
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the biomimetic cofactors of NMNH for cytochrome P450 BM3 based on binding conformation refinement†
Yao Liu‡
 a,
Yalong Cong‡a,
Chuanxi Zhangd,
Bohuan Fanga,
Yue Pana,
Qiangzi Lief,
Chun Youef,
Bei Gaod,
John Z. H. Zhangabc,
Tong Zhu*ab and
Lujia Zhang*ab
a,
Yalong Cong‡a,
Chuanxi Zhangd,
Bohuan Fanga,
Yue Pana,
Qiangzi Lief,
Chun Youef,
Bei Gaod,
John Z. H. Zhangabc,
Tong Zhu*ab and
Lujia Zhang*ab
aShanghai Engineering Research Center of Molecular Therapeutics & New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: tzhu@lps.ecnu.edu.cn; ljzhang@chem.ecnu.edu.cn
bNYU-ECNU Center for Computational Chemistry at NYU Shanghai, Shanghai 200062, China
cDepartment of Chemistry, New York University, New York, New York 10003, USA
dSchool of Biotechnology, East China University of Science and Technology, Shanghai 200237, China
eUniversity of Chinese Academy of Sciences, 19A Yuquan Road, Shijingshan District, Beijing 100049, P. R. China
fTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, 32 West 7th Avenue, Tianjin Airport Economic Area, Tianjin 300308, P. R. China
First published on 24th March 2021
Abstract
Cytochrome P450 BM3 (BM3) is an important oxidoreductase that is widely used in drug synthesis, chemical synthesis, and other industries. However, as BM3 unquestionably increases costs by consuming a natural cofactor that unstably provides electrons, an alternative biomimetic cofactor with simpler structures represented by nicotinamide mononucleotide (NMNH) has been utilized. Currently, few reports exist on artificially modified BM3 enzymes using NMNH, especially regarding theoretical simulation and calculation. With the cognition of the mechanism in mind, we propose a strategy that optimizes and refines catalytic conformation. Based on constrained molecular dynamics simulation, the distance between N-5 of FAD flavin and C-4 of NMNH is used as a cue for the determination of improved conformation, and the potential positive mutants are subsequently screened virtually in accordance with binding free energy requirements. As a result, the Kcat/KM values of the favorable mutant S848R increased to 205.38% compared to the wild-type BM3 with NMNH. These data indicate that our strategy can be applied for the specific utilization of biomimetic cofactors by oxidoreductases represented by BM3.
Introduction
Oxidoreductase, which catalyzes biological oxidation/reduction reactions, is widely used for industrial drug synthesis, chemical synthesis, and food processing, among other industries.1,2 Most oxidoreductases require natural cofactors in the catalyzation process,3 such as nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+), whose reduced forms are NADH and NADPH, respectively.4 However, these natural cofactors are structurally complex and unstable, which undoubtedly increases the cost and obstacles of industrial production.5 Nowadays, many biomimetic cofactors with simpler structures, such as nicotinamide mononucleotide (NMN+/NMNH),6 have been developed and utilized. However, most wild-type enzymes either cannot utilize biomimetic cofactors, including NMNH, or exhibit low activity when such cofactors are utilized.Cytochrome P450 BM3 (BM3, EC1.14.14.1) is a soluble monooxygenase derived from Bacillus megaterium.7–9 On the one hand, it is a typical 120 kDa natural fusion enzyme, with two parts comprising a 55 kDa P450 heme domain and a 65 kDa P450 reductase (CPR) domain, which contains both the FMN-binding domain (FMN domain) and the FAD/NADPH-binding domain (FAD domain).10 As in CPR, electrons pass from NADPH to FAD and thence to FMN before being shuttled to the heme iron.7 On the other hand, BM3 formerly hydroxylates long-chain fatty acids, amides, and alcohols at the ω-1, ω-2, and ω-3 positions.11 Due to its self-sufficiency in electron transfer, incomparable regioselectivity, and stereoselectivity, various researchers have employed protein engineering strategies to obtain BM3 mutants that exhibit specific substrate selectivity.12 Munro's group demonstrated that A82F, F87V, and F87V/A82F mutants of BM3 can catalyze diverse azole antifungal drugs more efficiently with voriconazole, omeprazole, and esomeprazole.13,14 The BM3 variant C400S designed by Arnold's group could complete NAD(P)H-driven cyclopropanation with high conversion in whole-cell catalysis.15 However, the wild-type (WT) BM3 consumes only a natural cofactor that provides electrons, which is a disadvantage. Consequently, research on biomimetic cofactors has exploded, and finding alternative biomimetic cofactors is the key to resolving this issue. Biomimetic cofactors, such as NMNH,6 N-4-methoxybenzyl-1,4-dihydronicotinamide16 and nicotinamide cytosine dinucleotide,17 have been confirmed to be used by oxidoreductase. Positive mutants were obtained by randomized and irrational strategies, and the mechanism of enzymes using biomimetic cofactors and further rational artificial modification strategies have not yet been explained in detail.
Currently, few studies have been conducted on artificially modified BM3 enzymes using NMNH, especially regarding theoretical simulation and calculation. Recently, our group has obtained several modified mutant strains of Gox2181, changing its exclusive preference for the cofactor NADPH into dual cofactor specificity for NADH and NADPH.18,19 Based on the cognition of the selective mechanism of cofactors, in this study, a variant of BM3 was found through virtual computer mutation strategies based on catalytic conformation—a variant which ultimately improves enzymatic activity towards NMNH. Our data further prove that the mutation of BM3 improves NMNH utilization activity. This rational design method largely completes the specific utilization of biomimetic cofactors by oxidoreductase.
Experimental
Preparation of protein structure
The crystal structure of the FAD/NADPH-binding domain (FAD domain) of P450 BM3 in the presence of the ligand NADP+ has been resolved (PDB ID: 4DQL).20 Given that the ribose-nicotinamide mononucleotide of NADPH is highly disordered, this structure (4DQL) does not contain the exact conformation of ribose nicotinamide.21 Fortunately, the crystal structure of NADPH-cytochrome P450 oxidoreductase (CYPOR) from rats contains detailed information on the ligand NADP+ (PDB ID: 1J9Z). The FAD domains from 4DQL and 1J9Z are very similar (approximately 49.0% sequence similarity). Therefore, the structure of NADPH was obtained by aligning 1J9Z to 4DQL. The extra R group of NADPH was then deleted to generate the structure of NMNH, as shown in Fig. 1(A) and (B).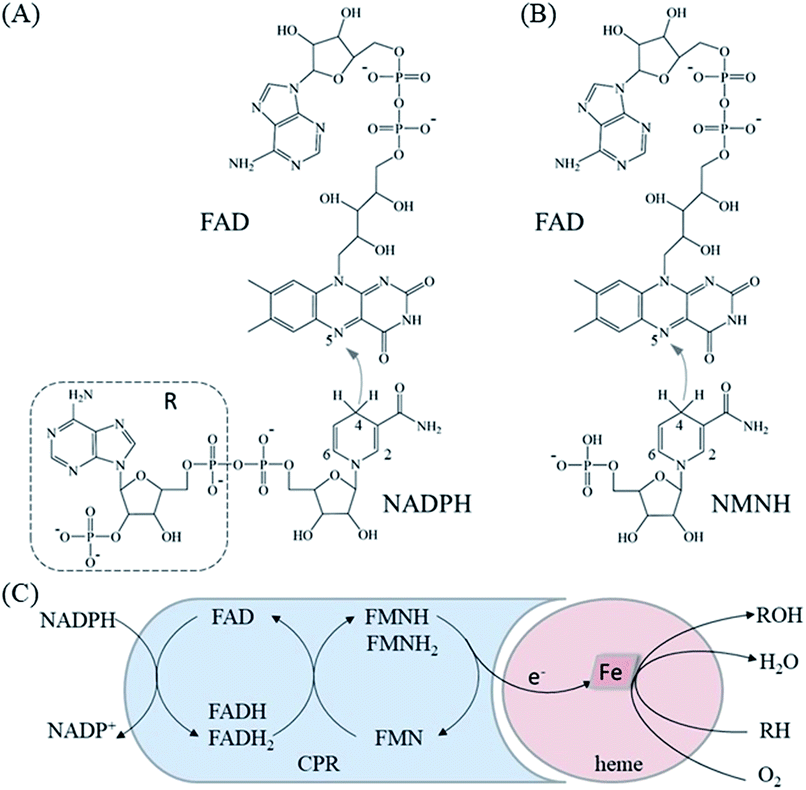 | ||
| Fig. 1 Mechanism of electron transfer by P450 BM3. (A) The 2D structure of NADPH and FAD and the specific direction of electron transfer. (B) The 2D structure of NMNH and FAD and the specific direction of electron transfer. (C) The approximate process of electron transfer between different domains in P450 BM3. | ||
MD simulation
The AM1-BCC22,23 method was used to calculate the charge of NADPH and NMNH. The ff14SB and GAFF2 force fields of AMBER18 (ref. 24) software were employed to provide parameters for protein and substrate, respectively. The TIP3P water box with a buffer distance of 10 Å (about 171![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 water molecules) was used as the solvent environment, and 17 sodium ions were added to maintain neutrality of the system. Next, energy minimization is carried out in two steps using the steepest descent and the conjugate gradient methods to remove bad contacts of atoms. In the first step, the solvent water molecules are optimized, and the solute molecules are constrained under a force of 500 kcal (mol Å2)−1. In the second step, the entire system is optimized without any constraints. Then, the system was slowly heated from 0 to 300 K for 300 ps under the weak constraint of 10 kcal (mol Å2)−1. The SHAKE25 algorithm was used to constrain all chemical bonds involving hydrogen atoms, and the particle mesh Ewald26 method was used to handle long-range electrostatic interactions. Finally, MD simulation was performed for 10 ns under the NPT ensemble.
000 water molecules) was used as the solvent environment, and 17 sodium ions were added to maintain neutrality of the system. Next, energy minimization is carried out in two steps using the steepest descent and the conjugate gradient methods to remove bad contacts of atoms. In the first step, the solvent water molecules are optimized, and the solute molecules are constrained under a force of 500 kcal (mol Å2)−1. In the second step, the entire system is optimized without any constraints. Then, the system was slowly heated from 0 to 300 K for 300 ps under the weak constraint of 10 kcal (mol Å2)−1. The SHAKE25 algorithm was used to constrain all chemical bonds involving hydrogen atoms, and the particle mesh Ewald26 method was used to handle long-range electrostatic interactions. Finally, MD simulation was performed for 10 ns under the NPT ensemble.
Computer virtual mutation
All residues (except the catalytic triad) within 8 Å of the substrate were considered by the computer virtual saturation mutation strategy to screen for favorable mutants. The leap module of AMBER18 was used to replace amino acid side chains to achieve amino acid mutations. The structure of the protein after mutation is optimized by the steepest descent and the conjugate gradient methods. During the optimization, the distance between N-5 of FAD and C-4 of NMNH atoms is constrained so that the substrate is in a catalytic conformation.27 The structure of the wild type is optimized using the same method. The molecular mechanics/generalized Born surface area (MM/GBSA)28,29 method is subsequently adopted to calculate the binding free energy of protein and substrates based on the optimized structure obtained after mutation. The free energy of binding is used as the objective function to select mutations with enhanced affinity.MM/GBSA method
The MM/GBSA method was used to calculate the binding free energy (ΔGbind) of protein and substrate molecules as follows:| ΔGbind = ΔGgas + ΔGsol | (1) |
| ΔGgas = ΔEele + ΔEvdW | (2) |
| ΔGsol = ΔGgb + ΔGnp | (3) |
| ΔGnp = γ × SASA + β | (4) |
Construction of strains and plasmid
Cytochrome P450 BM3 (GenBank: J04832.1) was amplified from the plasmid pET-28a(+) with an N-terminal His-tag. Site-directed mutagenesis, using WT BM3 as a template, was performed using PrimeSTAR Max DNA Polymerase (Takara) for the PCR process. All primers were synthesized by Generay Biotechnology (Shanghai, China), which was used for mutagenesis, as shown in Table S1.† According to the manufacturer's instructions of the Blunting Kination Ligation Kit (Takara), phosphorylation was carried out at 37 °C for 20 min, after which the phosphatase was deactivated by heat at 70 °C for 5 min, and ligation was allowed to take place for 60 min. Next, the ligation mixtures were used to transform E. coli DH5α cells to select positive clones. According to reports in the literature, the glucose dehydrogenase from Bacillus subtilis (Bs GDH, UniProt ID: A0A1B2ATD9_BACIU) of the mutant I195R6 was cloned into pET-22b(+) with an N-terminal His-tag, which was used to reduce NMN+ (Aladdin) and generate NMNH. The gene sequences of all the mutants were determined using Shanghai Sunny Biotechnology.Protein expression and purification
The WT/mutant plasmids were transformed into E. coli BL21 (DE3) for the heterogeneous expression of enzymes. Recombinant E. coli BL21 (DE3) cells were cultured in Luria–Bertani medium with 50 μg mL−1 kanamycin. After incubation at 37 °C and shaking at 220 rpm, cells were grown to an OD600 of 0.8–1.0. The cultures were then induced by adding IPTG to a final concentration of 0.1 mM at 20 °C. After a further 12 h of incubation, the cells were harvested by centrifugation at 8000g for 5 min at 4 °C.The cell pellet was resuspended in lysis buffer (50 mM Tris–HCl buffer, 10 mM imidazole, pH 7.5) and the cells were lysed by sonication. Cell debris was removed by centrifugation at 18000g for 15 min at 4 °C. The supernatant was filtered through a 0.22 μm filter. Next, the lysate was subjected to a HisTrap HP affinity column using an AKTA Purifier system. The WT/mutant sample was eluted with a gradient of 10–500 mM elution buffer (50 mM Tris–HCl buffer, 10–500 mM imidazole, pH 7.5), and the target protein was eluted with 100 mM elution buffer (50 mM Tris–HCl buffer, 100 mM imidazole, pH 7.5).
Enzyme purity was analyzed by 8% SDS-PAGE, and active fractions were also buffer exchanged into 50 mM Tris–HCl (pH 7.5). The enzyme was concentrated using Amicon Ultra (Millipore) with a 30 kDa molecular weight cut-off, and the three centrifugation steps were allowed to take place at 4800g at 4 °C for 20 min. The concentration of the target protein after purification was determined using the BCA Protein Quantification kit, which was purchased from Yeasen Biotechnology (Shanghai, China).
Enzyme activity and kinetics investigation
The enzyme activity was detected using a Multiskan Sky Microplate Spectrophotometer (Thermo Scientific). The enzyme activity reaction mixtures (0.1 mL) containing 0.75 mg mL−1 purified enzyme, 0.3 M glucose, 1 M NaCl, 2 mM NMNH, and 0.2 M PBS buffer (pH 7.5) were placed into a 96-well plate. The plates were pre-incubated at 30 °C for 2 min, then 80 μM cytochrome c (Sigma-Aldrich) was added, which was monitored at 550 nm (ε550 = 21.1 mM−1 cm−1). NMNH was prepared according to previously described methods.6 The activities of the WT/mutant enzymes are expressed as the difference in the ability to reduce cytochrome c between the experimental and control groups. One unit (U) of enzymatic activity was defined as the amount of enzyme required to catalyze the reduction of 1 nmol cytochrome c per min at 30 °C (pH 7.5). Enzyme activity data are shown as means with standard deviations.The method to determine kinetic parameters is similar to that for enzyme activity. In the reaction mixture, the enzyme concentration was 6.25 μM, and the concentration of cytochrome c varied in the range of 3.125 μM to 160 μM. Each set of data was tested in parallel at least in triplicate. The Kcat and Km values were fitted to Michaelis–Menten using non-linear regression (OriginPro 9.0).
Results and discussion
Structural comparison of NMNH and NADPH
Before MD simulations, the electron transport mechanism of BM3 and the difference between NMNH and NADPH should be understood in detail. As shown in Fig. 1(C), the first transfer of electrons in BM3 from NADPH to FAD form FADH2. Next, the electrons pass from FADH2 to FMN, leading to the simultaneous formation of FMNH and FADH semiquinone. The final step is to provide electrons from FMNH semiquinone to iron in the heme domain.7,33,34 Fig. 1(A) and (B) show the structures of FAD, NMNH, and NADPH. The structural difference between NMNH and NADPH is the lack of the R part, but both have a nicotinamide moiety in common. The nicotinamide C-4 of NMNH or NADPH donates a proton to the isoalloxazine N-5 of FAD. It was found that a shorter distance between N-5 (FAD) and C-4 (NMNH or NADPH) atoms can provide a better conformation.Both NMNH and NADPH can be catalyzed by wild enzymes, but they have a lower preference for NMNH. To study the reasons for different preferences between NMNH and NADPH, MD simulations of FAD domain–NMNH and FAD domain–NADPH were performed for 10 ns. Given the stability of the protein crystal structure and cofactors, the root mean square deviation (RMSD) of the protein binding pocket backbone atoms, FAD, NMNH, and NADPH relative to the initial structure was calculated to measure the overall stability of the structure. As shown in Fig. 2, the RMSD of the FAD domain–NMNH and FAD domain–NADPH simulations tended to be stable, without major fluctuations, which indicates that the simulations have reached convergence within 10 ns and can be used for further analysis.
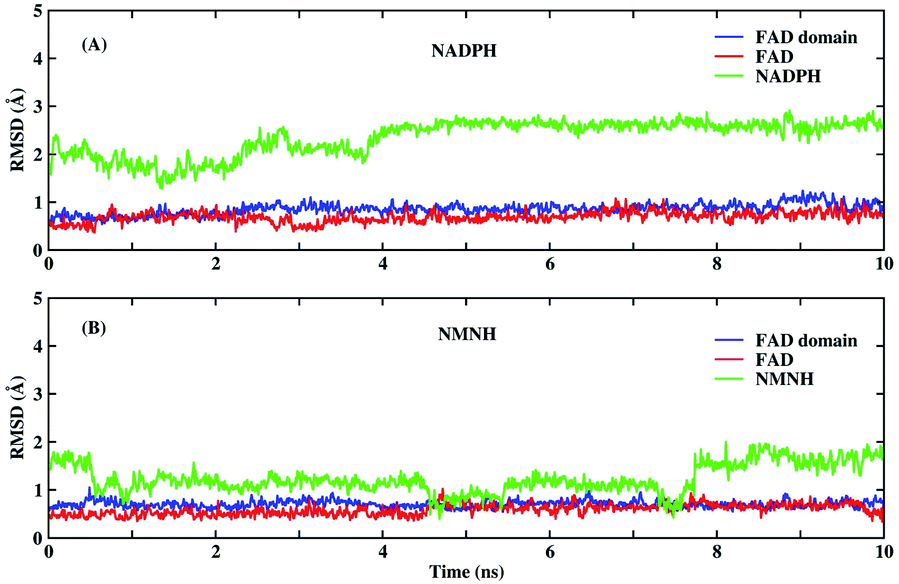 | ||
| Fig. 2 The root mean square deviation (RMSD) of the backbone of the FAD domain (wild-type) binding pocket, FAD, NMNH and NADPH. (A) The RMSD of FAD domain–NADPH simulations. (B) The RMSD of FAD domain–NMNH simulations. | ||
According to the electron transport mechanism, the closer the distance between the N-5 and C-4 atoms, the greater the probability of the electron transfer. When the distance is large, electrons cannot be transferred from N-5 to C-4, and the catalytic reaction cannot occur. Therefore, the distance between the N-5 and C-4 atoms is an important factor that determines the influence of the catalytic reaction. The distances and their frequency distributions during the FAD domain–NMNH and FAD domain–NADPH simulation are plotted in Fig. 3. It can be found that the distance of the FAD domain–NMNH system is evidently farther than that of the FAD domain–NADPH, which causes the electron transfer of the FAD domain–NMNH system to be more difficult. This phenomenon explains the low catalytic efficiency of the BM3 with NMNH cofactor. At the same time, it shows that distance is a prerequisite for catalysis, which can be used as a research basis for constructing rational designs that improve catalytic activity.
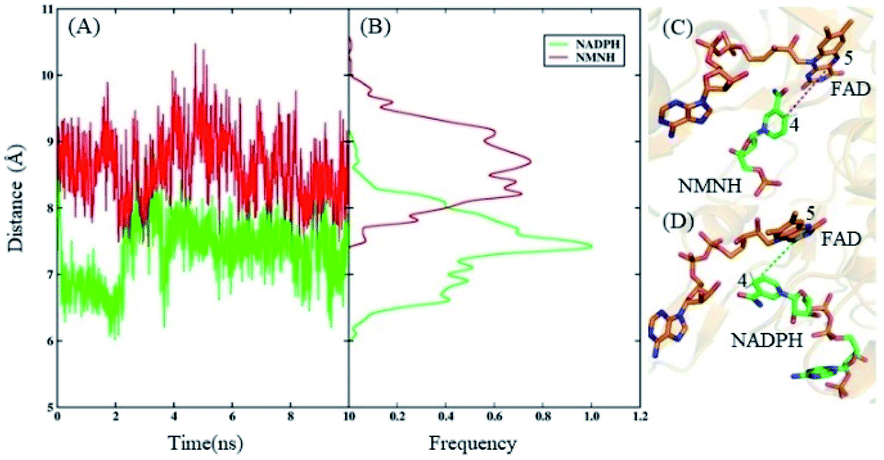 | ||
| Fig. 3 Unconstrained dynamic simulations. (A) The distance of N-5 and C-4 atoms during the FAD domain–NMNH and FAD domain–NADPH simulation. (B) The frequency distribution of N-5 and C-4 atoms during the FAD domain–NMNH and FAD domain–NADPH simulation. (C) The lowest potential energy structure of NMNH and FAD during the FAD domain–NMNH simulations. (D) The lowest potential energy structure of NADPH and FAD during the FAD domain–NADPH simulations. | ||
In addition, a longer time (100 ns) MD simulation on FAD domain–NMNH complex was performed to verify the convergence of simulation and the reliability of the results. The RMSD and distance between the N-5 and C-4 atoms were calculated and presented in Fig. S4.† As can be seen, there is no significant difference between the average distance of 100 ns and 10 ns simulation. This result demonstrates the convergence of our simulation and the reliability of the results.
Screening of mutants
The structural comparison found that the distance between the N-5 (FAD) and C-4 (NMNH) atoms is a prerequisite for catalysis. To improve the catalytic activity of BM3 with NMNH cofactor, the distance of N-5 (FAD) and C-4 (NMNH) is used as a constraint condition, and the binding free energy of BM3 and NMNH is used as an objective function to select favorable mutants. All residues (49 with the exception of the catalytic triad) within 8 Å near the NMNH performed the computer virtual saturation mutation. The binding free energies of 931 single point mutants are presented in Table S2.† Based on the principle of the scattered mutation points, ten mutations with lower binding energy, as shown in Table 1, were selected for further experimental verification.| ID | WT | Mutant | ΔGbind | ΔΔGbindb |
|---|---|---|---|---|
| a All energies are in kcal mol−1.b ΔΔGbind = ΔGbind(mut) − ΔGbind(WT). | ||||
| WT | −41.54 | 0.00 | ||
| 1001 | GLY | ARG | −46.18 | −4.64 |
| 943 | ASP | ARG | −45.00 | −3.46 |
| 1001 | GLY | GLN | −44.20 | −2.58 |
| 907 | GLY | PRO | −43.83 | −2.30 |
| 848 | SER | ARG | −43.35 | −1.81 |
| 866 | SER | ARG | −43.22 | −1.68 |
| 907 | GLY | THR | −43.21 | −1.68 |
| 905 | GLY | THR | −42.49 | −0.95 |
| 943 | ASP | MET | −42.44 | −0.90 |
| 1002 | ASP | ASN | −42.05 | −0.52 |
Analysis of expression and purification
According to the results of theoretical calculations and simulations, ten mutations that may increase the enzyme activity of WT BM3 for cytochrome c were screened. According to experimental procedures, WT and mutant proteins have been constructed and expressed. Fig. S1† summarizes the results of the expression procedure. Except for D943M, the molecular mass of the other nine mutations all showed a protein band at 120 kDa on SDS-PAGE, which verifies soluble protein expression. Then, nine mutations were purified by HisTrap HP, and the purity of the target protein reached 95% (Fig. S2†), preparing for the next determination of enzyme activity and kinetic parameters.Analysis of enzyme activity and kinetics
To complete the detection of enzyme activity more accurately and quickly, we use the Multiskan Sky Microplate Spectrophotometer to detect the ability of the mutants to reduce substrate cytochrome c at 550 nm with NMNH cofactor. It was found that only four mutants were active, namely D943R, G905T, S848R, and D1002N. Detailed data of enzyme activity are shown in Fig. 4. Among them, S848R has greater enzyme activity towards NMNH cofactor than WT BM3, and relative enzyme activities for WT BM3 and S848R were 486.42 U mg−1 and 1332.88 U mg−1, indicating that the ability of reduced cytochrome c of this mutant is 2.74-fold of the wild type. Compared to WT BM3, the enzyme activity of the mutants D943R, G905T, and D1002N was decreased by 4.46%, 16.15%, and 54.25%, respectively. Five other mutants, S866R, G907T, G907P, G1001R, and G1001Q, did not demonstrate any activity. It is likely that the mutation site destroys the catalytic active center, leading to lost activity. Using NMNH cofactor, we successfully screened the mutant S848R, which has higher enzyme activity than WT BM3.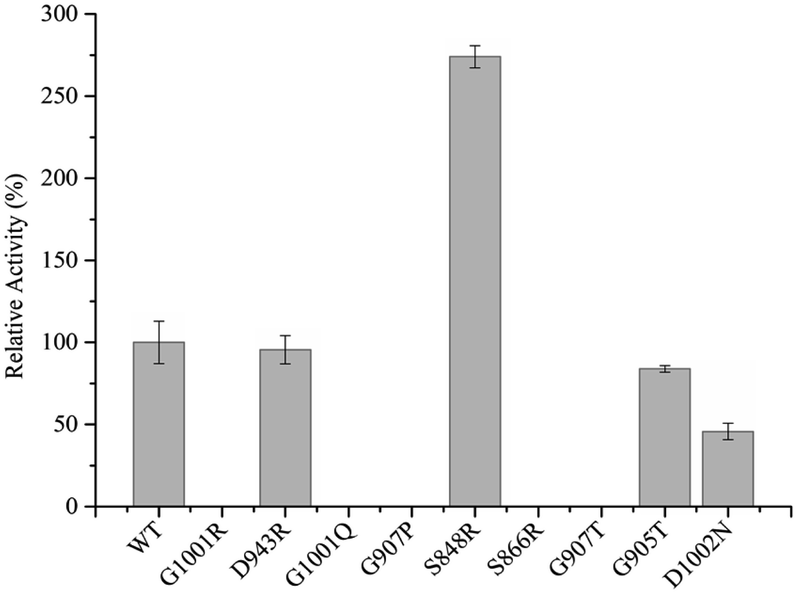 | ||
| Fig. 4 Relative activity of WT BM3 and variants. | ||
Enzyme kinetic parameters of WT BM3 and variant S848R were measured for substrate cytochrome c. Kinetic data of purified proteins are shown in Table 2. The Km value of the mutant S848R was 15.20 μM, which was lower than that of the WT (23.09 μM). This indicated that S848R showed a higher affinity with NMNH cofactor. The Kcat value of the mutant S848R was 87.08 min−1 and more than 1.35-fold higher than that of the WT BM3 (64.34 min−1). Moreover, compared to the WT enzyme, the Kcat/KM values of the mutant S848R increased to 205.38%. The kinetic investigations of cytochrome c reduction with NMNH cofactor based on Michaelis–Menten are shown in Fig. S3.† These data indicate that the BM3 mutant of S848R exhibits higher catalytic efficiency with NMNH, and the increase in catalytic efficiency is due to improvement in the Kcat value and affinity.
| Enzyme | Vmax (nM min−1) | Km (μM) | Kcat (min−1) | Kcat/Km (min−1 μM−1) |
|---|---|---|---|---|
| WT BM3 | 414.22 ± 18.05 | 23.09 ± 3.08 | 64.34 ± 1.75 | 2.79 |
| S848R | 525.87 ± 41.41 | 15.20 ± 3.73 | 87.08 ± 2.91 | 5.73 |
Mechanism of high catalytic efficiency
Experiments found that the S848 mutant caused an increase in catalytic efficiency, while D943R, G905T, and D1002N led to a decrease. To explore the detailed mechanism of difference in catalytic efficiency, MD simulations of WT, S848R, D943R, G905T, and D1002N were performed in parallel under the same parameter settings. The RMSD of the protein binding pocket backbone atoms, FAD, and NMNH in the four mutant simulations were calculated and are shown in Fig. S5.† It shown that the 10 ns simulation for these four mutants reached convergence. The distances between N-5 (FAD) and C-4 (NMNH) atoms and average values as a function of time in the five simulations are plotted in Fig. 5. The average distances of N-5 (FAD) and C-4 (NMNH) atoms in the WT system was 8.61 Å, and the average distance in the S848R mutant was reduced to 6.81 Å, and the average distances in the D943R, G905T, and D1002N mutants increased to 9.06, 8.93, and 11.27 Å, respectively. The order of distance and the order of relative enzyme activity are essentially equivalent, which shows that the distance of N-5 (FAD) and C-4 (NMNH) atoms is a suitable basis to measure enzyme activity.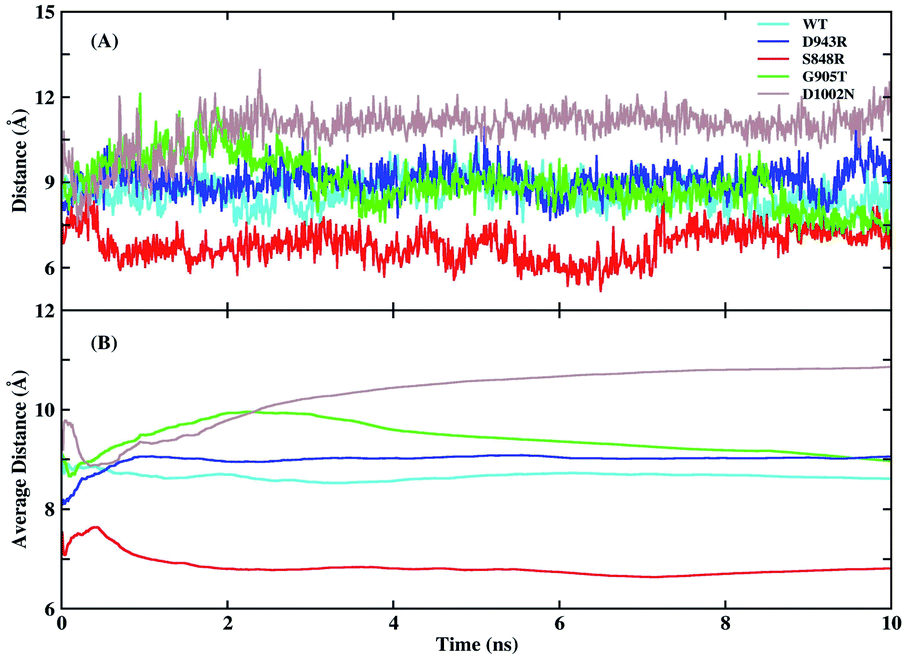 | ||
| Fig. 5 Distances of N-5 and C-4 atoms and average values as function of time in the five simulations. (A) Distances of N-5 and C-4 atoms as function of time in the five simulations. (B) The average distances of N-5 and C-4 atoms as function of time in the five simulations. | ||
For the S848R mutant with high catalytic efficiency, the reason for the closer distance of N-5 (FAD) and C-4 (NMNH) atoms is further explored. The binding free energy between BM3 and NMNH cofactor is calculated using the MM/GBSA method and is shown in Table 3. The binding free energies of the WT and S848R mutant to NMNH are −12.18 and −13.98 kcal mol−1, respectively, and the S848R mutation resulted in a higher affinity between BM3 and NMNH, which is consistent with the experimental Km observation. In addition, residue decomposition is performed to calculate the contribution residues S/R848 in terms of binding free energy by the decomposition model of AMBER18. As shown in Table 3, the contribution of S848 in WT is −2.72 kcal mol−1, and that of R848 in the S848R mutant is −4.38 kcal mol−1, indicating that mutation of residue 848 directly leads to an increase in affinity with BM3. In addition to the energy term, electrostatic interactions and vdW interactions both contribute to the improvement of binding affinity.
| System | ΔEele | ΔEvdW | ΔGGB | ΔGNP | ΔG | |
|---|---|---|---|---|---|---|
| BM3-NMNH | WT | 44.54 ± 2.52 | −17.33 ± 0.32 | −37.05 ± 2.04 | −2.32 ± 0.02 | −12.18 ± 0.48 |
| S848R | 33.02 ± 2.30 | −23.57 ± 0.29 | −20.88 ± 2.07 | −2.56 ± 0.02 | −13.98 ± 0.48 | |
| S/R848-NMNH | WT | −5.53 ± 0.19 | −1.09 ± 0.08 | 4.11 ± 0.12 | −0.21 ± 0.00 | −2.72 ± 0.09 |
| S848R | −33.84 ± 0.64 | −2.63 ± 0.07 | 32.42 ± 0.48 | −0.33 ± 0.01 | −4.38 ± 0.18 |
Besides, the root mean square fluctuation (RMSF) of protein backbone and NMNH are calculated as show Fig. 6 to explore the impact of S848R mutation on the flexibility of protein structure. In Fig. 6, the RMSF of WT and S848R are very similar overall, however, the RMSF of S848R is significantly smaller than that of WT, which indicates that the S848R mutation makes the structure more stable. Special residues and ligands (848, FAD and NMNH) are highlighted in Fig. 6. Their RMSF is relatively small, which shows that they are the key residue/ligand and can stably play a catalytic role. At the same time, for these three key residues/ligands, the RMSF of S848R is smaller than that of WT. The results of RMSF analysis show S848R mutation can make key residue and ligand (848, FAD and NMNH) and even the entire protein more stable to improve catalytic efficiency.
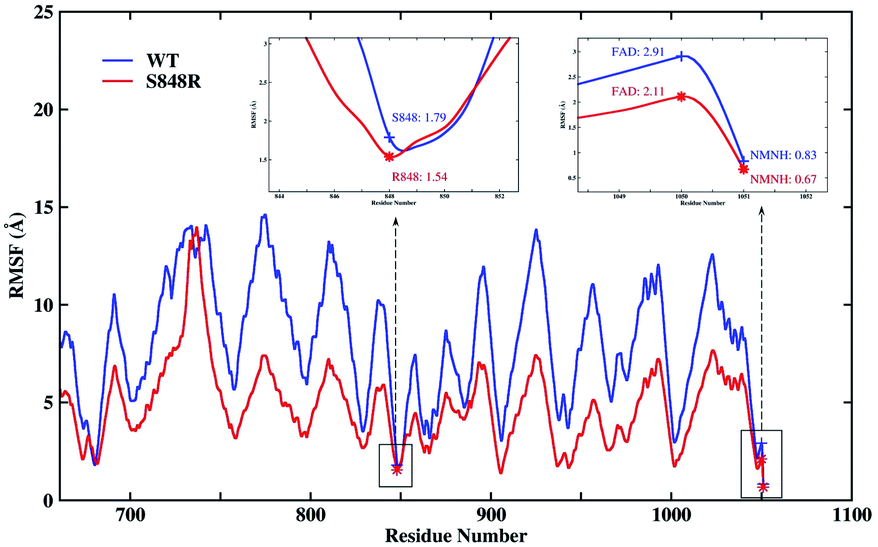 | ||
| Fig. 6 The RMSF of protein backbone and NMNH in the WT and S848R system. Special residues and ligands (848, FAD and NMNH) are high-lighted. | ||
To more intuitively explain the origin of the increased affinity caused by mutations, the lowest potential energy structures of the WT and S848R mutants are plotted in Fig. 7. After structural comparison of wild and S848R mutants, the residue mutates from a short chain residue to a long chain residue, and a longer side chain leads to stronger vdW interactions. On the other hand, the R848 residue with a positively charged group can form a salt bridge with the negatively charged phosphate group on NMNH, which leads to stronger electrostatic interactions. The difference in the above structure results in a higher affinity of the S848R mutant for NMNH, and it also simultaneously shortens the distance between N-5 (FAD) and C-4 (NMNH) atoms, rendering catalysis easier and increasing the Kcat value.
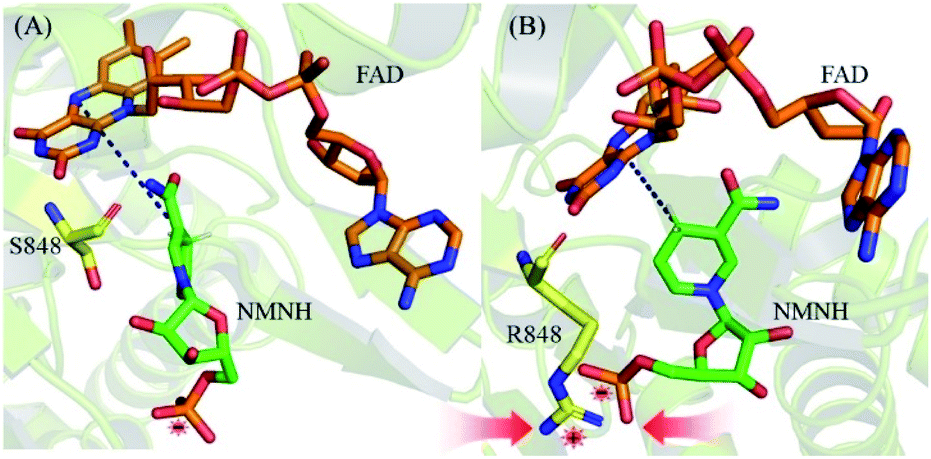 | ||
| Fig. 7 The lowest potential energy structures. (A) The lowest potential energy structures of WT. (B) The lowest potential energy structures of S848R mutant. | ||
Conclusions
The use of biomimetic cofactors is a promising method for the economic feasibility of oxidoreductase in industrial production. In this study, the oxidoreductase of P450 BM3 was designed by theoretical simulation and calculation strategies that improve enzyme activity with cofactor NMNH. During the optimization of the MD simulation, the distance between N-5 of FAD and C-4 of NMNH is used as a cue to evaluate improved conformation.The positive mutant S848R was successfully screened. The enzyme activity of S848R is 2.74-fold of the wild type, and the Kcat/KM values increased by 205.38% with NMNH cofactor compared to the wild type. Based on the electron transfer mechanism, the utilization of biomimetic cofactors by oxidoreductase can be effectively improved by optimizing the binding conformation. Through MD simulation and computer virtual mutation strategies, ten mutations with better binding efficiency under the perfect conformation were selected for further experimental verification, of which one mutation was not expressed correctly. Among the nine successfully expressed mutants, five exhibited no enzyme activity and three demonstrated decreased enzyme activity. Eventually, a variant that increased enzyme activity was selected. The success rate of the binding conformation refinement strategy reached 10%. This strategy can be further optimized to become more automated. Furthermore, our strategy presents important cues and can be applied for the specific utilization of various biomimetic cofactors by oxidoreductases.
Author contributions
LJ and TZ designed and controlled the research. YL performed all the experiment and wrote the most of manuscript. YC performed all the calculations and wrote the related manuscript. CZ, YP and QL assisted in experiment. JZHZ, BG, BF and CY revised this research and manuscript. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2020YFA0908400), National Natural Science Foundation of China (grant no. U1805235, 31571786), the Innovation Program of Shanghai Municipal Education Commission (201701070005E00020), and NYU-ECNU Center for Computational Chemistry at NYU Shanghai.Notes and references
- S. W. May and S. R. Padgette, Nat. Biotechnol., 1983, 1, 677–686 CrossRef CAS.
- H. Younus, Oxidoreductases: Overview and Practical Applications, Springer International Publishing, Cham, 2019 Search PubMed.
- S. W. May, Curr. Opin. Biotechnol., 1999, 10, 370–375 CrossRef CAS PubMed.
- A. M. Chanique and L. P. Parra, Front. Microbiol., 2018, 9, 194 CrossRef PubMed.
- C. You, R. Huang, X. Wei, Z. Zhu and Y. P. Zhang, Synthetic and Systems Biotechnology, 2017, 2, 208–218 CrossRef PubMed.
- W. B. Black, L. Zhang, W. S. Mak, S. Maxel, Y. Cui, E. King, B. Fong, A. Sanchez Martinez, J. B. Siegel and H. Li, Nat. Chem. Biol., 2020, 16, 87–94 CrossRef CAS PubMed.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- L. O. Narhi and A. J. Fulco, J. Biol. Chem., 1986, 261, 7160–7169 CrossRef CAS.
- T. K. Hyster and F. H. Arnold, Isr. J. Chem., 2015, 55, 14–20 CrossRef CAS.
- L. O. Narhi and A. J. Fulco, J. Biol. Chem., 1987, 262, 6683–6690 CrossRef CAS.
- Y. Miura and A. J. Fulco, Biochim. Biophys. Acta, Lipids Lipid Metab., 1975, 388, 305–317 CrossRef CAS.
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed.
- C. F. Butler, C. Peet, A. E. Mason, M. W. Voice, D. Leys and A. W. Munro, J. Biol. Chem., 2013, 288, 25387–25399 CrossRef CAS PubMed.
- L. N. Jeffreys, H. Poddar, M. Golovanova, C. W. Levy, H. M. Girvan, K. J. McLean, M. W. Voice, D. Leys and A. W. Munro, Sci. Rep., 2019, 9, 1577 CrossRef PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS PubMed.
- J. D. Ryan, R. H. Fish and D. S. Clark, ChemBioChem, 2008, 9, 2579–2582 CrossRef CAS PubMed.
- Y. Liu, Y. Feng, L. Wang, X. Guo, W. Liu, Q. Li, X. Wang, S. Xue and Z. K. Zhao, ACS Catal., 2019, 9, 1883–1887 CrossRef CAS.
- D. Cui, L. Zhang, S. Jiang, Z. Yao, B. Gao, J. Lin, Y. A. Yuan and D. Wei, FEBS J., 2015, 282, 2339–2351 CrossRef CAS PubMed.
- D. Cui, L. Zhang, Z. Yao, X. Liu, J. Lin, Y. A. Yuan and D. Wei, J. Biotechnol., 2013, 167, 386–392 CrossRef CAS PubMed.
- M. G. Joyce, I. S. Ekanem, O. Roitel, A. J. Dunford, R. Neeli, H. M. Girvan, G. J. Baker, R. A. Curtis, A. W. Munro and D. Leys, FEBS J., 2012, 279, 1694–1706 CrossRef CAS PubMed.
- M. Wang, D. L. Roberts, R. Paschke, T. M. Shea, B. S. Masters and J. J. Kim, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8411–8416 CrossRef CAS PubMed.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- D. A. Case, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, H. Gohlke, A. W. Goetz, D. Greene, R Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- X. Li, Y. Cong, M. Ma, Z.-N. You, B. Gao, J. Z. H. Zhang and L. Zhang, J. Agric. Food Chem., 2020, 68, 5129–5137 CrossRef CAS PubMed.
- D. Bashford and D. A. Case, Annu. Rev. Phys. Chem., 2000, 51, 129–152 CrossRef CAS PubMed.
- M. Feig, A. Onufriev, M. S. Lee, W. Im, D. A. Case and C. L. Brooks, J. Comput. Chem., 2004, 25, 265–284 CrossRef CAS PubMed.
- V. Tsui and D. A. Case, Biopolymers, 2000, 56, 275–291 CrossRef CAS PubMed.
- A. Onufriev, D. Bashford and D. A. Case, Proteins: Struct., Funct., Bioinf., 2004, 55, 383–394 CrossRef CAS PubMed.
- M. F. Sanner, A. J. Olson and J. C. Spehner, Biopolymers, 1996, 38, 305–320 CrossRef CAS PubMed.
- M. B. Murataliev, R. Feyereisen and F. A. Walker, Biochim. Biophys. Acta, 2004, 1698, 1–26 CrossRef CAS PubMed.
- O. Roitel, N. S. Scrutton and A. W. Munro, Biochemistry, 2003, 42, 10809–10821 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00352f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |