
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupporting effects of a N-doped carbon film electrode on an electrodeposited Ni@Ni(OH)2 core–shell nanocatalyst in accelerating electrocatalytic oxidation of oligosaccharides†
Shunsuke Shiba a,
Saki Ohtab,
Kazuya Ohtanib,
Shota Takahashib,
Dai Katoc and
Osamu Niwa*b
a,
Saki Ohtab,
Kazuya Ohtanib,
Shota Takahashib,
Dai Katoc and
Osamu Niwa*b
aDepartment of Materials Science and Biotechnology, Graduate School of Science and Engineering, Ehime University, 3 Bunkyo-cho, Matsuyama, Ehime 790-8577, Japan
bAdvanced Science Research Laboratory, Saitama Institute of Technology, 1690 Fusaiji, Fukaya, Saitama 369-0293, Japan. E-mail: niwa@sit.ac.jp; Fax: +81-48-585-6896; Tel: +81-48-585-6304
cHealth and Medical Research Institute, National Institute of Advanced Industrial Science and Technology, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8566, Japan
First published on 9th April 2021
Abstract
The supporting effect of a N-doped carbon film induced superior crystallinity in electrodeposited Ni@Ni(OH)2 core–shell nanoparticles. This improvement resulted in a much higher regeneration rate of catalytic sites (NiOOH), leading to higher oxidation currents of sugars. Also, the overpotential of the maltopentaose (G5) oxidation reaction decreased significantly, probably due to the effect of the electrostatic interaction between NPs and G5.
Nickel-based materials have been attracting wide interest due to their excellent electrocatalytic activity.1 In particular, electrochemically generated nickel oxyhydroxide (NiOOH) in alkaline electrolyte is electrocatalytically active toward oxidation of organic molecules such as urea,2 alcohols,3 sugars,4 and so on.5,6 These electrocatalytic reactions are very important for a variety of technologies including energy devices, electrosynthesis, and electroanalysis.
A well-established strategy to improve the electrocatalytic activity is to nanoscale a metal component.5 Recently, interest has grown in the supporting effect (matrix effect) as another novel strategy to boost electrocatalytic activity and stability. The supporting substrate's influence on the electrocatalytic reaction can be ascribed mainly to the charge transfer between metal and support (metal–support interaction), and/or to electrostatic interaction between a charged nanocatalyst and target molecules.7
In particular, metal–support interactions between noble metal nanomaterials (such as Pt,8–10 Pd,10,11 Ir12) and N-doped carbon support have been intensively studied mainly with the aim of improving fuel cell performance. More recently, those of Ni counterparts have begun to attract attention due to the strong coordination interaction between the Ni component and defects at the N-doped carbon, which prevent the Ni nanoparticles from detaching, and utilized to synthesize single atom electrocatalysts.13 Some reports have investigated the combination of Ni nanocatalysts and N-doped carbon support applied to the electrocatalytic oxidation of small organic molecules. However, they did not elucidate N-doping effects and, importantly, none of these studies reported on the overpotential reduction of electrocatalytic reaction, as far as we know.14–16
Here, we found that N-doping of carbon film can much improve electrocatalytic performance parameters such as the turnover rates and oxidation potentials of sugars. Electrodeposition was adopted to prepare Ni@Ni(OH)2 core–shell nanoparticles (Ni@Ni(OH)2-NP) with similar size distributions and morphologies on untreated (C) and N2 plasma-treated carbon (N–C) film electrodes. The Ni@Ni(OH)2-NPs on the N–C exhibited much higher rates of electrochemical NiOOH formation, resulting in high turnover rates of electrocatalytic oxidation of glucose and oligosaccharide (maltopentaose, denoted as G5). This, in turn, significantly increased the electrocatalytic oxidation current. Even more interestingly, we observed significantly decreased overpotential of the G5 electro–oxidation reaction when Ni@Ni(OH)2-NP/N–C was used in a strong alkaline solution (pH = 12.7).
To elucidate the chemical state and check the composition of the electrode surface, we performed XPS analysis and water droplet contact angle measurements, as summarized in Table 1. After N2 plasma treatment, the nitrogen concentration increased from 0.3 to 1.7 at%, indicating that the net 1.4 at% of N was successfully introduced to the carbon surface. In contrast, the atomic concentration of O increased from 5.7 to 12.2 at%, which may be derived from the air remaining in the chamber. As a result, the water droplet contact angle decreased significantly from 86.0 to 7.8 degrees (Fig. S1†), indicating much improved hydrophilicity by N2 plasma. The N 1s peak was deconvoluted to four major N-functionalities (Fig. S2† and Table 1). The highest fraction of pyrrolic-N (36%) was observed which is hydrogen-terminated atoms incorporated in the pentagonal ring. Almost the same content of amine/amide N (34%) was observed. The 26% of graphitic-N was also estimated. In this report, graphitic N means either noncharged substituted-N or positively charged quaternary-N,9,17 although some reports attribute these two functionalities to different binding energies, the descriptions of which are inconsistent among the reports.8,10 Compared with these three functionalities, the fraction of pyridinic-N is lower (6%) which is sp2-hybridized N bonded to two sp2 hybridized carbons.
| Atomic conc.% | N 1s peak deconvolution | |||||
|---|---|---|---|---|---|---|
| C | N–C | Functional group | B.E./eV | FWHM/eV | Peak area% | |
| C 1s | 91.4 | 82.9 | Pyridinic N | 398.5 | 1.1 | 6 |
| O 1s | 5.7 | 12.2 | Amine/amide N | 399.3 | 1.1 | 34 |
| N 1s | 0.3 | 1.7 | Pyrrolic N | 400.1 | 1.1 | 36 |
| Si 2p | N.D. | 1.2 | Graphitic N (substituted N or quaternary N) | 401.1 | 1.1 | 25 |
| Ar 2p | 2.6 | 1.9 | ||||
| Contact angle | 86.0° | 7.8° | ||||
Fig. 1 shows FE-SEM, HR-TEM, and HAADF-STEM-EDS images of the NPs electrodeposited on C and N–C film electrodes. As shown in Fig. 1a and e, deposited NPs had similar size distributions which was achieved by controlling the electrodeposition potential (Fig. S3†). All the observed NPs exhibited a core–shell morphology having an O-rich shell around the Ni-rich core. These results indicate that the electrochemical pretreatment of NPs, the 10th repeated potential sweeping in 0.1 M NaOH at 100 mV s−1, oxidized the Ni atoms only near the surface of the Ni NPs. The deconvoluted XPS spectra implied that the chemical states of NP shells are similar, and the surfaces of both films were composed mainly of Ni(OH)2 and NiO at ratios of 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 for Ni@Ni(OH)2-NP/C and 92:8 for Ni@Ni(OH)2-NP/N–C, respectively (Fig. S4 and Table S1†).18 The shell thicknesses of the Ni NPs electrodeposited on both electrodes showed similar values between 3–5 nm. Note that the Ni NPs on the C film have highly contrasted images composed of light gray and shaded black zones, as shown in Fig. 1b-1, 1c-1, 1d-1, and S5.† This indicated that the Ni NP have a polycrystalline structure with abundant grain boundaries. In contrast, the contrast is weaker in the case of Ni NPs on the N–C film electrode, indicating higher crystallinity of the Ni NPs. As described later, such a difference in crystallinity is very important for understanding the electrocatalytic behavior of Ni NPs.
:22 for Ni@Ni(OH)2-NP/C and 92:8 for Ni@Ni(OH)2-NP/N–C, respectively (Fig. S4 and Table S1†).18 The shell thicknesses of the Ni NPs electrodeposited on both electrodes showed similar values between 3–5 nm. Note that the Ni NPs on the C film have highly contrasted images composed of light gray and shaded black zones, as shown in Fig. 1b-1, 1c-1, 1d-1, and S5.† This indicated that the Ni NP have a polycrystalline structure with abundant grain boundaries. In contrast, the contrast is weaker in the case of Ni NPs on the N–C film electrode, indicating higher crystallinity of the Ni NPs. As described later, such a difference in crystallinity is very important for understanding the electrocatalytic behavior of Ni NPs.
 | ||
| Fig. 1 (a, e) FE-SEM (and histograms with average diameter), (b-1, c-1, d-1, f-1, g-1, h-1) HR-TEM, and (b-2, c-2, d-2, f-2, g-2, h-2) HAADF-STEM-EDS images of Ni@Ni(OH)2-NPs electrodeposited on C (upper) and N–C (bottom) films. | ||
The basic mechanisms of an electrocatalytic oxidation reaction at the nickel oxyhydroxide were reported by Fleischmann.19 Basically, an electrochemical reaction can be described as an electrochemical formation of NiOOH from Ni(OH)2 and the following chemical reaction between NiOOH and organic molecules.
| Ni(OH)2 + OH− = NiOOH + H2O + e− | (1) |
| NiOOH + organic molecules = Ni(OH)2 + oxidation products |
The electrocatalytic performance depends on both steps. Specifically, the rate of either the NiOOH formation reaction or the chemical reaction between NiOOH and target molecules determine the overall turnover rate. To estimate the rate of NiOOH formation reaction, we conducted cyclic voltammetry in 0.1 M NaOH aqueous solution (pH = 12.7) at different scan rates. As summarized in Fig. 2, each CV exhibits a couple of redox peaks, which correspond to the reaction as is eqn (1). A scan rate of 1 mV s−1 is sufficiently slow to allow the investigation of the NiOOH redox properties whether or not the NiOOH formation reaction is a limiting process. Such a slow scan rate provided similar sharp redox peak currents at both electrodes with similar redox potentials and charges, indicating that the amounts of electroactive Ni(OH)2 on both electrodes are almost the same. At a scan rate of 5 mV s−1, both electrodes show similar redox peak potentials; however, the Ni@Ni(OH)2-NP/C electrode exhibited smaller redox peak currents than those at 1 mV s−1. In addition, the oxidation peak current was tailed in the positive direction. These results indicate that the NiOOH formation reaction at the Ni@Ni(OH)2-NP/C electrode was slower and was not completed on the time scale of the CV. Further increasing the scan rate to 100 mV s−1 resulted in a positively shifted, broad, and smaller oxidation peak at the Ni@Ni(OH)2-NP/C electrode, while potential shifts of the oxidation and reduction peaks at the Ni@Ni(OH)2-NP/N–C electrode are much smaller. Fig. 2d shows the amounts of NiOOH formation, Γ, from these CVs according to the equation,
| Γ = Qc/nFA |
485 C mol−1), n is the number of electrons transferred in the redox process (n = 1), and A is the geometric area of the Ni@Ni(OH)2-NP/C and Ni@Ni(OH)2-NP/N–C electrodes. Obviously, the amounts of NiOOH formed during CVs were almost unchanged at the Ni@Ni(OH)2-NP/N–C electrode even when the scan rate was increased from 1 to 100 mV s−1. In contrast, those at the Ni@Ni(OH)2-NP/C electrode decreased significantly, by about 25%, despite the small increase in the scan rate from 1 to 5 mV s−1. These results indicate that the NiOOH formation reaction is much faster at the N–C film than at the C film despite similar NP size distributions and immobilization densities. This faster NiOOH formation reaction enables a high turnover rate. As shown in Fig. S5,† the Γ-normalized glucose amperograms indicate that the high turnover rate of Ni@Ni(OH)2-NP/N–C resulted in a larger electrocatalytic current and a wider dynamic range than those of Ni@Ni(OH)2-NP/C. These characteristics are also advantageous for oligosaccharide oxidation, as described later. Nam et al. reported that the NiOOH formation reaction from NiOx is faster than that from Ni(OH)2; however, deconvoluted Ni 2p3/2 spectra indicate similar Ni(OH)2/NiO ratios (Fig. S4†).
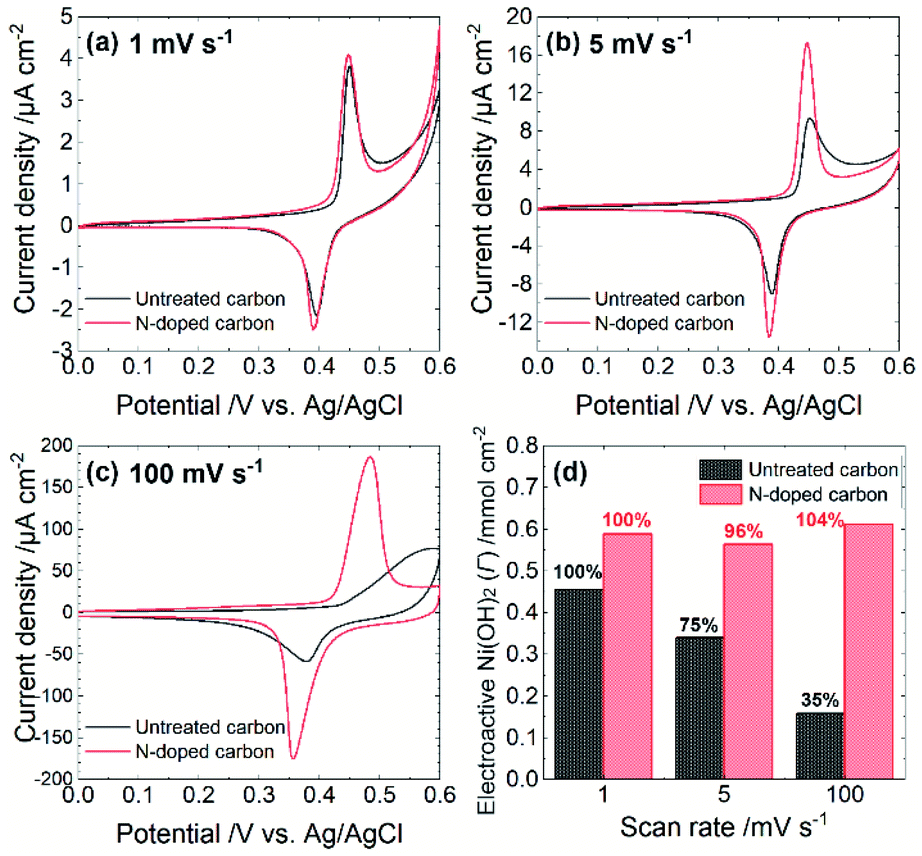 | ||
| Fig. 2 Cyclic voltammograms in a solution containing 0.1 M NaOH and 1 M sodium acetate (pH 12.7) obtained at scan rates of (a) 1, (b) 5, and (c) 100 mV s−1. (d) Dependence of scan rate on the amount of NiOOH formed during CV measurements. The Γ value obtained at 1 mV s−1 at each electrode is taken as 100%. | ||
We speculate that crystallinity is an important factor in describing the difference in the rate of NiOOH formation reaction. Since the electron conductivity of Ni(OH)2 is quite low (10−17 S cm−1),20 the electrochemical NiOOH formation reaction might occur at a part of the Ni(OH)2-shell attached or very close to the N–C surface. Subsequently, the formed NiOOH may diffuse throughout the Ni(OH)2-shell by oxidizing adjacent Ni(OH)2 (proton transfer from Ni(OH)2 to NiOOH). Therefore, proton conductivity is crucial for describing electrochemical behavior. As described above, Ni@Ni(OH)2-NPs on the C film exhibited polycrystalline-like structures, while those on the N–C film showed higher crystallinity. Based on these observations, abundant grain boundaries throughout the Ni oxide shells may exist from electrodeposition on the C film. Such defect sites may decrease proton conductivity, resulting in a slower NiOOH formation reaction. However, further detailed investigation is required to confirm the proposed mechanism.
To test the electrocatalytic activity of Ni@Ni(OH)2-NP/C and that of Ni@Ni(OH)2-NP/N–C, we performed linear sweep voltammetry (LSV) in the absence and presence of 300 μM maltopentaose (G5) as a typical oligosaccharide compound dissolved in the solvent with a different pH value (Fig. 3). We set the scan rate of 1 mV s−1 to provide sufficient time to form the fully oxidized NiOOH shell on the time scale of LSV, thus making it possible to properly estimate the electrocatalytic activity. Indeed, we confirmed that both electrodes exhibited almost the same background peak potentials, around 0.55 V for pH 12.0 and 0.45 V for pH 12.7, respectively (Fig. 3a, dotted lines). Interestingly, the difference in electrocatalytic activities between Ni@Ni(OH)2-NP/C and Ni@Ni(OH)2-NP/N–C depended on the pH of the solvent. When the pH was 12.0 (<predicted pKa of G5), the Ni@Ni(OH)2-NP/N–C electrode exhibited a larger current of G5 oxidation (Fig. 3a, red solid line) compared with that of the Ni@Ni(OH)2-NP/C electrode (Fig. 3a, black solid line). These results reflect the higher turnover rate (faster rate of NiOOH regeneration) of Ni@Ni(OH)2-NP on N–C. The onset potentials of the G5 oxidation current at both electrodes (Fig. 3a, solid lines) were almost the same as those of background currents (Fig. 3a, dotted lines). In contrast, when the pH was 12.7 (>predicted pKa of G5), unusual electrocatalytic properties were observed at both electrodes, which were triggered by G5 having a negative charge (predicted pKa = 12.4). Specifically, the electrocatalytic current started to increase sharply at +0.28 V at the Ni@Ni(OH)2-NP/N–C, whereas that at Ni@Ni(OH)2-NP/C started to increased gradually at +0.34 V. These onset potentials were 0.24 V and 0.08 V lower than those of NiOOH formation reactions at the respective electrodes. These results could be due to the slightly formed NiOOH at the low potential region, which cannot be detected as the current changes. Moreover, the onset potential of the G5 oxidation current at the Ni@Ni(OH)2-NP/N–C electrode is 60 mV lower than that of the Ni@Ni(OH)2-NP/C electrode. This 60 mV potential difference could be interpreted as electrostatic interaction between G5 and the electrode surface. As described above, the deconvoluted XPS N 1s spectra revealed that N–C has 0.4 at% of graphitic N. We think that the deconvoluted peak at 401.1 eV was attributed to quaternary N, which has a positive charge. Based on these data, we speculate that the distance between negatively charged G5 and Ni@Ni(OH)2-NP's surface on the positively charged quaternary N sites was reduced, resulting in decreased oxidation overpotential. In addition, the G5 concentration near the electrode surface could increase due to the same effect. As a result, electrocatalytic performance toward G5 oxidation was much improved at the Ni@Ni(OH)2-NP/N–C electrode, in addition to the ability to rapidly regenerate catalytic sites (NiOOH).
 | ||
| Fig. 3 Linear sweep voltammograms in the presence (solid line) and absence (dotted line) of 300 μM maltopentaose (G5) dissolved in (a) pH12.0 and (b) pH12.7 aqueous solution containing NaOH and 1 M sodium acetate. Ni@Ni(OH)2-NP electrodeposited on C (black) and N–C (red) films were used as working electrodes, respectively. The scan rate was 1 mV s−1. | ||
In summary, we report the impact of N-doping of carbon film on the nanostructure and electrocatalytic performance of electrodeposited Ni@Ni(OH)2-NP. N-doping both increased the turnover rate and decreased the overpotential toward sugar oxidation. The former effect may be derived from the high proton conductivity of the Ni(OH)2-shell with less grain boundary. The latter effect was observed when the pH was more than the pKa of G5 (12.4), suggesting electrostatic interaction between negatively charged G5 and positively charged NPs. Based on this insight, we think that positively charged quaternary-N modulates the charging state of Ni@Ni(OH)2-NPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 17H03081 and 20K21133.Notes and references
- Y. Miao, L. Ouyang, S. Zhou, L. Xu, Z. Yang, M. Xiao and R. Ouyang, Biosens. Bioelectron., 2014, 53, 428–439 CrossRef CAS.
- W. Yang, X. Yang, C. Hou, B. Li, H. Gao, J. Lin and X. Luo, Appl. Catal. B, 2019, 259 CAS.
- P. E. Sharel, D. Q. Liu, R. A. Lazenby, J. Sloan, M. Vidotti, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2016, 120, 16059–16068 CrossRef.
- S. Sedaghat, C. R. Piepenburg, A. Zareei, Z. Qi, S. Peana, H. Wang and R. Rahimi, ACS Appl. Nano Mater., 2020, 3, 5260–5270 CrossRef CAS.
- G. C. Sedenho, P. T. Lee, H. S. Toh, C. Salter, C. Johnston, N. R. Stradiotto and R. G. Compton, J. Phys. Chem. C, 2015, 119, 6896–6905 CrossRef CAS.
- Š. Trafela, J. Zavašnik, S. Šturm and K. Ž. Rožman, Electrochim. Acta, 2019, 309, 346–353 CrossRef.
- S. Wang, D. Yu, L. Dai, D. W. Chang and J. B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- H. Schmies, E. Hornberger, B. Anke, T. Jurzinsky, H. N. Nong, F. Dionigi, S. Kühl, J. Drnec, M. Lerch, C. Cremers and P. Strasser, Chem. Mater., 2018, 30, 7287–7295 CrossRef CAS.
- J. Melke, B. Peter, A. Habereder, J. Ziegler, C. Fasel, A. Nefedov, H. Sezen, C. Wöll, H. Ehrenberg and C. Roth, ACS Appl. Mater. Inter., 2016, 8, 82–90 CrossRef CAS PubMed.
- L. Perini, C. Durante, M. Favaro, V. Perazzolo, S. Agnoli, O. Schneider, G. Granozzi and A. Gennaro, ACS Appl. Mater. Inter., 2015, 7, 1170–1179 CrossRef CAS PubMed.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- X. Wu, B. Feng, W. Li, Y. Niu, Y. Yu, S. Lu, C. Zhong, P. Liu, Z. Tian, L. Chen, W. Hu and C. M. Li, Nano Energy, 2019, 62, 117–126 CrossRef CAS.
- M. Zhou, Y. Jiang, G. Wang, W. Wu, W. Chen, P. Yu, Y. Lin, J. Mao and L. Mao, Nat. Commun., 2020, 11 Search PubMed.
- I. S. Pieta, A. Lewalska-Graczyk, P. Pieta, G. Garbarino, G. Busca, M. Holdynski, G. Kalisz, A. Sroka-Bartnicka, R. Nowakowski, M. Naushad, M. B. Gawande and R. Zbořil, ACS Sustain. Chem. Eng., 2020, 8, 7244–7255 CrossRef.
- W. Shi, Q. Wang, F. Qin, J. Yu, M. Jia, H. Gao, Y. Zhang, Y. Zhao and G. Li, Electrochim. Acta, 2017, 232, 332–338 CrossRef CAS.
- J. Zhu, H. Yin, J. Gong, M. S. H. Al-Furjan and Q. Nie, J. Alloy. Compd., 2018, 748, 145–153 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- K. W. Nam, K. H. Kim, E. S. Lee, W. S. Yoon, X. Q. Yang and K. B. Kim, J. Power Sources, 2008, 182, 642–652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photo of water droplet contact angles, relationship between electrodeposition potential and NP size distribution, deconvoluted N 1s and Ni 2p2/3 spectra, low-magnification HR-TEM images, glucose amperograms. See DOI: 10.1039/d1ra01157j |
| This journal is © The Royal Society of Chemistry 2021 |