
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular self-associating amphiphiles (SSAs) as enhancers of antimicrobial agents towards Escherichia coli (E. coli)†
Jessica E. Boles,
Rebecca J. Ellaby,
Helena J. Shepherd and
Jennifer R. Hiscock*
and
Jennifer R. Hiscock*
School of Physical Sciences, University of Kent, Canterbury, CT2 7NH, UK. E-mail: J.R.Hiscock@Kent.ac.uk
First published on 3rd March 2021
Abstract
Supramolecular self-associating amphiphiles (SSAs) are a class of amphiphilic salt which have demonstrated antimicrobial activity against both Gram-positive and Gram-negative bacteria. Herein, we show that SSAs are also able to increase the efficacy of a range of currently used antimicrobial/therapeutic agents with a range of different chemical structures and modes of antimicrobial action against Gram-negative Escherichia coli, which include: octenidine (an antiseptic); ampicillin (an antibiotic); and cisplatin (a DNA chelating agent). Additionally, we show these effects to be dependent on the order of agent addition. Finally, through completion of a range of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 SSA: antimicrobial/therapeutic agent physicochemical studies we gain an understanding as to how the self-association events and resultant SSA aggregate structure are effected by the presence of these secondary molecular species.
:1 SSA: antimicrobial/therapeutic agent physicochemical studies we gain an understanding as to how the self-association events and resultant SSA aggregate structure are effected by the presence of these secondary molecular species.
Introduction
The discovery of antibiotics/antimicrobials has revolutionised healthcare, with these agents becoming a game changing weapon in the fight against bacterial infections.1 However, since the first legitimate use of these agents, bacteria have evolved resistance. The ongoing misuse of these same compounds within the clinical,2 veterinary3 and animal feedstuff sectors,4 has resulted in the development of widespread microbial resistance and the emergence of multi-drug-resistant bacterial strains.5 To date, antimicrobial resistance (AMR) has now been reported towards all antimicrobial agents currently available,6 including antiseptics such as octenidine6 and more ominously, the antibiotic of last resort, colistin.7 Additionally, a recently commissioned UK governmental report has predicted that by 2050, approximately 10 million people per year will die globally from the primary effects of AMR, overtaking those caused by cancer in 2014, ca. 8.2 million per year.8In light of the growing prevalence of AMR, the development of new molecular weapons to combat the threat of bacterial infection are of increasing importance.5 The development of such technologies include those driven by the supramolecular chemistry community.9 Developments in this area include work by Gunnlaugsson et al. who have used the aryl-pyridyl urea scaffold within supramolecular gelator design to produce a material which demonstrates antimicrobial activity against both Gram-positive methicillin resistant Staphylococcus aureus (MRSA) and Gram-negative Escherichia coli (E. coli).10 In addition, Hou et al. have demonstrated the ability of pillar[5]arenes to selectively insert into Gram-positive bacterial membranes, exhibiting efficient antimicrobial activity against Staphylococcus epidermidis.11 Furthermore, the supramolecular silica nanoplatform developed by Zink et al. has been shown to co-deliver the antibiotic ofloxacin and the antimicrobial peptide melittin for synergistic eradication of Pseudomonas aeruginosa PAO1 biofilms.12 Finally, Zhang et al. have demonstrated the enhanced antibacterial activity of silver nitrate when combined with PEGylated bisimidazolylbenzyl alcohol, against both MRSA and E. coli.13
Our own work in this area has focused on the development of a novel class of supramolecular, self-associating amphiphilic salts (SSAs), such as those exemplified by 1 (Fig. 1).14–21 Similar constructs developed by Faustino and co-workers have previously been shown to act as amphiphiles with enhanced surfactant properties, attributed to intermolecular hydrogen bonded self-association.22,23 Additionally, Supuran et al. have shown the promise of agents such as SSAs for development as therapeutics against Mycobacterium tuberculosis (TB).24 To date, members from this >70 compound SSA library have been shown to self-associate within the solution state, producing self-associated dimers, spherical aggregates and hydrogels.16 Additionally, SSAs have also demonstrated antimicrobial activity against Gram-positive MRSA and Gram-negative E. coli.14–16,20 This previous work has led us to hypothesise that SSA antimicrobial activity is linked to both molecular self-association and selective SSA:phospholipid complexation, resulting in molecular membrane permeation events.15,16,25,26 Additionally, not only have we shown SSAs to arrive at the external microbial membrane as self-associated spherical aggregates,16 but that these aggregates can incorporate small drug(like) guest species.17 The summation of this evidence has therefore led us to hypothesise that this class of compound has the potential to act as efficacy enhancers for other therapeutic agents. Herein, we characterise the SSA aggregates formed upon the co-formulation of 1 (Fig. 1) with examples of antiseptics, antibiotics, and other antimicrobial/cytotoxic agents (2–6). Additionally, we explore the ability of 1 to enhance the activity of 2–6 against the model Gram-negative bacteria, E. coli.
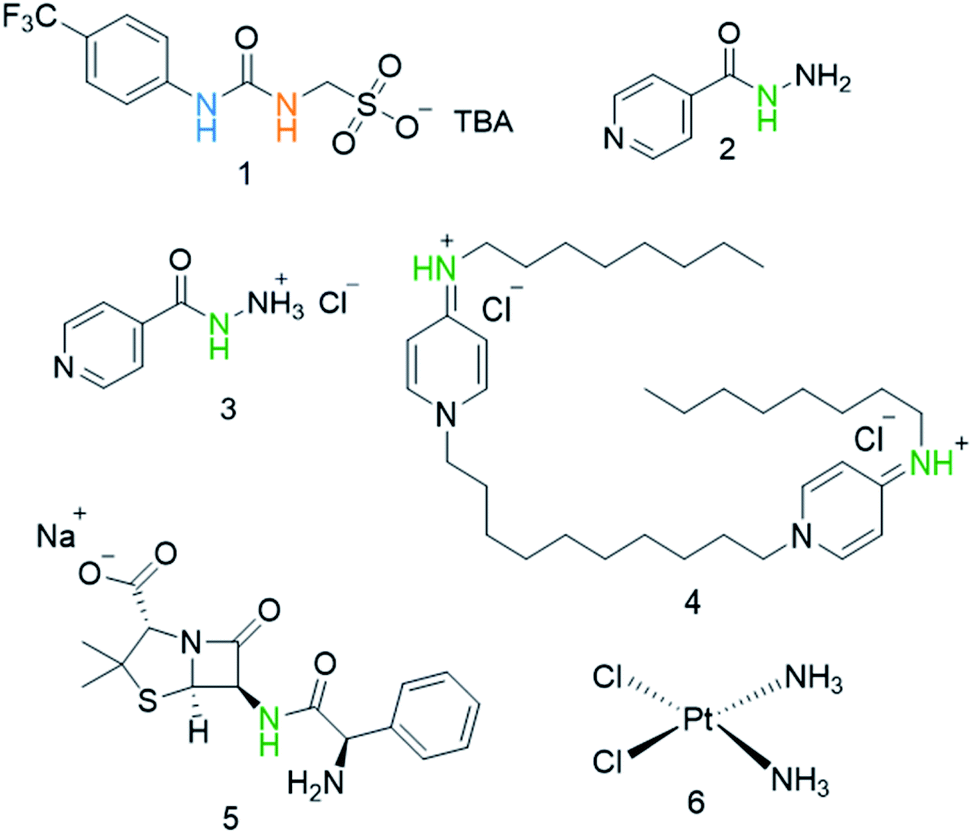 | ||
| Fig. 1 Chemical structures of SSA 1 and antimicrobial/therapeutic co-formulants 2–6. TBA = tetrabutylammonium. The NHs highlighted in green, blue and orange indicate the resonances monitored during 1H NMR dilution studies (Fig. 3). | ||
Results and discussion
Physicochemical characterisation of SSA co-formulations
A series of SSA co-formulation physicochemical studies, conducted in line with previous work,17 allowed us to understand the effects of the addition of 2–6 on the self-associative properties of SSA 1, when supplied in an 1:1 molecular ratio. The preparation of these 1:1 SSA: secondary compound mixtures produced co-formulations a–e, as detailed in Table 1. Here, 1 was synthesised in line with previously published procedures,27 whereas 2–6 were purchased from commercial sources and used without further purification.
:1 ratio, for co-formulations a–e
| Co-formulation | Molecular components | Co-formulation | Molecular components |
|---|---|---|---|
| a | 1 + 2 | d | 1 + 5 |
| b | 1 + 3 | e | 1 + 6 |
| c | 1 + 4 |
Due to the complexities associated with SSA self-association events, the physicochemical properties of 1 when co-formulated with 2–6 have been studied through a variety of complimentary methods that include: single crystal X-ray diffraction, quantitative 1H NMR, 1H NMR self-association constant determination, 1H NMR DOSY, dynamic light scattering (DLS), zeta potential studies, surface tension and critical micelle concentration (CMC) determination.† However, we have not attempted scanning/transmission electron microscopy (SEM/TEM) studies, as the self-associated aggregate structures produced by SSAs have previously been shown not to survive traditional SEM/TEM sample preparation methods.27
Slow evaporation of co-formulation a in acetone led to the generation of a crystal sample, suitable for single crystal X-ray diffraction analysis, Fig. 2.‡ Here 2 has undergone a reaction with the acetone, as previously described by Sarcevica et al.28 The product of this reaction, the cation shown in Fig. 2, has then formed a salt with the anionic component of 1. Here we see the formation of a hydrogen bonded urea-sulfonate anionic tape, similar to that observed previously for the pyridinium salt of this same anion.29 As observed for this analogous structure, the counter cation is also involved in competitive hydrogen bonded complexation events with the anionic sulfonate group, preventing sulfonate-urea dimer formation as observed for 1 only.29
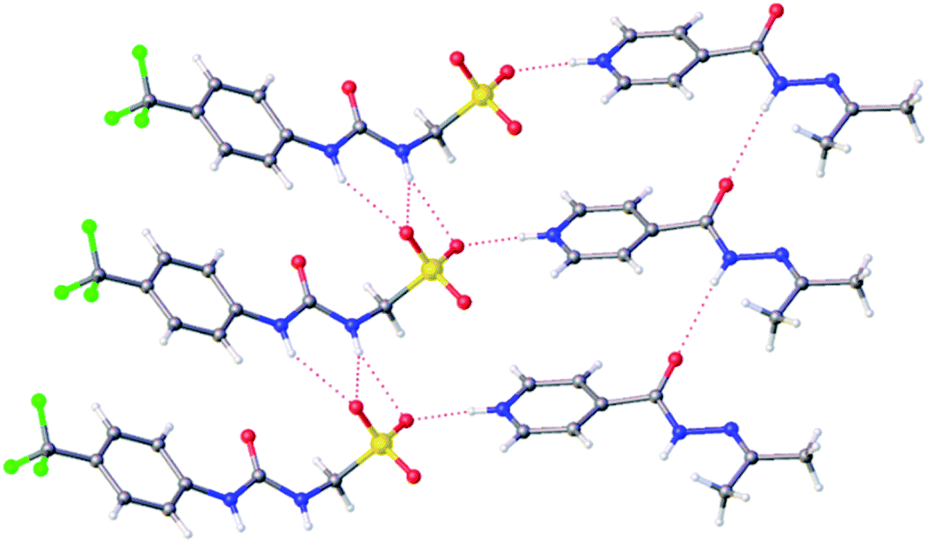 | ||
| Fig. 2 A single crystal X-ray structure obtained from a single crystal sample produced from a solution of co-formulation a in acetone. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulfur, white = hydrogen, green = fluorine, red dashed lines = hydrogen bonds.‡ | ||
SSA self-association events are known to be dependent on solvent environment, typically forming hydrogen bonded anionic dimers in DMSO, larger (≈100–550 nm in diameter) self-associated spherical aggregates in aqueous or aqueous ethanol (19:1 H2O:EtOH) solutions and self-associated hydrogel fibers in aqueous salt solutions.16 Initially, to enable characterisation of those SSA self-associated species present within the solution state, quantitative 1H NMR techniques are used to confirm the presence of larger self-associated species. Here comparative integration against an internal standard is used to calculate the proportion of a molecular component visible using standard solution state NMR techniques. The proportion of a molecular component that appears ‘lost’ from a solution is assumed to form larger higher-order self-associated structures with solid-like properties, thus rendering them NMR inactive. These studies are thus also able to enable the elucidation of the proportion of any secondary molecular substituents (such as 2–6) involved in the construction of these larger higher order species.
Those solutions which exhibit no apparent ‘loss’ of molecular components, through comparative integration with the appropriate internal standard, are taken forward for 1H NMR self-association constant determination studies. These studies enable us to quantify the strength of the hydrogen-bonded self-association events undertaken by the anionic SSA components, through completion of 1H NMR dilution studies and the subsequent fitting of these data and generation of self-association constants, using BindFit v0.5.30 The limitation of this method however, is that these models are limited to fitting one component, one dimensional, homogeneous aggregation events.31
A summary of the quantitative 1H NMR results obtained for 1 alone and when co-formulated as co-formulations a–e in a DMSO-d6 (1% DCM) and D2O (5% EtOH) solution at 112 mM and 5.56 mM respectively is given in Table 2. As with SSA 1 alone, co-formulations a–e showed no evidence for the formation of larger self-associated species with solid-like properties in a DMSO-d6 (1% DCM) solution.
| Co-formulation | Solvent system | Anion | Cation | Co-formulant |
|---|---|---|---|---|
| a Analogous quantitative 1H NMR control experiments conducted with 2–5 only in a D2O solution standardised with 5.0% ethanol at 5.56 mM confirmed the absence of any larger self-associated structures.b Values could not be determined due to peak overlap. | ||||
| 1 only19 | DMSO-d6 | 0 | 0 | n/a |
| D2O | 51 | 50 | n/a | |
| aa | DMSO-d6 | 0 | 0 | 0 |
| D2O | 56 | 57 | 50 | |
| ba | DMSO-d6 | 0 | 0 | 0 |
| D2O | 50 | 49 | 41 | |
| ca | DMSO-d6 | 0 | 0 | 0 |
| D2O | 53 | 29 | 64 | |
| da | DMSO-d6 | 0 | 0 | 0 |
| D2O | 48 | 55 | 62 | |
| e | DMSO-d6 | 0 | 0 | 0 |
| D2O | 65 | 83 | b | |
A complimentary series of 1H NMR DOSY studies confirmed that under these experimental conditions, the anionic and cationic components of the SSA and appropriate co-formulant 2–6, were all shown to exhibit different diffusion rates in a DMSO-d6/0.5% H2O solution, meaning that these molecular components are not strongly associated to one another. The hydrodynamic diameter (DH) of the molecular species present within these solutions, calculated using the Stokes–Einstein equation from the appropriate diffusion rate measurements, are listed in Table 3. The size of the species present also indicates that as previously observed for this class of compound, where self-association of the SSA's anionic component (1) exists, it is likely to adopt a dimeric binding mode.
To verify the presence of any hydrogen bonded self-association events within a DMSO-d6/0.5% H2O solution at 298 K, 1H NMR dilution studies were performed. From these data collected, the change in chemical shift for those NH resonances within the SSA anion and co-formulant were plotted with respect to concentration (Fig. 3). With these data fitted to the dimerization (EK) model using Bindfit v0.5,30 a Kdim of 2.7 M−1 was calculated for 1 only.19 A similar Kdim of 2.1 M−1 was calculated for the SSA anion in co-formulation a. This is not surprising as comparing the change in chemical shift for those NH resonances of the SSA anion and co-formulant 2, we observed a downfield change in chemical shift for the urea NH's of the anion but no discernible change for those NH resonances associated with 2, meaning that it is unlikely this co-formulant is involved within the SSA anion self-association events (Fig. 3a).
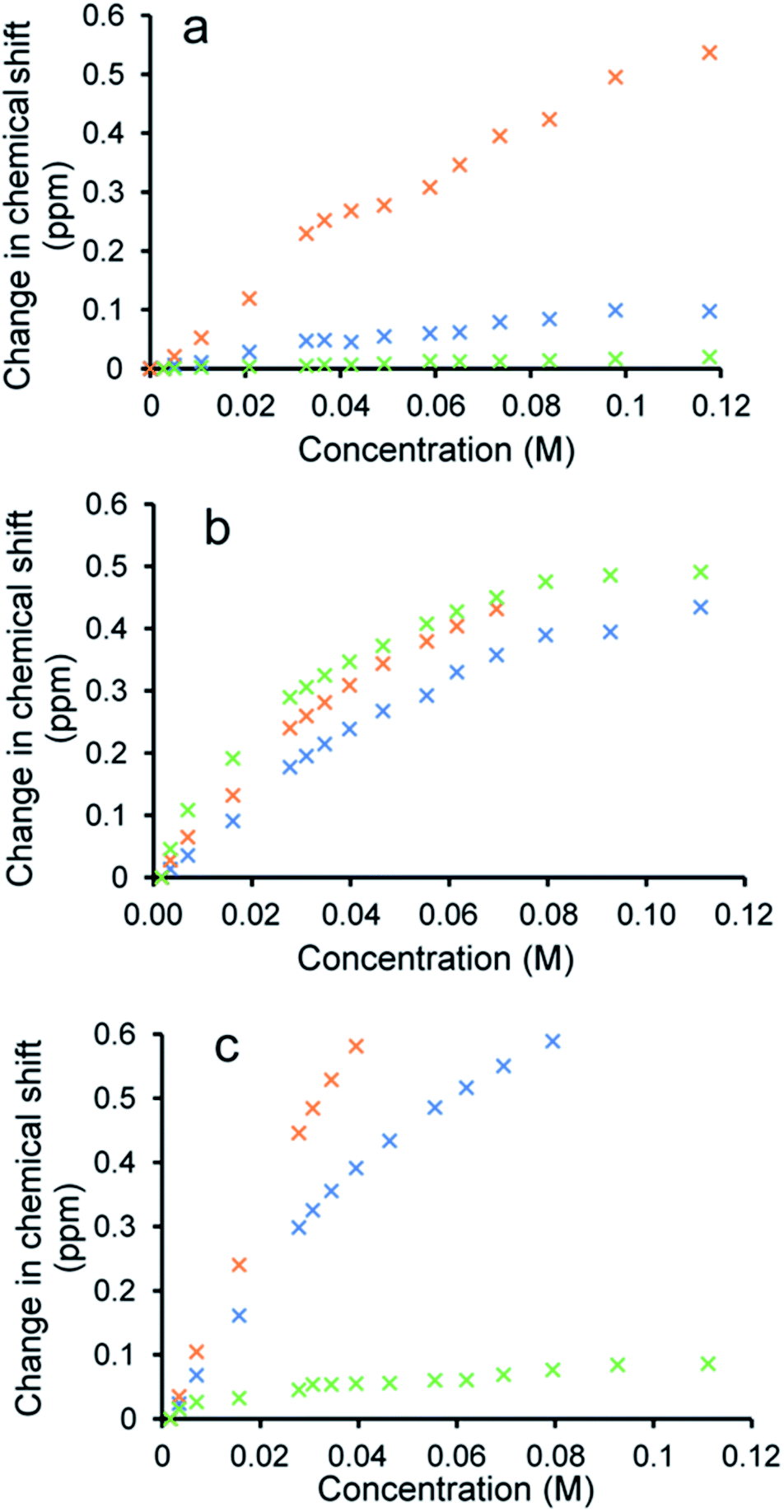 | ||
| Fig. 3 Graphs illustrating the 1H NMR down-field change in chemical shift of SSA (1) urea and appropriate co-formulant NH resonances in DMSO-d6/0.5% H2O (298 K) for: (a) co-formulation a; (b) co-formulation c; and (c) co-formulation d. The relevant NH's plotted within these graphs have been highlighted in the appropriate colour within Fig. 1. | ||
Fitting the analogous 1H NMR dilution data obtained for the SSA component of co-formulations b and e to the same dimerization isotherm resulted in an increased Kdim of 4.4 and 3.7 M−1 respectively. Unfortunately, in this instance we were unable to observe the position of the NH resonances associated with co-formulants 3 and 6. Compound 6 is a metal complex also capable of competing with any SSA self-association events. However, 3 exists as an HCl salt, therefore introducing Cl− ions into the solution. It is likely that these ions will also compete with the SSA sulfonate unit to coordinate with the urea functionality and therefore, this complex self-associative event would mean that these data are no longer appropriate to fit to this binding model, so this dimerization value should be treated with caution.
When comparing the change in chemical shift values for the different components of co-formulation c, the NH resonances of the SSA anion and co-formulant (4) were found to exhibit similar changes in chemical shift with respect to concentration (Fig. 3b). This leads us to believe that co-formulant 4, is also involved in molecular association/self-association events within this solution. This means that once again the Kdim reported in Table 2 should be treated with caution due to the possible complex nature of these self-association events. It is also hypothesised that complex self-association events also occur with co-formulation d (Fig. 3c). Here the co-formulant contains a carboxylate functionality, carboxylate anions are known to form stronger complexes with a urea functionality than the sulfonate anion,32 therefore it is possible that this ion is preferentially coordinating with the anionic component of the SSA. Although the Kdim calculated in this instance is similar to that of 1 alone.
Moving from a DMSO-d6 solution into an aqueous D2O:EtOH 19:1 solution at an SSA/co-formulation concentration of 5.56 mM at 298 K, our quantitative 1H NMR control experiment (Table 2) containing 1 only shows 50% of this SSA to become incorporated into larger self-associated structures. The proportion of SSA (1) to become incorporated when supplied as co-formulation b, remains the same as with the SSA only however, 41% of 3 also becomes NMR silent. We hypothesise that this is because this proportion of 3 is now incorporated into the larger self-associated aggregates of 1. Interestingly, the presence of 2 (co-formulation a) results in approximately 5–7% more of the SSA becoming incorporated into these larger self-associated aggregates, along with 50% of the available co-formulant. We hypothesise that this may be due to the lack of competitive Cl− and H+ ions, alongside the stabilising effects of uncharged, comparatively hydrophobic isoniazid (2) incorporation within the extended aggregate structure.
The cationic component of 4 in co-formulation c is amphiphilic in nature and in the presence of this SSA (1) 64% of this amphiphilic cation is incorporated into these larger SSA structures, replacing a portion of the SSA cation, as now 53% of the SSA anion remains incorporated into the extended aggregate structure whereas, only 29% of the SSA TBA counter cation is now utilised within aggregate formation. The reverse is true for co-formulation d. Here the carboxylate anion is incorporated into the SSA self-associated aggregate (62%), replacing a proportion of the SSA's anionic component (48%) in comparison to the SSA counter cation (55%). Interestingly, except for co-formulations a and e, the proportion of SSA anion to be incorporated into those self-associated aggregate species appears to be maintained at ≈50%.
To further characterise the formation of these larger self-associated aggregates, CMC values were also determined for co-formulations a–e and, compared to a control solution containing 1 only. These results have been summarised in Table 4. Here, co-formulation b exhibits a similar CMC value (11.21 mM) to that of 1 only (10.39 mM) however, co-formulations a, c and e, did exhibit decreased CMC values of 6.76 mM, 5.58 mM and 3.29 mM respectively. We hypothesise that this may be due to the presence of the amphiphilic/comparatively non-polar agents within these co-formulations. Interestingly the CMC obtained for co-formulation d was found to have increased significantly from that of 1 only, from 10.38 mM to 16.24 mM. It is believed that this is due to the presence of the carboxylate moiety within the structure of 5. As suggested by the results of the 1H NMR dilution studies, this compound perturbs the self-associative interaction of the SSA anion. Here we predict that those same competitive association events are responsible for this increase in CMC. It is therefore plausible that the slight increase in CMC observed for co-formulation b may also be due to competitive molecular association events. This hypothesis is further supported by the results of comparative dynamic light scatting and zeta potential studies (Table 4). Here a value of −76 mV was recorded for the control solution of 1 only at 5.56 mM. This value was found to decrease through the addition of 3 and 5 to −22 mV and −3 mV for co-formulations b and d respectively while, the stability of those larger aggregate structures formed with co-formulation a were found to exhibit a similar stability to 1 alone.
| Co-formulation | DH (nm) | Polydispersity (%) | Zeta potential (mV) | CMC (mM) |
|---|---|---|---|---|
| a * = experiments were performed at 0.56 mM due to solubility issues. | ||||
| 1 alone19 | 164 | 24 (±1.20) | −76 | 10.39 |
| 1* alone19 | 142 | 25 (±0.68) | −66 | n/a |
| a | 200 | 20 (±1.56) | −72 | 6.76 |
| b | 209 | 17 (±1.65) | −22 | 11.21 |
| c* | 240 | 25 (±0.75) | +76 | 5.58 |
| d | 136 | 13 (±1.26) | −3 | 16.24 |
| e* | 160 | 21 (±21.51) | −42 | 3.29 |
To explore the effects of the presence of an SSA on the efficacy of different currently available antimicrobial/therapeutic agents with different modes of action, E. coli was chosen as a model Gram-negative organism. Additionally, the effects of the order of SSA or antimicrobial/therapeutic agent addition was also investigated. Here either the SSA and antimicrobial agent was added simultaneously as the co-formulation (a–e) or, the cells were incubated firstly; with the SSA only for 10 minutes, followed by the addition of the antimicrobial agent and secondly; by incubating first with the antimicrobial agent for 10 minutes followed by the addition of the SSA. In this instance the concentrations of both the SSA (1) and antimicrobial agents (2–6) were chosen through completion of microbial susceptibility studies. These studies were conducted to ensure that both the SSA and antimicrobial/therapeutic agent were at a concentration where some inhibition of bacterial growth, and therefore antimicrobial efficacy was being imparted on the E. coli.† The results of these studies have been summarised for convenience within Fig. 4.
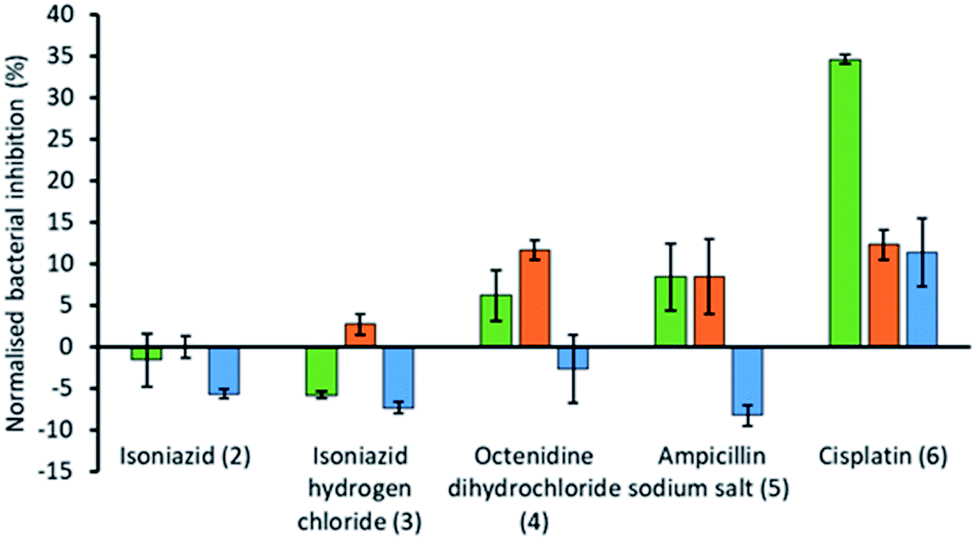 | ||
| Fig. 4 Bar chart showing the increase/decrease in efficacy of the antimicrobial/therapeutic agent (2–6) when supplied as a co-therapy in the presence of SSA 1. These results have been normalised against the combined bacterial growth inhibition effects of 1 and co-formulants 2–6 alone. OD600 measurements were obtained at 1100 minutes against E. coli DH10B in the presence of SSA 1 (1.5 mM) and either isoniazid (2) (1.45 μM), isoniazid hydrogen chloride (3) (1.73 μM), octenidine dihydrochloride (4) (0.29 μM), ampicillin sodium salt (5) (0.89 μM) or cisplatin (6) (16.6 μM). Each co-formulated system was studied where the: (i) SSA was pre-incubated with the E. coli for 10 min before the antimicrobial agent was added (green); (ii) antimicrobial agent was pre-incubated with the E. coli for 10 min before the SSA was added (orange); (iii) SSA and antimicrobial agent was added in co-formulation without any prior incubation (blue). The concentration of 2–6 alone was found to impede bacterial growth by <30% over 1100 min. | ||
The antimicrobial/therapeutic agents were chosen for this study not only to be diverse in chemical structure but also diverse in antimicrobial mode of action. These agents include: (i) the antibiotic ampicillin (5) which inhibits the synthesis of the peptidoglycan bacterial cell wall;33 (ii) octenidine (4) which acts to disrupt the cell membrane,34 and is thought to therefore act in a similar fashion to the SSAs themselves, at least in part; (iii) cisplatin (6), which is more commonly considered an anticancer agent, but still promotes bacterial cell death through DNA cross-linking within the cell, preventing/blocking DNA replication;35 (iv) two variations of isoniazid (2 and 3), which is traditionally used to treat mycobacteria infections through inhibition of mycolic acid synthesis.36
From these studies we have shown that when E. coli undergoes combination therapy with both an antimicrobial agent and an SSA, the presence of the SSA is able to enhance the activity of the antimicrobial agent in the cases of octenidine (4), ampicillin (5) and cisplatin (6). However, interestingly the degree of antimicrobial efficacy enhancement was found to be dependent on the order in which those agents were supplied as part of the appropriate combination therapy to be studied. When the SSA and antimicrobial agent were supplied to the E. coli as a co-formulation (a–e), with all co-formulations apart from co-formulation e there is an antagonistic effect, where bacterial growth is decreased when compared to the additive effects of the single SSA or antimicrobial agent alone. However, the reverse is true for co-formulation e where there is an agonistic effect. This is hypothesised to be because co-formulation e retains similar properties to that of the SSA alone at comparative concentrations. As we have previously shown a decrease in CMC value of an SSA was found to correlate with the MIC50 value obtained for a series of 50 SSAs against this same strain of bacteria, where the CMC value is ca. ≥11 mM.15 Here the presence of cisplatin (6), supplied in a 1:1 ratio with SSA 1 was shown to lower the CMC from 10.39 mM to 3.29 mM, which we suggest could increase SSA antimicrobial activity in line with these previous observations. Additionally, the size and stability of those self-associated structures produced in the presence of cisplatin (6) also retain some similarity (Table 4).
The presence of an SSA was not found to increase the efficacy of isoniazid, either when supplied as the neutral compound (2), or as the hydrochloric acid salt (3), with the exception of preincubation of the hydrochloric acid salt with the bacteria. However, this increase in efficacy is very small. The greatest increase in antimicrobial efficacy was observed for 1 with cisplatin (6). Here an enhancement of 34.6%, 12.3% and 11.4% in bacterial growth inhibition was observed with prior incubation of the bacteria with SSA (1), prior incubation of the bacteria with cisplatin (6) and the SSA co-formulated with the cisplatin before addition, respectively (Fig. 4). Thus, the greatest enhancement in antimicrobial efficacy was observed when the cells were incubated with the SSA, prior to the addition of the cisplatin (6).
Additionally, SSA 1 was also confirmed to slightly enhance the efficacy of both ampicillin (5) and octenidine (4), which impart antimicrobial effects through disruption of either the cell wall (ampicillin) or cell membrane (octenidine). However, these enhancement effects were only observed when the two agents were supplied to the cells as a combination therapy (one after another) and not when added simultaneously as a co-formulation. From evidence collected from our physicochemical analysis we hypothesise that this is because these antimicrobial agents interfere with SSA self-association, as observed within the scope of our 1H NMR dilution study experiments (Fig. 3b and c). In disrupting SSA self-association and membrane interaction events, we believe that this decreases the proportion of therapeutic agents to arrive at the surface of the cell in an active form, due to competitive multicomponent interactions between antimicrobial/therapeutic agents at the cell surface.
Conclusions
As one of the greatest threats to human health remains the rise of antimicrobial resistant bacterial infections, particularly those caused by Gram-negative bacteria,37 new technologies are required to combat this threat. Here, we have shown that the self-associated aggregates formed by SSA 1 are capable of interacting with antimicrobial/therapeutic agents currently in use. However, of particular interest are the observed differences in SSA/antimicrobial combination therapy over co-formulation efficacy brought about by changing the order of agent addition to bacteria.Here, we have confirmed that 1 is able to increase the efficacy of cisplatin (6) (a DNA chelating agent), ampicillin (5) (an antibiotic that disrupts cell wall synthesis) and octenidine (4) (a membrane disrupting antiseptic). However, in the case of 4 and 5 an antagonistic effect was observed when these agents were added as a co-formulation. The summation of the evidence presented here in leads us to conclude that SSA 1 acts most effectively as an antimicrobial efficacy enhancer when supplied as a combination therapy, through a prior 10 minute incubation period, before the addition of the antimicrobial/therapeutic agent. We also hypothesise, through data provided from a range of complimentary SSA co-formulation physicochemical studies, that the degree of antimicrobial effect maybe due to a combination of either destructive or constructive molecular interaction events at the cell surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the University of Kent and the Royal Society (RGS\R2\180336) for funding. J. R. Hiscock thanks the UKRI (MR/T020415/1) for funding. We also thank D. P. Mulvihill (University of Kent) for his assistance with the microbiology aspects of this work. J. E. Boles thanks the University of Kent and Public Health England for PhD studentship funding.Notes and references
- J. Bérdy, J. Antibiot., 2012, 65, 385–395 CrossRef PubMed
.
- P. Davey, C. A. Marwick, C. L. Scott, E. Charani, K. Mcneil, E. Brown, I. M. Gould, C. R. Ramsay and S. Michie, Cochrane Database Syst. Rev., 2017, 1–315 Search PubMed
- P. S. Morley, M. D. Apley, T. E. Besser, D. P. Burney, P. J. Fedorka-Cray, M. G. Papich, J. L. Traub-Dargatz and J. S. Weese, J. Vet. Intern. Med., 2005, 19, 617–629 CrossRef PubMed
- B. M. Marshall and S. B. Levy, Clin. Microbiol. Rev., 2011, 24, 718–733 CrossRef CAS PubMed
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed
- L. J. Bock, C. K. Hind, J. M. Sutton and M. E. Wand, Lett. Appl. Microbiol., 2018, 66, 368–377 CrossRef CAS PubMed
- B. Mott, D. Payne, M. Riley, A. Rubio, S. Therapeutics, L. Silver, L. Silver Consulting, J. Silverman, J. Sutcliffe, T. Pharmaceuticals, A. Tomaras, R. Tommasi, E. Therapeutics and D. Zurawski, A report from A Scientific Roadmap for Antibiotic Discovery, 2016 Search PubMed
- S. Davies, J. Farrar, J. Rex, L. J. White, R. Murry and J. O'Neill, The Review on Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations, ed. H. Government, 2014, pp. 1–14 Search PubMed
- X. Li, H. Bai, Y. Yang, J. Yoon, S. Wang and X. Zhang, Adv. Mater., 2018, 31, 1805092 CrossRef PubMed
- K. Pandurangan, J. A. Kitchen, S. Blasco, F. Paradisi and T. Gunnlaugsson, Chem. Commun., 2014, 50, 10819–10822 RSC
- M. Zhang, P.-P. Zhu, P. Xin, W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2017, 56, 2999–3003 CrossRef CAS PubMed
- Q. Yu, T. Deng, F. C. Lin, B. Zhang and J. I. Zink, ACS Nano, 2020, 14, 5926–5937 CrossRef CAS PubMed
- F. Wu, D. He, L. Chen, F. Liu, H. Huang, J. Dai, S. Zhang and J. You, RSC Adv., 2018, 8, 20829–20835 RSC
- K. K. L. Ng, M. Dimitrovski, J. E. Boles, R. J. Ellaby, L. J. White and J. R. Hiscock, Supramol. Chem., 2020, 412–424 Search PubMed
- N. Allen, L. J. White, J. E. Boles, G. T. Williams, D. F. Chu, R. J. Ellaby, H. J. Shepherd, K. K. L. Ng, L. R. Blackholly, B. Wilson, D. P. Mulvihill and J. R. Hiscock, ChemMedChem, 2020, 15, 2193–2205 CrossRef CAS PubMed
- L. White, J. E. Boles, N. Allen, L. Alesbrook, J. M. Sutton, C. Hind, K. Hilton, L. R. Blackholly, R. Ellaby, G. Williams, D. P. Mulvihill and J. R. Hiscock, J. Mater. Chem. B, 2020, 8, 4694–4700 RSC
- L. J. White, J. E. Boles, K. L. F. Hilton, R. J. Ellaby and J. R. Hiscock, Molecules, 2020, 25, 4126 CrossRef CAS PubMed
- L. J. White, N. J. Wells, L. R. Blackholly, H. J. Shepherd, B. Wilson, G. P. Bustone, T. J. Runacres and J. R. Hiscock, Chem. Sci., 2017, 8, 7620–7630 RSC
- L. J. White, S. N. Tyuleva, B. Wilson, H. J. Shepherd, K. K. L. Ng, S. J. Holder, E. R. Clark and J. R. Hiscock, Chem.–Eur. J., 2018, 24, 7761–7773 CrossRef CAS PubMed
- S. N. Tyuleva, N. Allen, L. J. White, A. Pépés, H. J. Shepherd, P. J. Saines, R. J. Ellaby, D. P. Mulvihill and J. R. Hiscock, Chem. Commun., 2019, 55, 95–98 RSC
- T. L. Gumbs, L. J. White, N. J. Wells, H. J. Shepherd and J. R. Hiscock, Supramol. Chem., 2018, 30, 42–51 CrossRef CAS
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Phys. Chem. B, 2009, 113, 977–982 CrossRef CAS PubMed
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Colloid Interface Sci., 2010, 351, 472–477 CrossRef CAS PubMed
- A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, J. Enzyme Inhib., 2001, 16, 425–432 CrossRef CAS PubMed
- G. Townshend, G. S. Thompson, L. J. White, J. R. Hiscock and J. L. Ortega-Roldan, Chem. Commun., 2020, 56, 4015–4018 RSC
- E. Medina-Carmona, L. Varela, A. C. Hendry, G. S. Thompson, L. J. White, J. E. Boles, J. R. Hiscock and J. L. Ortega-Roldan, Chem. Commun., 2020, 56, 11665–11668 RSC
- J. R. Hiscock, G. P. Bustone, B. Wilson, K. E. Belsey and L. R. Blackholly, Soft Matter, 2016, 12, 4221–4228 RSC
- I. Sarcevica, L. Orola, M. V. Veidis and S. Belyakov, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 392–395 CrossRef CAS PubMed
- L. R. Blackholly, H. J. Shepherd and J. R. Hiscock, CrystEngComm, 2016, 18, 7021–7028 RSC
- Supramolecular.org, Binding Constant Calculators|Supramolecular, http://supramolecular.org/, accessed 1 June 2020 Search PubMed.
- L. K. S. Von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC
- P. A. Gale, J. R. Hiscock, N. Lalaoui, M. E. Light, N. J. Wells and M. Wenzel, Org. Biomol. Chem., 2012, 10, 5909–5915 RSC
- D. Kaushik, M. Mohan, D. M. Borade and O. C. Swami, J. Clin. Diagn. Res., 2014, 8, ME01 CAS
- N. Malanovic, A. Ön, G. Pabst, A. Zellner and K. Lohner, Int. J. Antimicrob. Agents, 2020, 56, 106146 CrossRef CAS PubMed
- N. Chowdhury, T. L. Wood, M. Martínez-Vázquez, R. García-Contreras and T. K. Wood, Biotechnol. Bioeng., 2016, 113, 1984–1992 CrossRef CAS PubMed
- G. S. Timmins and V. Deretic, Mol. Microbiol., 2006, 62, 1220–1227 CrossRef CAS PubMed
- Z. Breijyeh, B. Jubeh and R. Karaman, Molecules, 2020, 25, 2888 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting experimental methods and data (synthesis, zeta potential, antimicrobial studies, quantitative NMR, dilution study data, 1H NMR DOSY, tensiometry and critical micelle concentration determination). CCDC 1999018. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00998b |
| ‡ A suitable crystal was selected and mounted on a Rigaku Oxford Diffraction Supernova diffractometer. Data were collected using Cu Kα radiation at 100 K. The structure was solved with the ShelXS38 via direct methods and refined with ShelXL39 on least squares minimisation. Olex2 (ref. 40) was used as an interface to all ShelX programs. CCDC deposition number for the structure shown in Fig. 2 = 1999018. |
| This journal is © The Royal Society of Chemistry 2021 |