
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly robust, novel aluminum counter cation-based monophosphate tungsten bronze electro-catalysts for oxygen evolution in acidic solution†
Md. Imran Hossain a,
Tapas Debnathb,
M. Yousuf Ali Mollaha,
Md. Abu Bin Hasan Susana and
Md. Mominul Islam*a
a,
Tapas Debnathb,
M. Yousuf Ali Mollaha,
Md. Abu Bin Hasan Susana and
Md. Mominul Islam*a
aDepartment of Chemistry, Faculty of Science, University of Dhaka, Dhaka 1000, Bangladesh. E-mail: mominul@du.ac.bd
bDepartment of Theoretical and Computational Chemistry, Faculty of Science, University of Dhaka, Dhaka, Bangladesh
First published on 12th March 2021
Abstract
This study describes the successful synthesis of novel bronze with a low tungsten oxidation state for the efficient electro-catalytic oxidation of water. An extraordinarily robust monophosphate tungsten bronze (MPTB)-modified graphite anode was successfully fabricated for the oxygen evolution reaction (OER) at a thermodynamic potential of 1.23 V in H2SO4 acidic solution. Several Al, Cr and Fe counter-cation-based MPTBs were synthesized by the solution combustion method. Novel Al-based MPTBs calcined at 700 °C in O2 (AlO7) showed almost zero onset overpotential, high current density, high turnover frequency for OER and steady catalysis in repeated use even after 30 weeks. The orthorhombic AlO7 comprising crystallites of 9.89 nm and an indirect band gap (1.89 eV), is an unusually stable MPTB that contains 98% W5+ state stabilized with the Al3+ counter cation. The catalysis decreases as the ratio of W5+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W6+ in MPTBs decreases and [410] and [601] facets play main roles in the first H2O association and nucleophilic attack of the second H2O molecule on the catalyst surface. Thus, MPTBs can be non-noble metal anode materials for robust acidic H2O electrolyzers.
:W6+ in MPTBs decreases and [410] and [601] facets play main roles in the first H2O association and nucleophilic attack of the second H2O molecule on the catalyst surface. Thus, MPTBs can be non-noble metal anode materials for robust acidic H2O electrolyzers.
1. Introduction
The modern world demands clean and renewable forms of energy to overcome the current environmental crisis.1 The generation of energy from renewable sources and the development of efficient storage and safe distribution protocols are the most attractive domains for current research. Energy sectors consistently pursue innovative batteries, capacitors, supercapacitors, etc., to store electrical energy and these systems require huge storage chemicals that are limited, create secondary waste, pollute the environment and are toxic to living beings. In the H2O ↔ H2 cycle, H2 gas is a high density clean energy carrier. The H2O ↔ H2 cycle has been considered an energy storage alternative to batteries and capacitors. Moreover, H2 gas fuel is used in the H2–O2 fuel cell, but the green production of H2 gas from different sources, including H2O, is still costly, which actually prolongs the flourishing of fuel cell technology and keeps it primitive.The formation of O2 and H2 gases from H2O involves:
| 2H2O(l) → O2(g) + 4H+(aq) + 4e− at anode | (1) |
| 2H+(aq) + 2e− → H2(g) at cathode | (2) |
The thermodynamic potential of overall water splitting is 1.23 V versus the normal hydrogen electrode (NHE). In practice, an excess potential called overpotential (η) has to be applied to regulate cell reactions. The overall potential (ηop) for water splitting can be described as2
| ηop = 1.23 + ηa + |ηc| + ηΩ | (3) |
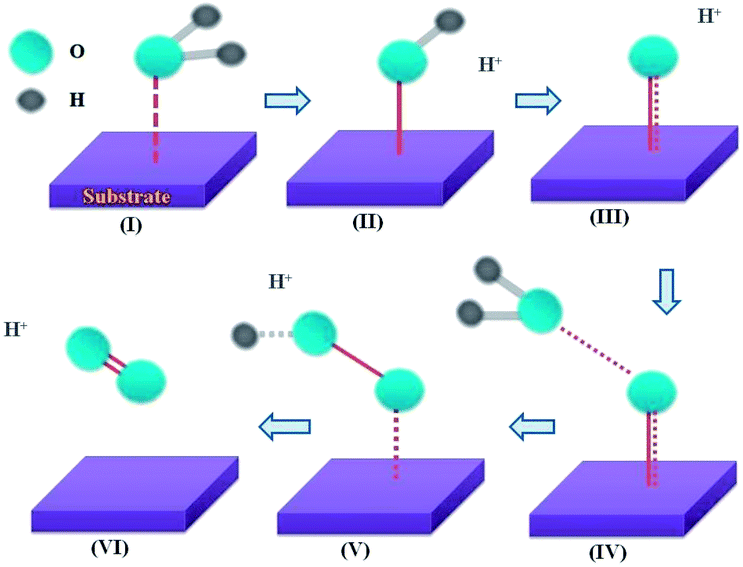 | ||
| Scheme 1 General mechanism for OER at catalytic surface. Different stages of OER at the electrode surface: (I) water association, (II) first proton-coupled electron transfer (PCET), (III) second PCET, (IV) nucleophilic attack, (V) third PCET and (VI) fourth PCET and evolution of O2. | ||
The effective activation of molecules that can minimize these intrinsic barriers takes place through their binding, with suitable conformations, to the active sites of a heterogeneous catalyst, from which the intermediate species should not be desorbed until the product is formed. To improve the activity of electro-catalysts, two general strategies have been applied: (i) active site engineering and (ii) conductivity optimization. The former is based on the particle size, morphology, intrinsic and lattice defects, and crystallinity that maximize the density of accessible active sites.11 The second is improved by doping with hetero-atoms which may minimize the intrinsic activation energy (charge-transfer resistance) and ηa in a composite type catalyst.11,12 Substantial efforts have been devoted to designing efficient OER catalysts other than precious metal-based ones, e.g., Pt, RuO2, and IrO2, which are known to show superior catalytic activities but have no real possibility to scale up to meet global demand due to their scarcity.13,14 The ηa of OER studied on a FeOx adsorbed indium-tin oxide electrode in phosphate buffer was found to be 580 mV.15 Iron dopants have been found to play a critical role in enhancing the activity of Ni-based electrocatalyts.16 Nano-sized layered aluminum or zinc-manganese oxides have been found to show efficient OER activity in the presence of cerium(IV) ammonium nitrate as a non-oxo transfer oxidant, wherein aluminum or zinc ions have been shown to play a key role in the catalysis.17 The redox-active center of Ru4(γ-SiW10)2 immobilized on multi-walled carbon nanotubes decorated with poly(amidoamine) ammonium dendrimers showed greatly improved catalysis in OER and performed multiple functions: (i) the co-existence of tungsten (W) and ruthenium (Ru) sites enhanced the hydrophilic character of the catalyst, creating a suitable environment for H2O adsorption, (ii) the charged nitrogen and oxygen residues assisted H2O diffusion into the molecular cavity and (iii) the coordination and hydrogen bonding interactions stabilized OER intermediates until product formation.18 Blasco et al. reported that the enhanced catalytic activity of polyoxometalates (POMs) with cesium or barium counter-cations for OER occurred at an ηa of 189 mV vs. NHE in acidic solution.19 These catalysts suffer from a stability problem in acidic solution. The earth abundant and inexpensive catalysts perform well in alkaline or neutral media2 in which |ηc| is high to regulate the H2 evolution (eqn (2)) that eventually increases the ηop (eqn (3)).
Here we report, for the first time, the synthesis and use of extraordinarily stable monophosphate tungsten bronze (MPTB) catalysts that perform OER in acidic media at the thermodynamic potential. MPTBs are polyanion clusters with structural diversity in which the oxometal polyhedra of MOx (in the present case, WOx and x = 5, 6), are the basic units and the polyanions act as Lewis acid or base under different conditions.18 The MPTB family was chosen because of their low dimensionality20 and their polyanions make it easy to selectively incorporate the desired counter-cations close to their active sites to regulate and stabilize the oxidation state (i.e., x = either 5 or 6) of the central metal (e.g., W). In this study, three series of MPTBs based on Al, Cr and Fe counter cations were prepared through solution combustion synthesis (SCS) and were calcined at different temperatures in either O2 or N2 atmosphere. Approximately zero onset ηa, high current density at low ηa, high turnover frequency (TOF) and robust electro-catalytic stability in repeated performances of OER in H2SO4 solution were achieved with the Al-based MPTB deposited on a graphite electrode. The reason for the persistent catalytic activity of the studied MPTBs was revealed by analyzing the catalysts with state-of-the-art techniques.
2. Experimental
2.1. Materials
The analytical grade chemicals and materials, such as potassium chloride (KCl, Sigma, Germany), potassium bromide (KBr, Sigma-Aldrich, Germany), sulphuric acid (H2SO4, Merck, Germany), ethanol (C2H5OH, Merck, Germany), poly(vinylidene fluoride) (PVdF, Sigma-Aldrich, Germany), tungstic acid (H2WO4, Sigma, Japan), ammonium dihydrogen phosphate (NH4H2PO4, Sigma, Japan), chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O, Merck, Germany), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, Merck, Germany), aluminum nitrate nonahydrate (Al(NO3)3·9H2O, Merck, Germany) as a source of trivalent cations, glycerin (Sigma-Aldrich, Germany), and graphite rods (Alfa Aesar, USA), were used without further purification. Distilled water was passed through HPLC grade water purification systems (BOECO, BOE 8082060, Germany) to yield de-ionized water (conductivity: 0.055 μS cm−1 at 25.0 °C) for use throughout the experiments.2.2. Syntheses of catalysts
MPTBs were prepared by the SCS method, which involves uniform preheating of the reaction mixture prior to self-ignition.21,22 Appropriate amounts of tungstic acid (H2WO4) and metal (Al, Cr and Fe) nitrates were dissolved in a minimum amount of aqueous ammonia. Glycerin and ammonium dihydrogen phosphate, which acted as a fuel and oxidizer, respectively, were added in a ratio of 1:8 in all runs.22,23 The colored, solid mass was treated in a tube furnace (ESI†) and the conditions of synthesis and abbreviated names of the catalysts can be seen in the captions of figures and in Table S1.†
2.3. Deposition of prepared catalysts on electrode surface
The prepared MPTBs were deposited on a graphite disk electrode (diameter ca. 0.615 cm). Each mixture of a particular MPTB and PVdF binder was prepared with the appropriate amount of these compounds (Table S7†). Typically, MPTB and ca. 15% binder were dispersed in ethanol. These mixtures were sonicated in an ultrasonic bath for 15 min and the dispersed solid mass was deposited on a flat, polished graphite electrode by the solution casting method in which the dispersion was added dropwise to the surface of electrode. The ethanol evaporated within 5 min. A thin layer of MPTB, as indicated by the characteristic color, was obtained by heat treatment at 100 °C in an oven. The thickness of the fabricated MPTB on the graphite electrode was determined by spectrometer (Model: AvaSpec-ULS2048-USB2, Avantes, Netherlands) where white light interference and the pattern were translated into optical thickness. The reference spectrum was taken from a clean bare graphite electrode and a spectrum of a fabricated graphite electrode was obtained. The average thickness of these thin films was around 160 nm as found from the difference of these two spectra.2.4. Electrochemical measurements
A single compartment three-electrode cell was used for the electrochemical measurements in which the data were recorded using a computer-controlled electrochemical analyzer system (Model: CHI 760E, USA). The electrochemical measurements were conducted in 0.08 M H2SO4 electrolytic solution using a modified graphite electrode as the working electrode, a spiral Pt wire electrode as the counter electrode and a silver|silver chloride|saturated KCl solution [Ag|AgCl|KCl (sat.)] as the reference electrode. Linear sweep voltammograms (LSVs) measured at various experimental conditions were analyzed to evaluate the catalytic performance of MPTBs for OER in 0.08 M H2SO4 solution. The ηa was determined as the potential corresponding to the intersection point of the tangent lines drawn from the rising current and baseline current of an LSV. The applied potential was converted to ηa in the NHE scale19 by the mathematical manipulations described in the ESI.† The dependence of j on ηa can be depicted by Tafel plots. The overpotential is generally logarithmically related to j and its linear portion is given as the Tafel equation (eqn (4)) where (η > 0.05 V)2,42|
η = a + blogj
| (4) |
The value of TOF was determined from the experimental j and charge (Q) according to2
| TOF = jη/(4 × Q) | (5) |
3. Results and discussion
3.1. Material characterizations
MPTBs were subjected to systematic, comprehensive analyses as described. The typical SEM images (Fig. 1) present the characteristic stony surface of AlO7 and AlN7 particles; the particles of AlO7 are larger in size than those of AlN7. Detailed morphological analyses of the synthesized MPTBs are given in the ESI (Table S2†). Particle counts for scales of both 10 and 100 nm are higher for AlN7 than AlO7. The Fe-based MPTB showed its characteristic spongy surface (Fig. S3†). To reveal the composition and chemical state of W and other elements in MPTBs, FTIR and XPS analyses were carried out for AlO7 and AlN7 (Fig. 2). | ||
| Fig. 1 (a and b), SEM images of AlO7 and AlN7, respectively. | ||
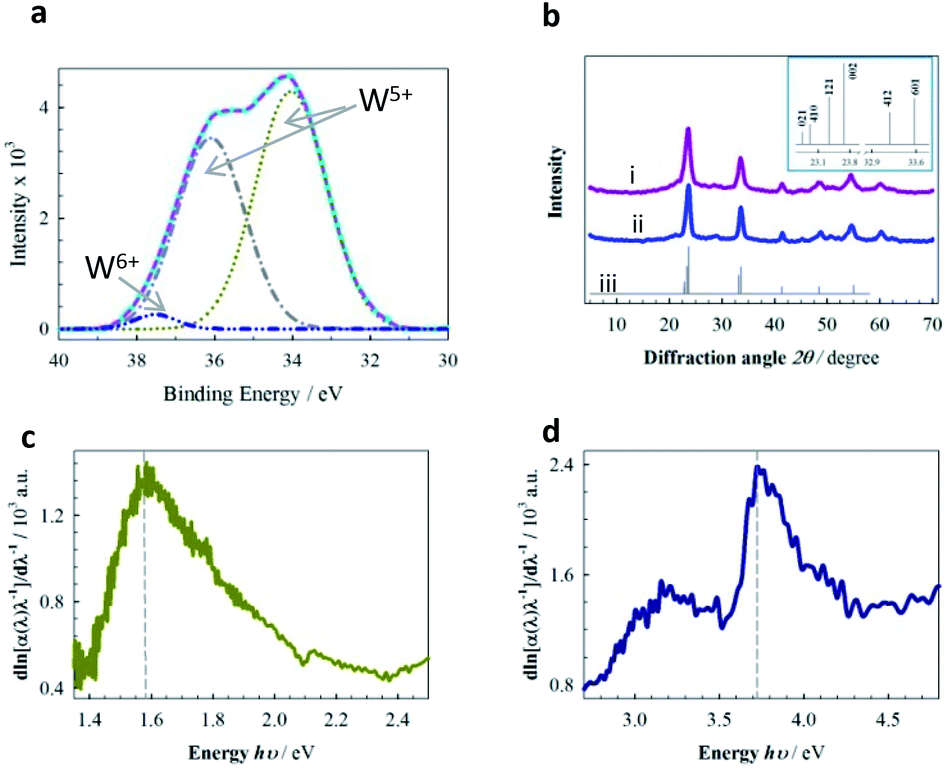 | ||
| Fig. 2 (a) XPS spectra of W 4f for AlO7. (b) XRD patterns of (i) AlN7, (ii) AlO7 and (iii) calculated refined response. Inset represents lower 2θ zone of calculated XRD response with the Miller indices. (c and d) Derivative response obtained from the experimental reflectance data of AlO7 and AlN7, respectively, using the DASF method.30 AlO7 and AlN7 refer to Al-based MPTBs calcined at 700 °C in O2 and N2 atmospheres, respectively. | ||
Intense bands at 600–659 cm−1 for the characteristic stretching vibration of W–O–W and a broad band at 1100 cm−1 for the stretching vibration of Al–O were observed in the FTIR spectrum of AlO7 (Fig. S1†).24 Similarly, the characteristic bands for the stretching vibrations of Cr–O and Fe–O in the FTIR spectra of Cr- and Fe-based MPTBs were noted (see ESI†). Only Al-based MPTBs were purposely subjected to further characterization. Fig. 2a represents the typical de-convoluted XPS spectrum of tungsten 4f in AlO7. Asymmetric doublets assignable to 34.03 and 36.10 eV for W5+ of 4f7/2 and 4f5/2, respectively, were observed, along with a weak peak at 37.52 eV for 4f5/2 of the W6+ oxidation state (Fig. 2a).25 Characteristic peaks at 133.5 and ca. 530.0 eV for phosphorus 2p in phosphate and 1s of lattice oxygen, respectively, confirm the formation of MPTB (Fig. S7†).26,27 UV-visible reflectance spectra of AlO7 and AlN7 are shown in Fig. S4.† One prominent absorption band is observed in the region of 200 to 500 nm. These spectral analyses also support the presence of W with different oxidation states in which W5+ is significantly greater (ca. 98%) than W6+ (ca. 2%) in AlO7. In contrast, the amount of W5+ in AlN7 decreased to ca. 60% (Fig. S4 and Table S4†). The Al3+ counter-cation in MPTB donates its electrons to the vacant t2g orbital and stabilizes W5+ states.28,29 The W5+ state of bronze continuously converts to stable W6+ in an open environment, leading to a characteristic color change. It is noteworthy that no such color change of MPTBs, especially for the Al-series, was observed, indicating that we succeeded for the first time in synthesizing MPTBs with a stable W5+ state with the assistance of Al3+ counter cations that actually play the key role in the catalysis of OER, vide infra.
The derivation of absorption spectrum fitting (DASF) method30 was applied to gain more insight into the band gap energy (Eg) and the nature of the transition in AlO7 and AlN7. The Kubelka–Munk method31 was initially applied to determine the absorption coefficient. The systematic calculation of Eg is described in Table S3 and Fig. S5, S6. † The transition is indirect, meaning there is a misalignment of crystal momentum between the minimum energy level of the conduction band and the maximum energy level of the valence band. However, the Eg of AlO7 (1.58 eV) is less than one-half that of AlN7 (3.72 eV) (Fig. 2c and d).
This can be supported by considering the presence of a high percentage of the Al3+ state in AlO7 (Fig. S7b†), which lowers the Eg and hence increases conductivity, as identified by electrochemical impedance spectroscopy (Fig. S12†). The band gap of AlN7 at an Eg of 3.16 eV is due to the increase in quantum confinement due to the agglomeration of particles.32
The XRD diffraction lines shown in Fig. 2b indicate that AlO7 and AlN7 are crystalline in nature and the crystal system of these MPTBs is orthorhombic (ESI†).33 WO3 is normally cubic polymorph, which is thermodynamically unstable.34 In 1989, Siedle et al. 34 speculated that if WO3 could be specially doped with Al, the degree of inversion of Al3+ in octahedral sites would take place at the available O2− lattices30 and hence enhance the stability of the W5+ state (Fig. 2a). The calculated Miller indices of AlO7 are higher and the high-index facets, such as [410] and [601], are the surfaces of the lattice (Table S6†) where the atoms are located further apart. These atoms are ready to bond to more potential neighbors if the crystal continues to grow and this leads to a higher surface free energy than in the low-index facets, showing high catalytic activity.35,36 The crystallite sizes of AlO7 were determined to be larger (9.89 nm) than those of AlN7 (7.45 nm), supporting the SEM observations (ESI†).
3.2. Electrocatalytic performance of MPTBs
The catalytic performances of three series of MPTBs deposited on graphite electrodes for OER were tested in 0.08 M H2SO4 solution. Fig. 3a presents the cyclic voltammograms (CVs) recorded; no significant peak could be distinguished except for the anodic shoulder at 1.185 V vs. NHE for OER. LSVs of OER recorded with three representative MPTBs are compared in Fig. 3b. Similar LSVs for OER were also measured for all compounds under consideration. From the measured LSV responses, the ηonset, Tafel slope and TOF were evaluated and are summarized in Fig. 4.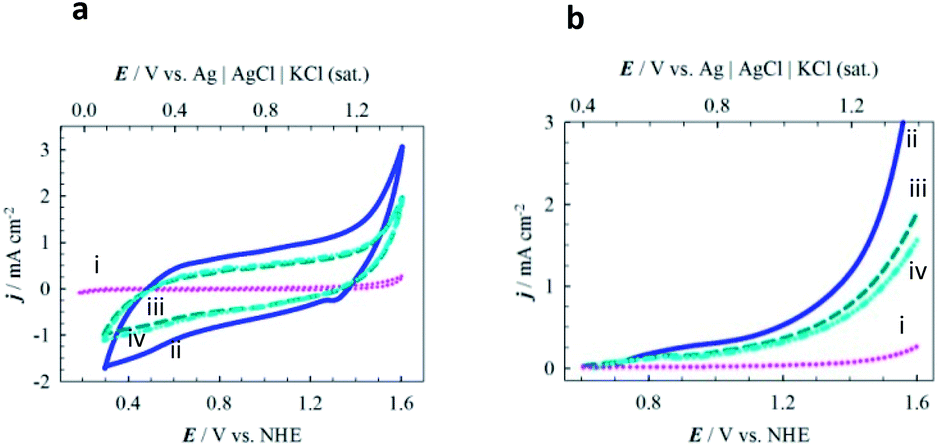 | ||
| Fig. 3 Comparative electrochemical behavior of MPTB-modified graphite electrodes in acidic media. (a) CVs and (b) background-corrected LSVs of OER measured with (i) bare, (ii) AlO7-, (iii) CrO7- and (iv) FeO8-modified graphite electrodes in 0.08 M H2SO4 solution at a potential scan rate of 5 mV s−1. In AlO7, CrO7 and FeO8 MPTB catalysts, Al, Cr and Fe refer to the counter-cations of Al3+, Cr3+ and Fe3+, the digits 7 and 8 refer to the calcination temperatures of 700 and 800 °C, respectively, and O stands for a calcination atmosphere of O2. | ||
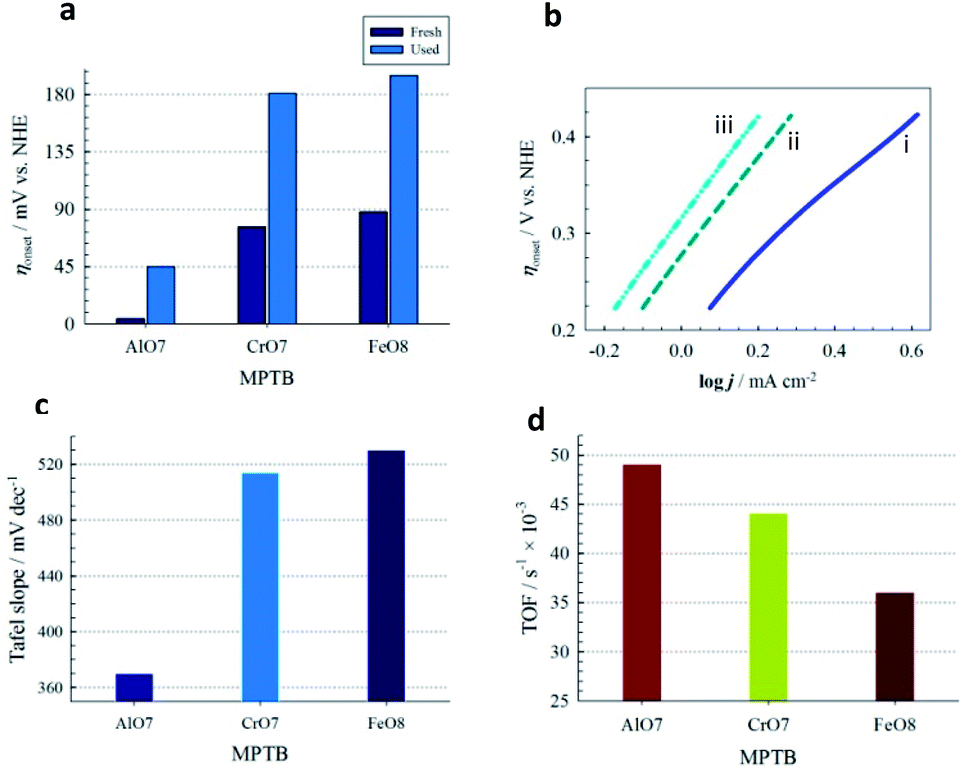 | ||
| Fig. 4 (a) Comparison of ηonset determined with freshly fabricated and tested (for continuous OER at a constant potential of 1.51 V vs. NHE for 18 h) electrodes. (b) Tafel plot: (i) AlO7, (ii) CrO7 and (iii) FeO8. (c) Tafel slope. (d) TOF of OER. | ||
At a glance, AlO7 shows the lowest ηonset of 4 mV for OER among the other MPTBs studied. Importantly, this ηonset is almost zero, that is, the OER started at its thermodynamic potential. A similar observation for AlN7 can also be seen. The ηonset generally increases with Al-series MPTB samples calcined at temperatures lower or higher than 700 °C. In addition, ηonset was found to slightly increase to 45 mV after continuous performance of OER for 18 h. The corresponding Tafel slope and TOF for AlO7 were determined to be 370 mV dec−1 and 0.049 s−1, respectively.
The role of Al in MPTBs for this catalysis in OER was further investigated by replacing Al3+ with Cr3+ and Fe3+. As can be seen in Table S8† and Fig. 3 and 4, the ηonset values with CrO7, CrO8, FeO7 and FeO8 are higher than those of the corresponding series of Al-based MPTBs. In this case, the Tafel slopes are also much higher than those obtained with Al-based MPTBs. Moreover, the TOFs are significantly low. Therefore, Al-based MPTBs are superior in all respects to the other two series of MPTBs studied. Other important characteristics of an OER catalyst are the ηa corresponding to the current density (j) of 10 mA cm−2 (η10), which is the maximum efficiency of a solar cell reported so far,37,38 and the stability. The η10 for AlO7 was 620 mV vs. NHE (Fig. 5a). In addition, the chronoamperometric j–t response (Fig. 5b) clearly evidenced that AlO7 is highly stable, with about a 2-times higher limiting j than those obtained with CrO7 and FeO8. Blasco-Ahicart et al. investigated OER in 1.0 M H2SO4 solution with cobalt oxides and cobalt-based POMs with counter-cations of cesium and barium (Ba[Co-POM]) and compared the results of catalytic performance with state-of-the-art IrO2 catalyst.19 IrO2 exhibited an ηonset of ca. 340 mV and η10 of ca. 500 mV. The lowest ηonset of ca. 90 mV and η10 of ca. 360 mV with a Tafel slope of 97 mV dec−1 were observed with Ba[Co-POM] incorporated in a carbon paste electrode.19 They reported a decreasing trend in j for the OER measured at an ηa of 250 mV for 24 h. In contrast, the stability of OER studied at an ηa of 324 mV exhibited that the AlO7 catalyst is highly stable and, interestingly, the current increases with time. Almost no change in the OER response was noticed after keeping the AlO7 modified electrode in the measured solution for 30 weeks. This suggests that the prepared MPTB is highly stable in an open environment.
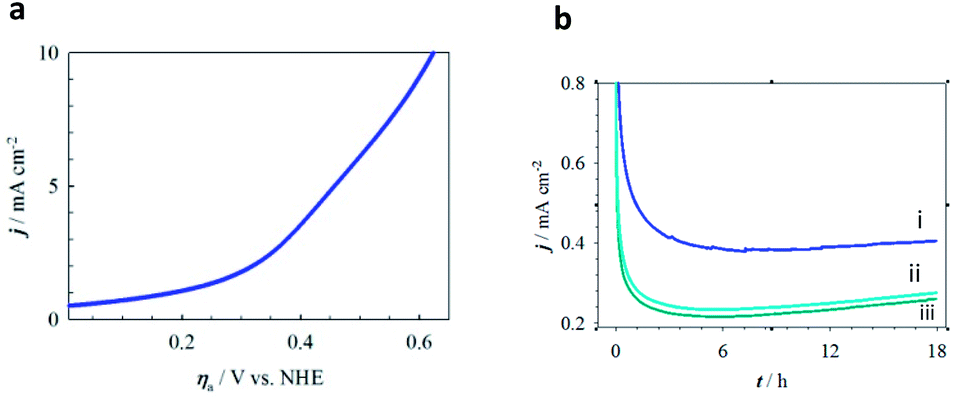 | ||
| Fig. 5 Electrocatalytic stability for OER at MPTB-modified graphite electrodes in acidic media. (a) LSV of AlO7 in 0.08 M H2SO4 solution at a scan rate of 5 mV s−1. (b) Chronoamperometric data for (i) AlO7, (ii) CrO7 and (iii) FeO8 at a constant potential of ηa = 324 mV vs. NHE. | ||
Three main concerns, namely the origin of the low ηonset, the extraordinary stability of MPTBs and the pathway of OER, are addressed here. The solution resistance and charge-transfer resistance were found to be negligibly small, at 17.2 and 37.8 Ω, respectively. In addition, the electro-conductivity of AlO7 which can be manifested by a low Eg (Table S3†) should be high. Such an enhanced electrical conductivity of AlO7 would likely be facilitated through the formation of a two-dimensional (2D) layer-type structure. A relatively low content of PO43− in AlO7 (Fig. S8†) is suitable for the stability of the observed orthorhombic phase and suggests the formation of a 2D layered structure (Fig. S9†).28 In addition, the observation of a large number of oxygen vacancies in AlO7 and a low Eg also supports the high conductivity of AlO7.39 All of these combined together minimize the ηonset of OER at AlO7 to almost zero. The reason for the increasing trend of j with time, shown in Fig. 5b, is ambiguous, but may be associated with the enhancement of catalytic activity via the spin-exchange phenomenon.40 The MPTBs studied, especially the Al-based series, are paramagnetic in nature,41 possessing an unpaired electron that may assist the inversion of the singlet state of O2, formed as an intermediate during OER, to a stable triple state of O2 (stage V, Scheme 1). This spin inversion occurs simultaneously by flipping the spin of the electron in the eg orbital of the W5+ ion in AlO7 to be parallel,40 which is considered to enhance the catalytic activity.
The catalytic current is proportional to the reaction rate over the electrode surface that is potential dependent42 and the catalytic OER consists of number of elementary steps (Scheme 1). The activity and reaction mechanism of the electrocatalyst can be revealed by examining the sensitivity of the current against applied potential (Fig. 4c) and their slopes (Tafel slope), as compared in Fig. 4c. A low Tafel slope suggests the surface-adsorbed species produced in the early stage of OER remains predominant, while a high Tafel slope indicates the occurrence of competition between a chemical and an electron-transfer limiting step during OER.19 In Fig. 6, a hypothetical pathway for the MPTB-catalyzed OER is proposed. The adsorption of H2O would occur on the active metal (W5+) sites with a higher oxidation state.43 Thus, the eg orbital of the W5+ ion of AlO7 participates in σ-bonding with a surface H2O molecule44,45 to enhance H2O association (stage I, Scheme 1).
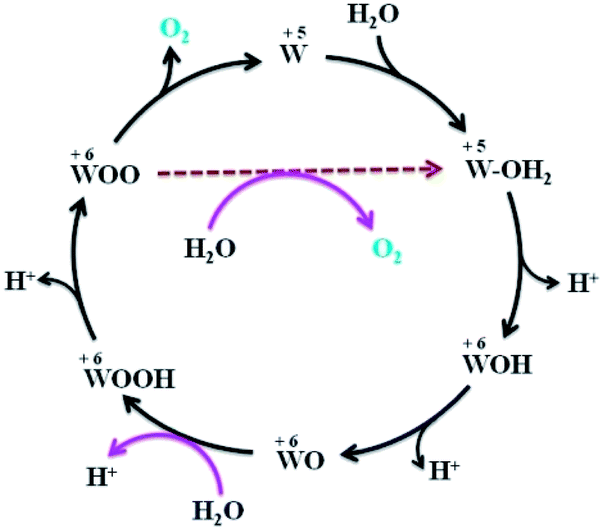 | ||
| Fig. 6 Probable electrochemical OER mechanism at MPTB in acidic media.46 | ||
The coordinated tungsten hydroxide starts to form through the first PCET reaction. The second PCET occurs to form {W–O–H} and another molecule of H2O takes part in a third PCET process to form {W–O–O–H}. In the following fourth PCET mechanism, O2 gas is evolved and these steps continue in a cyclic manner. Since the rate-determining step faces the highest kinetics hurdle, to attain a high j, i.e., to achieve an acceptable rate of OER, a higher potential must be applied. Formation of WOOH is the slowest step (i.e., the rate-determining step) because the thermodynamic energy barrier for surface oxygen coupling (stage IV, Scheme 1) is possibly larger than that of the associative reaction (stage I, Scheme 1).47 The Al-based MPTBs definitely enhance H2O association (stage I), but their direct role in surface oxygen coupling involving nucleophilic attack is also a burgeoning question that may be clarified by quantum mechanical calculations and computer modeling.
4. Conclusions
Highly stable Al3+ counter-cation-supported W5+ state-enriched MPTB and Cr3+ and Fe3+ counter-cation-based MPTBs were prepared by SCS method and characterized by FTIR, SEM imaging, XPS, reflectance and XRD techniques. The band gaps of the Al-based MPTB are indirect in nature and the values of Eg decrease by changing the calcination atmosphere from N2 to O2 due to the formation of the 2D-layered structure of the orthorhombic phase of AlO7. The catalytic performances of all MPTBs towards electrochemical OER varied widely as established by comparing ηonset, Tafel slope and TOF and were revealed to be associated with the ratios of the W5+:W6+ states in the MPTBs, which entirely depend on the counter-cations48 and calcination49 conditions. The robustness of AlO7 in catalysis is rationally achieved due to the unusual stability of W5+ in MPTB created by the incorporation of Al3+, as explained earlier.34 The eg orbital of W5+, particularly of the high-index facets of [410] and [601], in AlO7 may participate in σ-bonding with a surface H2O molecule44,45 to enhance the H2O association step and the nucleophilic attack of a second H2O molecule on the catalyst surface. Such self-coordination of H2O to the surface of the catalyst reasonably reduces the associated kinetic hurdles, offering OER at a low ηa. Thus, the developed anode is promising for the design of a cost-effective, efficient electrolyzer for large scale production of H2 fuel from H2O.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to Dr Altaf Hussain, retired professor of the Department of Chemistry, University of Dhaka (DU) and currently working as an academic advisor in Bangabandhu Sheikh Mujibur Rahman Maritime University, Bangladesh for his supports in designing and synthesizing bronze compounds. Authors thank Dr S. M. A. Hakim Siddiki, Institute of Catalysis, Hokkaido University, Japan for supporting the XPS analysis. Authors are also grateful to Dr Mangir Murshed, Solid State Chemical Spectroscopy Division, University of Bremen, Germany for discussing about DASF method used for calculation of band gap energy and Dr Subrata Roy, Department of Chemistry, Jagannath University, Bangladesh for sharing his expertise in XRD analysis of tungsten-bronze compounds. The instrumental supports of CARS, DU used for characterizing MPTB samples are also greatly acknowledged. MMI acknowledges the financial support as a special allocation for research of the Ministry of Science and Technology, Bangladesh. MIH acknowledges the financial supports in the form of fellowships of the University Grants Commission, Bangladesh and the Bose Centre for Advanced Study and Research in Natural Sciences, DU.References
- J. H. Bentley, Procedia Social and Behavioral Sciences, 2013, 77, 108–115 CrossRef.
- M. M. Islam and M. S. Miran, in Innovations in Engineered Porous Materials for Energy Generation and Storage Applications, ed. R. Rajagopalan and A. Balakrishnan, CRC Press, Boca Raton, 2018, pp. 267–288 Search PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- L. G. Bloor, P. I. Molina, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2014, 136, 3304–3311 CrossRef CAS PubMed.
- B. C. M. Martindale and E. Reisner, Adv. Energy Mater., 2016, 6, 1–9 Search PubMed.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- J. M. Gonçalves, T. A. Matias, K. C. F. Toledo and K. Araki, Adv. Inorg. Chem., 2019, 74, 241–303 CrossRef.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- R. D. L. Smith, B. Sporinova, R. D. Fagan, S. Trudel and C. P. Berlinguette, Chem. Mater., 2014, 26, 1654–1659 CrossRef CAS.
- E. Pizzolato, S. Scaramuzza, F. Carraro, A. Sartori, S. Agnoli, V. Amendola, M. Bonchio and A. Sartorel, J. Energy Chem., 2016, 25, 246–250 CrossRef.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. M. Najafpour, B. Pashaei and S. Nayeri, Dalton Trans., 2012, 41, 7134–7140 RSC.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- M. Blasco-Ahicart, J. Soriano-Lopez, J. J. Carbo, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef CAS PubMed.
- P. Roussel, O. Pérez and P. Labbé, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 603–632 CrossRef CAS PubMed.
- A. Kopp Alves, C. P. Bergmann and F. A. Berutti, Novel Synthesis and Characterization of Nanostructured Materials, Springer, Berlin, 2013, p. 11 Search PubMed.
- A. S. Mukasyan, P. Epstein and P. Dinka, Proc. Combust. Inst., 2007, 31, 1789–1795 CrossRef.
- T. Mimani and K. C. Patil, Mater. Phys. Mech., 2001, 4, 134–137 CAS.
- R. Romero Toledo, V. Ruiz Santoyo, C. D. Moncada Sánchez and M. Martínes Rosales, Nova Scientia, 2018, 10, 83 CrossRef.
- Y. Liu, S. Shrestha and W. E. Mustain, ACS Catal., 2012, 2, 456–463 CrossRef CAS.
- S. Lee, M. Choi and J. Kim, IEEE Trans. Magn., 2017, 53, 1–5 Search PubMed.
- S. Du, C. Zhang, P. Jiang and Y. Leng, ACS Appl. Nano Mater., 2019, 2, 7432–7440 CrossRef CAS.
- M. Greenblatt, Int. J. Mod. Phys. B, 1993, 7, 3937–3971 CrossRef CAS.
- W. Grünert, E. S. Shpiro, R. Feldhaus, K. Anders, G. V. Antoshin and K. M. Minachev, J. Catal., 1987, 107, 522–534 CrossRef.
- L. I. Granone, A. C. Ulpe, L. Robben, S. Klimke, M. Jahns, F. Renz, T. M. Gesing, T. Bredow, R. Dillert and D. W. Bahnemann, Phys. Chem. Chem. Phys., 2018, 20, 28267–28278 RSC.
- D. P. Joseph and C. Venkateswaran, J. At., Mol., Opt. Phys., 2011, 2011, 1–7 CrossRef.
- T. Edvinsson and R. Soc, R. Soc. Open Sci., 2018, 5, 1–17 Search PubMed.
- D. A. Woodcock, P. Lightfoot and C. Ritter, J. Solid State Chem., 2000, 149, 92–98 CrossRef CAS.
- A. R. Siedle, M. L. Brostrom, T. E. Wood, D. C. Koskenmaki, B. Montez and E. Oldfield, J. Am. Chem. Soc., 1989, 111, 1665–1669 CrossRef CAS.
- Y. Sun, S. Zhang, W. H. Zhang and Z. Y. Li, Chin. J. Chem. Phys., 2018, 31, 485–491 CrossRef CAS.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1–35 Search PubMed.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- R. R. Rakhimov and A. I. Aleksandrov, J. Phys. Chem. B, 2000, 104, 10973–10977 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- J. Carrasco, A. Hodgson and A. Michaelides, Nat. Mater., 2012, 11, 667–674 CrossRef CAS.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe and N. Nishiyama, et al., Nat. Commun., 2015, 6, 1–6 Search PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- M. Schilling and S. Luber, in Advances in Inorganic Chemistry, ed. R. V. Eldik and C. D. Hubbard, Academic Press, Cambridge, 2019, vol. 74, pp. 61–114 Search PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin and M. Mavrikakis, et al., J. Catal., 2002, 209, 275–278 CrossRef.
- S. C. Roy, W. Assenmacher, T. Linden, L. Esser, W. Mader and R. Glaum, Zeitschrift für Naturforschung B, 2016, 71, 543–552 CrossRef CAS.
- G. Leftheriotis, S. Papaefthimiou, P. Yianoulis and A. Siokou, Thin Solid Films, 2001, 384, 298–306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00699a |
| This journal is © The Royal Society of Chemistry 2021 |