
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient and scalable synthesis of 2,4-di-N-acetyl-L-altrose (L-2,4-Alt-diNAc)†
Anna Niedzwiecka,
Carita Sequeira,
Ping Zhang and
Chang-Chun Ling *
*
Department of Chemistry, University of Calgary, 2500 University Dr NW, Calgary, AB T2N 1N4, Canada. E-mail: ccling@ucalgary.ca
First published on 19th March 2021
Abstract
Bacterial nonulosonic acids such as pseudaminic acids and others constitute a family of 9-carbon monosaccharides that contain a common 3-deoxy-2-ketoacid fragment but differ in their stereochemistries at 5 stereogenic centers between C-4 to C-8. Their unique structures make them attractive targets for use as antigens in vaccinations to combat drug-resistant bacterial infections and their challenging stereochemistries have attracted considerable attention from chemists. In this work we report the development of an improved synthesis for 2,4-di-N-acetyl-L-altrose (L-2,4-Alt-diNAc), which is a key hexose required for the chemical and chemoenzymatic synthesis of pseudaminic acids. Using L-fucose as a starting material, our synthesis overcomes several pitfalls in previously reported syntheses.
Introduction
Bacterial infection is becoming an increasing cause of mortality and morbidity world-wide as traditional antibiotics continuously lose their effectiveness to pathogen resistance mechanisms.1 There are currently very few antimicrobial drug candidates undergoing clinical trials,2 creating an urgent need for alternative antibacterial strategies. The vaccination approach has demonstrated long-lasting beneficial effects, while resistance to it has rarely been observed.3 The success of a vaccine critically depends on the choice of proper antigens. Pseudaminic acid (Pse, 1, Fig. 1), along with legionaminic acid (2) and its 8- or 4-epimers (3–4), are members of a class of unique 9-carbon sugars called nonulosonic acids, commonly distinguished by their 3-deoxy-2-ketoacid fragment.4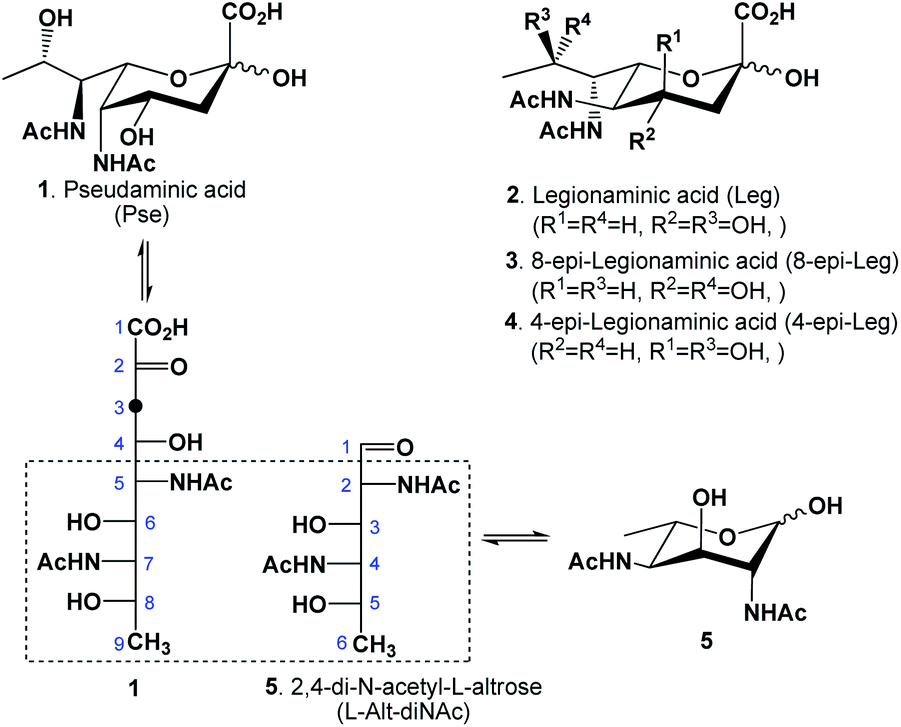 | ||
| Fig. 1 Structure of pseudaminic acid and its biosynthetic precursor L-Alt-diNAc (5). | ||
Pse and its derivatives are structural components of flagella and pili,5,6 exclusive to several Gram-positive bacteria: among them, Pseudomonas aeruginosa is the main cause of mortality for people suffering from cystic fibrosis, and Acinetobacter baumannii is a common cause of ventilator associated pneumonia (VAP).4 These pathogens thrive in mucous-rich environments, relying on flagella and pili as efficient motility systems for their virulence.5,7,8 Studies have shown that mutants with the inability to incorporate Pse into their cell structures lose mobility, thus their ability to colonize and infect hosts.9 In addition to their structural significance, these types of carbohydrates could function as interesting antigens for immunogenic vaccine-induced antibodies because of their unique L-altro-configuration between C5–C9, combined with their acylamido substitution at both 5,7-positions and their 9-deoxygenation10,11 (Fig. 1).
In nature, the acyl groups attached to the two amido groups could either be identical (commonly acetyl) or different, depending on the bacterial strain.4 For example, P. aeruginosa O7a produces a version of Pse that has a formamido group at C-7 and an acetamido group at C-5, while the related O9a strain produces another version with a (R)-3-hydroxybutanamido at C-7 and the same acetamido group at C-5; for Vibrio cholera O:2, another variant of Pse with the C-5 amido substituted by an acetamidoyl group is produced. In the biosynthesis of Pse, the intermediate 2,4-dideoxy-2,4-diacetamido-L-altrose (L-Alt-diNAc, 5) is enzymatically extended via an aldol condensation with phosphoenolpyruvate (PEP).12,13 Analogously, suitably protected L-Alt-diNAc substrates have also been used for the chemical synthesis of Pse, typically by reaction with methyl 2-(bromomethyl)acrylate via indium-mediated allylation,14 followed by an ozonolysis to cleave the alkene, revealing the desired 3-keto functionality. There have to date been several chemical syntheses of Pse reported in literature with varying success; some suffered from long reaction sequences, others from low overall yield, or difficulty in scaling up. For example, in 2001, Zähringer reported15 a synthesis from L-rhamnose which took advantage of its 6-deoxygenation and L-configuration. Although the use of L-sugar16 represented an advantage because of a configurational match at the C-5 position, the conversion to a 2-epimer of L-Alt-diNAc was achieved in a long and rather tedious reaction sequence (16 steps), as the introduction of the two acetamido groups required two double inversions, which was inefficient. Before that, in early 1970, Liav and Sharon reported17 a synthesis of L-Alt-diNAc from β-L-fucopyranoside, by first converting it to a 2,3-anhydro-4-O-mesyl-β-L-talopyranoside intermediate that then underwent a nucleophilic attack by azide; unfortunately, this reaction resulted in a mixture that included the 2,4-diazido-2,4,6-trideoxy-β-L-altropyranoside in very poor yield (16%) and 2,4-diazido-2,4,6-trideoxy-β-L-idopyranoside, also in very poor yield (23%). Ito and coworkers later reported an improved synthesis of Pse18 via the L-Alt-diNAc intermediate from the readily available N-acetyl-D-glucosamine starting material. Their synthesis benefited from the initial configurations of the acetamido group at the C-2 position and the hydroxyl group at C-3 of the hexose, which matched those of the desired compound. The reaction sequence in their case was also lengthy (14 steps to L-Alt-diNAc) due to the required 6-deoxygenation, the inversion of configurations at both C-4 and C-5 centers and the introduction of an acetamido group to C-4. More recently Li and coworkers19 established an elegant de novo synthesis of L-Alt-diNAc and Pse from L-threonine: their synthesis included an initial multistep transformation of L-threonine to a suitably protected 2-amino-2,4-dideoxy-L-erythrose that underwent a key aldol condensation step with glycine thioester isonitrile to generate a diastereomeric mixture with the desired L-altro-thioester isolated as a major product. Although Li's synthesis of L-Alt-diNAc was quite efficient, the synthesis included a C–C bond-forming step necessitating subsequent separations of the formed diastereomers. To eliminate C–C bond formation, one other strategy was reported using the readily available 9-carbon N-acetylneuraminic acid (Neu5Ac) as a starting material.20–24 Although attractive, there were complications associated with this strategy, as it generally entailed stereo- and regioselective conversions at several key stereocenters, resulting in low overall yields. Here we wish to report an improved synthesis of L-Alt-diNAc (5) from L-fucose (6).
Results and discussion
The hexose L-fucose (6) was chosen as an ideal starting material because it is conveniently deoxygenated at the C-6 position and has the same L-configuration at C-5 as the target compound, while the configurations at the remaining three centers, C-2, C-3 and C-4, could be selectively inverted. This tactic allowed for the introduction an acetamido group for both C-2 and C-4 positions, which could be done via nucleophilic substitution using a nitrogen-based nucleophile such as azide. Unlike Liav and Sharon's approach, where a β-L-talopyranoside intermediate that simultaneously contained a 2,3-anhydro and 4-O-mesyl group was utilized, our strategy took advantage of the significant difference in reactivity between secondary sulfonyl esters in a polysulfonylated pyranoside during a nucleophilic substitution. Previously, McGeary et al. reported25,26 that nucleophiles such as benzoate or azide could be used to regioselectively displace the 4-tosylate from 2,3,4-tritosylated pyranosides of L-arabinose, D-ribose, D-lyxose and D-xylose. As shown in Scheme 1, our strategy involved using a di-O-mesylated substrate such as the benzyl 2-O-acetyl-3,4-di-O-mesyl-α-L-fucopyranoside (11) as the key substrate for the reaction with sodium azide; this was predicted to afford regioselectively the C-4-monosubstituted product (12), which would then undergo further transformations to form a 2,3-anhydro-4-azido-α-L-allopyranoside (25) that would advantageously invert the configuration at C-3 center. Following the Fürst–Plattner rules, another ring opening of the 2,3-anhydro functionality by an azide nucleophile was expected to regioselectively yield the desired 2,4-diazido-2,4,6-trideoxy-α-L-altropyranoside (26). Obtaining the final compound (5) was devised by a reduction of both azides, followed by an in situ N-acetylation and lastly a deprotection of the anomeric benzyl group.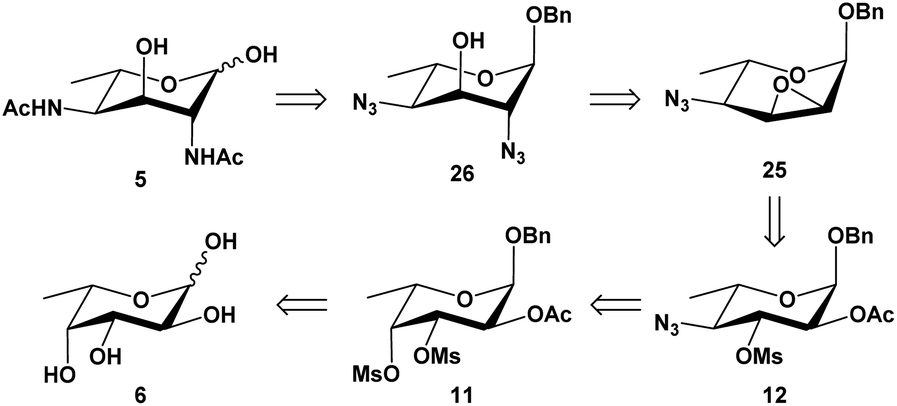 | ||
| Scheme 1 Retrosynthesis of L-Alt-diNAc (5). | ||
Our synthetic route to compound 12 from L-fucose (6) is presented in Scheme 2. The synthesis of the 2-O-acetyl-3,4-O-isopropylidenated intermediate 1027 and its β-anomeric glycoside 1417 had previously been reported, and so we focused on optimizing the experimental procedures to allow the synthesis in a multigram scale. Starting with L-fucose 6 (30 g), a Fischer glycosylation (90 °C, 24 hours) with benzyl alcohol (150 mL, 5 mL g−1 L-fucose) in the presence of HCl (generated in situ by adding acetyl chloride to the benzyl alcohol) gave the desired α-isomer 7 as a majority due to anomeric effect. The benzyl α-fucopyranoside (7) was isolated in almost pure form by a simple precipitation in hexanes (65% yield, contaminated with <5% of β-anomer 8). The pure α-isomer 7 was isolated by a recrystallization in ethyl acetate and hexanes, and its 3,4-cis-diol was regioselectively protected via an isopropylidenation to afford compound 9 in excellent yield (93%). After a simple acetylation, compound 10 was isolated in quantitative yield. Alternatively, the crude α/β-fucopyranoside (7 and 8) mixture obtained from the initial precipitation could be used directly for the 3,4-O-isopropylidenation, followed by an O-2-acetylation and purification by column chromatography, affording 10 in pure form in 34% yield over 3 steps from L-fucose (6). Although the above methods were suitable for preparing the α-anomer 10, it was found that most of the formed β-fucopyranoside precursor 8 was lost during the post-Fisher glycosylation precipitation. In order to obtain the analogous β-anomer 14, we developed a one-pot synthesis from 6 by carrying out a Fisher glycosylation using a much reduced amount of benzyl alcohol (2 mL g−1 L-fucose) and a trace amount of p-toluenesulfonic acid as catalyst. After heating to 130–140 °C with azeotropic removal of water, the reaction mixture was cooled to room temperature; then, instead of carrying out a precipitation, the reaction mixture was directly subjected to an in situ 3,4-O-isopropylidenation with an excess amount of 2,2-dimethoxypropane. The crude reaction mixture was concentrated and acetylated using acetic anhydride and pyridine. After a purification on column chromatography on silica gel, the desired α-glycoside 10 and its β-anomer 14 were isolated in 48% and 20% yield respectively, over three steps from L-fucose. Reducing the amount of benzyl alcohol in our improved protocols thus allowed for a higher-yielding one-pot synthesis of both α/β-fucopyranosides (10 and 14).
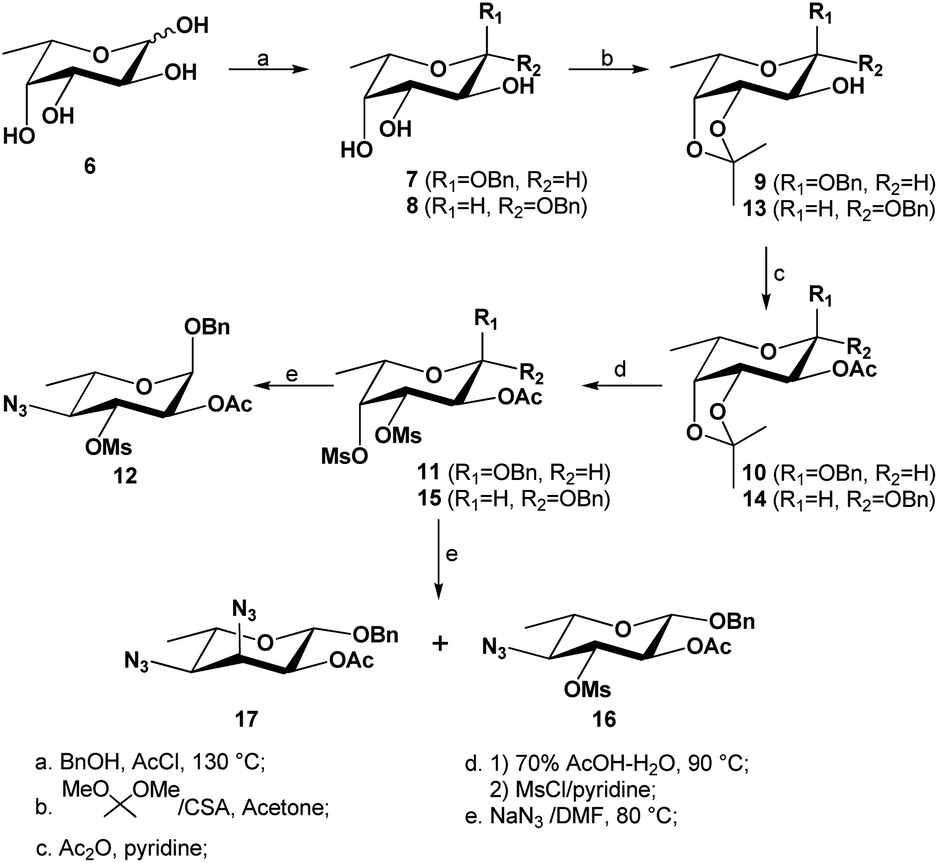 | ||
| Scheme 2 Synthesis of 3,4-di-O-mesylated precursors 11 and 15 and their subsequent substitutions with azide nucleophile. | ||
Deprotection of the isopropylidene from the α-fucopyranoside 10 was achieved in 85% aqueous acetic acid at 80 °C, exposing the two hydroxyl groups at C-3 and C-4 centers (Scheme 2). To avoid possible O-deacetylation at C-2, the hydrolysis was carefully monitored and stopped once the desired product was formed (∼2 hours). The free 3,4-O-diol intermediate was not isolated but subjected to a dimesylation without difficulty, using an excess amount of methanesulfonyl chloride (2.5 equivalents per OH group) in pyridine, giving compound 11 in 90% yield over two steps. The structure of the 3,4-O-dimesylate 11 was confirmed with the appearance of two distinct singlets at 3.12 and 3.19 ppm, integrated to three protons each (2 × Ms's). The β-glycoside 14 was likewise hydrolyzed, and dimesylated afterwards as above, to afford the corresponding 3,4-di-O-mesylated β-fucopyranoside 15.
As shown in Scheme 2, when compound 11 was heated in DMF with an excess amount of sodium azide (∼5.2 equivalents) at 90 °C for 40 hours and subsequently at 100 °C for 4 hours, the monosubstituted product 12 was obtained in almost quantitative yield; despite the use of a large excess of nucleophile, no disubstituted product was isolated. This azide substitution was carried out in a ∼20 gram scale without losing the selectivity nor the productivity (98% yield). The 4-O-mesylate of compound 11 was confirmed to have been selectively displaced by 1H NMR spectrum, because according to 1H–1H COSY spectrum, H-4 was found to shift upfield from 5.04 ppm (compound 11) to 3.37 ppm (compound 12); furthermore, the coupling pattern of H-4 of formed product 12 was observed to be a doublet of doublets with two large coupling constants (9.7, 9.9 Hz), confirming that the C-4 center had an axial hydrogen and equatorial azide. Interestingly, when the analogous 3,4-di-O-mesylate of the β-fucopyranoside (15) was heated to 90 °C for 40 hours, two products were obtained after a purification by column chromatography on silica gel: the major was found to be mono-substituted (60% yield), and the minor was found to be di-substituted (29% yield). The structure of major product was confirmed by 1H NMR spectrum to be the 4-monosubstituted product 16, as one mesylate was still present and determined to attach to C-3 center; furthermore, the H-4 resonance was found to have shifted upfield to ∼3.35 ppm, confirming the regioselective displacement of the O-4-mesylate with an azide. The structure of the minor compound was determined to be the 3,4-diazide 17, as no mesylate signal was observed in the 1H NMR spectrum of the compound, and both H-3 (4.34 ppm) and H-4 (3.23 ppm, J = 3.1, 9.4 Hz) were observed to have shifted upfield.
It was interesting to observe a significant difference in regioselectivity during azide substitutions between the 3,4-di-O-mesylated α/β-fucopyranoside (11 and 15). For the α-fucopyranoside 11, the substitution of the 3-O-mesylate was completely inhibited, while for the β-fucopyranoside 15, the 3-O-mesylate was partially exchanged. This phenomenon was attributed to the dipole–dipole interactions (Fig. 2) explained the Richardson–Hough rules,28 recently updated by Hale et al.29
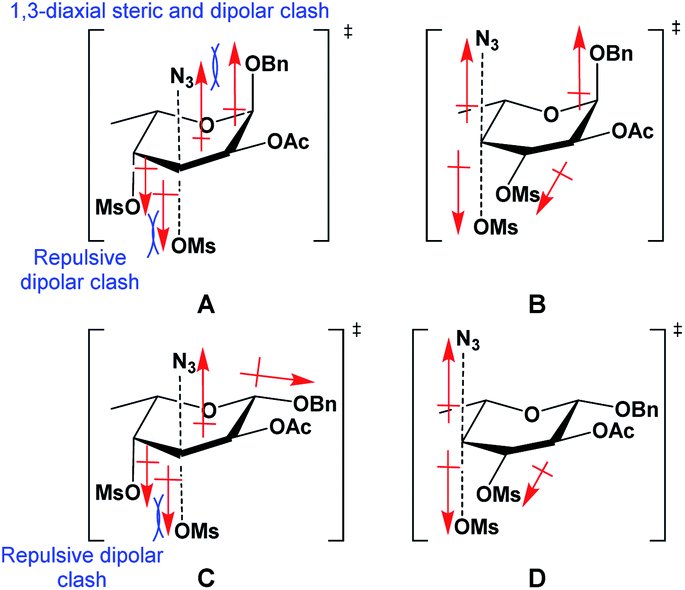 | ||
| Fig. 2 Transition states formed from the SN2 displacement of 3- and 4-mesylates of α-fucopyranoside 11 (structures A and B) and β-fucopyranoside 15 (structures C and D), by an azide. | ||
As depicted in Fig. 2, for α-fucopyranoside 11, the SN2 displacement of mesylates on pyranosides by azide undergoes two possible transition states (TS); the attack on the 3-O-sulfonate (structure A) causes a repulsive alignment between the dipole of the partially bonded departing C-3⋯OMs and the permanent dipole of axial C-4⋯OMs, in addition to a 1,3-diaxial dipole clash between the C-1⋯OBn dipole and the partially bonded incoming C-3⋯N3 dipole, making the substitution of C-3-mesylate difficult. On the other hand, the SN2 substitution of 4-O-mesylate can be illustrated by a TS (structure B) with misaligned dipoles between the C-3 and C-4 mesylates, where no hindering repulsion effect is present, making the SN2 displacement of 4-mesylate favorable. In the case of SN2 attack on the 3-O- and 4-O-mesylates of β-fucopyranoside 15, the unfavorable dipole interactions for the substitutions at both positions are markedly reduced in the TS. The attack on 3-O-mesylate (structure C) is thus made more likely by the elimination of the 1,3-diaxial clash with the incoming C-3⋯N3 dipole, while the bottom-face dipole repulsions remain, and in structure D, there are no dipole alignments whatsoever in the TS: this explains why both compounds 16 and 17 were formed, but in different yields.
To illustrate the influence of size of aglycone on the nucleophilic substitutions at C-3 and C-4 centers, we also prepared the 3,4-dimesylates of both α/β-anomers of methyl L-fucopyranosides 1930 and 22 from L-fucose 6 (Scheme 3). As can be seen, when the same reaction was performed on the methyl 2-O-acetyl-3,4-di-O-mesyl α-L-fucopyranoside 19, the mono-substituted product 20 was isolated in 68% yield; no 3,4-disubstituted product was observed. However, when the same reaction was carried out with the corresponding 3,4-dimesylate 23 of β-L-fucopyranoside, the monosubstituted product 24 was isolated in 24% yield while the 3,4-disubstituted product 25 was isolated in majority (52% yield). These results confirm that both the steric hindrance and strong dipole/dipole repulsions in the TS play a significant role in this type of reaction.
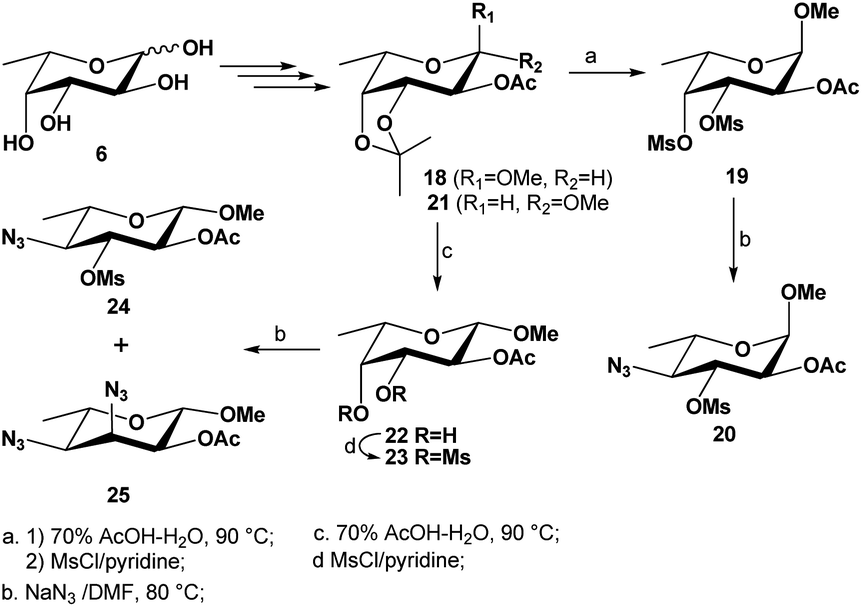 | ||
| Scheme 3 Synthesis of 3,4-di-O-mesylated precursors 19 and 23 and their subsequent substitutions with azide nucleophile. | ||
With large amounts of benzyl 2-O-acetyl-4-azido-4,6-dideoxy-3-O-mesyl-α-L-fucopyranoside 12 at our disposal, a regio- and stereo-selective epoxidation was elegantly performed via transesterification in anhydrous methanol in the presence of sodium methoxide to remove the O-2-acetyl group, followed by an in situ intramolecular ring closure to displace the 3-O-mesylate (Scheme 4): the configuration at the C-3 center was thus inverted. The oxirane formation was found to be much slower than expected at room temperature, which required overnight stirring (∼20 hours) to complete, while heating to 55 °C gave the desired compound 26 in large amounts (>10 g) and in good yields (89%) within a few hours. The inversion was confirmed by 1H NMR spectrum through the change in coupling constants of the H-3 signal (doublet of doublets), from two large J coupling constants to two small J coupling constants (J = 1.6, 4.1 Hz, H-3); furthermore, both H-2 and H-3 signals were observed to exhibit upfield shifts at 3.49 ppm and 3.55 ppm, respectively.
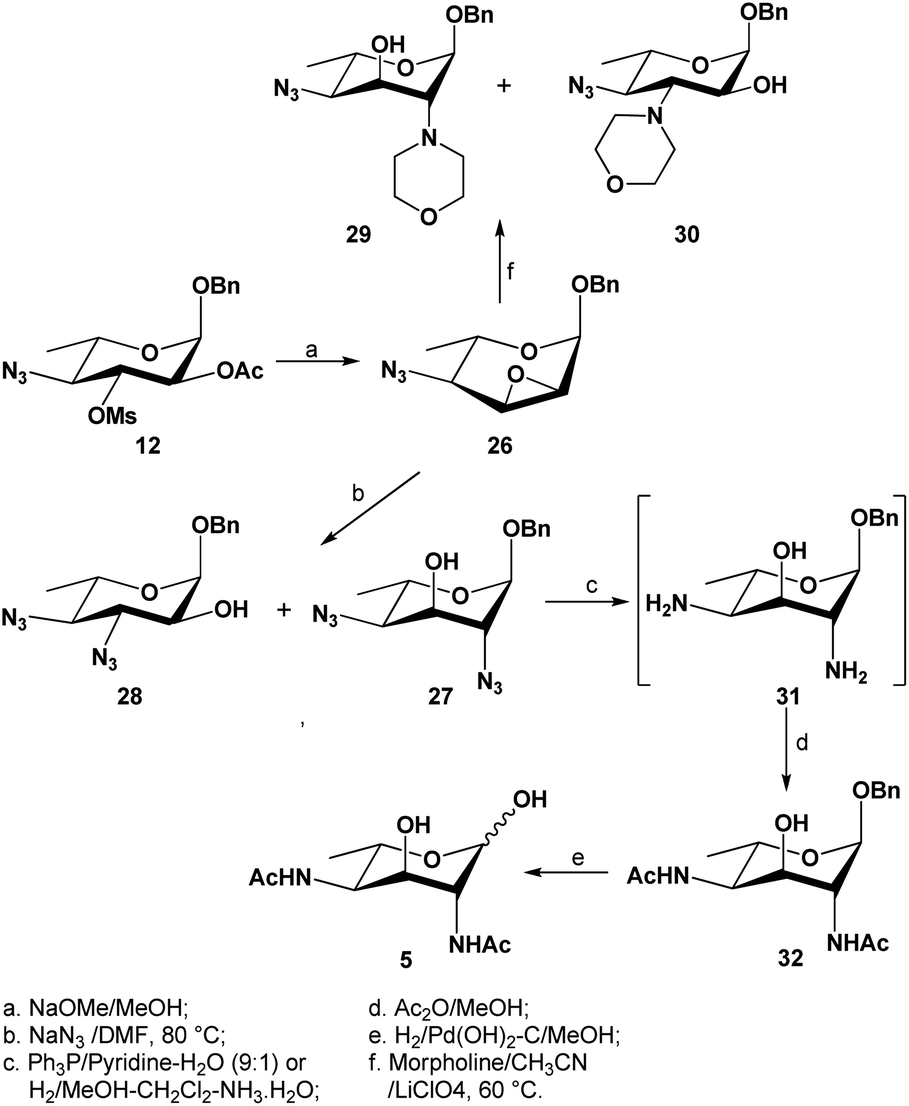 | ||
| Scheme 4 Base-mediated epoxidation of compound 12, subsequent ring opening with nucleophiles and final conversion of 2,4-diazido-α-L-altropyranoside 26 to L-Alt-diAc 5. | ||
Following the successful synthesis of 4-azido-L-allo-epoxide 26, we proceeded to investigate the oxirane ring opening using sodium azide as the nucleophile (Scheme 4). It was anticipated that the desired 2,4-diazido-α-L-altropyranoside (27) would be the major isomer from a trans-diaxial opening following the well-established Fürst–Plattner rules, where the undesired 3,4-diazido-α-L-glucopyranoside (28) would be formed in minority due to its unfavorable boat-like TS (Scheme 5).31 However, when the reaction was performed under typical conditions, with 5 equivalents of sodium azide in anhydrous DMF at 90 °C, both compounds 27 and 28 were formed with very poor selectivity (27/28: 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, entry 1 in Table 1).32 A change of solvent to ethanol did not increase selectivity, despite an anticipated activation by the media engaging in hydrogen-bonding with the oxirane ring (entry 2). These results are in sharp contrast with those obtained with conformationally restricted pyranosyl substrates that bear a fused 4,6-O-benzylidene ring: in those cases, excellent regio and stereoselectivity are normally achieved.33–38 In our case, compound 26 is a non-rigid system that undergoes rapid conformational exchanges between the OH5 and 5HO conformations, especially at higher temperatures, giving the observed results.
:3, entry 1 in Table 1).32 A change of solvent to ethanol did not increase selectivity, despite an anticipated activation by the media engaging in hydrogen-bonding with the oxirane ring (entry 2). These results are in sharp contrast with those obtained with conformationally restricted pyranosyl substrates that bear a fused 4,6-O-benzylidene ring: in those cases, excellent regio and stereoselectivity are normally achieved.33–38 In our case, compound 26 is a non-rigid system that undergoes rapid conformational exchanges between the OH5 and 5HO conformations, especially at higher temperatures, giving the observed results.
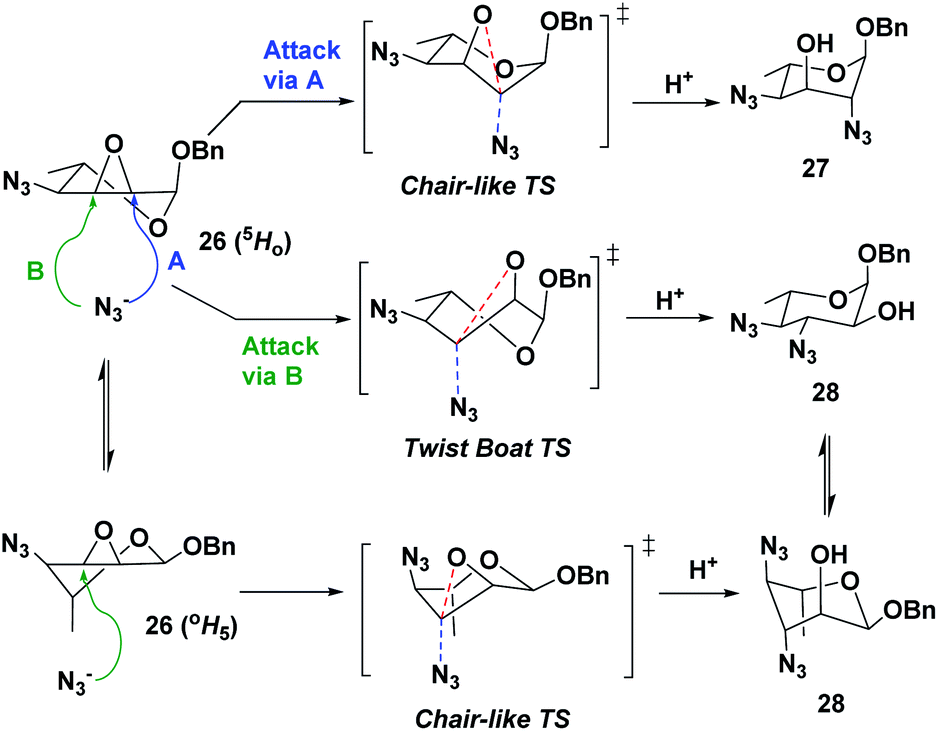 | ||
| Scheme 5 Enhanced flexibility between the 5HO and OH5 conformations of compound 26 results in poor regio- and stereo-selectivity during azide-mediated opening of the epoxide. | ||
| Entry | Amount of 26 | Solvent | NaN3 equivalents | Catalyst | Temp/time | Ratioa 27:28 |
Yielda |
|---|---|---|---|---|---|---|---|
| a Measured by integration in crude 1H NMR spectra, comparing integration of anomeric peaks, unless specified otherwise.b Actual yield % of desired regioisomer (27) from products purified by silica gel column chromatography.c Measured by TLC. | |||||||
| 1 | 2.30 g | DMF | 5 | — | 90 °C, 24 hours | 4:3 |
<50%b |
| 2 | 20 mg | EtOH (95%) | 4 | — | 90 °C, 2 days | 1:1 |
50% |
| 3 | 20 mg | DMF | 4 | NH4Cl (3.5 eq.) | 90 °C, 2 days | 3:2 |
55% |
| 4 | 20 mg | EtOH | 8 | CeCl3 (2 eq.) | 85 °C, 2 days | 4:3 |
25% |
| 5 | 20 mg | MeCN | 2 | CeCl3 (2 eq.) | 85 °C, 2 days | 5:3 |
32% |
| 6 | 20 mg | MeCN | 8 | Mg(ClO4)2 (2 eq.) | 85 °C, 2 days | — | — |
| 7 | 20 mg | MeCN–H2O (9:1) |
8 | Fe(ClO4)3 H2O (2 eq.) | 85 °C, 2 days | — | — |
| 8 | 100 mg | MeCN | 4 | LiCF3SO3 (2 eq.) | 85 °C, 2 days | 5:1 |
70% |
| 9 | 97 mg | MeCN | 5 | LiCF3SO3 (6 eq.) | 80 °C, 2 days | 5:1 |
77%b |
| 10 | 20 mg | MeCN | 4 | LiClO4 (2 eq.) | 90 °C, 2 days | 3:1 |
70% |
| 11 | 100 mg | MeCN | 4 | LiClO4 (2 eq.) | 85 °C, 2 days | 5:1 |
80% |
| 12 | 1.0 g | MeCN | 4 | LiClO4 (3 eq.) | 85 °C, 2 days | 5:1 |
62%b |
| 13 | 1.0 g | MeCN | 4 | LiClO4 (4 eq.) | 85 °C, 2 days | 5:1 |
70%b |
| 14 | 20 mg | EtCN | 4 | LiClO4 (4 eq.) | 100 °C, 1 day | 4:1 |
60% |
| 15 | 20 mg | MeCN | 4 | LiClO4 (2 eq.)/12-crown-4 | 85 °C, 2 days | 4:1 |
75% |
| 16 | 21 mg | MeCN | 4 | LiClO4 (2 eq.)/15-crown-5 | 90 °C, 2 days | 3:1 |
60% |
| 17 | 21 mg | MeCN | 4 | LiClO4 (2 eq.)/TBACl (10 mol%) | 90 °C, 2 days | 5:1 |
80% |
| 18 | 5.53 g | MeCN | 4 | LiClO4 (4 eq.) | 85 °C, 5 days | 5:1c |
62%b |
The lack of the C-6 hydroxy handle that allows for the formation of a rigid bicyclic system prompted us to investigate other approaches to address our challenge. It has been reported that using either a Brønsted39,40 or a Lewis acid41–47 to coordinate with the ring oxygen of the epoxide could improve reactivity and selectivity in the desired ring opening reaction. We believed that a strong coordination with the oxirane would slow down ring flipping, while the added polarization would likely make the molecule preferentially adopt the 5HO conformation, thus cancelling its dipole moments (Fig. 3).
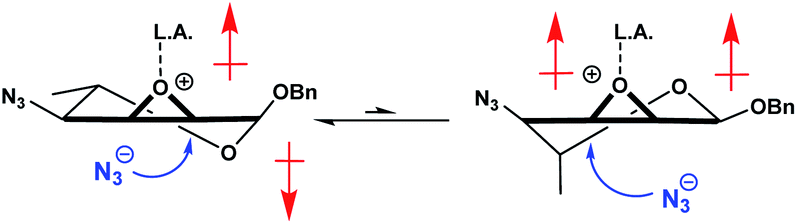 | ||
| Fig. 3 Coordination of the oxirane could lead to reduced conformational exchange, favoring the 5HO conformer due to cancellation of dipole moments. | ||
It has been reported that ammonium chloride39,40 could assist the ring opening of a 2-acetamido-3,4-anhydro-β-D-allopyranoside, providing the trans-diaxially opened product in >70% yield.39 Our attempt to use this catalyst to promote the ring opening of compound 26 in DMF, however resulted in a very slight increase in selectivity (27/28: 3:2, entry 3 in Table 1). Trimethylsilyl azide has been used in the synthesis of oseltamivir,48 and others have successfully used cerium(III) chloride,43 magnesium(II) perchlorate,47 zinc(II) perchlorate,41,42 ferric perchlorate44 and Amberlyst 15 (ref. 49) for the regiochemical control of oxirane openings with amino- or azido-nucleophiles; we have tried some of these metal-catalysed conditions (entries 4–7), with little success. Fortunately, there have also been reports of soluble lithium salts such as lithium perchlorate45,46 that improved the regioselective opening of epoxides in carbohydrates; we therefore investigated the oxirane opening of compound 26 using either lithium triflate (2 or 6 equiv., entries 8–9) or lithium perchlorate (entries 10–14) in either acetonitrile (entries 8–13) or propionitrile (entry 14) at 85–100 °C. To our delight, we observed a significant improvement in selectivity in these trials, with the best ratio of 27/28 to be 5:1 (entries 8–9, 11–13), with a much-improved yield as well (60–80% of the desired altropyranoside). While the change of solvent from acetonitrile to propionitrile improved reaction time, because the reaction in the latter was carried out at a higher temperature, it did not help with regioselectivity. We also found that using either lithium triflate or lithium perchlorate gave similar results. The addition of crown ethers such as 12-crown-4 (entry 15) or 15-crown-5 (entry 16) decreased selectivity, presumably due to a competitive binding of crown ethers to lithium cation that led to a decreased coordination of lithium to the oxirane: these results confirmed the importance of lithium coordination.
In an attempt to improve reaction times, we performed a trial by adding tetra-n-butylammonium chloride (10 mol%) as a phase catalyst (entry 17): the yield was not significantly improved in the 48 hour timeframe, despite an anticipated increase of solubility of the sodium azide. We concluded that the best condition to obtain compound 27 from compound 26 was to use 4 equivalents of sodium azide and 4 equivalents of lithium perchlorate as an additive in anhydrous acetronitrile at 85 °C. Indeed, under such optimized conditions, we were able to carry out a large-scale conversion of compound 26 (5.53 g) to compound 27 in 62% yield. Another oxirane opening of compound 26 was performed with morpholine as a nitrogen-based nucleophile using similar conditions; the reaction afforded the 2-morpholino-substituted α-L-altropyranoside 29 (63% yield) and the 3-morpholino-substituted α-L-glucopyranoside 30 (15% yield). This result was comparable to the azide-mediated opening of 2,3-epoxide, despite the increased reagent solubility.
The structures of the two diazido products 27 and 28 were confirmed through 1H–1H gCOSY and 1H–13C gHSQC NMR correlation experiments as well as through O-acetylation. For compound 27, the 13C signals for the two carbons attached to an azide were observed at 60.8 ppm (C-2) and 61.3 ppm (C-4), while the other carbon with a hydroxyl group attached to was seen at 69.4 ppm (C-3). The shielding effect was also corroborated in the 1H NMR, where signals for H-2 and H-4 moved significantly upfield (3.85 ppm and 3.24 ppm respectively). Similarly, for compound 28, the 13C signals for the two carbons attached to an azide were observed at 65.8 ppm (C-3) and 66.4 ppm (C-4), while the other carbon with a hydroxyl group attached to was observed at 71.9 ppm (C-2). Again, the shielding effect of the azide groups was seen in the 1H NMR with H-3 and H-4 shifted upfield (3.58 ppm and 2.91 ppm respectively).
Following the success of synthesizing 2,4-diazido-α-L-altropyranoside 27, the synthesis of L-2,4-Alt-diNAc 5 (Scheme 4) was continued by first reducing the 2,4-diazido functionalities in compound 27. Previously, this transformation had been achieved using lithium aluminium hydride as a reducing reagent, followed by a peracetylation17 to afford the corresponding fully acetylated product in very poor yields (38%). We thus investigated two alternative methods to achieve the conversion. First, a Staudinger reduction as a mild and chemoselective method to reduce the two azides was attempted: using triphenylphosphine as a reagent in a mixture of pyridine–water (9:1), the two azide functionalities were smoothly reduced at room temperature after 48 hours, and after concentrating the mixture, the crude 2,4-diamine (31) was chemoselectively N-acetylated in methanol to give the 2,4-di-N-acetylated compound 32 in 59% yield over two steps. The second method involved a controlled hydrogenolysis, where a few drops of ammonia were added as a mild poison against the palladium on charcoal catalyst to reduce its reactivity towards the O-benzyl group,50 thus selectively reducing the two azide functionalities. Indeed, when compound 27 was subjected to the latter conditions in a mixture of methanol-dichloromethane, the desired selectivity was achieved to afford crude diamine 31 which was further N-acetylated as above. This last method afforded the desired compound 32 in much better yield (81%). The formation of 32 was confirmed with the obvious appearance of the two doublets at 6.01 and 6.13 ppm, as well as the presence of two overlapping singlets at 2.02 ppm, attributed to the two NHAc protons. Finally, the anomeric benzyl group was removed using standard catalytic hydrogenolysis conditions to afford the targeted L-2,4-Alt-diNAc 5 in quantitative yield. 1H NMR spectrum of compound 5 showed the presence of both α- and β-pyranosyl forms. The electrospray high resolution mass spectrometry also confirmed the identify of L-2,4-Alt-diNAc 5 (calc'd m/z for C12H21N2O6 [M + H]+: 289.1396; found 289.1394).
The successful selective reduction of compound 27 encouraged us to study a complete catalytic hydrogenation of compound 27 in one step, including the O-benzyl functionality (Scheme 6). We predicted that the uncontrolled reduction would produce the reactive intermediate 33 that would then self-condense into a 1,4-iminofuranosyl ring (→34), further undergoing a reduction to afford the stable iminofuranoside 35. This was indeed found to be the case when compound 27 was subjected to a palladium-catalyzed hydrogenolysis in methanol in the presence of a few drops of acetic acid. A new polar product, presumably compound 35 was thus formed and after a full acetylation, the iminofuranoside 36 was isolated in 60% yield.
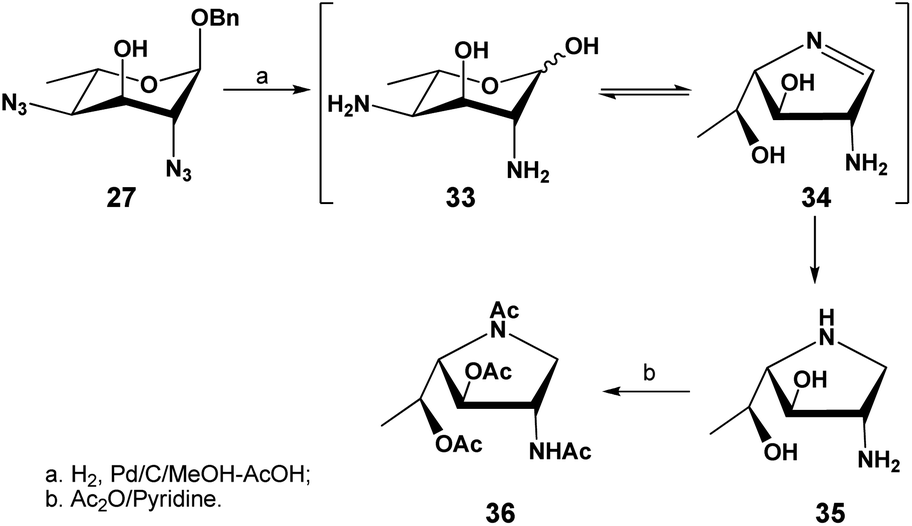 | ||
| Scheme 6 Conversion of compound 27 to the iminofuranosides 35 and 36 via a complete hydrogenation. | ||
In the 1H NMR spectrum of the isolated compound, a highly deshielded proton was observed at 6.70 ppm as a doublet, which was assigned to be the single amide proton. 4 singlets were observed at 2.12, 2.11, 2.08 and 2.03 ppm, corresponding to the 4 acetyl groups present in the molecule. Two highly deshielded protons were observed at 5.45 and 5.35 ppm, attributed to the H-5 and H-3 respectively and confirmed by 1H–1H gCOSY experiment; their large chemical shifts are consistent with an O-acetylation on O-5 and O-3, further supporting that O-5 is not involved in cyclization. Two mutually coupled protons (high orders) were observed at 4.24 ppm and 3.19 ppm, attributed to the two diastereotopic protons attached to C-1. These assignments were also confirmed by a 1H–13C 2D gHSQC correlation spectrum. The C-1 carbon was observed at a highly shielded region (52.9 ppm), confirming the attachment of C-1 to a less electronegative nitrogen.
Conclusions
In conclusion, we have developed a convenient and readily scalable synthesis toward the challenging L-2,4-Alt-diNAc (5) from L-fucose (6) in 10 steps (23% overall yields). Our synthesis has overcome some major bottlenecks in previously reported syntheses.17 Because of the shorter reaction sequence and much improved transformation in some key steps, our synthesis allows the preparation of key intermediates such as compounds 12, 26, 27 in gram quantities. By further combining with the indium-mediated allylation with ethyl 2-(bromomethyl)acrylate, already attempted by several other groups working in this area, Pse and analogs could be prepared in a straightforward manner.Experimental section
General methods
All commercial reagents were used as supplied unless otherwise stated. Thin layer chromatography was performed on Silica Gel 60-F254 (E. Merck, Darmstadt) with detection by fluorescence, charring with 5% H2SO4 (aq), or a ceric ammonium molybdate solution. Column chromatography was performed on Silica Gel 60 (Silicycle, Ontario) and solvent gradients given refer to stepped gradients and concentrations are reported as % v/v. Organic solutions were concentrated and/or evaporated to dry under vacuum in a water bath (<60 °C). Optical rotations were determined in a 5 cm cell at 20 ± 2 °C; [α]20D values are given in units of 10−1 deg cm2 g−1. NMR spectra were recorded on Bruker spectrometers at 400 MHz, and the first-order chemical shifts of 1H and 13C (DEPT-Q) are reported in δ (ppm) and referenced to residual CHCl3 (δH 7.24, δC 77.23, CDCl3), or residual HDO (δH 4.79, external acetone δC 29.9, D2O), residual CD2HOD (δH 3.31, δC 49.1, CD3OD) 1H and 13C NMR spectra were assigned with the assistance of 2D gCOSY and 2D gHSQC experiments. High-resolution ESI-QTOF mass spectra were recorded on an Agilent 6520 Accurate Mass Quadrupole Time-of-Flight LC/MS spectrometer. All the data were obtained with the assistance of the analytical services of the Department of Chemistry, University of Calgary.Method 1. To a mixture of benzyl α-L-fucopyranoside 7 (5.27 g, 20.7 mmol) in acetone (30 mL), was added 2,2-dimethoxypropane (10.0 mL), and 10-camphorsulfonic acid (CSA, 120 mg), and the mixture was heated to reflux for 0.5 h. The mixture was then neutralized with triethylamine (∼2 mL) and concentrated under reduced pressure to afford a yellow oil. The mixture was dissolved in a 10
:1 mixture of methanol and water (220 mL) and heated to reflux for one hour, and concentrated under reduced pressure. The mixture was dissolved in ethyl acetate (50 mL), and the obtained organic solution was washed successively with brine (50 mL), 2 N HCl (50 mL) and saturated aqueous NaHCO3 (50 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residue was purified by column chromatography on silica gel using a gradient of 5 → 10% ethyl acetate in hexanes as an eluent, to yield the desired compound (9) as a colourless syrup (5.70 g, 93% yield). 1H NMR (400 MHz, CDCl3): δH 7.41–7.30 (m, 5H, ArH), 4.95 (d, 1H, J = 3.9 Hz, H-1), 4.80 (d, 1H, J = 11.8 Hz, CHaHbPh), 4.59 (d, 1H, J = 11.8 Hz, CHaHbPh), 4.24 (t, 1H, J = 6.2 Hz, H-3), 4.17 (dq, 1H, J = 6.6 Hz, J = 2.3 Hz, H-5), 4.07 (dd, 1H, J = 6.1 Hz, J = 2.3 Hz, H-4), 3.84 (dt, 1H, J = 6.7 Hz, J = 4.0 Hz, H-2), 2.28 (d, 1H, J = 6.9 Hz, OH), 1.53 (s, 3H, Me_ISP), 1.37 (s, 3H, Me_ISP), 1.32 (d, 3H, J = 6.6 Hz, H-6). 13C NMR data are in agreement with previously reported.27
Method 2. To a mixture of the crude precipitate that contained the benzyl α/β-L-fucopyranosides (7/8, 18.2 g, 62.6 mmol) in acetone (50.0 mL) and 2,2-dimethoxypropane (50 mL), was added CSA (200 mg), and the solution was heated to reflux for 24 hours. The mixture was then neutralized with triethylamine, and solution was concentrated under reduced pressure. The yellow syrup was purified by column chromatography on silica gel using a gradient of 5 → 10% ethyl acetate–hexanes as an eluent to yield the pure β-fucopyranoside 13 (3.89 g, 20% yield) and also the α-fucopyranoside 9 (7.17 g, 37% yield), as colourless syrups. Data for compound 13: Rf 0.39 (40% ethyl acetate–hexanes). 1H NMR (400 MHz, CDCl3): δH 7.41–7.23 (m, 5H, ArH), 4.93 (d, J = 11.6 Hz, 1H, CHaHbPh), 4.58 (d, J = 11.7 Hz, 1H, CHaHbPh), 4.22 (d, J = 8.3 Hz, 1H, H-1), 4.01 (dd, J = 7.2, 5.5 Hz, 1H, H-3), 3.97 (dd, J = 5.5, 2.2 Hz, 1H, H-4), 3.82 (qd, J = 6.6, 2.2 Hz, 1H, H-5), 3.58 (ddd, J = 8.2, 7.2, 2.5 Hz, 1H, H-2), 2.80 (d, J = 2.6 Hz, 1H, OH-2), 1.53 (s, 3H, CH3_ISP), 1.44 (d, J = 6.6 Hz, 3H, H-6), 1.35 (s, 3H, CH3_ISP). 13C NMR data are in agreement with previously reported.
General method. A solution of benzyl 2,3-anhydro-4-azido-4,6-dideoxy-α-L-allopyranoside (26), sodium azide and catalyst in the specified solvent (Table 1) was heated to the specified temperature for the specified amount for time. The solvent was then evaporated under reduced pressure. The residue was extracted into ethyl acetate, and the organic solution was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. 1H NMR experiment was used to directly determine the conversion rates and product ratios (27/28). For some experiments, the obtained residue was purified by column chromatography on silica gel using 2.5% ethyl acetate–hexanes as the eluent to yield the desired compounds 27 and 28 respectively. Data for compound 27: Rf 0.43 (20% ethyl acetate–hexanes, twice). [α]25D −116.1 (c 0.43, CHCl3). 1H NMR (400 MHz, CDCl3): δH 7.45–7.31 (m, 5H, ArH), 4.89 (s, 1H, H-1), 4.78 (d, 1H, J = 11.7 Hz, CHaHbPh), 4.59 (d, 1H, J = 11.8 Hz, CHaHbPh), 4.13–4.02 (m, 2H, H-3 and H-5), 3.85 (dd, 1H, J = 4.0 Hz, 1.9 Hz, H-2), 3.45 (d, 1H, J = 9.8 Hz, OH), 3.23 (dd, 1H, J = 9.8, 3.1 Hz, H-4), 1.38 (d, 3H, J = 6.3 Hz, H-6). 13C NMR (101 MHz, CDCl3): δC 135.8 (ArC), 128.7 (ArC), 128.5 (ArC), 128.3 (ArC), 96.9 (C-1), 70.3 (CHaHbPh), 69.4 (C-3), 63.1 (C-5), 61.3 (C-4), 60.8 (C-2), 18.1 (C-6). HRMS (ESI-TOF) calc'd m/z for C13H16N6O3 [M + NH4]+: 322.1626; found 322.1622. Data for compound 28: Rf 0.37 (20% ethyl acetate–hexanes, twice). [α]25D −330.0 (c 1.7, CHCl3). 1H NMR (400 MHz, CDCl3): δH 7.43–7.32 (m, 5H, ArH), 4.93 (d, J = 3.1 Hz, 1H, H-1), 4.75 (d, J = 11.7 Hz,1H, CHaHbPh), 4.55 (d, J = 11.7 Hz, 1H, CHaHbPh), 3.69–3.57 (m, 3H, H-2, H-4, and H-5), 2.92 (high order t, J = 9.9, 9.9 Hz, 1H, H-3) 2.17 (high order d, J = 10.4 Hz, 1H, OH-2), 1.30 (d, J = 6.2 Hz, 3H, H-6). 13C NMR (101 MHz, CDCl3): δC 128.7 (ArC), 128.4 (ArC), 128.2 (ArC), 96.8 (C-1), 71.9 (C-2), 70.1 (CHaHbPh), 66.6 (C-5), 66.4 (C-3), 65.8 (C-4), 18.2 (C-6). HRMS (ESI-QTOF) calc'd m/z for C13H16N6O3 [M + NH4]+: 322.1626; found 322.1617.
Method 1. To a solution of 2,4-diazido compound 27 (80 mg, 0.26 mmol) in a 9
:1 mixture of pyridine–H2O (2.0 mL) was added triphenylphosphine (275 mg, 1.05 mmol, 4 equiv.), and the mixture was stirred at ambient temperature for 48 hours. The solution was concentrated under reduced pressure. The residue (31) was redissolved in methanol (2.0 mL), and acetic anhydride (250 μL) was added. After stirring for 4 hours, the solution was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using a gradient of 30 → 40% acetone–toluene as the eluent to afford the desired Alt-2,4-DiNHAc compound 32 (51.7 mg, 59% yield).
Method 2. To a solution of 2,4-diazido compound 27 (206 mg, 0.68 mmol) in dichloromethane (5.0 mL) and methanol (5.0 mL), was added concentrated ammonia (30%, 4 drops), and palladium on charcoal (5%, 28 mg) was added; the mixture was purged with hydrogen and stirred continuously under an atmosphere of hydrogen for 5 hours. The reaction solution was filtered over a 0.22 μM membrane disk, and the solution was concentrated under reduced pressure. The residue was redissolved in methanol (5.0 mL), and acetic anhydride (0.5 mL) was added. After stirring for 2 hours, the reaction solution was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel as above to afford compound 32 in pure form (184 mg, 81% yield). Rf 0.55 (10% MeOH–CH2Cl2). [α]25D −74.8 (c 0.7, CHCl3). 1H NMR (400 MHz, CDCl3): δH 7.41–7.26 (m, 5H, Ph), 6.45 (d, J = 8.8 Hz, 1H, NHAc-C2), 6.25 (d, J = 9.2 Hz, 1H, NHAc-C4), 4.79 (br s, 1H, H-1), 4.69 (d, J = 11.8 Hz, 1H, CHaHbPh), 4.52 (d, J = 11.8 Hz, 1H, CHaHbPh), 4.42 (dd, J = 8.8, 2.8 Hz, 1H, H-2), 4.03 (ddd, J = 10.1, 10.1, 2.9 Hz, 1H, H-4), 3.89–3.78 (m, 2H, H-5 + OH-3), 3.73 (m, 1H, H-3), 1.98 (s, 6H, 2 × Ac), 1.24 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (101 MHz, CDCl3): δC 170.0 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 169.7 (CO), 136.1 (Ph), 128.7 (Ph), 128.3 (Ph), 128.1 (Ph), 98.3 (C-1), 69.9 (CHaHbPh), 69.1 (C-3), 63.9 (C-5), 50.1 (C-2), 49.7 (C-4), 23.4 (Ac), 23.2 (Ac), 17.8 (C-6). HRMS (ESI-QTOF) calc'd m/z for C17H25N2O5 [M + H]+: 337.1751; found 337.1758.
:1 mixture of CH2Cl2–MeOH (5.0 mL) was added ten drops of water and acetic acid (12 μL, 0.20 mmol) and Pd/C on charcoal (42 mg), and the flask was purged with an atmosphere of hydrogen, and kept under a positive hydrogen pressure with stirring for 48 hours. The solids were filtered off using a 0.22 mM membrane filter, and the solution was concentrated under reduced pressure. The crude syrup was acetylated using a mixture of acetic anhydride (1.0 mL) and pyridine (1.0 mL). After stirring overnight, the reaction solution was concentrated and co-evaporated with toluene (3 × 5 mL). The crude residue was purified by column chromatography on silica gel using 2% methanol–dichloromethane to afford compound 27 as a syrupy product (19 mg, 60% yield). Rf 0.55 (10% MeOH–CH2Cl2). [α]25D −9.85 (c 1.3, MeOH). 1H NMR (400 MHz, CDCl3): δH 6.70 (d, J = 4.7 Hz, 1H, NHAc), 5.45 (qd, J = 6.6, 4.1 Hz, 1H, H-5), 5.35 (dd, J = 4.7, 3.9 Hz, 1H, H-3), 4.30–4.23 (m, 2H, H-1a + H-2), 4.22 (dd, J = 4.1, 4.1 Hz, H-4), 3.19 (high order dd, J = 10.3, 14.2 Hz, 1H, H-1b), 2.12 (s, 3H, Ac), 2.11 (s, 3H, Ac), 2.08 (s, 3H, Ac), 2.03 (s, 3H, Ac), 1.28 (d, J = 6.6 Hz, 3H, H-6). 13C NMR (101 MHz, CDCl3): δC 171.0 (Ac), 170.5 (Ac), 170.0 (Ac), 169.7 (Ac), 75.9 (C-3), 69.4 (C-5), 63.7 (C-4), 55.0 (C-2), 52.9 (C-1), 23.1 (Ac), 22.3 (Ac), 21.4 (Ac), 20.9 (Ac), 17.0 (C-6). HRMS (ESI-QTOF) calc'd m/z for C14H22N2O6 [M + Na]+: 337.1376; found 337.1365.
O), 169.7 (CO), 136.1 (Ph), 128.7 (Ph), 128.3 (Ph), 128.1 (Ph), 98.3 (C-1), 69.9 (CHaHbPh), 69.1 (C-3), 63.9 (C-5), 50.1 (C-2), 49.7 (C-4), 23.4 (Ac), 23.2 (Ac), 17.8 (C-6). HRMS (ESI-QTOF) calc'd m/z for C17H25N2O5 [M + H]+: 337.1751; found 337.1758.
:1 mixture of CH2Cl2–MeOH (5.0 mL) was added ten drops of water and acetic acid (12 μL, 0.20 mmol) and Pd/C on charcoal (42 mg), and the flask was purged with an atmosphere of hydrogen, and kept under a positive hydrogen pressure with stirring for 48 hours. The solids were filtered off using a 0.22 mM membrane filter, and the solution was concentrated under reduced pressure. The crude syrup was acetylated using a mixture of acetic anhydride (1.0 mL) and pyridine (1.0 mL). After stirring overnight, the reaction solution was concentrated and co-evaporated with toluene (3 × 5 mL). The crude residue was purified by column chromatography on silica gel using 2% methanol–dichloromethane to afford compound 27 as a syrupy product (19 mg, 60% yield). Rf 0.55 (10% MeOH–CH2Cl2). [α]25D −9.85 (c 1.3, MeOH). 1H NMR (400 MHz, CDCl3): δH 6.70 (d, J = 4.7 Hz, 1H, NHAc), 5.45 (qd, J = 6.6, 4.1 Hz, 1H, H-5), 5.35 (dd, J = 4.7, 3.9 Hz, 1H, H-3), 4.30–4.23 (m, 2H, H-1a + H-2), 4.22 (dd, J = 4.1, 4.1 Hz, H-4), 3.19 (high order dd, J = 10.3, 14.2 Hz, 1H, H-1b), 2.12 (s, 3H, Ac), 2.11 (s, 3H, Ac), 2.08 (s, 3H, Ac), 2.03 (s, 3H, Ac), 1.28 (d, J = 6.6 Hz, 3H, H-6). 13C NMR (101 MHz, CDCl3): δC 171.0 (Ac), 170.5 (Ac), 170.0 (Ac), 169.7 (Ac), 75.9 (C-3), 69.4 (C-5), 63.7 (C-4), 55.0 (C-2), 52.9 (C-1), 23.1 (Ac), 22.3 (Ac), 21.4 (Ac), 20.9 (Ac), 17.0 (C-6). HRMS (ESI-QTOF) calc'd m/z for C14H22N2O6 [M + Na]+: 337.1376; found 337.1365.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada as well as the Canadian Foundation for Innovation and the Government of Alberta for their support of this project. C. S. thanks the University of Calgary for an Eyes High graduate scholarship.Notes and references
- K. U. Jansen, C. Knirsch and A. S. Anderson, Nat. Med., 2018, 24, 10–19 CrossRef CAS PubMed.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed.
- D. A. Kennedy and A. F. Read, Proc. R. Soc. B, 2017, 284, 20162562 CrossRef PubMed.
- M. Zunk and M. J. Kiefel, RSC Adv., 2014, 4, 3413–3421 RSC.
- S. M. Logan, Microbiology, 2006, 152, 1249–1262 CrossRef CAS PubMed.
- S. L. Howard, A. Jagannathan, E. C. Soo, J. P. M. Hui, A. J. Aubry, I. Ahmed, A. Karlyshev, J. F. Kelly, M. A. Jones, M. P. Stevens, S. M. Logan and B. W. Wren, Infect. Immun., 2009, 77, 2544–2556 CrossRef CAS PubMed.
- J. Horzempa, C. R. Dean, J. B. Goldberg and P. Castric, J. Bacteriol., 2006, 188, 4244–4252 CrossRef CAS PubMed.
- S. R. Haseley and S. G. Wilkinson, Eur. J. Biochem., 1997, 250, 617–623 CrossRef CAS PubMed.
- P. Guerry, C. P. Ewing, M. Schirm, M. Lorenzo, J. Kelly, D. Pattarini, G. Majam, P. Thibault and S. Logan, Mol. Microbiol., 2006, 60, 299–311 CrossRef CAS PubMed.
- Y. A. Knirel, A. S. Shashkov, Y. E. Tsvetkov, P. E. Jansson and U. Zähringer, Adv. Carbohydr. Chem. Biochem., 2003, 58, 371–417 CrossRef CAS.
- S. Goon, J. F. Kelly, S. M. Logan, C. P. Ewing and P. Guerry, Mol. Microbiol., 2003, 50, 659–671 CrossRef CAS PubMed.
- W. K. Chou, S. Dick, W. W. Wakarchuk and M. E. Tanner, J. Biol. Chem., 2005, 280, 35922–35928 CrossRef CAS PubMed.
- R. Ménard, I. C. Schoenhofen, L. Tao, A. Aubry, P. Bouchard, C. W. Reid, P. Lachance, S. M. Twine, K. M. Fulton, Q. Cui, H. Hogues, E. O. Purisima, T. Sulea and S. M. Logan, Antimicrob. Agents Chemother., 2014, 58, 7430–7440 CrossRef PubMed.
- K. Pradhan and S. S. Kulkarni, Eur. J. Org. Chem., 2020, 2020, 6819–6830 CrossRef CAS.
- Y. E. Tsvetkov, A. S. Shashkov, Y. A. Knirel and U. Zähringer, Carbohydr. Res., 2001, 335, 221–243 CrossRef CAS PubMed.
- N. W. See, N. Wimmer, E. H. Krenske and V. Ferro, Eur. J. Org. Chem., 2021, 2021, 1575–1584 CrossRef CAS.
- A. Liav and N. Sharon, Carbohydr. Res., 1973, 30, 109–126 CrossRef CAS PubMed.
- Y. J. Lee, A. Kubota, A. Ishiwata and Y. Ito, Tetrahedron Lett., 2011, 52, 418–421 CrossRef CAS.
- H. Liu, Y. Zhang, R. Wei, G. Andolina and X. Li, J. Am. Chem. Soc., 2017, 139, 13420–13428 CrossRef CAS PubMed.
- M. Zunk, J. Williams, J. Carter and M. J. Kiefel, Org. Biomol. Chem., 2014, 12, 2918–2925 RSC.
- J. T. Williams, L. Corcilius, M. J. Kiefel and R. J. Payne, J. Org. Chem., 2016, 81, 2607–2611 CrossRef CAS PubMed.
- B. Dhakal, S. Buda and D. Crich, J. Org. Chem., 2016, 81, 10617–10630 CrossRef CAS PubMed.
- O. Popik, B. Dhakal and D. Crich, J. Org. Chem., 2017, 82, 6142–6152 CrossRef CAS PubMed.
- B. Dhakal and D. Crich, J. Am. Chem. Soc., 2018, 140, 15008–15015 CrossRef CAS PubMed.
- R. P. McGeary, S. Rasoul Amini, V. W. S. Tang and I. Toth, J. Org. Chem., 2004, 69, 2727–2730 CrossRef CAS PubMed.
- R. P. McGeary, K. Wright and I. Toth, J. Org. Chem., 2001, 66, 5102–5105 CrossRef CAS PubMed.
- H. M. Flowers, Carbohydr. Res., 1979, 74, 177–185 CrossRef CAS.
- L. Hough and A. C. Richardson, in Rodd's Chemistry of Carbon Compounds, ed. S. Coffey, Elsevier, Amsterdam, 2nd edn, 1964, pp. 67–595 Search PubMed.
- K. J. Hale, L. Hough, S. Manaviazar and A. Calabrese, Org. Lett., 2014, 16, 4838–4841 CrossRef CAS PubMed.
- U. Zehavi and N. Sharon, J. Org. Chem., 1972, 37, 2141–2145 CrossRef CAS PubMed.
- M. Miljković, in Carbohydrates: Synthesis, Mechanisms, and Stereoelectronic Effects, ed. M. Miljkovic, Springer, New York, NY, 2009, pp. 221–244 Search PubMed.
- D.-Q. Yuan, T. Tahara, W.-H. Chen, Y. Okabe, C. Yang, Y. Yagi, Y. Nogami, M. Fukudome and K. Fujita, J. Org. Chem., 2003, 68, 9456–9466 CrossRef CAS PubMed.
- M. T. C. Walvoort, G.-J. Moggré, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Org. Chem., 2011, 76, 7301–7315 CrossRef CAS PubMed.
- A. Medgyes, E. Farkas, A. Lipták and V. Pozsgay, Tetrahedron, 1997, 53, 4159–4178 CrossRef CAS.
- R. Hevey, A. Morland and C.-C. Ling, J. Org. Chem., 2012, 77, 6760–6772 CrossRef CAS PubMed.
- R. Hevey and C.-C. Ling, Org. Biomol. Chem., 2013, 11, 1887–1895 RSC.
- P. Zhang, R. Hevey and C.-C. Ling, J. Org. Chem., 2017, 82, 9662–9674 CrossRef CAS PubMed.
- R. Hevey and C.-C. Ling, Carbohydr. Res., 2017, 445, 65–74 CrossRef CAS PubMed.
- Y. Cai, C.-C. Ling and D. R. Bundle, J. Org. Chem., 2009, 74, 580–589 CrossRef CAS PubMed.
- H. Okazaki, K. Hanaya, M. Shoji, N. Hada and T. Sugai, Tetrahedron, 2013, 69, 7931–7935 CrossRef CAS.
- B. P. Shivani and A. K. Chakraborti, J. Org. Chem., 2007, 72, 3713–3722 CrossRef CAS PubMed.
- B. P. Shivani and A. K. Chakraborti, J. Mol. Catal. A: Chem., 2007, 263, 137–142 CrossRef.
- G. Sabitha, R. S. Babu, M. Rajkumar and J. S. Yadav, Org. Lett., 2002, 4, 343–345 CrossRef CAS PubMed.
- P. Salehi, B. Seddighi, M. Irandoost and F. K. Behbahani, Synth. Commun., 2000, 30, 2967–2973 CrossRef CAS.
- T. Nemcsok, Z. Rapi, P. Bagi and P. Bakó, Synth. Commun., 2019, 49, 2388–2400 CrossRef CAS.
- J. M. Vega-Pérez, J. I. Candela, M. Vega and F. Iglesias-Guerra, Carbohydr. Res., 1995, 279, C5–C8 CrossRef.
- M. Chini, P. Crotti and F. Macchia, Tetrahedron Lett., 1990, 31, 5641–5644 CrossRef CAS.
- Y. Fukuta, T. Mita, N. Fukuda, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 6312–6313 CrossRef CAS PubMed.
- V. P. K. Kondapi, O.-M. Soueidan, S. N. Hosseini, N. Jabari and F. G. West, Eur. J. Org. Chem., 2016, 2016, 1367–1379 CrossRef.
- H. Sajiki, Tetrahedron Lett., 1995, 36, 3465–3468 CrossRef CAS.
- H.-Y. L. Wang and G. A. O'Doherty, Chem. Commun., 2011, 47, 10251 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01070k |
| This journal is © The Royal Society of Chemistry 2021 |