DOI:
10.1039/D1RA01037A
(Paper)
RSC Adv., 2021,
11, 11595-11605
External electric field: a new catalytic strategy for the anti-Markovnikov hydrohydrazination of parent hydrazine†
Received
7th February 2021
, Accepted 3rd March 2021
First published on 22nd March 2021
Abstract
The anti-Markovnikov hydroamination reaction is considered to be a particular challenge, and one of the reactants, parent hydrazine, is also regarded as a troubling reagent. In this study, we first studied the hydrohydrazination of parent hydrazine via an effective and green catalyst—external electric field (EEF). The calculation results demonstrated that the anti-Markovnikov and Markovnikov pathways are competitive when there was no catalyst. EEF oriented along the negative direction of the X axis (Fx) accelerated the anti-Markovnikov addition reaction. Moreover, it lowered the barrier height of the first step by 16.0 kcal mol−1 (from 27.8 to 11.8 kcal mol−1) when the field strength was 180 (×10−4) au. Under the same conditions, the Markovnikov reaction pathway was inhibited, which means that EEF achieved the specificity of hydrohydrazination. The solvents are favorable for the first step addition reaction, particularly the synergy between solvents and Fx lowered the barrier heights by 8.3 (C6H6) and 10.7 (DMSO) kcal mol−1 for an Fx = −60 (×10−4) au. Besides, the introduction of the electron-withdrawing substituent (trifluoromethyl) is also a good strategy to catalyze hydrohydrazination, while the electron-donating group (methoxy) is unfavorable.
Introduction
Nitrogen-containing compounds constitute a useful family of natural products,1–3 such as agrochemicals, cosmetics, and pharmaceuticals, and are also important intermediates in numerous industrial processes.4 The inspection of the known synthesis methods, such as hydroamination reaction, which is the addition of the N–H bond across an unsaturated C–C bond, is a highly desirable atom-efficient strategy starting from readily accessible materials.5–14 Therein, the use of hydrazine as a donor of the N–H bond7,15–18 is quite appealing to generate potentially useful products.19,20 In 2002, Odom et al. reported the first example of the intermolecular hydrohydrazination of 1,1-disubstituted hydrazine to alkynes catalyzed by titanium.21 Moreover, increasing number of investigation on the hydrohydrazination of hydrazine were carried out using different metal catalysts.9–11,21–34 Among these hydrohydrazination reactions, mono- and di-substituted hydrazines are widely employed. However, the parent hydrazine (H2NNH2) is used rarely despite the potential utility of such a reaction. The possible reasons for hampering the usage of parent hydrazine are as follows: first, hydrazine readily forms Werner complexes, which are usually inert.35,36 Second, H2NNH2 is known as a strong reducing reagent,37,38 which can induce the formation of inactive metal (0) particles.39 Lastly, the resulting products can undergo a metal-mediated N–N bond cleavage to form undesired by-products.40–42 In 2010, the disclosed palladium-catalyzed cross-coupling of aryl chlorides and tosylates with hydrazine29 is the only example of the transition-metal-catalyzed functionalization of H2NNH2. A few years later, gold-catalyzed hydrohydrazination of alkynes with parent hydrazine was reported.30–33
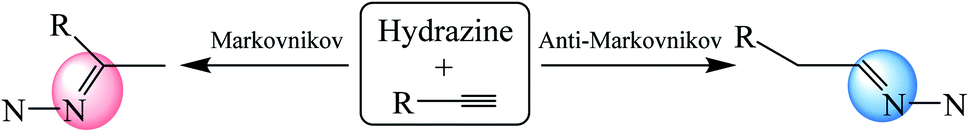
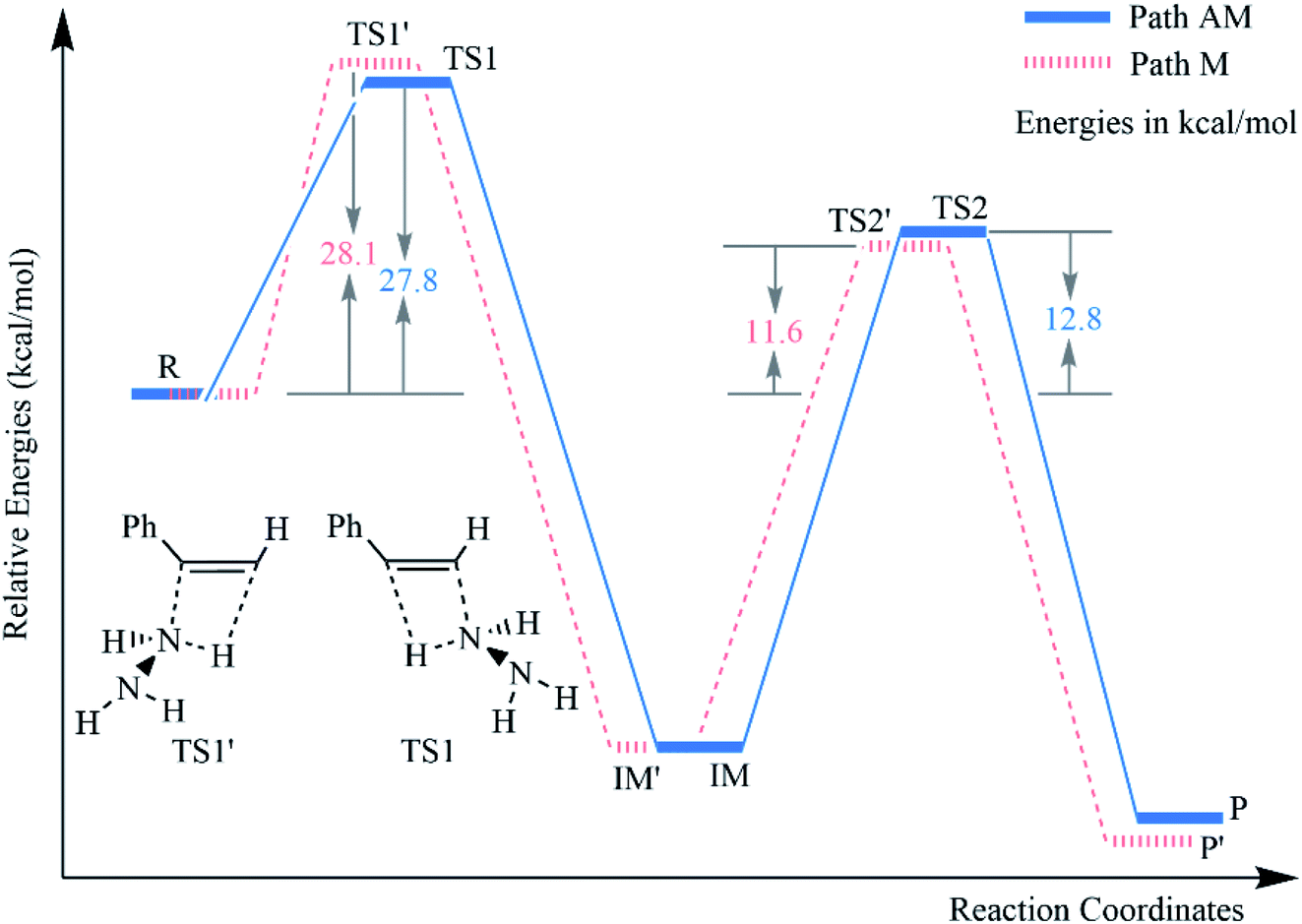
In general, the hydroamination of a terminal unsaturated C–C bond can take place by two possible pathways, namely43–49 the Markovnikov (path M) and the anti-Markovnikov (path AM) addition (Scheme 1). The anti-Markovnikov hydroamination is considered to be a particular challenge because it requires the nucleophilic amine to attack the less electrophilic primary carbon and generate the new [M]–C bond at the more sterically encumbered carbon when the metal catalyst was used.43 However, our calculations revealed that the respective barrier heights of the anti-Markovnikov and Markovnikov hydrohydrazination of phenylacetylene and parent hydrazine are 27.8 and 28.1 kcal mol−1, which are competitive when there was no metal complex, respectively (Fig. 1). It probably benefits from the steric effects in the metal-mediated anti-Markovnikov reaction being eliminated. Therefore, the challenging anti-Markovnikov hydrohydrazination of the parent hydrazine becomes a possible pathway. By analyzing the alkyne molecules, we found that reducing the charge of the terminal carbon may be an effective method to generate anti-Markovnikov products because it would make the nucleophilic amine easier to attack the primary carbon atom.
 |
| | Scheme 1 Two reaction pathways (Markovnikov: path M and anti-Markovnikov: path AM) for the hydroamination of a terminal alkyne with hydrazine (some of the hydrogen atoms in hydrazine are omitted). | |
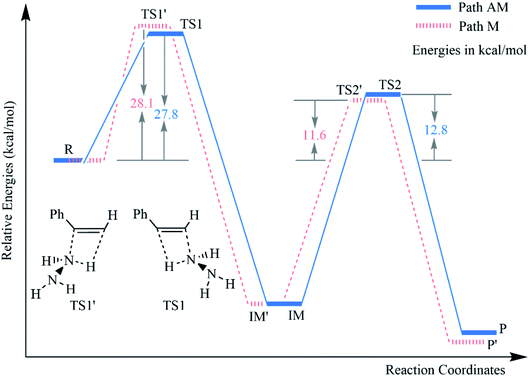 |
| | Fig. 1 Relative energy (kcal mol−1) profiles for the hydrohydrazination of phenylacetylene and parent hydrazine according to the two pathways. | |
The external electric field (EEF), which is well-known as a green and effective metal-free catalyst, is emerging into one's insight.50–53 This “invisible catalyst”, by now, has been investigated by numerous theoretical studies54–65 and further confirmed by experiments for the Diels–Alder reaction in 2016.66 These investigations manifested that EEF not only influence the atom charge but also catalyzes the chemical reactions. Moreover, using EEF to replace the metal-mediated catalyst could reduce the costs of preparing ligands and metal complexes. Inspired by these reports, we studied the hydrohydrazination of phenylacetylene and parent hydrazine by EEF as a catalyst. The effects of solvents on hydrohydrazination were also taken into account, which are usually considered in experiments. Besides, the local electric field was studied as well, in which the electron-withdrawing and electron-donating groups were used. Another terminal alkyne, namely 1-hexyne, was probed to prove that the obtained conclusion is not occasional. This study conquers the difficulties associated with the usage of parent hydrazine and opens an avenue for extensive applications from theory point of view, and may further provide a new method for the experiments.
Computational details
All the studies of the hydrohydrazination of phenylacetylene and parent hydrazine, including geometry optimizations and energy calculations, were carried out using the Gaussian 09 program.67 The geometries of the reactants (Rs), transition states (TSs), intermediates (IMs), and products (Ps) were fully optimized at the level of M06/6-31G(d)68 because this functional has been proved previously in the hydroamination reactions.69–73 All of the stationary points were also calculated by frequency calculations at the same level of theory to confirm that all transition states had only one imaginary frequency, while the others had no imaginary frequency. Furthermore, the transition states were analyzed by intrinsic reaction coordinate (IRC)74–76 calculations with the aim of testing the TSs correctly connecting the former and the later stationary points. The single-point energies of all the structures were calculated using M06/6-311++G(d,p). Therefore, the corresponding energies are labelled as M06/6-311++G(d,p)//M06/6-31G(d). Considering the hydrohydrazination reactions are generally performed in solvents, we also examined the solvent effects using the solvation model density (SMD) model77 with C6H6 (ε = 2.3) and DMSO (ε = 48.9) solvents. The substituent effects were also considered in this study. The “Field = M ± N” keyword was used in calculations to probe the effects of external electric fields on hydrohydrazination, which defines the direction and magnitude of EEF in Gaussian 09. In this study, the directions of the used EEF are shown in Scheme 2. The EEF oriented along the C1–C2 triple bond was defined as Fx, with its positive direction pointing from C1 to C2 atom.
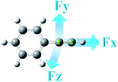 |
| | Scheme 2 Directions (denoted as Fx, Fy, and Fz) of the applied external electric field in the hydrohydrazination of phenylacetylene and parent hydrazine. | |
Besides, we also employed the M062X method, which combined the other two basis sets (6-311G(d,p) and 6-311++G(d,p)) to prove our calculations. The optimized geometries of every stationary point are similar to those of the previous method. From the relative energies listed in ESI Tables S2 and S3,† we could also obtain the same conclusions as the M06 functional results. Therefore, the calculation results in our study are convincing.
Results and discussions
Anti-Markovnikov hydrohydrazination of phenylacetylene
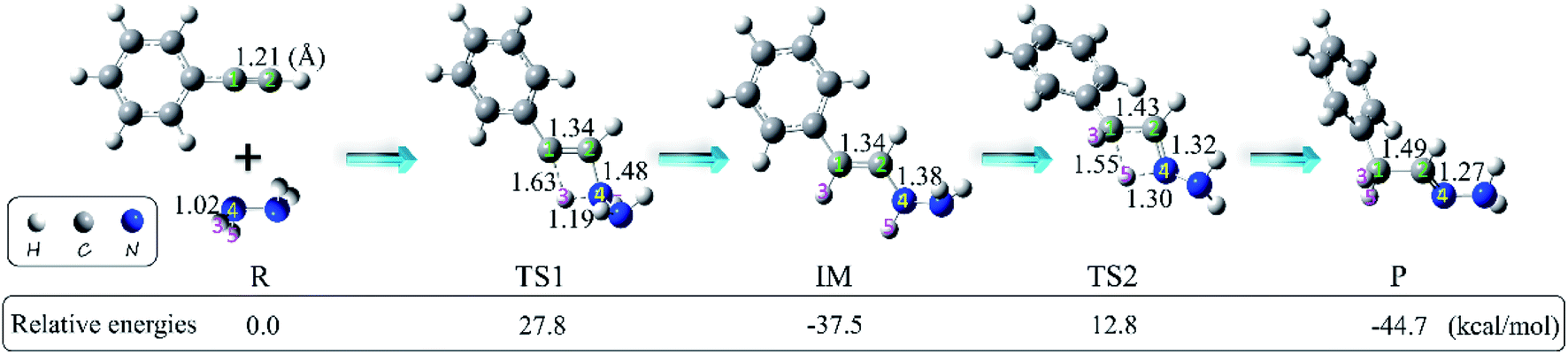
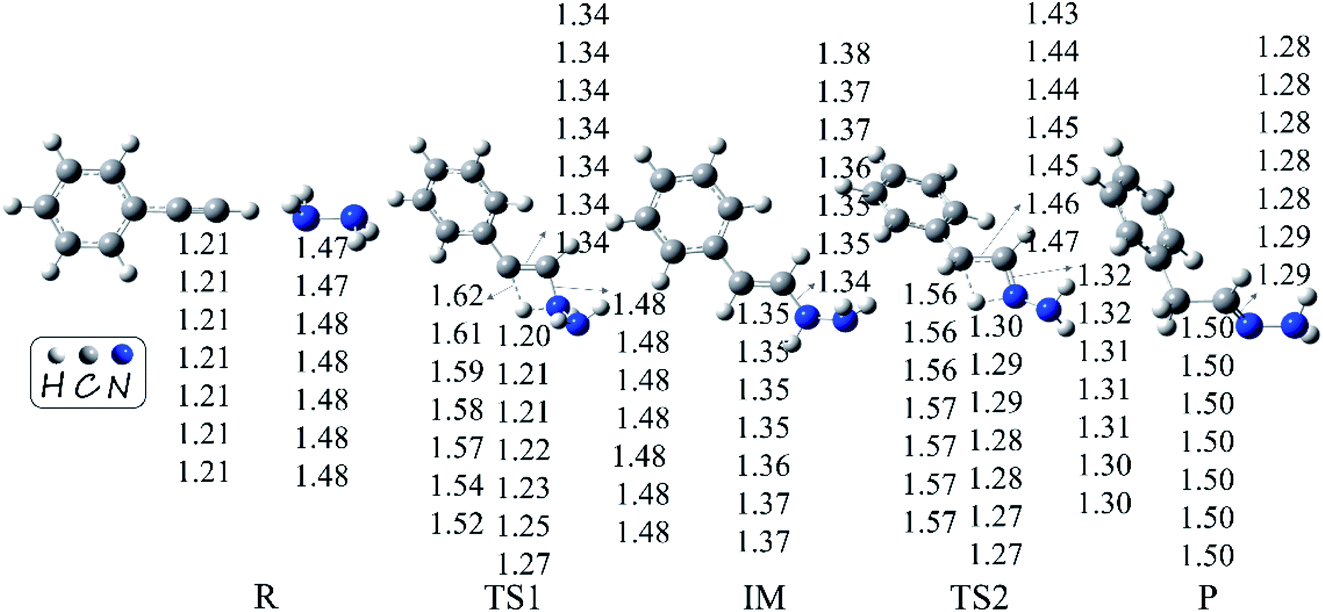
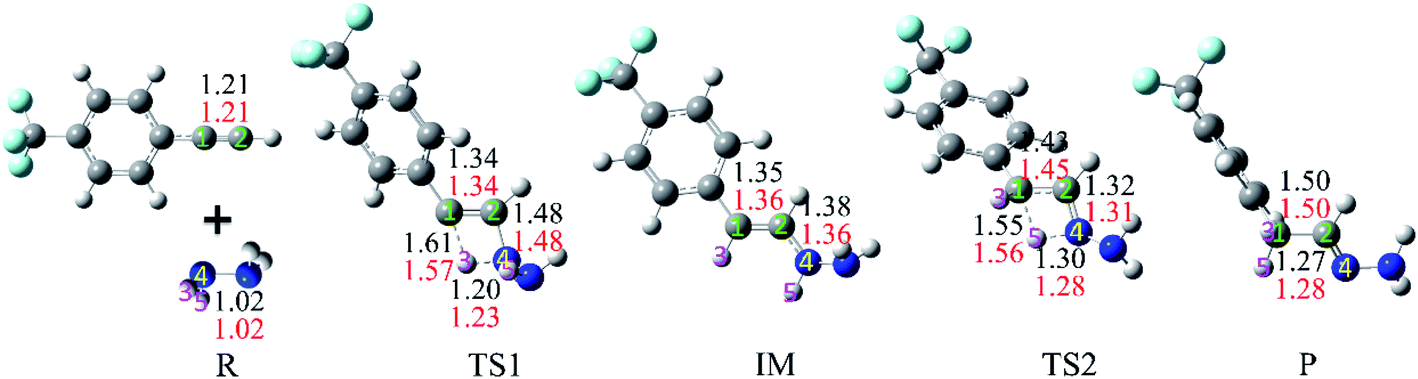
We first explored the anti-Markovnikov hydrohydrazination of phenylacetylene with parent hydrazine. The optimized geometries of the reactants (R), transition states (TS1 and TS2), intermediate (IM), and product (P) along the potential energy surface with their key bond parameters and relative energies are shown in Fig. 2.
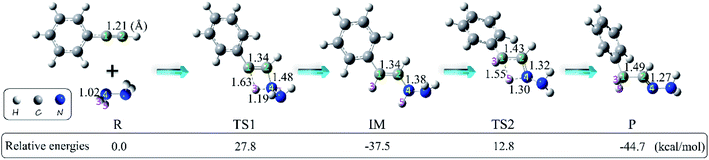 |
| | Fig. 2 Geometries (bond lengths are given in Å) and relative energies (in kcal mol−1) of the stationary points in the hydrohydrazination of phenylacetylene with parent hydrazine. | |
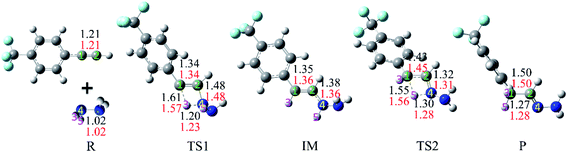
From Fig. 2, it can be seen that when the hydrazine molecule approached the phenylacetylene molecule, a four-membered ring (C1–C2–N4–H3) structure formed, which is the first transition state (TS1) of the anti-Markovnikov hydrohydrazination. The only imaginary frequency of TS1 is 1560.5i cm−1. In TS1, one of the nitrogen atoms (N4) in hydrazine interacted with the terminal carbon atom (C2) of phenylacetylene, accompanied by the formation of a new C2–N4 bond. One of the hydrogen atoms (H3), attached to the nitrogen atom (N4), reacted with the internal carbon atom (C1) in phenylacetylene, and the new C1–H3 bond took its shape. Simultaneously, both the bond lengths of C1–C2 and N4–H3 in TS1 were elongated, implying that the C1–C2 triple bond was impaired, and the N4–H3 bond tended to be ruptured. By inspecting the relative energy of TS1, we could conclude that the barrier height of the first step of this hydrohydrazination is 27.8 kcal mol−1. As the interactions between phenylacetylene and hydrazine were further strengthened, the N4–H3 bond broke completely. Then, the intermediate (IM) of the addition reaction formed, which is also a product of the first addition of N–H to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond. The relative energy of IM is −37.5 kcal mol−1. Next, the other hydrogen atom (H5) attached to the nitrogen atom (N4) interacted with the internal carbon atom (C1) again, and the second transition state (TS2) of the hydrohydrazination was generated. The sole imaginary frequency of TS2 is 1642.9i cm−1, which connected the intermediate and the product correctly. It can be seen that TS2 is also a four-membered ring structure (C1–C2–N4–H5). Similar to that of TS1, the C1–C2 bond was further impaired, and the distance of N4–H5 was also elongated, tending to break down. Moreover, a new chemical bond, C1–H5, began to form. The interactions between C2 and N4 atoms were further enhanced, along with the corresponding single bond becoming its transitional double bond. From Fig. 2, one can see that the relative energy of TS2 is 12.8 kcal mol−1. Thus, the second addition process needs to overcome a higher barrier of 50.3 kcal mol−1. Ultimately, the N4–H5 bond ruptured, and the final product hydrazone formed.
C bond. The relative energy of IM is −37.5 kcal mol−1. Next, the other hydrogen atom (H5) attached to the nitrogen atom (N4) interacted with the internal carbon atom (C1) again, and the second transition state (TS2) of the hydrohydrazination was generated. The sole imaginary frequency of TS2 is 1642.9i cm−1, which connected the intermediate and the product correctly. It can be seen that TS2 is also a four-membered ring structure (C1–C2–N4–H5). Similar to that of TS1, the C1–C2 bond was further impaired, and the distance of N4–H5 was also elongated, tending to break down. Moreover, a new chemical bond, C1–H5, began to form. The interactions between C2 and N4 atoms were further enhanced, along with the corresponding single bond becoming its transitional double bond. From Fig. 2, one can see that the relative energy of TS2 is 12.8 kcal mol−1. Thus, the second addition process needs to overcome a higher barrier of 50.3 kcal mol−1. Ultimately, the N4–H5 bond ruptured, and the final product hydrazone formed.
Hydrohydrazination under external electric field
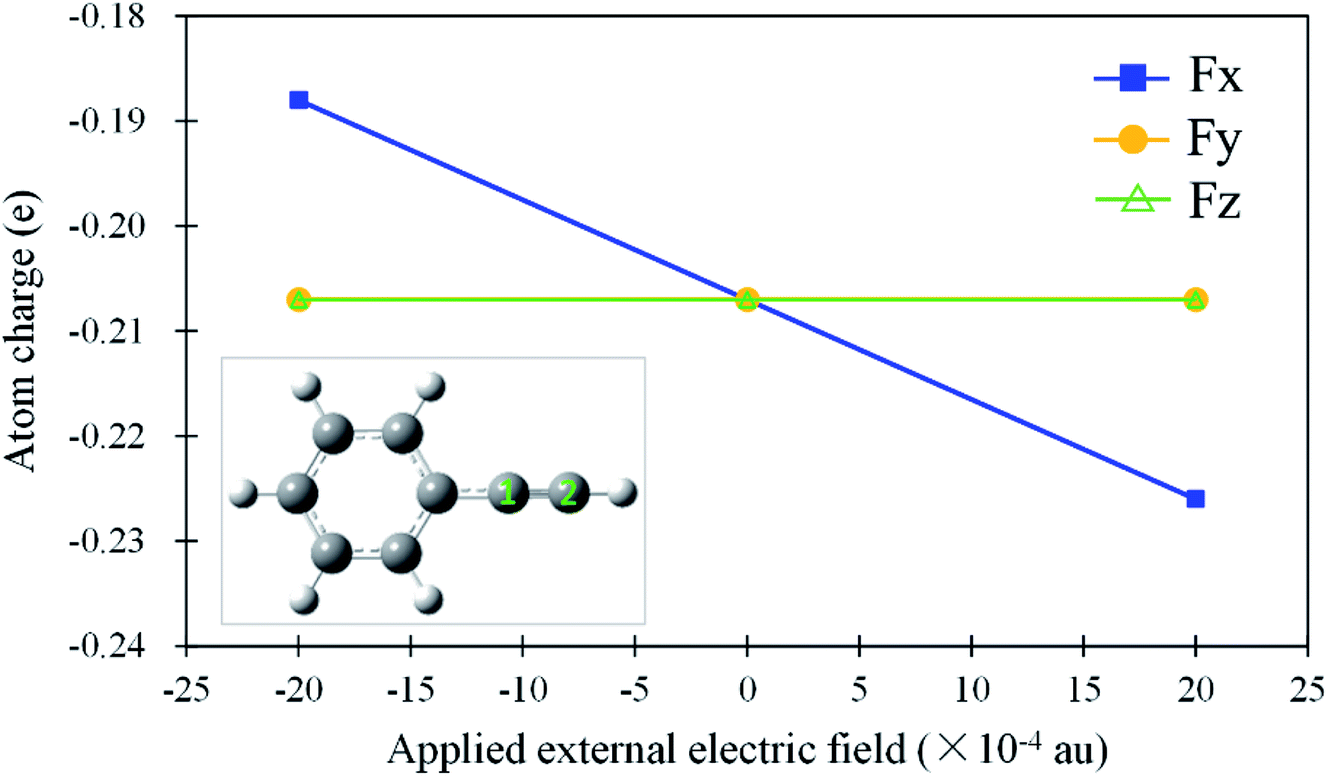
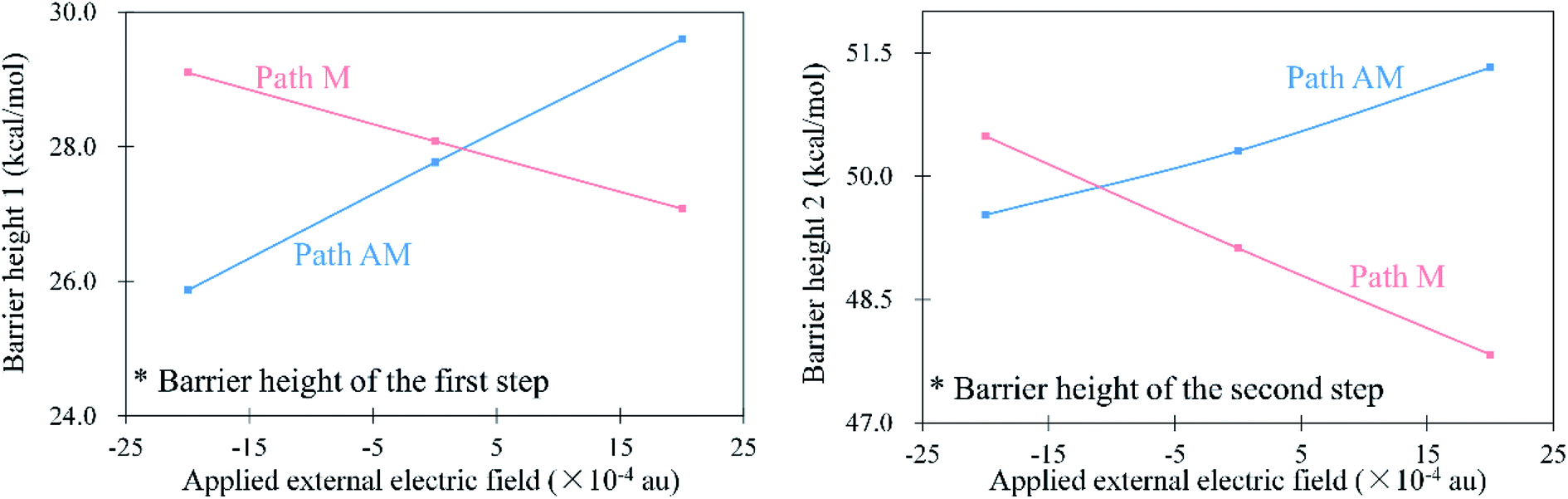
As mentioned above, the anti-Markovnikov and Markovnikov hydrohydrazination pathways are competitive. Therefore, reducing the barrier heights of the anti-Markovnikov addition is a good strategy to achieve the specific reaction. Based on the fact that the external electric field could change atom charges and catalyze reactions, we first tested the variations of natural bond orbital (NBO) charges on carbon atoms in phenylacetylene under different directions of EEF. The calculated results are shown in Fig. 3. From Fig. 3, it can be seen that EEF oriented along Y and Z axes (Scheme 2) induced little influence on the charges of the C2 atom, while Fx had an obvious effect on it. Therefore, we believe that the EEF oriented along the X-axis is most likely to catalyze the addition reaction. We also calculated the relative energies of anti-Markovnikov hydrohydrazination by the action of all directions of EEF. The results (ESI, Table S5†) also verified that Fx could catalyze the reaction much more. On the other hand, it can be seen from Fig. 3 that the charge of the C2 atom decreased with the increase in the external electric field oriented along the negative X-axis. This demonstrated that the nitrogen atoms in hydrazine are more likely to react with the C2 atom, which means the anti-Markovnikov channel is accelerated. On the contrary, the charge of the C2 atom increases gradually when the direction of Fx is flipped, which means that the anti-Markovnikov addition is unfavorable. Consequently, we believe that the negative Fx could accelerate the anti-Markovnikov hydrohydrazination, while the positive Fx inhibits this pathway. In order to prove our assumption, we calculated the effects of both the positive and negative Fx acting on the hydrohydrazination according to path AM and path M. The obtained results in Fig. 4 clearly revealed that the negative Fx decreased the barrier height of path AM and increased the barrier height of path M. In addition, the same direction of Fx has an opposite influence on the two pathways, which also illustrates that it might achieve reaction selectivity. In the following, we focused our study on the effects of the negative Fx for the anti-Markovnikov hydrohydrazination of parent hydrazine.
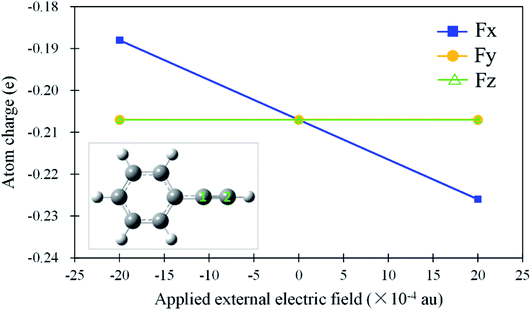 |
| | Fig. 3 Variations of the natural bond orbital (NBO) charges (e) on the C2 atom in phenylacetylene under different directions of the external electric field (×10−4 au). | |
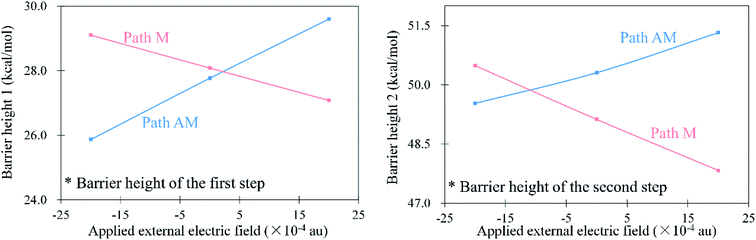 |
| | Fig. 4 Barrier heights (kcal mol−1) of the hydrohydrazination according to path AM and path M under the positive and negative Fx (×10−4 au). | |
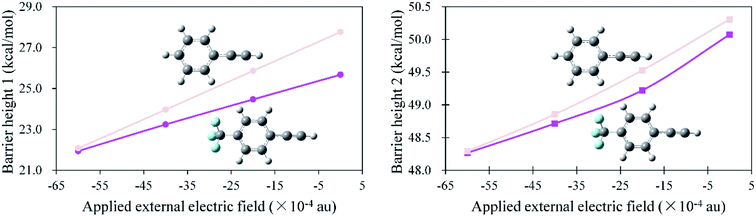
We re-optimized all the stationary points along the potential energy surfaces at different negative Fx field strengths and re-calculated their relative energies. The geometries and energies are shown in Fig. 5 and Table 1, respectively. From Fig. 5, it can be seen that the geometries of the stationary points changed very little under the negative Fx. By inspecting their relative energies from Table 1, one can conclude that the barrier heights lowered gradually with the increase in field strength. To be specific, for the first step, the barrier height is reduced by 5.7 kcal mol−1 when Fx = −60 (×10−4) au, equivalent to the rate enhancements by several orders of magnitude. More excitingly, it was lowered by 16.0 kcal mol−1 as the field strength was enhanced to 180 (×10−4) au. For the second step, the influence of negative Fx is also effective but not as large as in the first step. It can be seen that the barrier height of the second addition is lowered by 4.8 kcal mol−1 when Fx = −180 (×10−4) au. Therefore, the negative Fx had an effective influence on the anti-Markovnikov hydrohydrazination of phenylacetylene with parent hydrazine, particularly for the first step. Why the barrier heights are gradually lowered with the increase in negative Fx, and why the second step had a smaller variation than the first step? We analyzed the frontier molecular orbitals of ‘R’ (phenylacetylene) and ‘IM’ (intermediate) under different negative Fx, in which ‘R’ and ‘IM’ were separated from their corresponding optimized transition states. In addition, the absolute energies of the two transition states under negative Fx were also calculated. These results are summarized in Table 2. From the energies point of view, we can conclude that the stronger the field strength, the smaller the lowest unoccupied molecular orbital (LUMO) energy of ‘R’. Moreover, the total energy of TS1 is lowered as the field strength is increased. Therefore, the smaller LUMO energy of ‘R’ and the smaller absolute energy of TS1 made the addition reaction occur easily. Alternatively, the barrier height of the hydrohydrazination for the first step is reduced. For the second step, the total energy of TS2 is also reduced with the rising field strength. As a consequence, the barrier height of hydrohydrazination for the second step is lowered. However, the LUMO energy of ‘IM’ is increased when the field strength is increased, which is different from the first step. Thus, the enhancement of LUMO energy of ‘IM’ is unfavorable for the second addition. Thus, the second barrier height of hydrohydrazination had a smaller change than the first barrier height.
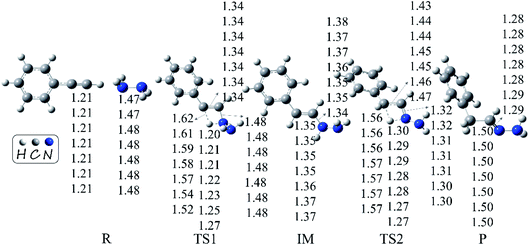 |
| | Fig. 5 Geometries (bond lengths are given in Å) of the stationary points in the hydrohydrazination of phenylacetylene with parent hydrazine under different negative Fx field strengths (×10−4 au). The parameters are listed with the increase in the field strength, which are −20, −40, −60, −80, −100, −150, and −180 (×10−4) au. | |
Table 1 Relative energies (kcal mol−1) of the stationary points in the hydrohydrazination of phenylacetylene with parent hydrazine under different negative Fx field strengths (×10−4 au)
| |
0 |
−20 |
−40 |
−60 |
−80 |
−100 |
−150 |
−180 |
| R |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
| TS1 |
27.8 |
25.9 |
24.0 |
22.1 |
20.3 |
18.5 |
14.3 |
11.8 |
| IM |
−37.5 |
−38.3 |
−39.6 |
−40.9 |
−42.5 |
−44.1 |
−48.4 |
−51.4 |
| TS2 |
12.8 |
11.2 |
9.3 |
7.3 |
5.3 |
3.2 |
−2.4 |
−5.9 |
| P |
−44.7 |
−45.3 |
−46.0 |
−46.7 |
−47.3 |
−47.9 |
−50.3 |
−51.6 |
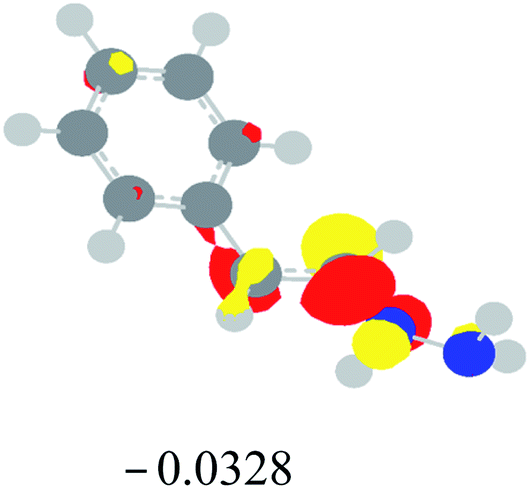
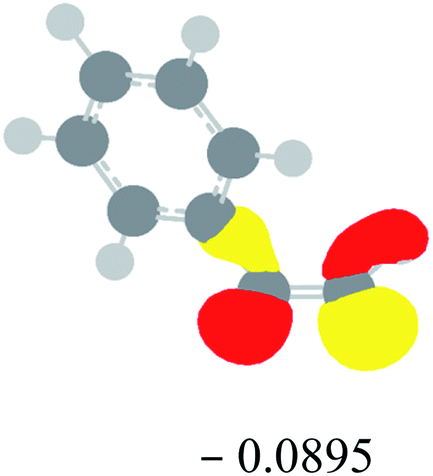
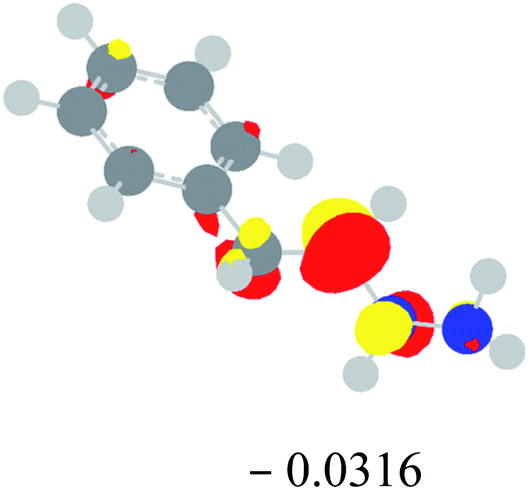
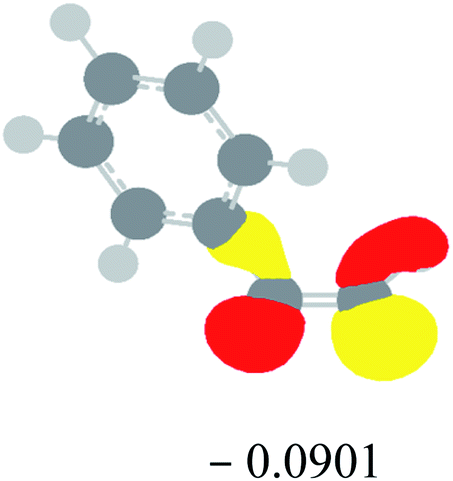
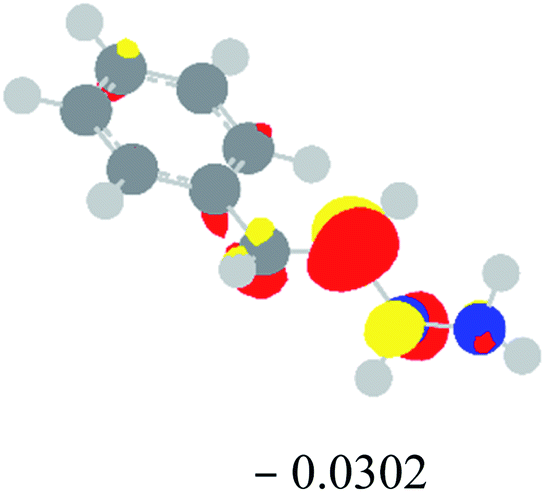
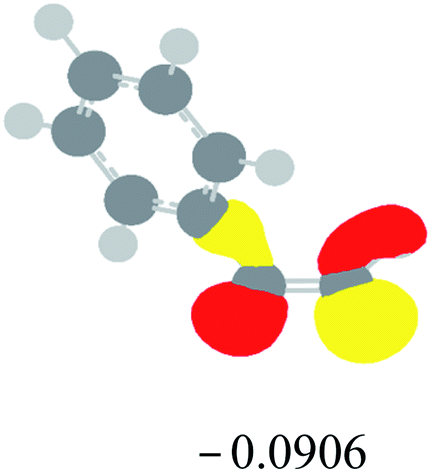
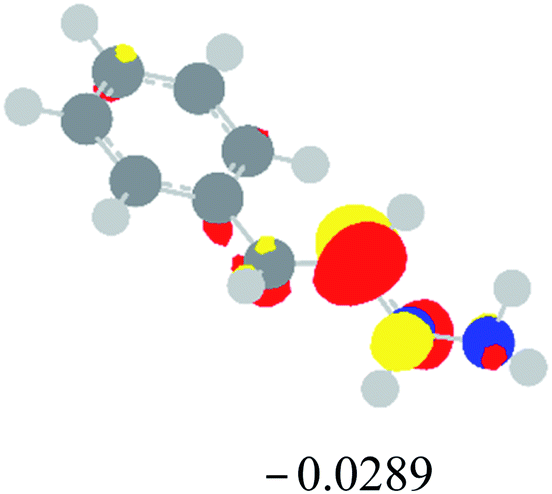
Table 2 Images of the lowest unoccupied molecular orbital (LUMO, isovalue = 0.08) of ‘R’ (phenylacetylene) and ‘IM’ (intermediate) and their energies (eV) and the absolute energies (in au) of the transition states (TS1 and TS2) under different negative Fx (×10−4 au)
| Fx, (×10−4 au) |
LUMO‘R’, ELUMO-‘R’ (eV) |
ETS1 (au) |
LUMO‘IM’, ELUMO-‘IM’ (eV) |
ETS2 (au) |
| 0 |
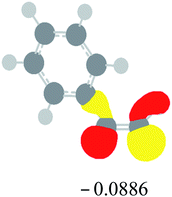 |
−419.8401 |
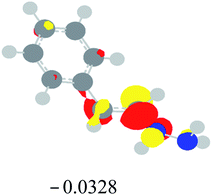 |
−419.8639 |
| −20 |
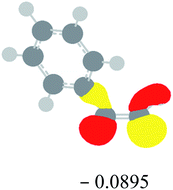 |
−419.8429 |
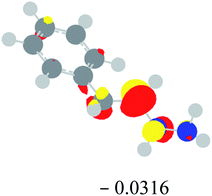 |
−419.8663 |
| −40 |
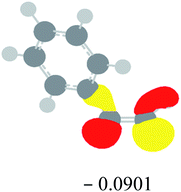 |
−419.8464 |
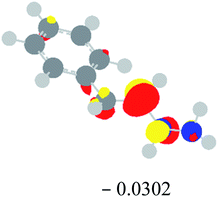 |
−419.8698 |
| −60 |
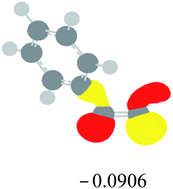 |
−419.8505 |
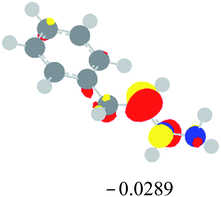 |
−419.8740 |
Solvent effects
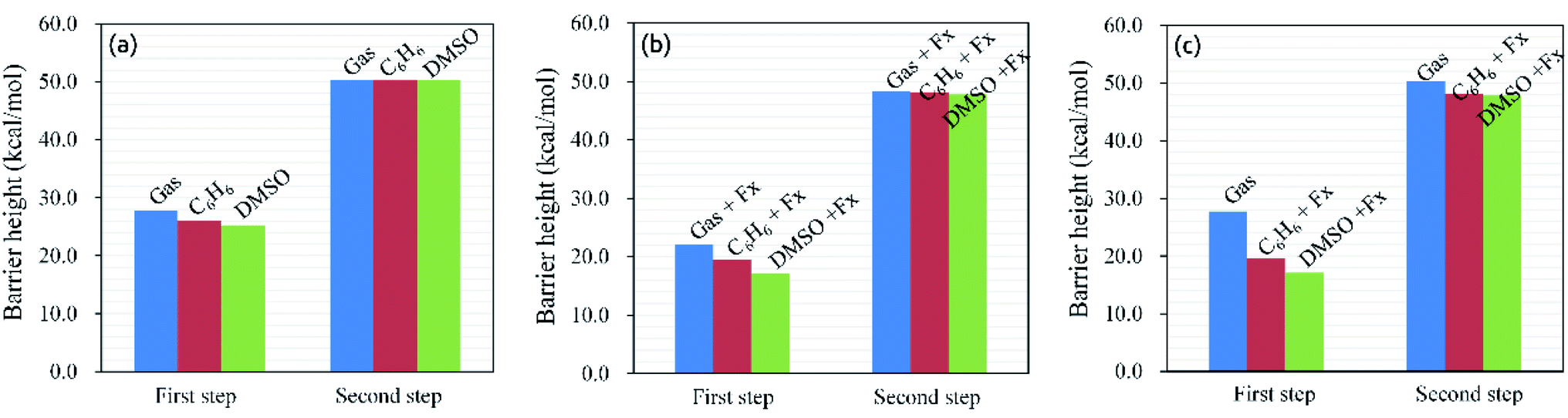
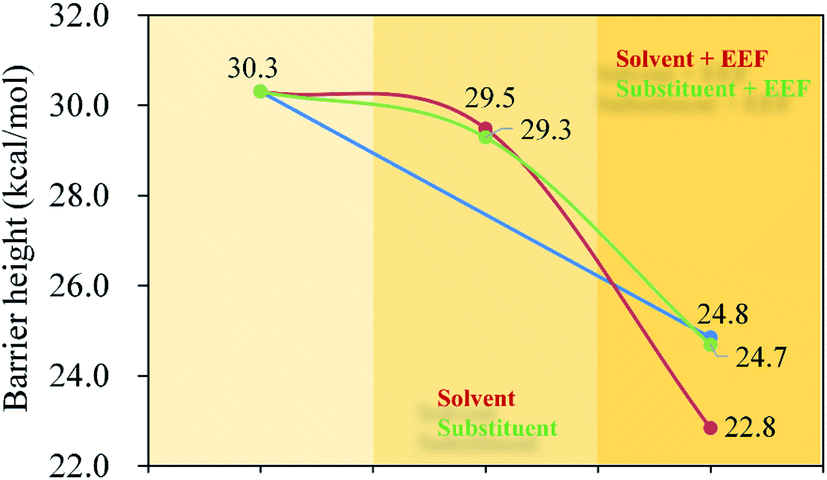
All the stationary points were re-optimized by the SMD model with C6H6 and DMSO solvents (ESI, Fig. S2†). Furthermore, the solvent effects of the hydrohydrazination of phenylacetylene with parent hydrazine were also considered under the negative Fx. The calculated reaction barrier heights are shown in Fig. 6. On the one hand, we can see that the application of solvents reduced the barrier height of the first step irrespective of the presence of negative Fx (for Fig. 6a, without EEF, and for Fig. 6b, with EEF), which indicates that the solvents are favorable for the first step of the reaction, while it had few influences on the second step. On the other hand, the combinations of the negative Fx and solvents significantly influenced the reaction barrier heights for the first step compared with that in vacuum (Fig. 6c). For example, the barrier heights were lowered 8.3 (C6H6) and 10.7 (DMSO) kcal mol−1 (ESI, Table S8†) for the first step with Fx = −60 (×10−4) au. We considered that the decrease in the barrier heights caused by the solvents also induced the redistribution of the atom charges, which is similar to the effect of the external electric field. Thus, the introduction of solvents catalyzed the hydrohydrazination, and the synergistic effects between solvents and Fx accelerated the reaction much more.
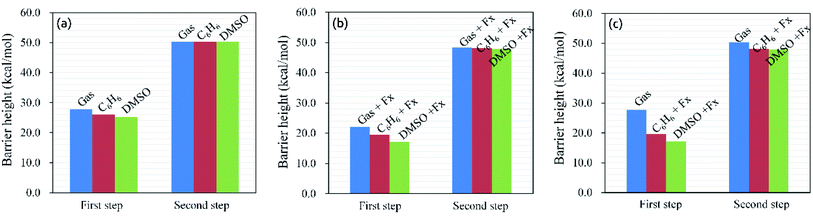 |
| | Fig. 6 Barrier heights (kcal mol−1) of hydrohydrazination for the first and second steps in the gas phase, C6H6 and DMSO solvents, and negative Fx (×10−4 au). (a) In solvents; (b) in solvents and negative Fx; (c) in the gas phase (the blue column) and in solvents and negative Fx (red and green columns). | |
Substituent effects and its synergism with EEF
The intramolecular local electric field is also an effective method to change the distribution of atom charges. The applications of substituents, including electron-withdrawing and electron-donating groups, are the common strategy in experiments. In this study, the electron-withdrawing substituent (trifluoromethyl) and electron-donating group (methoxy) are selected to study their influences on the hydrohydrazination, which are often used in experiments.30 All the structures were fully optimized, and their relative energies were calculated. The geometries of stationary points and their energy results are listed in Fig. 7, S3,† and Table 3, respectively. The introduction of the trifluoromethyl substituent reduced the barrier heights of the first and second steps by 2.1 and 0.2 kcal mol−1, respectively. However, the introduction of a methoxy substituent increased the barrier heights of the first and second step by 2.9 and 0.3 kcal mol−1, respectively. Therefore, we regarded the application of the electron-withdrawing substituent as an effective method to reduce the energy barrier heights.
 |
| | Fig. 7 Geometries (bond lengths are given in Å) of the stationary points in the hydrohydrazination of trifluoromethyl phenylacetylene with parent hydrazine (values in red were calculated with an external electric field, Fx = −60 × 10−4 au). | |
Table 3 Relative energies (in kcal mol−1) of the stationary points in the hydrohydrazination of the substituted phenylacetylene with parent hydrazine
| |
Trifluoromethyl |
Methoxy |
| R |
0.0 |
0.0 |
| TS1 |
25.7 |
30.7 |
| IM |
−39.2 |
−36.9 |
| TS2 |
10.9 |
13.7 |
| P |
−45.4 |
−41.6 |
Furthermore, we used negative Fx in hydrohydrazination in the presence of the trifluoromethyl substituent. The calculated results are exhibited in Fig. 8. It can be seen that the negative Fx further accelerated the addition reactions in the presence of an electron-withdrawing substituent. Besides, the combination of trifluoromethyl phenylacetylene and negative Fx lowered the barrier heights much more than that of phenylacetylene, particularly when the field strength is not large. Therefore, applying the substituted phenylacetylene combined with a small EEF is a promising method for catalyzing anti-Markovnikov hydrohydrazination.
 |
| | Fig. 8 Barrier heights (kcal mol−1) for the hydrohydrazination of phenylacetylene and trifluoromethyl phenylacetylene with parent hydrazine under negative Fx (× 10−4 au). | |
Hydrohydrazination of 1-hexyne
In order to test that the above-mentioned conclusions are not occasional, the hydrohydrazination of aliphatic 1-hexyne with parent hydrazine was also investigated. The C6H6 solvent as well as the substituted alkyne (6-trifluoromethyl-1-hexyne) were considered simultaneously. The obtained geometries and barrier heights of the first step are shown in Fig. 9 and 10, respectively. It can be seen that (i) the reaction mode of the hydrohydrazination of 1-hexyne is similar to that of phenylacetylene; (ii) the application of EEF lowered the barrier height of the first step of the reaction; (iii) whether the solvent effect or the introduction of an electron-withdrawing substituent accelerated the hydrohydrazination; (iv) the combination of solvent or substituent and EEF further decreased the reaction barrier heights. All the phenomena are consistent with the hydrohydrazination of phenylacetylene with parent hydrazine.
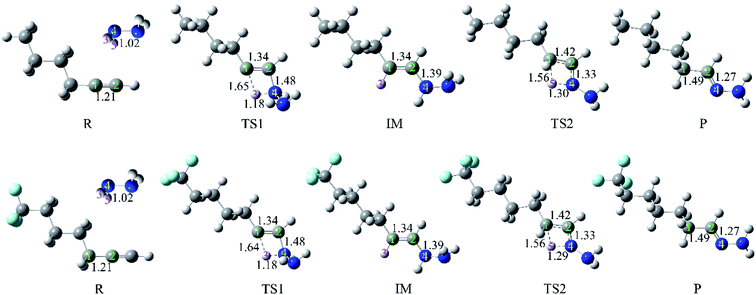 |
| | Fig. 9 Geometries (bond lengths are given in Å) of the stationary points in hydrohydrazination of 1-hexyne (6-trifluoromethyl-1-hexyne) with parent hydrazine. | |
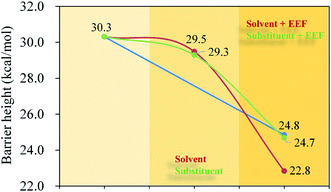 |
| | Fig. 10 Barrier heights (kcal mol−1) of the hydrohydrazination of 1-hexyne for the first step in the gas phase, C6H6 solvent, and external electric field (Fx = −60 × 10−4 au) and the substituted 1-hexyne. | |
Conclusions
The effective and green external electric field (EEF) catalyzed anti-Markovnikov hydrohydrazination of parent hydrazine was first studied in the present study using the M06 functional. There are two possible pathways (path M and path AM) in the hydrohydrazination reactions, which are competitive with each other when there was no metal catalyst. Once EEF was introduced, the anti-Markovnikov addition pathway was accelerated when its direction oriented along the negative Fx, which would achieve the selectivity of hydrohydrazination of the parent hydrazine. Furthermore, the larger the field strength of the negative Fx, the lower the barrier heights. The solvents reduced the barrier heights of the first step, but had little effects on the second step. The introduction of substituents in phenylacetylene demonstrated that the electron-withdrawing substituent (trifluoromethyl) could accelerate the reactions. In summary, the best strategy for accelerating this hydrohydrazination is by combining solvent or substituent and external electric field. Hopefully, this study would provide a new guidance for the extensive applications of hydrohydrazination of parent hydrazine in experiments, particularly for the first step of addition reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from the Doctoral Research Foundation of the Shandong University of Technology. H.-L. X. acknowledges the Fundamental Research Funds for the Central Universities 2412018ZD008.
References
- J. W. Daly, H. M. Garraffo and T. F. Spande, The Alkaloids: Chemistry and Pharmacology, ed, G. A. Cordell, Academic Press, San Diego, 1993, vol. 43 Search PubMed.
- A. G. King and J. Meinwald, Review of the Defensive Chemistry of Coccinellids, Chem. Rev., 1996, 96, 1105–1122, DOI:10.1021/cr950242v.
- D. O'Hagan, Pyrrole, Pyrrolidine, Pyridine, Piperidine and Tropane Alkaloids, Nat. Prod. Rep., 2000, 17, 435–446, 10.1039/a707613d.
- S. A. Lawrence, Amines: Synthesis, Properties, and Applications, Cambridge University Press, New York, 2004, vol. 1 Search PubMed.
- X. Zeng, Recent Advances in Catalytic Sequential Reactions Involving Hydroelement Addition to Carbon–Carbon Multiple Bonds, Chem. Rev., 2013, 113, 6864–6900, DOI:10.1021/cr400082n.
- X. Zhu, Z. Li, C. Jin, L. Xu, Q. Wu and W. Su, Mechanically Activated Synthesis of 1,3,5-Triaryl-2-Pyrazolines by High Speed Ball Milling, Green Chem., 2009, 11, 163–165, 10.1039/b816788e.
- B. Lee, S. H. Kang, D. Kang, K. H. Lee, J. Cho, W. Nam, O. H. Han and N. H. Hur, Isolation and Structural Characterization of the Elusive 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Adduct of Hydrazine and Carbon Dioxide, Chem. Commun., 2011, 47, 11219–11221, 10.1039/c1cc14542h.
:1 Adduct of Hydrazine and Carbon Dioxide, Chem. Commun., 2011, 47, 11219–11221, 10.1039/c1cc14542h. - S. C. Ensign, E. P. Vanable, G. D. Kortman, L. J. Weir and K. L. Hull, Anti-Markovnikov Hydroamination of Homoallylic Amines, J. Am. Chem. Soc., 2015, 137, 13748–13751, DOI:10.1021/jacs.5b08500.
- J. Waser and E. M. Carreira, Catalytic Hydrohydrazination of a Wide Range of Alkenes with a Simple Mn Complex, Angew. Chem., Int. Ed., 2004, 43, 4099–4102, DOI:10.1002/anie.200460811.
- Y. Li, Y. Shi and A. L. Odom, Titanium Hydrazido and Imido Complexes: Synthesis, Structure, Reactivity, and Relevance to Alkyne Hydroamination, J. Am. Chem. Soc., 2004, 126, 1794–1803, DOI:10.1021/ja038320g.
- J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins, J. Am. Chem. Soc., 2006, 128, 11693–11712, DOI:10.1021/ja062355+.
- A. M. Johns, Z. Liu and J. F. Hartwig, Primary Tert- and Sec-Allylamines via Palladium-Catalyzed Hydroamination and Allylic Substitution with Hydrazine and Hydroxylamine Derivatives, Angew. Chem., Int. Ed., 2007, 46, 7259–7261, DOI:10.1002/anie.200701899.
- N. Hazari and P. Mountford, Reactions and Applications of Titanium Imi-do Complexes, Acc. Chem. Res., 2005, 38, 839–849, DOI:10.1021/ar030244z.
- J. M. Hoover, A. Dipasquale, J. M. Mayer and F. E. Michael, Platinum-Cata-lyzed Intramolecular Hydrohydrazination: Evidence for Alkene Insertion into a Pt–n Bond, J. Am. Chem. Soc., 2010, 132, 5043–5053, DOI:10.1021/ja906563z.
- N. T. Patil and V. Singh, Synthesis of 1,3,5-Trisubstituted Pyrazolines via Zn(II)-Catalyzed Double Hydroamination of Enynes with Aryl Hydrazines, Chem. Commun., 2011, 47, 11116–11118, 10.1039/c1cc14850h.
- P. H. Cebrowski, J. G. Roveda, J. Moran, S. I. Gorelsky and A. M. Beauchemin, Intermolecular Cope-Type Hydroamination of Alkynes Using Hydrazines, Chem. Commun., 2008, 8, 492–493, 10.1039/b715022a.
- Q. Sha and Y. Wei, Base and Solvent Mediated Decomposition of Tosylhydrazones: Highly Selective Synthesis of N-Alkyl Substituted Hydrazones, Dialkylidenehydrazines, and Oximes, Tetrahedron, 2013, 69, 3829–3835, DOI:10.1016/j.tet.2013.03.055.
- M. N. Zhao, H. Liang, Z. H. Ren and Z. H. Guan, Copper-Catalyzed N–N Bond Formation by Homocoupling of Ketoximes via n–o Bond Cleavage: Facile, Mild, and Efficient Synthesis of Azines, Synthesis, 2012, 44, 1501–1506, DOI:10.1055/s-0031-1290779.
- M. Abid and A. Azam, Synthesis and Antiamoebic Activities of 1-N-Substituted Cyclised Pyrazoline Analogues of Thiosemicarbazones, Bioorg. Med. Chem., 2005, 13, 2213–2220, DOI:10.1016/j.bmc.2004.12.050.
- T. Higashi, R. Kikuchi, K. Miura, K. Shimada and H. O. H. Hiyamzu, NII-Electronic Library Service, Biol. Pharm. Bull., 1999, 22, 767–769 CrossRef CAS PubMed.
- C. Cao, Y. Shi and A. L. Odom, Intermolecular Alkyne Hydroaminations Involving 1,1-Disubstituted Hydrazines, Org. Lett., 2002, 4, 2853–2856, DOI:10.1021/ol0201052.
- I. A. Sayyed, K. Alex, A. Tillack, N. Schwarz, D. Michalik and M. Beller, A Convenient and General Method for the Synthesis of Indole-2,3-Dicarboxylates and 2-Arylindole-3-Carboxylates, Eur. J. Org. Chem., 2007, 27, 4525–4528, DOI:10.1002/ejoc.200700310.
- A. Tillack, H. Jiao, I. G. Castro, C. G. Hartung and M. Beller, A General Study of [(η5-Cp′)2Ti(η2-Me3SiC2SiMe3)]-Catalyzed Hydroamination of Terminal Alkynes: Regioselective Formation of Markovnikov and Anti-Markovnikov Products and Mechanistic Explanation (CpC5H5, C5H4Et, C5Me5), Chem.–Eur. J., 2004, 10, 2409–2420, DOI:10.1002/chem.200305674.
- S. Banerjee, E. Barnea and A. L. Odom, Titanium-Catalyzed Hydrohydrazination with Monosubstituted Hydrazines: Catalyst Design, Synthesis, and Reactivity, Organometallics, 2008, 27, 1005–1014, DOI:10.1021/om700852v.
- F. Loiseau, C. Clavette, M. Raymond, J. G. Roveda, A. Burrell and A. M. Beauchemin, Improved Cope-Type Hydroamination Reactivity of Hydrazi-ne Derivatives, Chem. Commun., 2011, 47, 562–564, 10.1039/c0cc02403a.
- K. Alex, A. Tillack, N. Schwarz and M. Beller, Zinc-Promoted Hydrohydrazination of Terminal Alkynes: An Efficient Domino Synthesis of Indoles, Angew. Chem., Int. Ed., 2008, 47, 2304–2307, DOI:10.1002/anie.200703823.
- K. Alex, A. Tillack, N. Schwarz and M. Beller, Zinc-Catalyzed Synthesis of Pyrazolines and Pyrazoles via Hydrohydrazination, Org. Lett., 2008, 10, 2377–2379, DOI:10.1021/ol800592s.
- S. L. Dabb and B. A. Messerle, Rh(I) and Ir(I) Catalysed Intermolecular Hydroamination with Substituted Hydrazines, Dalton Trans., 2008, 45, 6368–6371, 10.1039/b814591a.
- R. J. Lundgren and M. Stradiotto, Palladium-Catalyzed Cross-Coupling of Aryl Chlorides and Tosylates with Hydrazine, Angew. Chem., Int. Ed., 2010, 49, 8686–8690, DOI:10.1002/anie.201003764.
- D. R. Tolentino, L. Jin, M. Melaimi and G. Bertrand, Mesoionic Carbene-Gold(I) Catalyzed Bis-Hydrohydrazination of Alkynes with Parent Hydrazine, Chem.–Asian J., 2015, 10, 2139–2142, DOI:10.1002/asia.201403408.
- R. Kinjo, B. Donnadieu and G. Bertrand, Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine, Angew. Chem., Int. Ed., 2011, 50, 5560–5563, DOI:10.1002/anie.201100740.
- R. Kinjo, B. Donnadieu and G. Bertrand, ChemInform Abstract: Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine, J. Cheminf., 2011, 42 DOI:10.1002/chin.201141035.
- M. J. Lopezgomez, D. Martin and G. Bertrand, Anti-Bredt N-Heterocyclic Carbene: An Efficient Ligand for The Gold(I)-Catalyzed Hydroamination of Terminal Alkynes with Parent Hydrazine, Chem. Commun., 2013, 49, 4483–4485, 10.1039/c3cc41279b.
- E. Mizushima, T. Hayashi and M. Tanaka, Au(I)-Catalyzed Highly Efficient Intermolecular Hydroamination of Alkynes, Org. Lett., 2003, 5, 3349–3352, DOI:10.1021/ol0353159.
- J. L. Klinkenberg and J. F. Hartwig, Catalytic Organometallic Reactions of Ammonia, Angew. Chem., Int. Ed., 2011, 50, 86–95, DOI:10.1002/anie.201002354.
- J. I. Van Der Vlugt, Advances in Selective Activation and Application of Ammonia in Homogeneous Catalysis, Chem. Soc. Rev., 2010, 39, 2302–2322, 10.1039/b925794m.
- A. Furst, R. C. Berlo and S. Hooton, Hydrazine as a Reducing Agent for Organic Compounds (Catalytic Hydrazine Reductions), Chem. Rev., 1965, 65, 51–68, DOI:10.1021/cr60233a002.
- B. A. Roden and A. El-Awa, Hydrazine, Encyclopedia of Reagents for Organic Synthesis, 2014, DOI:10.1002/047084289X.rh027.pub2.
- J. P. Chen and L. L. Lim, Key Factors in Chemical Reduction by Hydrazine for Recovery of Precious Metals, Chemosphere, 2002, 49, 363–370, DOI:10.1016/s0045-6535(02)00305-3.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Metal-Organic Frameworks (MOFs) as Heterogeneous Catalysts for the Chemoselective Reduction of Carbon–Carbon Multiple Bonds with Hydrazine, Adv. Synth. Catal., 2009, 351, 2271–2276, DOI:10.1002/adsc.200900362.
- F. Alonso, G. Radivoy and M. Yus, Reduction of Hydrazines, Azo and Azoxy Compounds, and Amine N-Oxides with the NiCl2·2H2O-Li-DTBB (Cat.) Combination, Tetrahedron, 2000, 56, 8673–8678, DOI:10.1016/s0040-4020(00)00797-3.
- K. S. Lee, Y. K. Lim and C. G. Cho, Ag+ Mediated Deaminations of N-Boc Aryl Hydrazines for the Efficient Synthesis of N-Boc Aryl Amines, Tetrahedron Lett., 2002, 43, 7463–7464, DOI:10.1016/s0040-4039(02)01800-2.
- J. Guin, C. Mück-Lichtenfeld, S. Grimme and A. Studer, Radical Transfer Hydroamination with Aminated Cyclohexadienes Using Polarity Reversal Catalysis: Scope and Limitations, J. Am. Chem. Soc., 2007, 129, 4498–4503, DOI:10.1021/ja0692581.
- X. Shen and S. L. Buchwald, Rhodium-Catalyzed Asymmetric Intramolecular Hydroamination of Unactivated Alkenes, Angew. Chem., Int. Ed., 2010, 49, 564–567, DOI:10.1002/anie.200905402.
- D. Banerjee, K. Junge and M. Beller, Palladium-Catalysed Regioselective Hydroamination of 1,3-Dienes: Synthesis of Allylic Amines, Org. Chem. Front., 2014, 1, 368–372, 10.1039/c4qo00023d.
- C. S. Sevov, J. Zhou and J. F. Hartwig, Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles, J. Am. Chem. Soc., 2014, 136, 3200–3207, DOI:10.1021/ja412116d.
- L. J. Gooßen, L. Huang, M. Arndt, K. Gooßen and H. Heydt, Late Transition Metal-Catalyzed Hydroamination and Hydroamidation, Chem. Rev., 2015, 115, 2596–2697, DOI:10.1021/cr300389u.
- Q. A. Chen, Z. Chen and V. M. Dong, Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines, J. Am. Chem. Soc., 2015, 137, 8392–8395, DOI:10.1021/jacs.5b05200.
- J. C. Timmerman, B. D. Robertson, R. A. Widenhoefer, J. C. Timmerman and B. D. Robertson, Gold-Catalyzed Intermolecular Anti-Markovnikov Hydroamination of Alkylidenecyclopropanes, Angew. Chem., Int. Ed., 2015, 54, 2251–2254, DOI:10.1002/anie.201410871.
- S. Ciampi, N. Darwish, H. M. Aitken, I. Díez-Pérez and M. L. Coote, Harnessing Electrostatic Catalysis in Single Molecule, Electrochemical and Chemical Systems: A Rapidly Growing Experimental Tool Box, Chem. Soc. Rev., 2018, 47, 5146–5164, 10.1039/c8cs00352a.
- V. V. Welborn, L. Ruiz Pestana and T. Head-Gordon, Computational Optimization of Electric Fields for Better Catalysis Design, Nat. Catal., 2018, 1, 649–655, DOI:10.1038/s41929-018-0109-2.
- S. Shaik, R. Ramanan, D. Danovich and D. Mandal, Structure and Reactivity/Selectivity Control by Oriented-External Electric Fields, Chem. Soc. Rev., 2018, 47, 5125–5145, 10.1039/c8cs00354h.
- S. Shaik, D. Mandal and R. Ramanan, Oriented Electric Fields as Future Smart Reagents in Chemistry, Nat. Chem., 2016, 8, 1091–1098, DOI:10.1038/nchem.2651.
- S. Shaik, S. P. de Visser and D. Kumar, External Electric Field Will Control the Selectivity of Enzymatic-Like Bond Activations, J. Am. Chem. Soc., 2004, 126, 11746–11749, DOI:10.1021/ja047432k.
- H. Hirao, H. Chen, M. A. Carvajal, Y. Wang and S. Shaik, Effect of External Electric Fields on the C–H Bond Activation Reactivity of Nonheme Iron–Oxo Reagents, J. Am. Chem. Soc., 2008, 130, 3319–3327, DOI:10.1021/ja070903t.
- W. Lai, H. Chen, K.-B. Cho and S. Shaik, External Electric Field Can Control the Catalytic Cycle of Cytochrome P450Cam: A QM/MM Study, J. Phys. Chem. Lett., 2010, 1, 2082–2087, DOI:10.1021/jz100695n.
- R. Meir, H. Chen, W. Lai and S. Shaik, Oriented Electric Fields Accelerate Diels-Alder Reactions and Control the Endo/Exo Selectivity, ChemPhysChem, 2010, 11, 301–310, DOI:10.1002/cphc.200900848.
- R. Ramanan, D. Danovich, D. Mandal and S. Shaik, Catalysis of Methyl Transfer Reactions by Oriented External Electric Fields: Are Gold-Thiolate Linkers Innocent?, J. Am. Chem. Soc., 2018, 140, 4354–4362, DOI:10.1021/jacs.8b00192.
- M. X. Zhang, F. Y. Zhang, H. L. Xu and Z. M. Su, The Regulation of Hydroboration of Olefins by Oriented External Electric Field, New J. Chem., 2018, 18402–18408, 10.1039/c8nj04720k.
- Z. F. Wang, D. Danovich, R. Ramanan and S. Shaik, Oriented-External Electric Fields Create Absolute Enantioselectivity in Diels-Alder Reactions: The Importance of the Molecular Dipole Moment, J. Am. Chem. Soc., 2018, 140, 13350–13359, DOI:10.1021/jacs.8b08233.
- M. X. Zhang and H. L. Xu, Possible B–C Bonding in the Hydroboration of Benzonitrile by an External Electric Field, Phys. Chem. Chem. Phys., 2019, 21, 18–21, 10.1039/c8cp06704j.
- M. X. Zhang and H. L. Xu, A Greener Catalyst for Hydroboration of Imines—External Electric Field Modify the Reaction Mechanism, J. Comput. Chem., 2019, 40, 1772–1779, DOI:10.1002/jcc.25830.
- C. Yeh, T. M. Pham, S. Nachimuthu and J. Jiang, Effect of External Elec-tric Field on Methane Conversion on IrO2(110) Surface: A Density Functional Theory Study, ACS Catal., 2019, 9, 8230–8242, DOI:10.1021/acscatal.9b01910.
- K. Bhattacharyya, S. Karmakara and A. Datta, External electric field control: driving the reactivity of metal-free azide–alkyne click reactions, Phys. Chem. Chem. Phys., 2017, 19, 22482–22486 RSC.
- A. K. Jissy and A. Datta, Effect of External Electric Field on H-Bonding and π-Stacking Interactions in Guanine Aggregates, ChemPhysChem, 2012, 13, 4163–4172 CrossRef CAS PubMed.
- A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Electrostatic Catalysis of a Diels-Alder Reaction, Nature, 2016, 531, 88–91, DOI:10.1038/nature16989.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function, Theor. Chem. Acc., 2008, 120, 215–241, DOI:10.1007/s00214-007-0310-x.
- A. Couce-Rios, A. Lledós, I. Fernández and G. Ujaque, Origin of the Anti-Markovnikov Hydroamination of Alkenes Catalyzed by L-Au(I) Complexes: Coordination Mode Determines Regioselectivity, ACS Catal., 2019, 9, 848–858, DOI:10.1021/acscatal.8b03843.
- C. Lepori, P. Gómez-Orellana, A. Ouharzoune, R. Guillot, A. Lledós, G. Ujaque and J. Hannedouche, Well-Defined β-Diketiminatocobalt(II) Complexes for Alkene Cyclohydroamination of Primary Amines, ACS Catal., 2018, 8, 4446–4451, DOI:10.1021/acscatal.8b00631.
- A. Couce-Rios, A. Lledós and G. Ujaque, The Origin of Anti-Markovnikov Regioselectivity in Alkene Hydroamination Reactions Catalyzed by [Rh(DPEphos)]+, Chem.–Eur. J., 2016, 22, 9311–9320, DOI:10.1002/chem.201504645.
- A. E. Strom, D. Balcells and J. F. Hartwig, Synthetic and Computational Studies on the Rhodium-Catalyzed Hydroamination of Aminoalkenes, ACS Catal., 2016, 6, 5651–5665, DOI:10.1021/acscatal.6b01320.
- A. Couce-Rios, G. Kovács, G. Ujaque and A. Lledós, Hydroamination of C–C Multiple Bonds with Hydrazine Catalyzed by N-Heterocyclic Carbene-Gold(I) Complexes: Substrate and Ligand Effects, ACS Catal., 2015, 5, 815–829, DOI:10.1021/cs501705b.
- C. Gonzalez and H. B. Schlegel, Improved Algorithms for Reaction Path Following: Higher-Order Implicit Algorithms, J. Chem. Phys., 1991, 95, 5853–5860, DOI:10.1063/1.461606.
- C. Gonzalez and H. Bernhard Schlegel, An Improved Algorithm for Reaction Path Following, J. Chem. Phys., 1989, 90, 2154–2161, DOI:10.1063/1.456010.
- K. Fukui, The Path of Chemical Reactions – The IRC Approach, Acc. Chem. Res., 1981, 14, 363–368, DOI:10.1021/ar00072a001.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396, DOI:10.1021/jp810292n.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries, relative energies, NBO charges, IRC plots, Cartesian coordinates. See DOI: 10.1039/d1ra01037a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Wen-Zuo Lib,
Hong-Liang Xu
*a,
Wen-Zuo Lib,
Hong-Liang Xu