DOI:
10.1039/D1RA01151K
(Paper)
RSC Adv., 2021,
11, 12218-12226
Sonochemical synthesis of a trinuclear Cu(II) complex with open coordination sites for the differentiable optical detection of volatile amines†
Received
11th February 2021
, Accepted 11th March 2021
First published on 26th March 2021
Abstract
A discrete trinuclear Cu(II) complex, namely, [Cu3(pzdc)2(dpyam)2(H2O)4] (1) (H3pzdc = pyrazole-3,5-dicarboxylic acid, dpyam = 2,2′-dipyridylamine) was simply synthesized by the sonochemical process and structurally characterized. The single-crystal X-ray diffraction analysis revealed that three adjacent Cu(II) centers are linked via two bridging pzdc ligands to form a trinuclear Cu(II) unit. Each trinuclear Cu(II) unit contains open coordination sites with two trigonal bipyramidal Cu(II) centers and one elongated octahedral geometry. Moreover, the open coordination site of 1 was occupied by a small molecule, leading to the guest-induced structural transformation with chromism that was verified by FT-IR, UV-vis diffuse reflectance spectra, elemental analysis, PXRD, and SEM techniques. Compound 1 exhibits color change along with structural transformation in methanol media and after the dehydration process. Also, 1 shows different color responses after exposure to different amine vapors. In addition, compound 1 was conveniently deposited onto a filter paper by a sonochemical method used as a portable test strip for the discriminative qualitative detection of amines.
Introduction
Lewis acidic copper(II) center is one of the d9 transition metal ions that exhibits a diversity of colors1,2 and has a remarkable variety of coordination numbers and geometries due to the Jahn–Teller distortion.3–5 The Cu(II) center is an ideal candidate for the preparation of multinuclear complexes due to its variable coordination geometry, which can be combined with a variety of ligands.6,7 Generally, a polynuclear complex is held into macromolecules by non-covalent interactions such as hydrogen bonding, hydrophobic, π–π, and electrostatic interactions.8–10 In addition, some Cu(II) multinuclear complexes contain an open coordination site to interact with Lewis basic molecules.11,12 Interactions between the unsaturated Cu(II) unit and guest molecules usually affect the alteration in the coordination environment of Cu(II) core as well as the molecular packing, leading to chromotropic phenomena.13,14 Since Lewis basic amines are poisonous and corrosive compounds, they can be easily absorbed and endanger human safety and the environment.15,16 There are many techniques for amine detection such as high-performance liquid chromatography, gas chromatography coupled with mass spectrometry, fluorometric analysis, and electrochemical systems. However, these techniques not only require skilled operation and long analysis time but are also complicated, expensive, and not readily available on-site in most cases.17–20 Several chromic materials that can change color in response to amines are more convenient, fast, and can be identified on-site. As ligands, many amine molecules display good coordination ability with metal centers and excellent hydrogen donor/acceptor sites within the crystal lattice. In metal complexes, ammonia, methylamine, and ethylamine have been found in the form of coordination and lattice molecules and also construct N–H⋯X and Y–H⋯N bonds (X = H-acceptor and Y = H-donor) to stabilize the crystal structure.21–26 Ethylenediamine favors coordination with the metal center in a chelating bidentate mode.27,28 Therefore, the introduction of different amines with various sizes and shapes into the unsaturated polynuclear Cu(II) unit potentially affects the changes in the coordination environment and structural packing, resulting in the distinguishable detection of amines by naked eyes. The detection of amines has been commonly found in the category of coordination polymers, whereas zero-dimensional complexes are rarely detected due to their low chemical stability.29–31 However, the supramolecular packing of complexes is overly sensitive due to numerous non-covalent interactions that can easily respond to external stimuli, leading to change in physical properties. Thus, the stimulus-responsiveness associated with the structural transformation of supramolecular packing that could be monitored to denote the physical condition of the material is beneficial for the chemical sensor.
Herein, a new trinuclear Cu(II) complex, namely, [Cu3(pzdc)2(dpyam)2(H2O)4] (1) (H3pzdc = pyrazole-3,5-dicarboxylic acid, dpyam = 2,2′-dipyridylamine), for the detection of amine vapors was prepared on the basis of following aspects: (i) the versatile H3pzdc ligand exhibits diverse coordination modes so the structural alteration in the molecular packing can easily occur when it is stimulated by amines; (ii) H3pzdc has six potential interaction sites including four carboxylic oxygen atoms and two pyrazoly nitrogen atoms.32,33 These potentially generate intermolecular hydrogen bonding with amines and the interactions with the complexes are potentially influenced by various types and sizes of amines; (iii) the acidic Cu(II) center favors the binding with Lewis basic amines; (iv) the guest-induced chromism in the polynuclear Cu(II) complex can easily occur after binding with amines through the variation in the versatile distorted Cu(II) environment. As expected, the trinuclear Cu(II) complex demonstrated reversible chromic behavior induced by different types of volatile amines along with reversible structural transformation. In order to develop a convenient test strip for qualitative volatile amines' screening, the trinuclear Cu(II) complex was deposited onto a filter paper (denoted as 1-paper) by a simple sonochemical method to be used as a portable test strip.
Experimental section
Materials and physical measurements
All chemicals and solvents were commercially available and used as received. The FT-IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrophotometer in the range of 4000–380 cm−1 using KBr disks. A PerkinElmer 2400 Series II CHNS/O analyzer was used for elemental analyses (C, H, and N). Thermogravimetric analyses (TGA) were performed in the range of 35–750 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1 using a PerkinElmer Pyris Diamond thermogravimetric/differential thermal analyzer. The powder X-ray diffraction (PXRD) patterns were obtained in the range from 5 to 50° with 0.026 per step in 2θ angles on a PANalytical EMPYREAN with monochromatic CuKα radiation. The UV-vis diffuse reflectance spectra were recorded on a PerkinElmer Lambda2S spectrophotometer (400–1100 nm). Desktop scanning electron microscopes SNE-4500M with an acceleration voltage of 20 kV were used to obtain scanning electron microscopy (SEM) images. The SEM samples were prepared by placing a sample on a carbon conductive tape and then the samples were coated with a thin film of gold before being measured.
Preparation of [Cu3(pzdc)2(dpyam)2(H2O)4] (1)
Sonochemical synthesis of 1. The mixture solution containing Cu(NO3)2·3H2O (0.5 mmol, 121 mg), 2,2′-dipyridylamine (0.5 mmol, 86 mg), and pyrazole-3,5-dicarboxylic acid (0.5 mmol, 87 mg) in water, N,N-dimethylformamide (DMF), and ethanol (EtOH) (9 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 v/v) was sonicated for 30 minutes, then the green polycrystalline powder of 1 was obtained. Yield: 107 mg (70%) based on copper salt. Anal. Calcd for Cu3C30H28N10O12: C, 39.54; H, 3.10; N, 15.37. Found: C, 38.64; H, 3.17; N, 15.26%. FT-IR peaks (KBr, cm−1): 3199br (ν(OH)), 1640m (ν(C
:1:1 v/v) was sonicated for 30 minutes, then the green polycrystalline powder of 1 was obtained. Yield: 107 mg (70%) based on copper salt. Anal. Calcd for Cu3C30H28N10O12: C, 39.54; H, 3.10; N, 15.37. Found: C, 38.64; H, 3.17; N, 15.26%. FT-IR peaks (KBr, cm−1): 3199br (ν(OH)), 1640m (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)), 1593s (νas(OCO)), 1580s, 1514w, 1473s, 1433w, 1392w (νs(OCO)), 1329s, 1293m, 1231w, 1151w, 1058w, 1016w, 839w, 772m. UV-vis (diffuse reflectance, cm−1): 14409 and 11263.
N)), 1593s (νas(OCO)), 1580s, 1514w, 1473s, 1433w, 1392w (νs(OCO)), 1329s, 1293m, 1231w, 1151w, 1058w, 1016w, 839w, 772m. UV-vis (diffuse reflectance, cm−1): 14409 and 11263.
Synthesis of single crystals of 1. Pyrazole-3,5-dicarboxylic acid (0.5 mmol, 87 mg) in EtOH (3 mL) was carefully layered over the mixture solution containing Cu(NO3)2·3H2O (0.5 mmol, 121 mg) and 2,2′-dipyridylamine (0.5 mmol, 86 mg) in water and DMF (6 mL, 1:1 v/v) in 15 mL of glass vial. Then, the vial was sealed and allowed to stand undisturbed at room temperature. The green single crystals of 1 were obtained after 2 weeks. Yield: 30 mg (49%) based on copper salt. Anal. Calcd for Cu3C30H28N10O12: C, 39.54; H, 3.10; N, 15.37. Found: C, 40.42; H, 3.13; N, 15.55%. FT-IR peaks (KBr, cm−1): 3198br (ν(OH)), 1643m (ν(CN)), 1593s (νas(OCO)), 1583s, 1516w, 1479s, 1436w, 1393w (νs(OCO)), 1335s, 1295m, 1234w, 1152w, 1059w, 1017w, 840w, 775m. UV-vis (diffuse reflectance, cm−1): 14430 and 11263.
Optical detection of volatile amines
A 0.5 mL glass vial containing compound 1 (10 mg) was placed separately in a 10 mL glass vial containing a concentrated amine solution (3 mL) (ammonia solution 28%, methylamine (MA) solution 40%, ethylamine (EA) solution 70%, ethylenediamine (EDA), aniline (AL), benzylamine (BA), diethylamine (DEA), and triethylamine (TEA)) and then sealed at room temperature. The products were characterized by FT-IR and UV-vis diffuse reflectance spectroscopy, SEM, and PXRD techniques.
X-ray crystallographic study
The crystallographic study for 1 was carried out on a Bruker D8 Quest PHOTON 100 with a graphite-monochromated MoKα radiation at 298 K. The data frame were collected and processed using the SAINT routine in the APEX2 program.34,35 The diffracted absorption can be gathered by SADABS.36 The crystal structure was solved by the SHELXTL program via intrinsic phasing and refined by full-matrix least-squares on F2.37,38 All non-hydrogen atoms refined anisotropically and hydrogen atoms were located at geometrically calculated positions and refined isotropically in a riding manner. The crystallographic data, selected bond lengths, and angles for compound 1 are depicted in Tables 1 and 2.
Table 1 Crystallographic data for 1
| Compound 1 |
| R = ∑‖F0| − |Fc‖/∑|F0|. Rw = {∑[w(|F0| − |Fc|)]2/∑[w|F0|2]}1/2. |
| Formula |
Cu3C30H28N10O12 |
| Molecular weight |
911.24 |
| T (K) |
298(2) |
| Crystal system |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
7.5789(18) |
| b (Å) |
9.937(2) |
| c (Å) |
11.529(3) |
| α (deg) |
106.045(6) |
| β (deg) |
104.318(7) |
| γ (deg) |
99.588(6) |
| V (Å3) |
782.4(3) |
| Z |
1 |
| ρcald (g cm−3) |
1.934 |
| μ (Mo Kα) (mm−1) |
2.107 |
| Data collected |
2359 |
| Unique data (Rint) |
1465(0.1164) |
| R1a/wR2b [I > 2σ(I)] |
0.0894/0.2246 |
| R1a/wR2b [all data] |
0.1535/0.2598 |
| GOF |
1.071 |
| Max./min. electron density (e Å−3) |
1.072/−1.168 |
Table 2 The selected bond lengths (Å) and angles (°) for 1a
| Compound 1 |
| Symmetry code for 1: (i) 1 − x, 1 − y, 1 − z. |
| Cu1–N5i |
2.003(9) |
O3–Cu1–O3i |
180.00 |
| Cu1–N5 |
2.003(9) |
N5i–Cu1–O6 |
89.65(4) |
| Cu1–O3 |
1.989(7) |
N5–Cu1–O6 |
90.35(4) |
| Cu1–O3i |
1.989(7) |
O3–Cu1–O6 |
94.80(3) |
| Cu1–O6 |
2.519(7) |
O3i–Cu1–O6 |
85.20(3) |
| Cu1–O6i |
2.519(7) |
N1–Cu2–N4 |
168.6(4) |
| Cu2–N1 |
2.018(9) |
N1–Cu2–N2 |
87.8(4) |
| Cu2–N4 |
1.997(9) |
N4–Cu2–N2 |
101.9(4) |
| Cu2–N2 |
2.044(11) |
N1–Cu2–O1 |
90.7(3) |
| Cu2–O1 |
2.111(8) |
N4–Cu2–O1 |
79.5(3) |
| Cu2–O5 |
2.088(10) |
N2–Cu2–O1 |
120.3(4) |
| N5i–Cu1–N5 |
180.0(3) |
N1–Cu2–O5 |
92.2(4) |
| N5i–Cu1–O3 |
97.2(3) |
N4–Cu2–O5 |
88.5(4) |
| N5–Cu1–O3 |
82.8(3) |
N2–Cu2–O5 |
116.9(4) |
| N5i–Cu1–O3i |
82.8(3) |
O1–Cu2–O5 |
122.8(4) |
| N5–Cu1–O3i |
97.2(3) |
|
|
Results and discussion
Synthesis and characterization
Compound 1 was prepared at room temperature by two methods including the layering technique and the sonochemical method. Sonochemical synthesis was simply performed in a short time with a high yield. An obvious difference in the size of the crystalline products prepared from two methods was observed. According to the SEM images, the crystal size of 1 is about 30 μm and 6 μm for the layering technique and the sonochemical method, respectively (Fig. 1). To prove the phase purity of the synthesized samples, powder X-ray diffraction (PXRD) was performed (Fig. S1†). Both the experimental patterns are identical to that of the simulated 1 from the single-crystal X-ray data, indicating the purity of the crystalline phase of the synthesized products. The FT-IR spectrum of 1 (Fig. S2†) reveals a broad peak in the 3000–3500 cm−1 region, which is attributed to the stretching vibrations of the N–H and O–H groups from dpyam and water molecules, respectively.39–41 The medium peak at about 1640 cm−1 is attributed to the stretching vibration of the pyridine ring.42,43 The peaks at approximately 1600–1400 cm−1 are ascribed to the asymmetric and symmetric stretching vibrations of the carboxylate group for the pzdc ligand.44,45
 |
| | Fig. 1 SEM images of 1 prepared by (a) the layering method and (b) the sonochemical method. | |
The UV-vis diffuse reflectance spectrum of 1 (Fig. S3†) was performed. Compound 1 exhibits broad absorption bands at about 14400 and 11200 cm−1, which are attributed to distorted octahedral and trigonal bipyramidal geometries for Cu(II), respectively.46,47 The thermal stability of 1 was examined by thermogravimetric analysis (TGA) under an N2 atmosphere in the range of 35–750 °C. As shown in Fig. S4,† the TG curve displays a weight loss of 7.92% from 130 to 250 °C, corresponding to the release of four coordinated water molecules (calcd 7.91%), then its framework begins decomposing at 270 °C.
Crystal structure of [Cu3(pzdc)2(dpyam)2(H2O)4] (1)
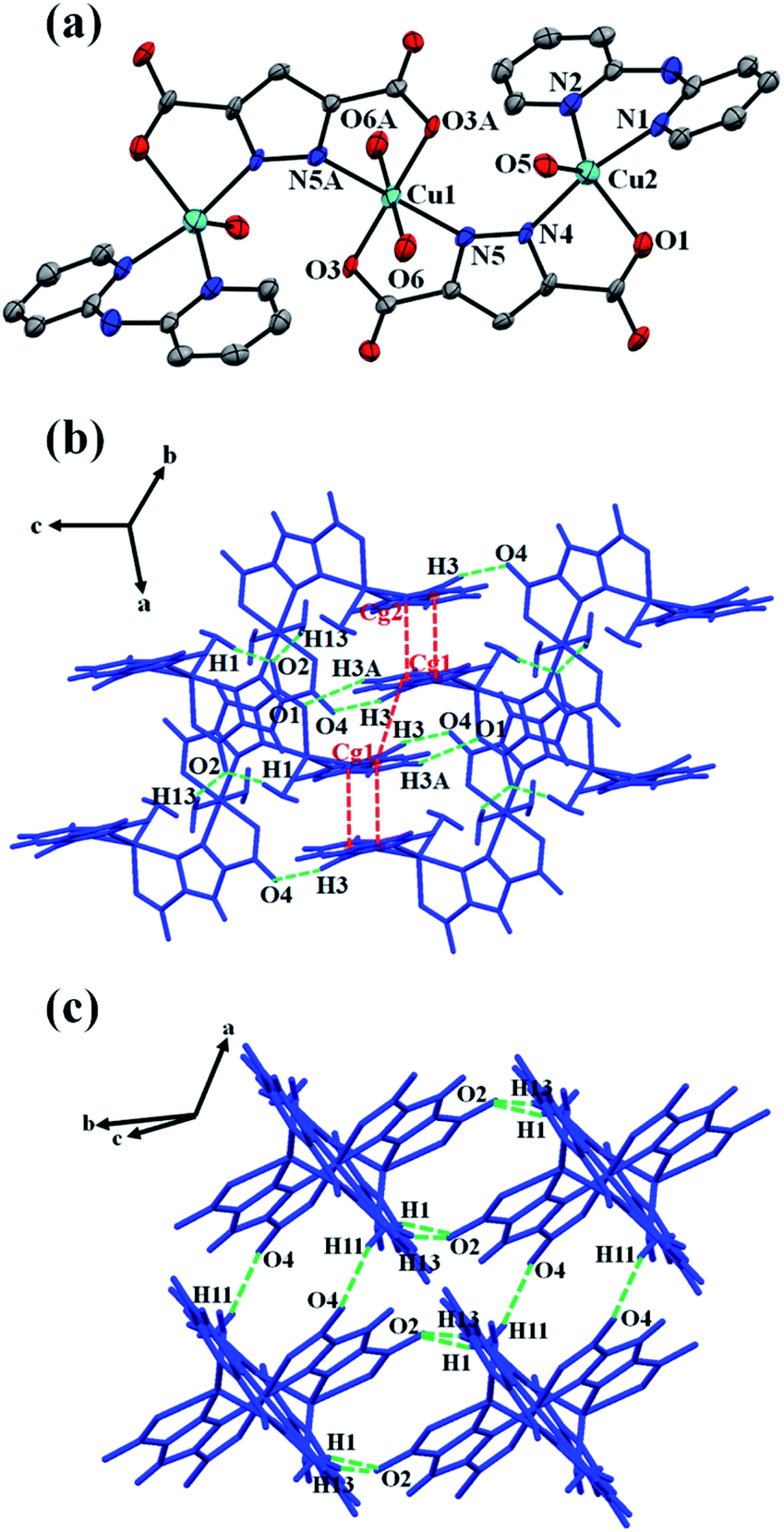
Single crystal X-ray diffraction analysis reveals that the asymmetric unit of 1 consists of two crystallographically independent Cu(II) ions (Cu1, Cu2), one pzdc3− ligand, one chelating dpyam ligand, and two coordinated water molecules. Each Cu1 ion lies on a crystallographic inversion center, showing a distorted octahedral geometry (Fig. 2a). The coordination sphere of the central Cu1 is completed by two carboxylate oxygen atoms (O3, O3A) and two pyrazole nitrogen atoms (N5, N5A) from two different pzdc3− ligands in the basal plane, and the axial position is occupied by two oxygen atoms (O6, O6A) from two coordinated water molecules. The terminal Cu2 shows distorted trigonal bipyramidal geometry (τ = 0.76, Addison's parameter τ = 0 for square pyramid and τ = 1 for trigonal bipyramid).48,49 The basal plane is composed of N2 from dpyam ligand, O1 from carboxylate group, and O5 from coordinated water molecule. The axial position is occupied by N1 from the dpyam ligand and the N4 of the pyrazole ring from pzdc3− ligand. The Cu–N and Cu–O distances fall in the range of 1.989(7)–2.111(8) Å. The elongated axial Cu1–O distance is 2.519(7) Å, indicating the presence of a common Jahn–Teller effect in the Cu(II) ion.50,51 Two pzdc3− ligands bridge Cu1 and Cu2 ions in a μ2-η2N,O, η2N′,O′ coordination mode, constructing the trinuclear Cu(II) unit. The Cu1⋯Cu2 distance via μ2-pzdc3− is 4.404(2) Å. The 3D supramolecular structure of 1 is constructed by intermolecular hydrogen bonding involving carboxylate oxygen atoms from pzdc3− as hydrogen acceptor and donor from dpyam ligand and water molecules (Table 3), and π–π stacking interactions between adjacent dpyam ligands with Cg⋯Cg separation of 3.707(8) and 3.614(8) Å, where Cg is the centroid of the pyridine's ring (Fig. 2b and c).
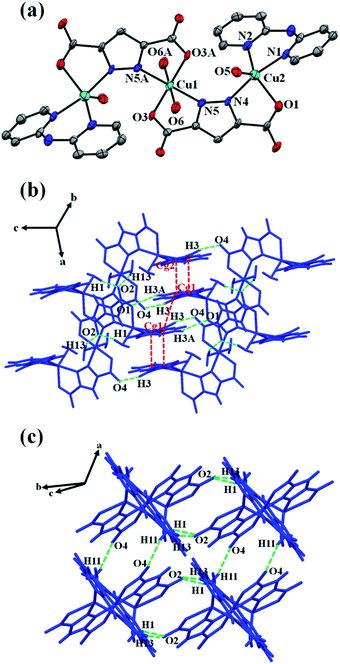 |
| | Fig. 2 (a) The trinuclear Cu(II) complex of 1 with atom labeling scheme. The ellipsoids are shown at 50% probability level. All hydrogen atoms are omitted for clarity (symmetry code: A = 1 − x, 1 − y, 1 − z). (b and c) 3D packing diagram of 1 built by hydrogen bonds and π–π stacking interactions. | |
Table 3 Intermolecular hydrogen bond length/Å and angles/° in 1a
| D–H⋯A |
d(D–H)/Å |
d(H⋯A)/Å |
d(D⋯A)/Å |
∠(DHA)/° |
| Symmetry code for 1: (i) x, y, 1 + z; (ii) 1 − x, −y, 1 − z; (iii) −1 + x, y, z; (iv) 1 − x, −y, 2 − z. |
| N3–H3⋯O4i |
0.86 |
2.06 |
2.895(14) |
165 |
| O5–H1⋯O2ii |
0.85 |
1.99 |
2.789(13) |
158 |
| O6–H11⋯O4iii |
0.90 |
2.09 |
2.946(14) |
157 |
| O6–H13⋯O2ii |
0.90 |
1.98 |
2.791(13) |
150 |
| C3–H3A⋯O1iv |
0.93 |
2.54 |
3.280(16) |
137 |
De-/rehydration induced reversible structural transformation
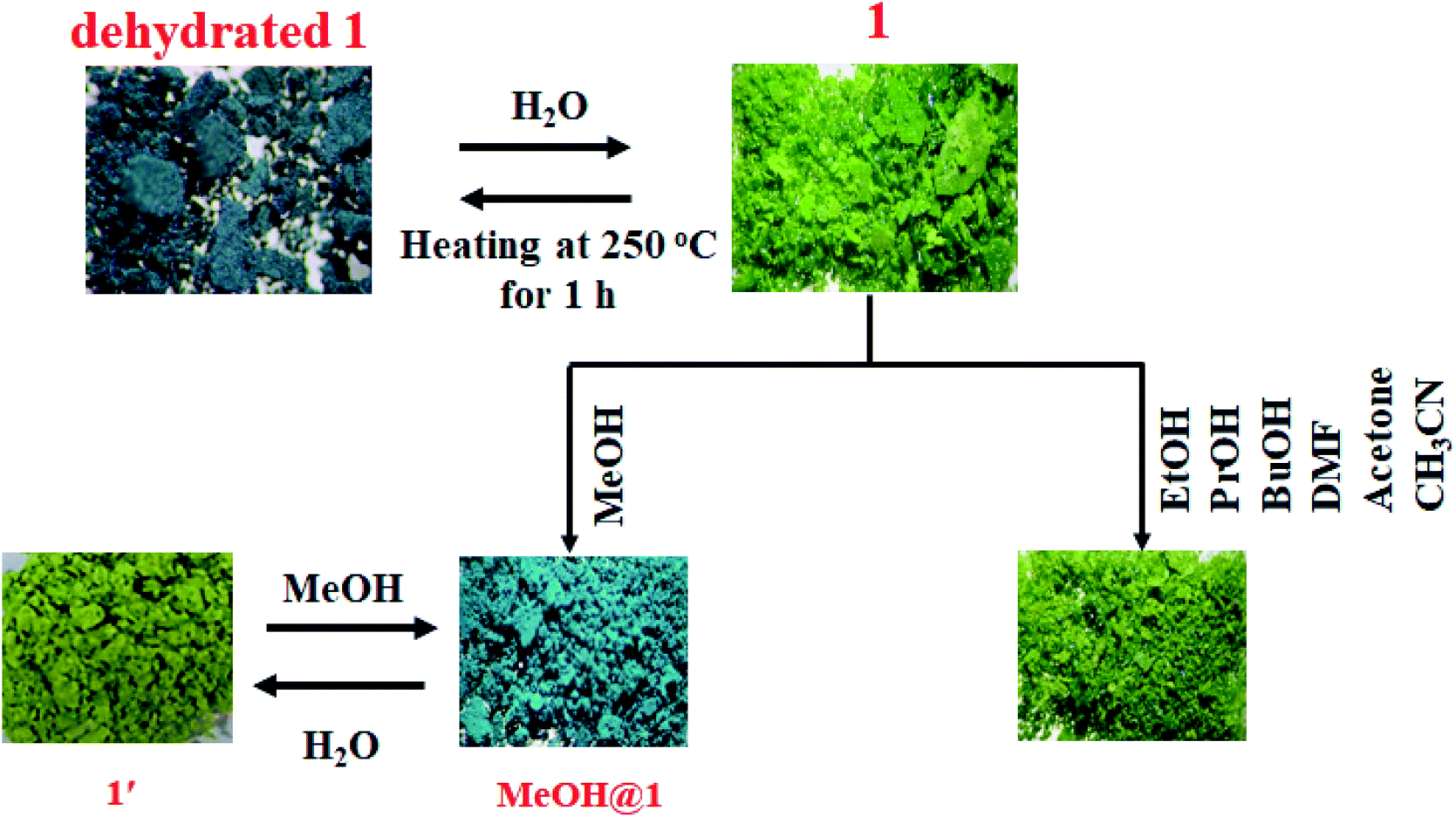
To study the water-induced structural transformation of 1, the green crystals of 1 were heated at 250 °C for 1 h (denoted as dehydrated 1). The green crystals of 1 turned dark cyan in color and lost their crystallinity (Fig. 3). The UV-vis diffuse reflectance spectrum of dehydrated 1 exhibits blue-shifted absorption broadband with λmax of about 14815 cm−1 which agrees with the alteration of color from green to dark cyan (Fig. S5†). The FT-IR spectrum of dehydrated 1 exhibits a strong broad peak at about 3000–3500 cm−1, corresponding to the vibration of N–H of dpyam.52,53 Compared with the FT-IR spectrum of 1, the peak at 428 cm−1 is assigned to ν(Cu–Owater) and it disappears after the dehydration process (Fig. S6†).54,55 Also, the TG curve was used to confirm that the coordinated water molecules are completely removed after heating. The dehydrated 1 is stable up to 270 °C (Fig. S7†). As shown in Table S1,† elemental analysis indicates that the chemical composition of dehydrated 1 corresponds to [Cu3(pzdc)2(dpyam)2] (Anal. Calcd for Cu3C30H20N10O8: C, 42.94; H, 2.40; N, 16.69%. Found: C, 42.88; H, 2.26; N, 16.38%).
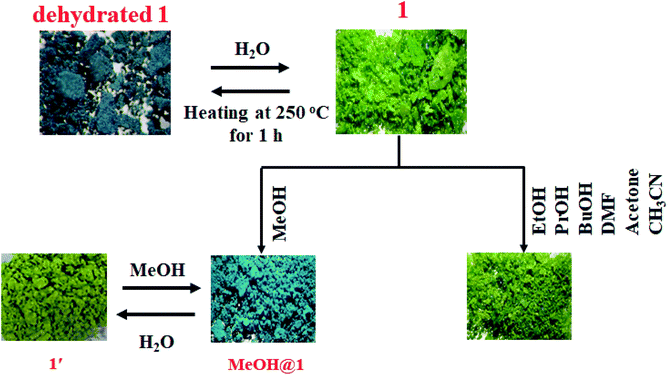 |
| | Fig. 3 Color responses of 1 induced by the dehydration and rehydration process and different solvents. | |
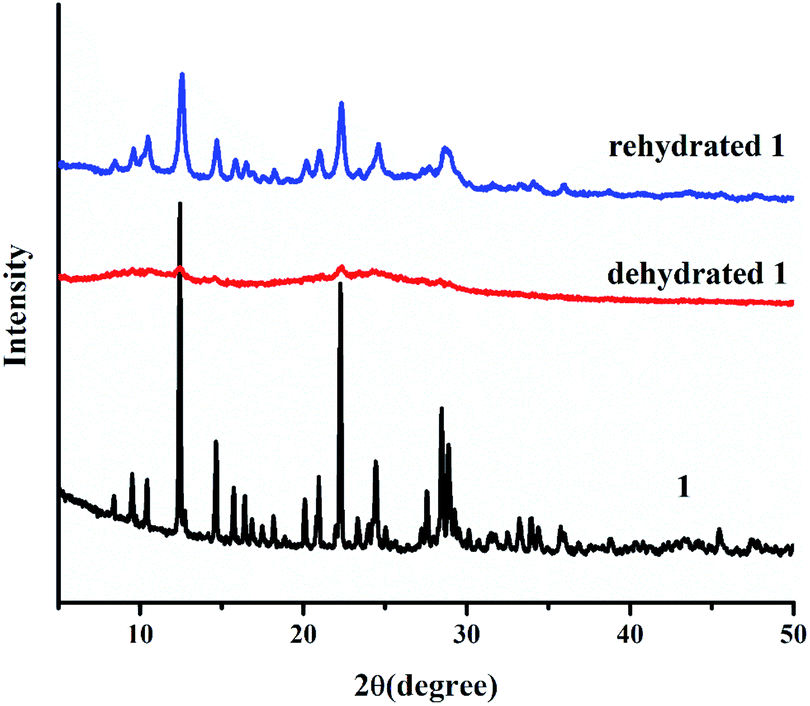
According to the PXRD pattern (Fig. 4), the dehydrated 1 significantly loses its crystallinity and becomes amorphous. Furthermore, the initial crystalline phase of 1 can be recovered by soaking dehydrated 1 in water for 2 days, as confirmed by FT-IR and UV-vis diffuse reflectance spectra, TGA, CHN, and PXRD. According to the above results, water molecules play a key role in the structural stability of 1 because the 3D supramolecular structure of 1 is constructed by hydrogen bonds between coordinated water molecules and organic ligands. Therefore, the packing structure of 1 collapsed after the loss of coordinated water molecules, leading to an amorphous phase. After the rehydration process, the water molecules can cause the transformation of the amorphous phase to the initial crystalline phase through intermolecular hydrogen bonding, which occurs through the dissolution process. This result indicates the flexibility of the supramolecular framework of 1.
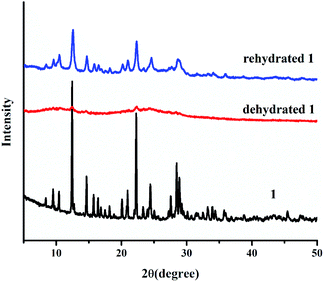 |
| | Fig. 4 PXRD patterns of 1 after the dehydration and rehydration process. | |
Solvent-induced structural transformations
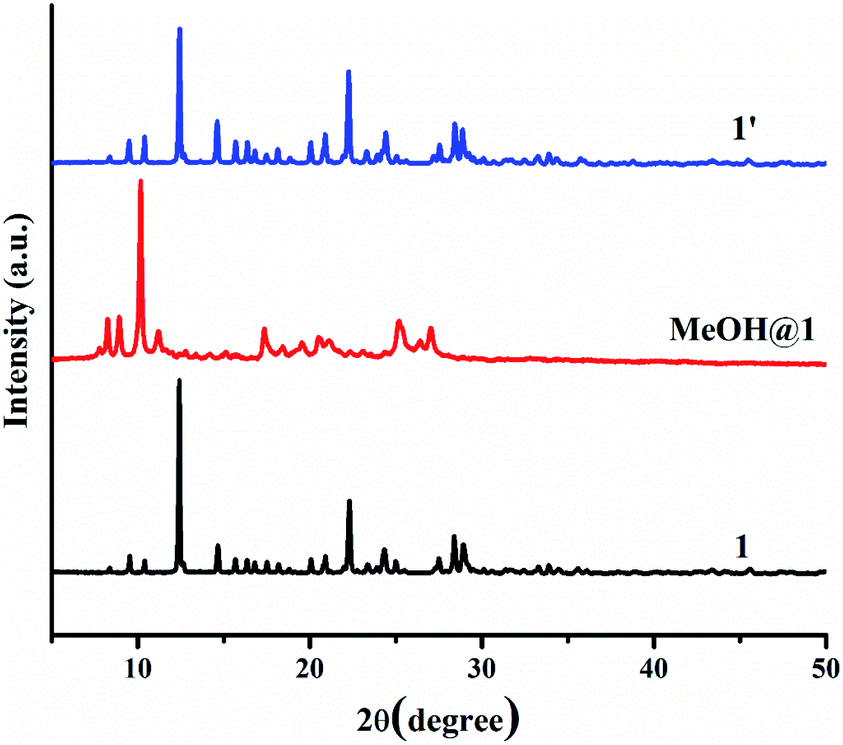
Two 5-coordinated Cu(II) centers in a trinuclear unit of 1 are weakly coordinated by a water molecule; therefore, these Cu(II) sites can be occupied or replaced by guest molecules, leading to the structural transformation and change of color. To explore the influence of the guest molecules on the change in the color of 1, the green crystals of 1 were immersed into different solvents (methanol (MeOH), ethanol (EtOH), propanol (PrOH), butanol (BuOH), dimethylformamide (DMF), acetone, and acetonitrile). The color of 1 was changed from green to greenish blue in MeOH media (denoted as MeOH@1) after 20 minutes. In comparison, compound 1 displays a negative response when exposed to methanol vapor. Compound 1 shows no significant color change in other solvents after 1 day. The FT-IR spectrum of MeOH@1 shows strong broad peak in the range of 3000–3550 cm−1, which are assigned to the stretching vibration of O–H from water and methanol. The appearance of vibrational peak at 1075 cm−1 corresponds to the ν(C–O) of methanol (Fig. S8†).56–58 The UV-vis diffuse reflectance spectrum of MeOH@1 shows the blue-shifted broadband with λmax of about 14250 cm−1, which agrees with the alteration in the color from green to greenish blue. This indicates that the coordination environment of the Cu(II) center is changed (Fig. S9†). The PXRD pattern of MeOH@1 significantly differs from that of the original 1, indicating guest-induced structural transformation (Fig. 5). In addition, the SEM images of 1 and MeOH@1 show the different morphologies and sizes that can confirm the structural transformation of 1 occurring through the dissolution process under methanol media. It is worth mentioning that 1 does not exhibit a change in the color and structural transformation upon contact with other common organic solvents, which corresponds to the PXRD patterns (Fig. S10†). Therefore, the structural transformation with solvochromism is selective toward methanol since methanol has a tiny size and strong coordination ability.59 Thus, it is easily accessible to the inner coordination sphere, causing the important structural change.60,61 Also, 5-coordinated Cu(II) centers in a trinuclear unit have vacant sites that are possibly possessed by methanol molecule. To study the reversibility, MeOH@1 was immersed in water for 6 h, after which the color of MeOH@1 was restored to the color of the initial 1 (denoted as 1′). The reversible structural transformation of MeOH@1 is confirmed by PXRD, SEM, and FT-IR as well as UV-vis diffuse reflectance spectrum (Fig. 5, 6, S8, and S9†).
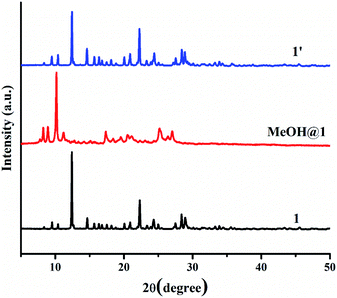 |
| | Fig. 5 PXRD patterns of 1, MeOH@1, and 1′. | |
 |
| | Fig. 6 SEM images of 1 and MeOH@1, and 1′. | |
Optical detection of volatile amines
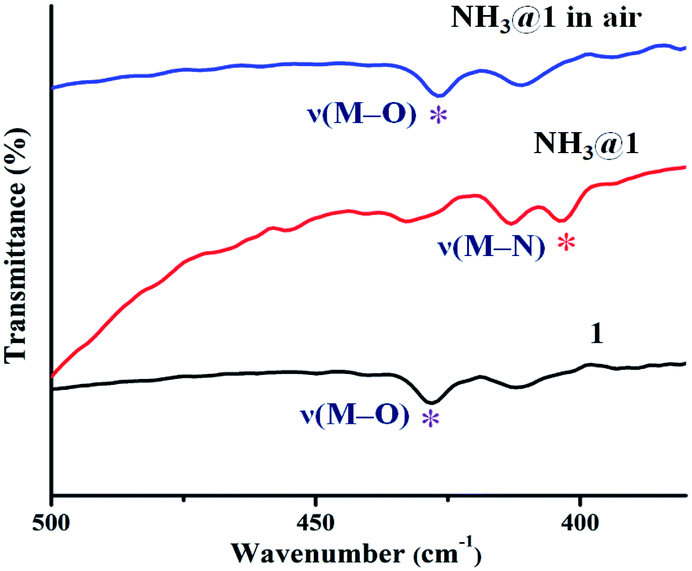
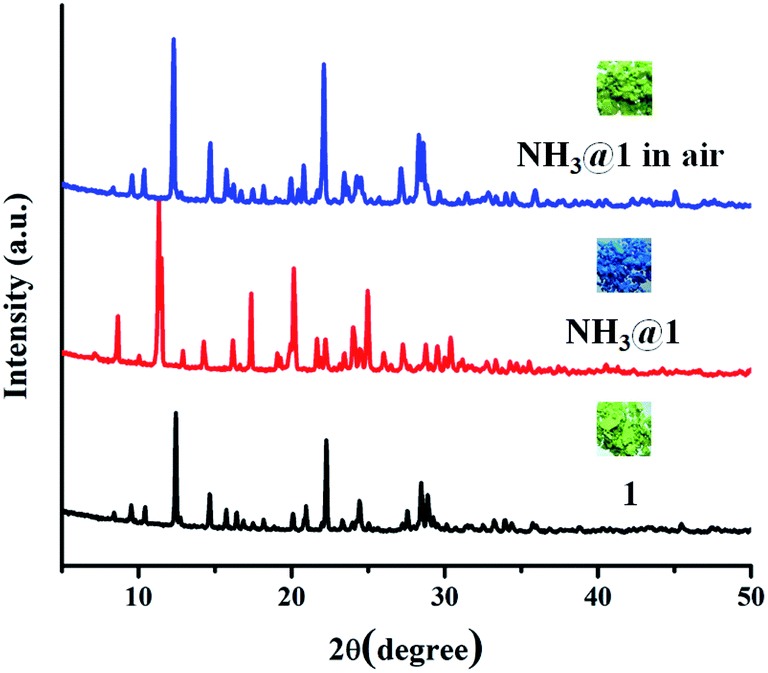
The green powder of 1 was exposed to different saturated amine vapors (Fig. S11†) including ammonia, primary amines (methylamine (MA), ethylamine (EA), and ethylenediamine (EDA), aniline (AL), and benzylamine (BA)), secondary amines (diethylamine, DEA) and tertiary amines (triethylamine, TEA). Compound 1 exhibits different responses to various amine vapors, as shown in Fig. 7. The green color of 1 became light blue completely upon exposure to ammonia within 30 min and turned indigo when exposing to methylamine within 45 min, became blue when exposing to ethylamine for 3 h, and turned violet upon exposure to ethylenediamine for 8 h. However, no appreciable color response occurs in other amines. To verify the selective vapochromic mechanism of 1, the FT-IR and UV-vis diffuse reflectance spectra, SEM, and PXRD patterns of the exposed samples were investigated. According to the FT-IR spectra, NH3@1, MA@1, EA@1, and EDA@1, the characteristic peak of ν(M–Owater) at 428 cm−1 disappears and new peaks for each product is grown at 403 cm−1 for NH3@1, 402 cm−1 for MA@1, 419 cm−1 for EA@1, and 403 cm−1 for EDA@1, which can be assigned to the vibration band of M–Namine (Fig. 8 and S12–S15†).54,62–64 The results indicate the coordination of amine molecules to Cu(II) centers. The UV-vis diffuse reflectance spectra of NH3@1, MA@1, EA@1, and EDA@1 (Fig. S12–S15†) show blue-shifted absorption broadband at about 14770–17180 cm−1, indicating that the coordination environment around the Cu(II) centers in 1 is changed, affecting the discoloration of 1. The PXRD patterns of the vapochromic products are different from that of original 1, implying guest-induced structural transformation (Fig. 9 and S12–S15†). The structural transformation of the amine-treated products was also characterized by SEM (Fig. 10). The products show the considerable differences in the morphology and size, further confirming the structural transformation of 1 via the dissolution process under the saturated ammonia, MA, EA, and EDA vapors. As described above, compound 1 shows different color responses to ammonia and primary amines (methylamine, ethylamine, and ethylenediamine), which possess relatively small alkyl groups, while other bulkier primary (aniline and benzylamine), secondary amines (diethylamine), and tertiary amines (triethylamine) give a negative response (Fig. S16†). It seems that the selective response is correlated with the molecular size, especially the size of the substituents around the N atom. The small amine molecules could interact with the structure of 1 through hydrogen bonds (N–H⋯O, N–H⋯N, O–H⋯N, and C–H⋯N) and weakly bind with Cu(II) ion within the internal coordination sphere, leading to structural transformation. Meanwhile, interactions between the structure of 1 and the bulkier amines are sterically restricted, especially tertiary amines that do not contain a hydrogen donor site. Moreover, the different vapochromic response times are potentially attributed to the different vapor pressures of amines (635 hPa (20 °C), 370 hPa (25 °C), 460 hPa (20 °C), 12 hPa (20 °C), 0.5 hPa (20 °C), 1.2 hPa (20 °C), 250 hPa (20 °C), and 69 hPa (20 °C) for ammonia, methylamine, ethylamine, ethylenediamine, aniline, benzylamine, diethylamine, and triethylamine, respectively).65 To study the reversibility, the amine-treated products can be switched to the initial green after standing in air for 2 days for NH3@1, and a week for MA@1 and EA@1. On the other hand, the recovery of EDA@1 cannot be achieved. The FT-IR and UV-vis diffuse reflectance spectra, and PXRD profiles of NH3@1 in air, and MA@1 in air, and EA@1 in air are identical with that of 1, confirming the reversible structural transformation. Moreover, the SEM images of NH3@1 in air, MA@1 in air, and EA@1 in air display the same morphology and size with that of original 1 (Fig. 10). Meanwhile, EDA@1 exhibits irreversible structural transformation due to the strong chelating ability of EDA.66–68
 |
| | Fig. 7 Photographs of 1 after exposure to different amine vapors. | |
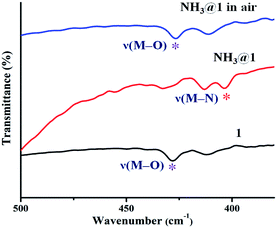 |
| | Fig. 8 FT-IR spectra of 1, NH3@1, and NH3@1 in air. | |
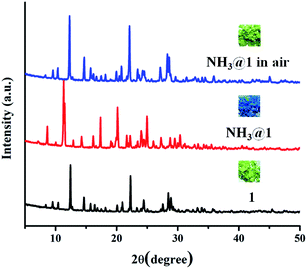 |
| | Fig. 9 PXRD patterns of 1, NH3@1, and NH3@1 in air. | |
 |
| | Fig. 10 SEM images of 1 (sonochemical method), NH3@1, NH3@1 in air, MA@1, MA@1 in air, EA@1, EA@1 in air, and EDA@1. | |
Test strip for volatile amine detection
To produce a convenient test strip for qualitative volatile amine detection, we prepared a test paper denoted as 1-paper. 1-paper can be easily prepared by adding filter paper into the mixture solution of Cu(NO3)2·3H2O, H3pzdc, and dpyam, followed by sonication for 30 min; the green 1-paper was obtained. According to the SEM image of 1-paper (Fig. S17†), the microcrystals of 1 were coated throughout the filter paper compared with the blank filter paper. As shown in Fig. 11, the green color of 1-paper became light blue, indigo, blue, and violet when exposed to saturated ammonia, methylamine, ethylamine, and ethylenediamine vapors for 10 min. On the other hand, 1-paper is still silent to sterically hindered amines including aniline, benzylamine, diethylamine, and triethylamine after 1 day. The change in the color of 1-paper correlates with the UV-vis diffuse reflectance spectra of amines@1-paper, which clearly exhibits blue-shifted absorption broadband (Fig. S18†). The results suggest that both 1-paper and 1 show similar selective response to ammonia, methylamine, ethylamine, and ethylenediamine. In addition, the vapochromic response time of 1-paper is faster than that of a bulk sample of 1 under saturated amine vapors because it has a higher surface area and better coverage on a filter paper. To study the reusability, NH3@1-paper, MA@1-paper, EA@1-paper, and EDA@1-paper were stood in air. The original green paper can be recovered after 1 day for NH3@1-paper and 5 days for MA@1-paper and EA@1-paper, while EDA@1-paper shows no reversibility. The UV-vis diffuse reflectance spectra of NH3@1-paper, MA@1-paper, and EA@1-paper in air are also reversible (Fig. S18†).
 |
| | Fig. 11 Photographs of 1-paper after exposure to different amine vapors. | |
Conclusions
The 3D supramolecular structure based on open coordination sites of trinuclear Cu(II) unit was synthesized and structurally characterized. Compound 1 exhibits color and structural changes as a result of the dehydration process and methanol media. The pristine state can be recovered by water-induced structural rearrangement. Also, 1 shows reversible differentiable detection and noticeable color responses to different types of saturated volatile amines, and the mechanism can be attributed to the guest-induced structural transformation. The selective and discriminative vapochromism towards amines is due to the different molecular sizes of amines, the steric hindrance around the N atom, and the coordination ability of amine. Furthermore, 1 was easily deposited onto filter paper by sonochemical synthesis for 30 min, which can be possibly used as a portable test paper for discriminative naked-eye detection of saturated amine vapors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Funding for this work is provided by The Thailand Research Fund: Grant No. RSA6180060, the Thailand Research Fund through the Royal Golden Jubilee (RGJ) PhD Program (Grant no. PHD/0056/2560), and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation.
References
- H. Golchoubian, G. Moayyedi and N. Reisi, Spectrochim. Acta, Part A, 2015, 138, 913–924 CrossRef CAS PubMed.
- W. Linert, Y. Fukuda and A. Camard, Coord. Chem. Rev., 2001, 218, 113–152 CrossRef CAS.
- X.-Y. Dong, M. Zhang, R.-B. Pei, Q. Wang, D.-H. Wei, S.-Q. Zang, Y. Fan and T. C. W. Mak, Angew. Chem., Int. Ed., 2016, 55, 2073–2077 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, S. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
- R. Nazari, H. Golchoubian and G. Bruno, J. Coord. Chem., 2018, 71, 2510–2525 CrossRef CAS.
- P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- J. D. Schneider, B. A. Smith, G. A. Williams, D. R. Powell, F. Perez, G. T. Rowe and L. Yang, Inorg. Chem., 2020, 59, 5433–5446 CrossRef CAS PubMed.
- A. A. Opalade, C. J. Gomez-Garcia and N. Gerasimchuk, Cryst. Growth Des., 2019, 19, 678–693 CrossRef CAS.
- X.-Y. Hu, T. Xiao, C. Lin, F. Huang and L. Wang, Acc. Chem. Res., 2014, 47, 2041–2051 CrossRef CAS PubMed.
- X.-Q. Wu, Y. Xie, J.-H. Liu, T. He, Y.-Z. Zhang, J. Yu, X.-J. Kong and J.-R. Li, J. Mater. Chem. A, 2019, 7, 25254–25257 RSC.
- H. Zhang, Y. Lu, Z.-m. Zhang and E.-b. Wang, Inorg. Chem. Commun., 2012, 17, 9–12 CrossRef CAS.
- R. Horikoshi, Y. Funasako, T. Yajima, T. Mochida, Y. Kobayashi and H. Kageyama, Polyhedron, 2013, 50, 66–74 CrossRef CAS.
- V. I. Nikolayenko, L. M. v. Wyk, O. Q. Munro and L. J. Barbour, Chem. Commun., 2018, 54, 6975–6978 RSC.
- Q. Sui, P. Li, N.-N. Yang, T. Gong, R. Bu and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2018, 10, 11056–11062 CrossRef CAS PubMed.
- Z.-P. Dong, J.-J. Zhao, P.-Y. Liu, Z.-L. Liu and Y.-Q. Wang, New J. Chem., 2019, 43, 9032–9038 RSC.
- H. M. Elbardisy, C. W. Foster, J. Marron, R. E. Mewis, O. B. Sutcliffe, T. S. Belal, W. Talaat, H. G. Daabees and C. E. Banks, ACS Omega, 2019, 4, 14439–14450 CrossRef CAS PubMed.
- D. S. Chormey, B. T. Zaman, E. Maltepe, Ç. Büyükpınar, A. E. Bulgurcuoğlu, F. Turak, F. A. Erulaş and S. Bakırdere, J. Anal. Chem., 2020, 75, 1330–1334 CrossRef.
- K. Debsharma, J. Santhi, B. Baire and E. Prasad, ACS Appl. Mater. Interfaces, 2019, 11, 48249–48260 CrossRef CAS PubMed.
- I. M. Apetrei and C. Apetrei, Sens. Actuators, B, 2016, 234, 371–379 CrossRef CAS.
- U. Cremer, S. Disch and U. Ruschewitz, Z. Anorg. Allg. Chem., 2004, 630, 2304–2310 CrossRef CAS.
- S.-Z. Zhan, X. Jiang, J. Zheng, X.-D. Huang, G.-H. Chen and D. Li, Dalton Trans., 2018, 47, 3679–3683 RSC.
- O. Kysliak and J. Beck, J. Solid State Chem., 2013, 203, 120–127 CrossRef CAS.
- W.-F. Liu, Z.-M. Su, Z. Jia and G.-Y. Yang, J. Cluster Sci., 2019, 30, 1139–1144 CrossRef CAS.
- J.-S. Zhou, J. Cai, L. Wang and S.-W. Ng, Dalton Trans., 2004, 1493–1497 RSC.
- K.-Y. Wang, S. Zhang, H.-W. Liu, L. Cheng and C. Wang, Inorg. Chem., 2019, 58, 12832–12842 CrossRef CAS PubMed.
- B.-D. Wu, Z.-N. Zhou, F.-G. Li, L. Yang, T.-L. Zhang and J.-G. Zhang, New J. Chem., 2013, 37, 646–653 RSC.
- A. Rodríguez-Diéguez, M. E. López-Viseras, J. E. Perea-Buceta, A. J. Mota and E. Colacio, Inorg. Chim. Acta, 2012, 385, 73–80 CrossRef.
- L. Li, Z.-M. Tu, Y. Hua, X.-N. Li, H.-Y. Wang and H. Zhang, Inorg. Chem. Front., 2019, 6, 3077–3082 RSC.
- X.-N. Li, L. Li, H.-Y. Wang, C. Fu, J.-W. Fu, Y.-N. Sun and H. Zhang, Dalton Trans., 2019, 48, 6558–6563 RSC.
- J. Suebphanpho, S. Wannapaiboon, S. Youngme and J. Boonmak, Cryst. Growth Des., 2020, 20, 7439–7449 CrossRef CAS.
- L. Pan, X. Huang and J. Li, J. Solid State Chem., 2000, 152, 236–246 CrossRef CAS.
- J.-H. Yang, S.-L. Zheng, X.-L. Yu and X.-M. Chen, Cryst. Growth Des., 2004, 4, 831–836 CrossRef CAS.
- APEX2, Bruker AXS Inc., Madison, WI, 2014 Search PubMed.
- SAINT 4.0 Software Reference Manual, Siemens Analytical X-Ray Systems Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- Y. Qiu, H. Deng, S. Yang, J. Mou, C. Daiguebonne, N. Kerbellec, O. Guillou and S. R. Batten, Inorg. Chem., 2009, 48, 3976–3981 CrossRef CAS PubMed.
- A. Gładysiak, T. N. Nguyen, J. A. R. Navarro, M. J. Rosseinsky and K. C. Stylianou, Chem.–Eur. J., 2017, 23, 13602–13606 CrossRef PubMed.
- S. Packiaraj, A. Pushpaveni, S. Govindarajan and J. M. Rawson, CrystEngComm, 2016, 18, 7978–7993 RSC.
- L. Chen, Z. Bai, L. Zhu, L. Zhang, Y. Cai, Y. Li, W. Liu, Y. Wang, L. Chen, J. Diwu, J. Wang, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32446–32451 CrossRef CAS PubMed.
- B. Tan, C. Chen, L.-X. Cai, Y.-J. Zhang, X.-Y. Huang and J. Zhang, Inorg. Chem., 2015, 54, 3456–3461 CrossRef CAS PubMed.
- Bhaskaran, M. Trivedi, A. K. Yadav, G. Singh, A. Kumar, G. Kumar, A. Husain and N. P. Rath, Dalton Trans., 2019, 48, 10078–10088 RSC.
- R. Rajak, M. Saraf and S. M. Mobin, Inorg. Chem., 2020, 59, 1642–1652 CrossRef CAS PubMed.
- J. Boonmak, S. Youngme, N. Chaichit, G. A. v. Albada and J. Reedijk, Cryst. Growth Des., 2009, 9, 3318–3326 CrossRef CAS.
- V. Shivaiah and S. K. Das, Inorg. Chem., 2005, 44, 8846–8854 CrossRef CAS.
- A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- F. Klongdee, J. Boonmak and S. Youngme, Dalton Trans., 2017, 46, 4806–4815 RSC.
- F. Klongdee, J. Boonmak, B. Moubaraki, K. S. Murray and S. Youngme, Polyhedron, 2017, 126, 8–16 CrossRef CAS.
- P. King, R. Clérac, C. E. Anson, C. Coulon and A. K. Powell, Inorg. Chem., 2003, 42, 3492–3500 CrossRef CAS PubMed.
- N. H. Murray, G. A. Farnum and R. L. LaDuca, Z. Anorg. Allg. Chem., 2013, 639, 2162–2170 CrossRef CAS.
- C. S. Devi, D. A. Kumar, S. S. Singh, N. Gabra, N. Deepika, Y. P. Kumar and S. Satyanarayana, Eur. J. Med. Chem., 2013, 64, 410–421 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons, Inc., 2009 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons, Inc., 2009 Search PubMed.
- C. Lucarelli, S. Galli, A. Maspero, A. Cimino, C. Bandinelli, A. Lolli, J. V. Ochoa, A. Vaccari, F. Cavani and S. Albonetti, J. Phys. Chem. C, 2016, 120, 15310–15321 CrossRef CAS.
- M.-r. Han, S.-d. Li, L. Ma, B. Yao, S.-S. Feng and M.-l. Zhu, Acta Crystallogr., Sect. C: Struct. Chem., 2019, 75, 1220–1227 CrossRef CAS PubMed.
- G. Boehm and M. Dwyer, J. Chem. Educ., 1981, 58, 809–811 CrossRef CAS.
- R. Díaz-Torres and S. Alvarez, Dalton Trans., 2011, 40, 10742–10750 RSC.
- G. Mehlana, G. Ramon and S. A. Bourne, CrystEngComm, 2013, 15, 9521–9529 RSC.
- M. Arıcı, O. Z. Yeşilel and M. Taş, Dalton Trans., 2015, 44, 1627–1635 RSC.
- M. S. Khan, M. Khalid, M. S. Ahmad, M. Shahid and M. Ahmad, J. Struct. Chem., 2019, 60, 1833–1841 CrossRef CAS.
- R. P. Sharma, S. Kumar, J. Kumar, P. Venugopalan and T. Aree, Polyhedron, 2017, 126, 245–251 CrossRef CAS.
- R. P. Sharma, A. Saini, S. Kumar, J. Kumar, P. Venugopalan, V. S. Gondil, S. Chhibber and T. Aree, Polyhedron, 2017, 123, 430–440 CrossRef CAS.
- W. Baden, Chemicals and Reagents, Merck KGaA, Darmstadt, 2011 Search PubMed.
- C. R. Maldonado, M. Quirós and J. M. Salas, Polyhedron, 2008, 27, 2779–2784 CrossRef CAS.
- L.-W. He, B.-Z. Lin, X.-Z. Liu, X.-F. Huang and Y.-L. Feng, Solid State Sci., 2008, 10, 237–243 CrossRef CAS.
- Z.-G. Han, Q.-H. Hao, Y.-N. Wang, J.-J. Wu and X.-L. Zhai, J. Cluster Sci., 2010, 21, 713–724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures for the FT-IR and UV-vis diffuse reflectance spectra, TGA curves, and PXRD pattern. CCDC 2062214. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01151k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*