with metals from either direction.
The Cu-MOF was heated to 100 °C for 24 h in vacuum and was then cooled to room temperature to obtain activated Cu-MOF, which could be observed as a green transparent solid (for images of activated Cu-MOF, see Fig. 2(d) and the SEM images of Cu-MOF and activated Cu-MOF see ESI†). Unfortunately, this substance could not be subjected to X-ray crystallography. However, the powder X-ray diffraction (PXRD) data showed that activated Cu-MOF maintained the same crystal structure as Cu-MOF (Fig. 2(b)). Next, the substance was subjected to thermogravimetric analysis. Cu-MOF remained stable up to a temperature of 190 °C when it began to lose weight rapidly. After a weight loss of approximately 16%, the rate of loss gradually slowed until a temperature of 250 °C was reached; subsequently, Cu-MOF continued to lose about 30% mass rapidly until it decomposed completely at 320 °C. Only one platform was observed from the thermogravimetric curves of Cu-MOF (Fig. 2(a)), indicating that the solvent molecules break away from the framework via heating. The activated Cu-MOF remained stable up to 250 °C and then abruptly lost weight and completely decomposed at 320 °C. No platform was observed in the thermogravimetric curve of activated Cu-MOF (Fig. 2(a)), indicating there are no solvent molecules in the framework. According to the crystal data (Table S3†) of Cu MOF, methanol accounted for 15.9% of the total weight, indicating that methanols were lost simultaneously. Further, Cu(II) exhibited a five-coordinate structure in the activated Cu-MOF, suggesting that no solvent or ligand coordinated with Cu(II) in the upper or lower directions of the plane on which binuclear SBUs were located.
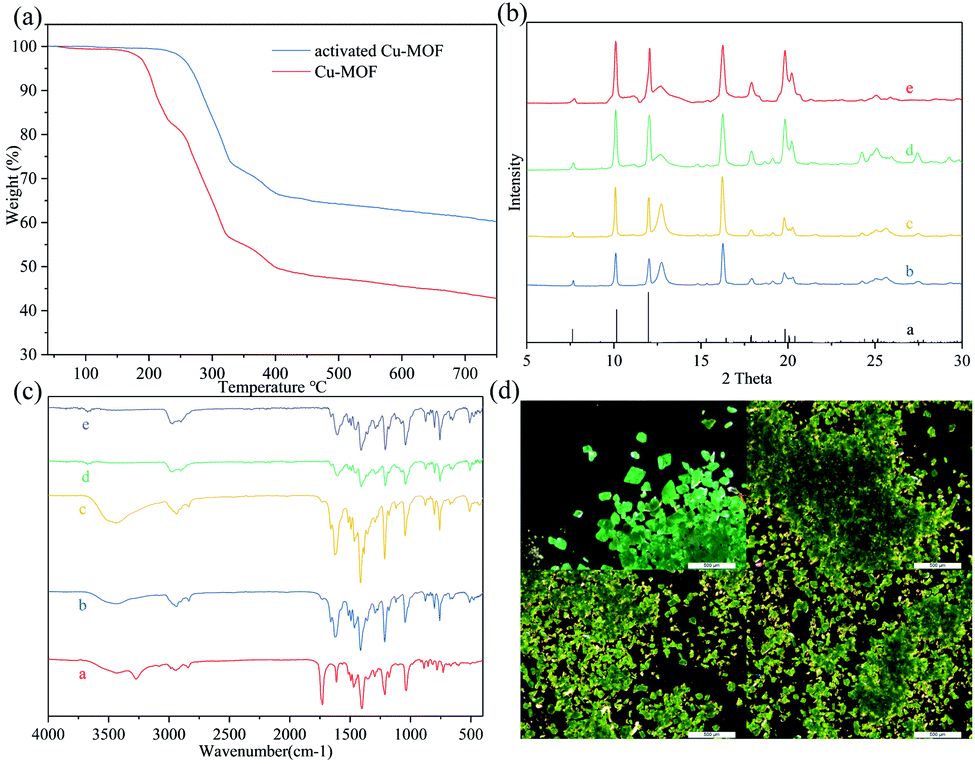 | ||
| Fig. 2 (a) Thermogravimetric curves of the Cu MOF and activated Cu MOF. (b) a, Simulated PXRD of the Cu MOF, b, measured PXRD of the Cu MOF, c, measured PXRD of the activated Cu MOF, d, measured PXRD of the activated Cu MOF after the dehydrogenative oxidation of alcohol cycle experiment, e, measured PXRD of the activated Cu MOF after N-arylation of the azole compound cycle experiment. (c) a, FT-IR spectra of the ligand, b, FT-IR spectra of the Cu MOF c, FT-IR spectra of the activated Cu MOF, d, FT-IR spectra of the activated Cu MOF after the dehydrogenative oxidation of alcohol cycle experiment, e, FT-IR spectra of the activated Cu MOF after the N-arylation of azole compound cycle experiment. (d) From left to right, top to bottom: images of the Cu MOF, images of the activated Cu MOF, images of the activated Cu MOF after the dehydrogenative oxidation of alcohol cycle experiment, images of the activated Cu MOF after the N-arylation of azole compounds cycle experiment. | ||
As shown in Table 1, the activated Cu MOF was applied as the catalyst for the dehydrogenative oxidation of alcohol. To optimize the reaction solvent, benzyl alcohol was used as the substrate, 0.25% mmol of the activated Cu MOF was used as the catalyst, and di-tert-butyl peroxide (DTBP) or tert-butyl hydroperoxide (TBHP) was used as the oxidant. The treatment was performed at 100 °C. When no solvent was added, the product was mainly self-esterified benzyl benzoate. Next, DMF was used as the solvent. In this case, the yield was the highest and the least amount of byproducts were obtained compared to DMSO and CH3CN. Hence, DMF was used as a solvent to optimize the type and amount of oxidant. First, yields were low when using N-methylmorpholine (NMM) and NaIO4 as oxidants. Next, TBHP was used as the oxidant; the obtained products consistently contained approximately 10% of the excessively oxidized benzoic acid. When DTBP was used as the oxidant, the yield increased initially when loaded with the oxidant and subsequently remained unchanged. Thus, optimal reaction conditions were obtained using DMF as the solvent and 2 equivalents of DTBP as the oxidant.
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Solvent | Yieldb (%) | ||
| 6a | 7 | 8 | |||
| a Reactions were conducted using benzyl alcohol (1.9 mmol), oxidant (3.8 mmol), activated Cu MOF (2 mg, 0.25 mmol%, counted as one copper), and solvent (3.0 mL) at 100 °C for 2.5 h.b Isolated yields.c Diphenylamine (3.8 mmol) was added.d Bromotrichloromethane (3.8 mmol) was added.e No catalyst was added.f No activated Cu MOF, but ligand (0.50 mmol%) was added.g No activated Cu MOF, but Cu(NO3)2·3H2O (0.25 mmol%) was added.h No activated Cu MOF, but MOF-199 (0.25 mmol%) was added. | |||||
| 1 | NMM (2 eq.) | DMF | 18 | 0 | 0 |
| 2 | NaIO4 (2 eq.) | DMF | 52 | 0 | 0 |
| 3 | TBHP (2 eq.) | — | 21 | 10 | 66 |
| 4 | TBHP (2 eq.) | DMF | 84 | 10 | 0 |
| 5 | TBHP (2 eq.) | DMSO | 71 | 10 | 10 |
| 6 | TBHP (2 eq.) | CH3CN | 13 | 10 | 0 |
| 7 | DTBP (2 eq.) | — | 19 | 0 | 77 |
| 8 | DTBP (1.5 eq.) | DMF | 79 | 0 | 0 |
| 9 | DTBP (1.8 eq.) | DMF | 87 | 0 | 0 |
| 10 | DTBP (2 eq.) | DMF | 95 | 0 | 0 |
| 11 | DTBP (3 eq.) | DMF | 95 | 0 | 0 |
| 12 | DTBP (2 eq.) | DMSO | 78 | 0 | 10 |
| 13 | DTBP (2 eq.) | CH3CN | 26 | 0 | 0 |
| 14c | DTBP (2 eq.) | DMF | <5 | 0 | 0 |
| 15d | DTBP (2 eq.) | DMF | <5 | 0 | 0 |
| 16e | DTBP (2 eq.) | DMF | 23 | 0 | 0 |
| 17f | DTBP (2 eq.) | DMF | 23 | 0 | 0 |
| 18g | DTBP (2 eq.) | DMF | 45 | 0 | 0 |
| 19h | DTBP (2 eq.) | DMF | 52 | 0 | 0 |
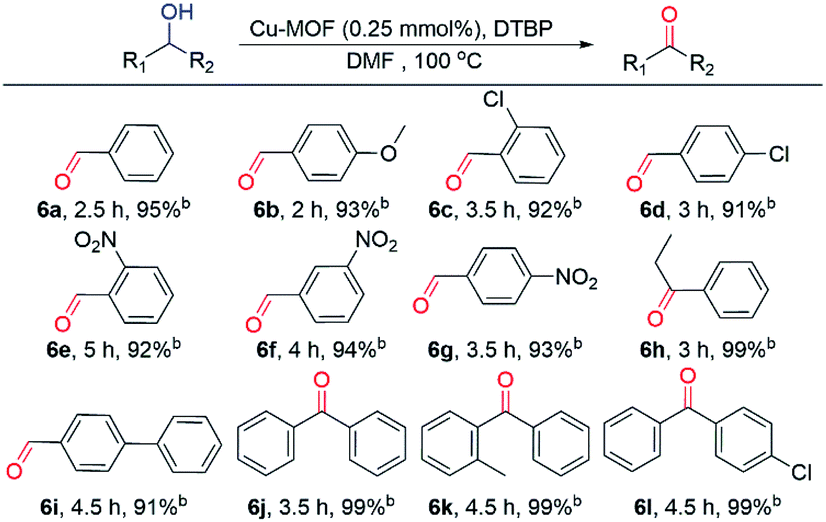
Under optimal reaction conditions, various primary and secondary alcohols were used as substrates to expand the scope of substrates (Table 2). Regardless of the alcohol that was used as the substrate, the type and position of the functional groups did not have any major impact on the final yield, which was always greater than 90%. Among the alcohols investigated, the increased reaction times can be attributed to the ortho position and electron-withdrawing groups. When secondary alcohols were used as substrates, the reaction time was slightly longer than that of primary alcohols and a higher separation yield was obtained. Therefore, the activated Cu MOF can be used as an efficient catalyst for the dehydrogenative oxidation of alcohols with low amounts of catalyst loading.
The recyclability of activated Cu-MOF was explored under optimal reaction conditions using benzyl alcohol as the substrate (Table 3). After the completion of the reaction, the solution was centrifuged. Further, the product was separated from the filtrate via column chromatography to calculate the yield. The precipitate was washed thrice with DMF (2 mL) and loaded to the next round of reaction as the catalyst. The amount of substrate remained the same during each cycle, and the active ingredient of the recovered catalyst was calculated by measuring the content of Cu via ICP-MS. Finally, a similar TOF could be observed after reusing the activated Cu-MOF for six cycles; the total TON of the six cycles reached 2286. Activated Cu-MOF could be recycled and reused as a catalyst for the dehydrogenative oxidation of alcohols.
|
||||
|---|---|---|---|---|
| Cycle | Yieldb (%) | Time (min) | Recovered Cu-MOF (mol%) | TOF (h−1) |
| a Reactions were carried out using benzyl alcohol (19.0 mmol), DTBP (38.0 mmol), activated Cu MOF (20 mg, 0.25 mmol% counted as one copper), and DMF (30.0 mL) at 100 °C.b Isolated yields. | ||||
| 1 | 95 | 150 | 99 | 152 |
| 2 | 94 | 150 | 98 | 152 |
| 3 | 94 | 150 | 97 | 154 |
| 4 | 93 | 150 | 97 | 154 |
| 5 | 91 | 150 | 96 | 150 |
| 6 | 92 | 150 | 96 | 153 |
| TON | 2286 | |||
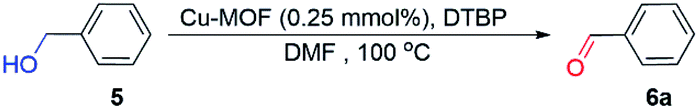
Under optimal reaction conditions, the benzaldehyde yield decreased to 23% when activated Cu-MOF were not loaded and decreased less than 5% in the presence of diphenylamine or bromotrichloromethane, which are renowned oxygen or carbon radical traps. The benzaldehyde yield with ligand loading only is the same as that without loading for activated Cu-MOF. This yield increased to 45% with Cu(NO3)2·3H2O loading only. The active center of the Cu MOF is Cu2+, which is consistent with a previous report.38 When compared with the classic 3D Cu-MOF, MOF-199,22,24 which is constructed of dinuclear Cu paddlewheel SBUs and carboxylic acid ligands, the catalytic efficiency of the 2D Cu-MOF synthesized in this work is considerably better under the same conditions. Thus, the Cu2+ of activated Cu-MOF promoted the reaction and the carbon and oxygen radicals participated in the process. Although the detailed mechanism remains unclear, a possible mechanism is proposed according to the results presented herein and related work, as shown in the Scheme 1.39,40 First, Cu(II) coordinates with DTBP to promote homolysis and produce the radical tBuO; then, the CuOHR1R2CH intermediate is generated via coordination with the R1R2CHOH substrate. With the disappearance of oxygen radicals and the generation of carbon radicals, protons transfer from the intermediate to tBuO, generating tBuOH and the new intermediate CuOHR1R2C. Another proton transfer from the new CuOHR1R2C˙ intermediate to tBuO·for obtaining tBuOH and the target product R1R2CO was accompanied by the rupture of the Cu–O coordination bond and the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds. At the same time, the catalyst returned to the activated Cu-MOF with a five-coordinate structure and continued to participate in the reaction. The presence of the activated Cu-MOF made it easier for the oxidant to homogenize to produce the radical and for the two protons in the substrate to be attacked by radicals via activation. In addition, it is easier for the substrate to concentrate around the metal center instead of the ligand because of the 2D split-level structure of the activated Cu-MOF and the existence of multiple large sterically hindered methoxy groups on it. Thus, the catalytic activity of the activated Cu MOF improves, whereas the amount of catalyst loading required decreases. Furthermore, the framework did not change during the reaction process, improving its recyclability (Table 4).
O double bonds. At the same time, the catalyst returned to the activated Cu-MOF with a five-coordinate structure and continued to participate in the reaction. The presence of the activated Cu-MOF made it easier for the oxidant to homogenize to produce the radical and for the two protons in the substrate to be attacked by radicals via activation. In addition, it is easier for the substrate to concentrate around the metal center instead of the ligand because of the 2D split-level structure of the activated Cu-MOF and the existence of multiple large sterically hindered methoxy groups on it. Thus, the catalytic activity of the activated Cu MOF improves, whereas the amount of catalyst loading required decreases. Furthermore, the framework did not change during the reaction process, improving its recyclability (Table 4).
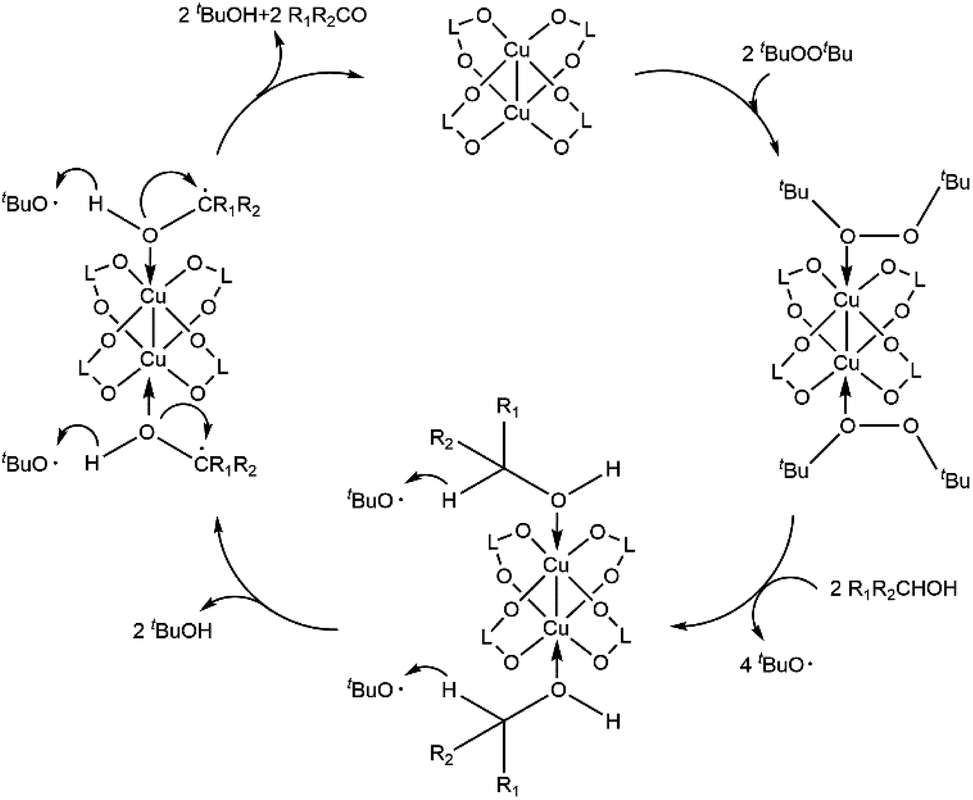 | ||
| Scheme 1 Proposed activation pathways for the dehydrogenative oxidation of alcohol catalyzed by the activated Cu MOF. R1 = alkyl, aryl, or H; R2 = aryl; L = ligand of activated Cu MOF. | ||
|
|||
|---|---|---|---|
| Entry | Solvent | Et3N | Yieldb (%) |
| a Reactions were conducted using imidazole (32 mg, 0.47 mmol), bromobenzene (89 mg, 0.57 mmol), Et3N, activated Cu MOF (2 mg, 1 mmol%, counted as one copper), and solvent (1.0 mL) at 120 °C for 12 h.b Isolated yields. | |||
| 1 | DMF | 2 eq. | 71 |
| 2 | NMP | 2 eq. | 55 |
| 3 | DMSO | 2 eq. | 62 |
| 4 | DMA | 1.5 eq. | 89 |
| 5 | DMA | 2 eq. | 97 |
| 6 | DMA | 3 eq. | 98 |
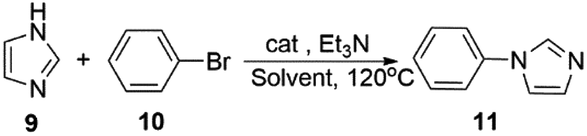
To expand the application range, we applied activated Cu-MOF to the N-arylation of azole compounds. To optimize the reaction solvent, bromobenzene and imidazole were used as substrates, triethylamine was used as the base, and 1.0 mmol% of the activated Cu MOF was used as the catalyst. The substrate conversion rate was the highest when DMA was used as the solvent. Then, we used DMA as the solvent to screen the amount of base. Finally, optimal reaction conditions were obtained with DMA as the solvent and two equivalents of triethylamine as the base (Table 5).
|
||||
|---|---|---|---|---|
| Entry | 9 | 10 | 11 | Yieldb (%) |
| a Reactions were conducted using azole (0.47 mmol), halohydrocarbon (0.57 mmol), Et3N (95 mg, 0.94 mmol), activated Cu MOF (2 mg, 1 mmol%, counted as one copper), and DMA (1.0 mL) at 120 °C for 12 h.b Isolated yields.c Reactions were completed in 7 h.d Reactions were completed in 9 h. | ||||
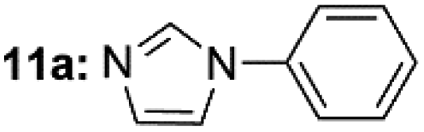
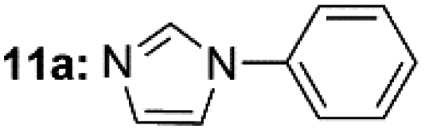
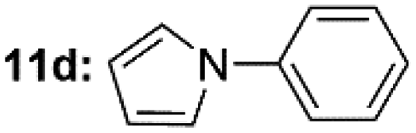
| 1c |  |
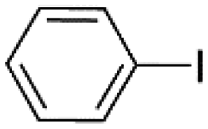 |
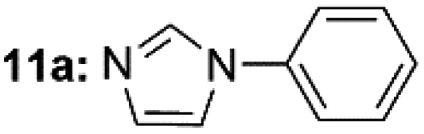 |
99 |
| 2 | 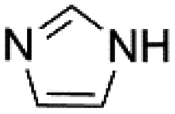 |
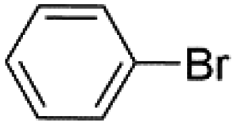 |
 |
97 |
| 3 | 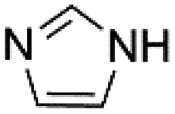 |
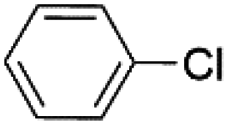 |
 |
26 |
| 4 | 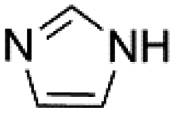 |
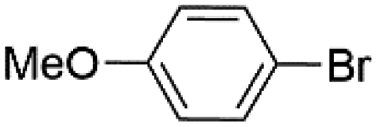 |
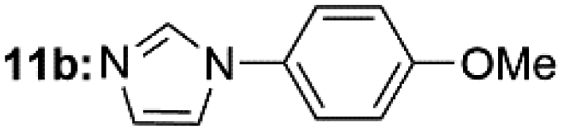 |
95 |
| 5 | 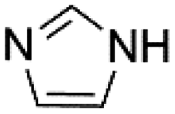 |
 |
 |
74 |
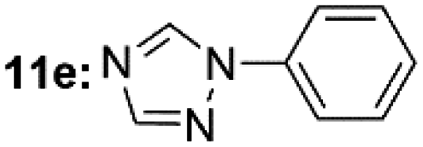
| 6d |  |
 |
 |
98 |
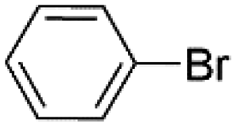
| 7 | 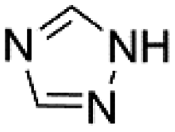 |
 |
 |
85 |
Under optimal reaction conditions, different halohydrocarbons and azole compounds were used as substrates for expanding the scope of substrates. When iodohydrocarbon was used as the substrate, the reaction could be completed in 7 h. The yields obtained after 12 h were 97% and 26% when bromohydrocarbon and chloroydrocarbon were used as the substrates, respectively. The nitro group on the halohydrocarbon passivated the benzene ring and increased the reaction time. The reaction could still proceed well when using different azole compounds. Therefore, the activated Cu MOF could catalyze the N-arylation reaction of azole compounds with low amounts of catalyst loading (Table 6).
|
|||
|---|---|---|---|
| Cycle | Yieldb (%) | Time (h) | Recovered Cu-MOF (mol%) |
| a Reactions were conducted using imidazole (160 mg, 2.4 mmol), bromobenzene (445 mg, 2.8 mmol), Et3N (475 mg, 4.7 mmol), activated Cu MOF (10 mg, 1 mmol%, counted as one copper), and DMA (5.0 mL) at 120 °C for 12 h.b Isolated yields. | |||
| 1 | 99 | 12 | 98 |
| 2 | 96 | 12 | 97 |
| 3 | 98 | 12 | 95 |
| TON | 296 | ||
1% mmol of the catalyst was loaded to verify the recyclability in other types of reactions. After three catalytic cycles, the activated Cu MOF is observed to still exhibit considerable catalytic activity.
Conclusions
In summary, we developed a recyclable heterogeneous catalyst that is a new type of Cu-MOF with 2D crystal structure constructed using Cu(NO3)2·3H2O and 2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarboxylic acid. The active Cu sites at which the substrates tend to be concentrated are better exposed and easier to approach because of the 2D split-layer crystal structure of the MOF. Therefore, activated Cu-MOF exhibited high conversion efficiencies for the dehydrogenative oxidation of alcohol and N-arylation of imidazole with low catalyst loading. Also, activated Cu-MOF can be separated from the reaction mixture via simple centrifugation. Further, they can be reused with only a negligible decrease in efficiency. The recovered catalyst was characterized via infrared spectroscopy and X-ray difraction. No obvious difference can be observed when compared with the fresh catalyst, which was confirmed by images.Author contributions
C. X. Liu designed the material and performed the experiments; C. X. Liu and Y. F. Wang wrote the paper; J. Cui and M. J. Zhang contributed to the supervision and editing; all authors contributed to the general discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Tianjin (17JCQNJC05500), P. R.China.Notes and references
- R. Banavali, M. J. Deetz and A. K. Schultz in Transition Metal Catalysts, ed. J. T. Puche and F. Albericio, Wiley-VCH, 2008 Search PubMed.
- L. M. D. Martins, A. Ribeiro, S. Carabineiro, J. Figueiredo and A. Pombeiro, Dalton Trans., 2016, 45, 6816 RSC.
- S. Kim, H. E. Lee, J. M. Suh, M. H. Lim and M. Kim, Inorg. Chem., 2020, 59, 17573 CrossRef CAS PubMed.
- Z. Wang, J. J. Feng, X. L. Li, R. Oh, D. D. Shi, O. Akdim, M. Xia, L. Zhao, X. Y. Huang and G. J. Zhang, J. Colloid Interface Sci., 2021, 588, 787 CrossRef CAS.
- I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691 CrossRef.
- J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1101 CrossRef CAS.
- L. Cicco, J. A. Hernández-Fernández, A. Salomone, P. Vitale, M. Ramos-Martín, J. González-Sabín, A. P. Soto, F. M. Perna, V. Capriati and J. García-Álvarez, Org. Biomol. Chem., 2021, 19, 1773 RSC.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 382 Search PubMed.
- F. Lang, D. Zewge, I. N. Houpis and R. P. Volante, Tetrahedron Lett., 2001, 42, 3251 CrossRef CAS.
- K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed.
- L. Esrafili, A. Morsali, F. D. Firuzabadi and P. Retailleau, ACS Appl. Mater. Interfaces, 2020, 12, 43115 CrossRef CAS PubMed.
- C. Gambarotti and H. Bjørsvik, Eur. J. Org. Chem., 2019, 6, 1405 CrossRef.
- A. Dhakshinamoorthy, A. Asiri and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922 RSC.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134 RSC.
- A. Chughtai, N. Ahmad, H. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC.
- S. Li Hou, J. Dong and B. Zhao, Adv. Mater., 2020, 32, 1806163 CrossRef PubMed.
- W. Gong, Y. Liu, H. Y. Li and Y. Cui, Coord. Chem. Rev., 2020, 420, 213400 CrossRef CAS.
- S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854 CrossRef CAS PubMed.
- A. Henschel, K. Gedrich, R. Kraehnertb and S. Kaskel, Chem. Commun., 2008, 35, 4192 RSC.
- A. Phan, A. U. Czaja, F. Gándara, C. B. Knobler and O. M. Yaghi, Inorg. Chem., 2011, 50, 7388 CrossRef CAS PubMed.
- K. Rui, G. Zhao, Y. Chen, Y. Lin, Q. Zhou, J. Chen, J. Zhu, W. Sun, W. Huang and S. X. Dou, Adv. Funct. Mater., 2018, 28, 1801554 CrossRef.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed.
- Z. H. Mai and D. X. Liu, Cryst. Growth Des., 2019, 19, 7439 CrossRef CAS.
- S. Marx, W. Kleist and A. Baiker, J. Catal., 2011, 281, 76 CrossRef CAS.
- N. Anbu and A. Dhakshinamoorthy, J. Ind. Eng. Chem., 2018, 65, 120 CrossRef CAS.
- P. V. Dau and S. M. Cohen, Inorg. Chem., 2015, 54, 3134 CrossRef CAS PubMed.
- L. Q. Wei and B. H. Ye, Inorg. Chem., 2019, 58, 4385 CrossRef CAS PubMed.
- R. Luo, H. Xu, H. X. Gu, X. Wang, Y. Xu, X. Shen, W. Bao and D. R. Zhu, CrystEngComm, 2014, 16, 784 RSC.
- J. Li, Y. Ren, C. Qi and H. Jiang, Eur. J. Inorg. Chem., 2017, 11, 1478 CrossRef.
- Y. Han, X. He, W. X. Yang, X. L. Luo, Y. Yu, W. Z. Tang, T. L. Yue and Z. H. Li, Food Chem., 2021, 345, 128839 CrossRef CAS.
- I. Boldog, L. Xing, A. Schulz and C. Janiak, C. R. Chim., 2012, 15, 866 CrossRef CAS.
- Y. Peng, Y. Li, Y. Ban and W. Yang, Angew. Chem. Int. Ed., 2017, 56, 9757 (Angew. Chem., 2017, 129, 9889) CrossRef CAS PubMed.
- J. Tang, M. K. Cai, G. G. Xie, S. X. Bao, S. J. Ding, X. X. Wang, J. Z. Tao and G. Q. Li, Chem.–Eur. J., 2020, 26, 4333 CrossRef CAS.
- A. Nuri, N. Vucetic, J. Smått, Y. Mansoori, J. Mikkola and D. Y. Murzin, Catal. Lett., 2019, 149, 1941 CrossRef CAS.
- A. Karmakar, L. M. D. R. S. Martins, S. Hazra, M. F. C. G. da Silva and A. J. L. Pombeiro, Cryst. Growth Des., 2016, 16, 1837 CrossRef CAS.
- B. Hans and V. Helmut, Chem. Ber., 1962, 95, 810 CrossRef.
- B. Kanakalakshmi, K. P. Mathai and S. Sethna, J. Indian Chem. Soc., 1966, 7, 469 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- Y. F. Zhu and Y. Y. Wei, Eur. J. Org. Chem., 2013, 21, 4503 CrossRef.
- N. M. R. Martins, K. T. Mahmudov, M. F. C. G. da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, New J. Chem., 2016, 40, 10071 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2053928. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00248a |
| This journal is © The Royal Society of Chemistry 2021 |