
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic aluminum complexes bearing novel spiro-phenanthrene-monoketone/OH derivatives: synthesis, characterization and the ring-opening polymerization of ε-caprolactone†
Erlin Yue a,
Furong Caob,
Jun Zhangc,
Wenjuan Zhang*b,
Youshu Jiangc,
Tongling Liangc and
Wen-Hua Sun*c
a,
Furong Caob,
Jun Zhangc,
Wenjuan Zhang*b,
Youshu Jiangc,
Tongling Liangc and
Wen-Hua Sun*c
aYan'an Key Laboratory of New Energy & New Functional Materials, Shaanxi Key Laboratory of Chemical Reaction Engineering, School of Chemistry and Chemical Engineering, Yan'an University, Yan'an 716000, China. E-mail: yueerlin@yau.edu.cn
bBeijing Key Laboratory of Clothing Materials R&D and Assessment, Beijing Engineering Research Center of Textile Nanofiber, School of Materials Science and Engineering, Beijing Institute of Fashion Technology, Beijing 100029, China. E-mail: zhangwj@bift.edu.cn
cKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn
First published on 8th April 2021
Abstract
A series of spiro-phenanthrene-monoketone/OH derivatives (L1–L6) were synthesized and fully characterized with 1H/13C NMR spectroscopy and elemental analyses. By treating ligands with AlMe3, oxygen-bridged binuclear aluminum complexes (Al1–Al6) were isolated and characterized by 1H/13C NMR spectroscopy. The molecular structures of ligands (L2, L4 and L5) and complex Al1 were determined by single crystal X-ray diffraction. In the presence of benzyl alcohol (BnOH), these aluminum complexes demonstrated high efficiency towards the ring-opening polymerization of ε-caprolactone (ε-CL), resulting in PCL in a linear manner with the BnO-end group. In addition, complexes Al1 and Al5 exhibited good catalytic activities even without BnOH. Moreover, complexes Al3 and Al6 with the bulkier substituent of iPr at the ortho-position of the arylamines demonstrated better catalytic activities than the analogs. Moreover, substituents on the backbone also affected catalytic behaviors.
Introduction
Biodegradable polyesters, such as polylactides (PLA) and polycaprolactone (PCL), have attracted much attention due to their biodegradability, biocompatibility and permeability, and have been extensive biomaterials in the pharmaceutical industries over the past few decades.1 These polyesters are commonly prepared by the ring-opening polymerization (ROP) of cyclic esters, such as ε-caprolactone (ε-CL) or lactide, and catalyzed by metal complexes as prevalent catalysts.2 Aluminum complex catalysts3 have appealed with good activities and being nicely characterized by NMR spectra in order to illustrate the coordination mechanism of the ROP of cyclic esters.4 Beyond ε-CL, the stereoselective ROP of rac-lactides was first achieved using the salen–Al complex catalyst.5 In general, the modifications of ligands with different substituents changed the steric and electronic environment around the metal in their aluminum complexes, thus finely tuning their activities and controllability of ROP.6 Attractive aluminum catalysts for ROP of cyclic esters were derived from simply N^O bidentate ligands, in which the six-membered ring N^O bidentate aluminum complexes showed less activity than their five-membered ring analogs.7 This was further confirmed by the DFT calculation, in which five-membered ring Al complexes reduced the steric repulsion and enhanced the activity of the ROP of ε-CL.7 However, there were less examples of five-membered coordinated ring Al complexes than popular six-membered ring N^O bidentate aluminum complexes.7–9The N^O bidentate aluminum complexes (A, Chart 1) with five-membered ring coordination were verified to be in monomeric or dimeric forms that depended on the bulkiness of Al–R,10 but usually formed dimeric ones (B, Chart 1) derived from 2-(1,3,5-dithiazinan-5-yl)ethanols,11 as well as other alcohols (C and D, Chart 1).12 Notably, complex D could catalyze the cycloaddition of CO2 with epoxies.12b The anilinotropone-based dimeric aluminum complexes (E) demonstrated high activities in the ring-opening polymerization (ROP) of rac-lactides with the assistance of BnOH.13 Subsequently, dimeric aluminum complexes (F) initiated the ROP of ε-CL14 with good controllability. Meanwhile, complexes G exhibited good activity for the ROP of cyclic esters, and reflected the polymerization mechanism through NMR measurements.15
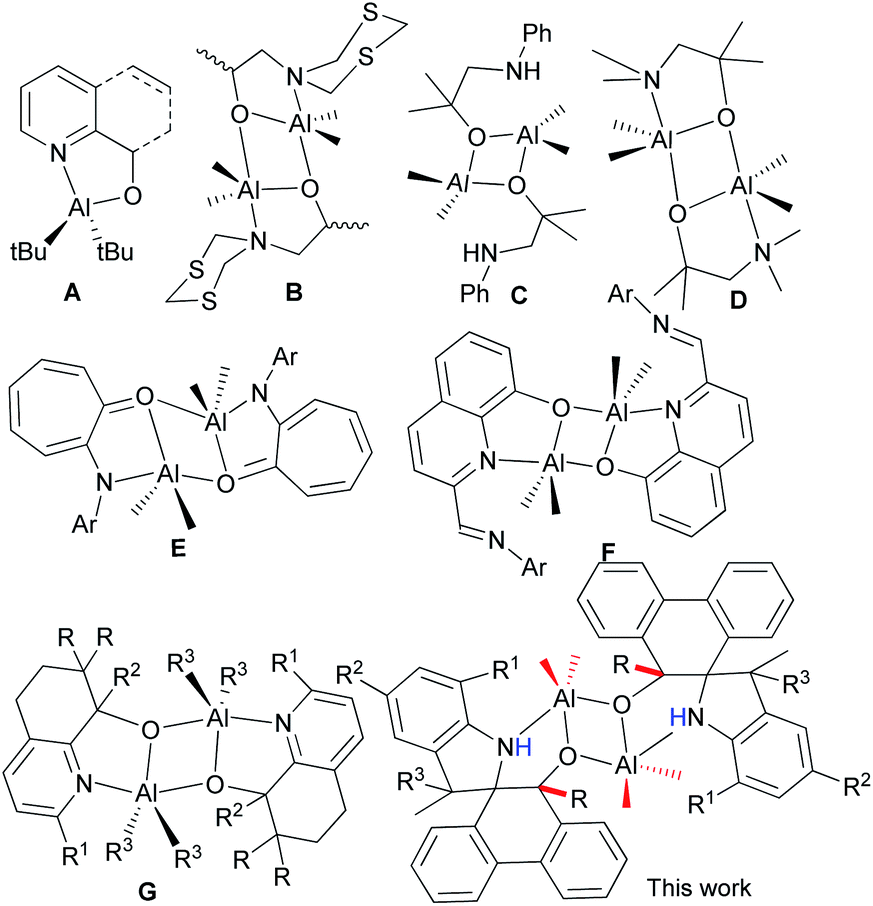 | ||
| Chart 1 N^O bidentate aluminum complexes. | ||
Phenanthrene-based ligands were widely used to coordinate with transition metals,16 and their metal complexes exhibited good activities for isoprene polymerization,17 as well as coupling reactions.18 However, there were a few examples with main group complexes, but with 9,10-diamido-phenanthrene only.19 Phenanthrenequinone derivatives react extensively with trimethylaluminum, forming novel dimeric aluminum complexes bearing spiro-phenanthrene derivatives. Interestingly, these complexes show high activities towards the ring-opening polymerization of ε-caprolactone. Herein, the synthesis and characterization of the title complexes are reported, along with their catalytic performances towards the ROP of ε-CL.
Results and discussion
Synthesis and characterization of the ligands (L1–L6) and aluminum complexes (Al1–Al6)
According to our previous work,18 refluxing the mixture of 9,10-phenanthrenequinone with various substituted anilines in the presence of p-toluenesulfonic acid (p-TsOH) in toluene afforded a series of novel spiro-phenanthrene-monoketone derivatives (L1–L3). Furthermore, the reduction reaction of L1–L3 with n-BuMgCl gave the corresponding spiro-phenanthrene-mono–OH compounds (L4–L6) (Scheme 1). Treatment of ligands L1–L6 with one equivalent of AlMe3 in toluene at −30 °C gave the dialkylaluminum complexes (Al1–Al6), respectively, in acceptable yields (Scheme 1). For the preparation of Al1–Al3, one methyl group from AlMe3 was added to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). For the preparation of Al1–Al6, the elimination of CH4 from the reaction between Al–Me and –OH occurred. All of these organic compounds and aluminum complexes were characterized by 1H/13C NMR spectroscopy and elementary analysis. Comparing the 1H/13C NMR spectra of the ligands, the disappearance of the –CO resonance of L1–L3 and the –OH resonance of the L4–L6 in those of the corresponding aluminum complexes indicated the formation of the aluminum complexes. It is noteworthy that the resonance of –NH for all aluminum complexes Al1–Al6 still remained, being similar to the reports of the –NH presence in aluminum complexes.8,12a Moreover, the crystal structures of the organic compounds L2, L4 and L5 and aluminum complex Al1 were determined by single crystal X-ray diffraction (Fig. 1–4).
O). For the preparation of Al1–Al6, the elimination of CH4 from the reaction between Al–Me and –OH occurred. All of these organic compounds and aluminum complexes were characterized by 1H/13C NMR spectroscopy and elementary analysis. Comparing the 1H/13C NMR spectra of the ligands, the disappearance of the –CO resonance of L1–L3 and the –OH resonance of the L4–L6 in those of the corresponding aluminum complexes indicated the formation of the aluminum complexes. It is noteworthy that the resonance of –NH for all aluminum complexes Al1–Al6 still remained, being similar to the reports of the –NH presence in aluminum complexes.8,12a Moreover, the crystal structures of the organic compounds L2, L4 and L5 and aluminum complex Al1 were determined by single crystal X-ray diffraction (Fig. 1–4).
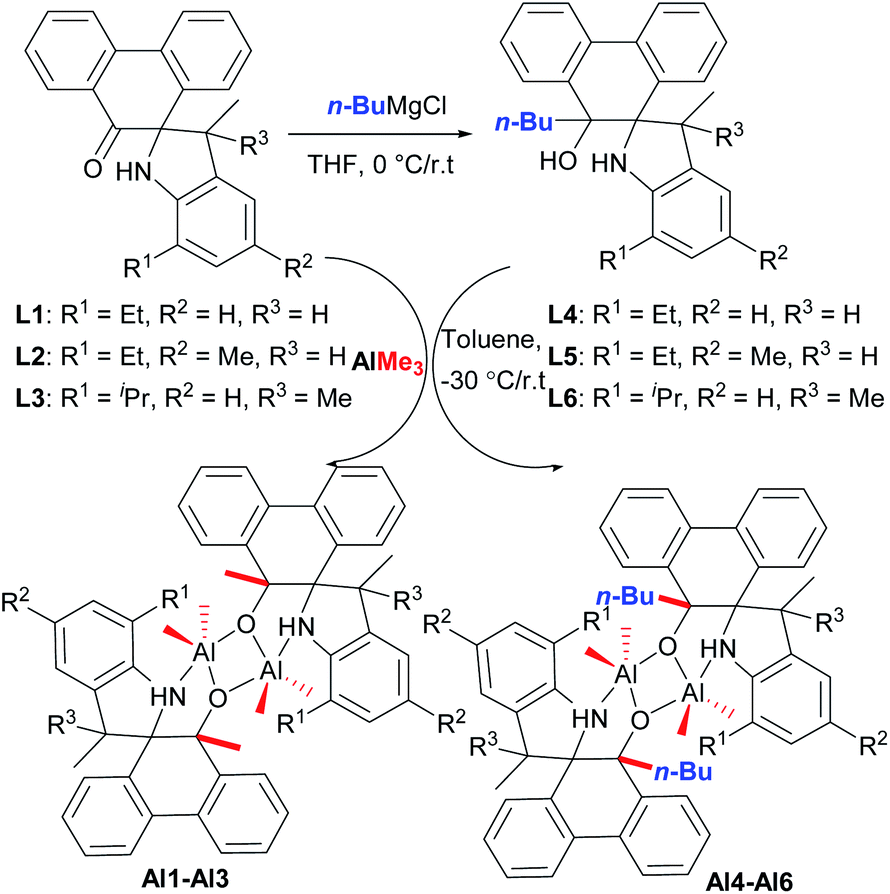 | ||
| Scheme 1 Synthesis of the spiro-phenanthrene-mono–OH derivatives L4–L6 and aluminum complexes Al1–Al6. | ||
Suitable crystals of the organic compounds L2, L4 and L5 were readily achieved from their respective heptane solution, and were determined by means of single crystal X-ray diffraction (see Fig. 1–3). Selected bond lengths and bond angles are given in Table 1.
| L2 | L4 | L5 | |
|---|---|---|---|
| Bond lengths (Å) | |||
| N1–C13 | 1.467 (4) | 1.487 (4) | 1.4815 (16) |
| N1–C18 | 1.395 (4) | 1.416 (4) | 1.4221 (18) |
| O1–C14 | 1.218 (4) | 1.427 (4) | 1.4228 (16) |
| C13–C12 | 1.515 (5) | 1.536 (4) | 1.5356 (18) |
| C13–C14 | 1.536 (4) | 1.557 (4) | 1.5513 (19) |
| C13–C15 | 1.591 (5) | 1.583 (4) | 1.5818 (18) |
| C15–C16 | 1.525 (5) | 1.537 (4) | 1.5304 (19) |
| C15–C17 | 1.522 (5) | 1.512 (4) | 1.506 (2) |
| C17–C18 | 1.391 (5) | 1.392 (4) | 1.3917 (19) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Bond angles (°) | |||
| C13–N1–C18 | 108.9 (3) | 107.7 (2) | 105.86 (10) |
| N1–C13–C12 | 112.8 (3) | 112.7 (2) | 112.06 (11) |
| N1–C13–C14 | 109.7 (3) | 109.7 (2) | 110.92 (10) |
| N1–C13–C15 | 102.8 (3) | 102.5 (2) | 102.75 (10) |
| N1–C18–C17 | 111.1 (3) | 110.3 (3) | 110.38 (12) |
| O1–C14–C5 | 122.4 (3) | 108.0 (2) | 107.02 (11) |
| O1–C14–C13 | 120.3 (3) | 110.3 (3) | 111.32 (11) |
Fig. 1 shows the structure of L2, in which the five-membered rings are each constructed by C13, C15, C17–C18 and N1, and the O1–C14 bond length of L2 is 1.218 (4) Å, indicating a typical double-bond character of CO. The O atom deviated from the plane of spiro-phenanthrene (C6, C2, C4, C10, C11, C8) with a distance of 0.505 Å. The plane of the aryl ring (C7, C21, C19) is almost perpendicular to the spiro-phenanthrene plane with a dihedral angle of 78.14°. From the packing model, there was hydrogen bonding of N–H⋯O between two adjacent molecules. In addition, the plane of spiro-phenanthrene is almost parallel to another phenanthrene from adjacent molecules with a small dihedral angle of 8.07°.
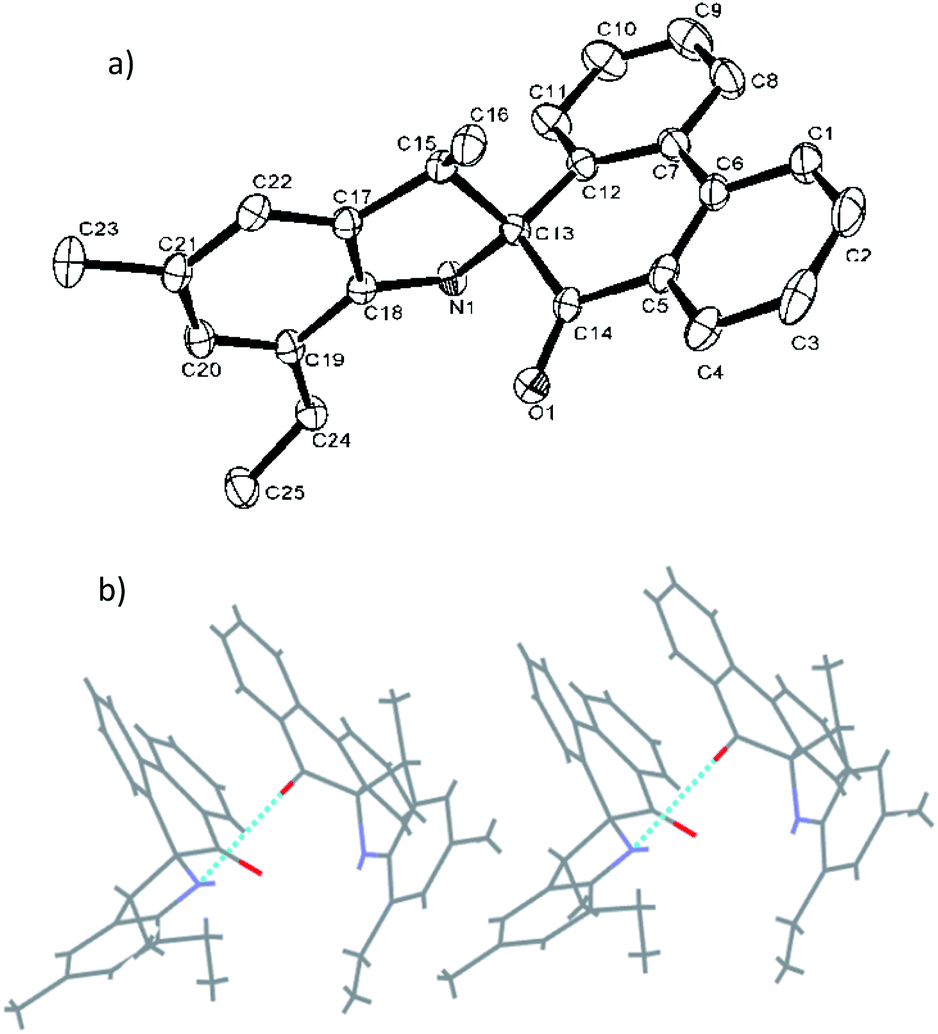 | ||
| Fig. 1 (a) ORTEP drawing of L2. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. (b) Hydrogen bonding motifs in L2. | ||
The structures of L4 and L5 are shown in Fig. 2 and 3 and they are similar to L2. Both possessed the five-membered rings that were constructed by C13, C15, C17–C18 and N1. However, the bond lengths of O1–C14 in L4 (1.427 (4) Å) and L5 (1.4228 (16) Å) are much longer than that of L2 (1.218 (4) Å), indicating the typical single bond characters. In addition, the bond length of N–C13, N–C18 in L4 (1.487 (4), 1.416 (4) Å), L5 (1.4815 (16), 1.4221 (18) Å) are much longer than that in L2 (1.467 (4), 1.395 (4) Å), indicating the influence of the alkyl group on C14. Due to the more distorted six-membered ring, the dihedral angle between the two phenyl rings of phenanthrene is about 19.7°, but these two phenyl rings are almost particular to the aryl ring with a dihedral angle of 76.69 and 84.1°, respectively. There were also intermolecular hydrogen bonds in the ligands L4 and L5, respectively, which came from O1–H1⋯N1. The motifs of hydrogen bonding are illustrated in Fig. S1 and S2.†
 | ||
| Fig. 2 ORTEP drawing of L4. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. | ||
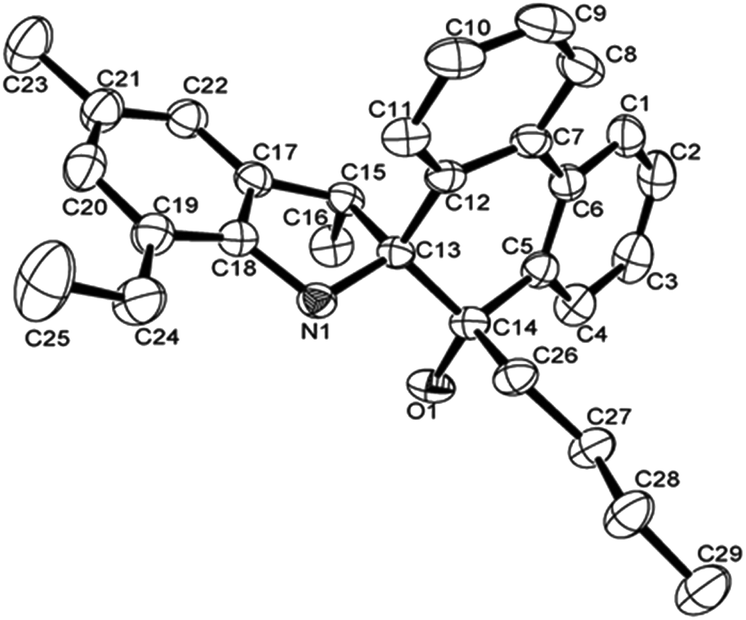 | ||
| Fig. 3 ORTEP drawing of L5. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. | ||
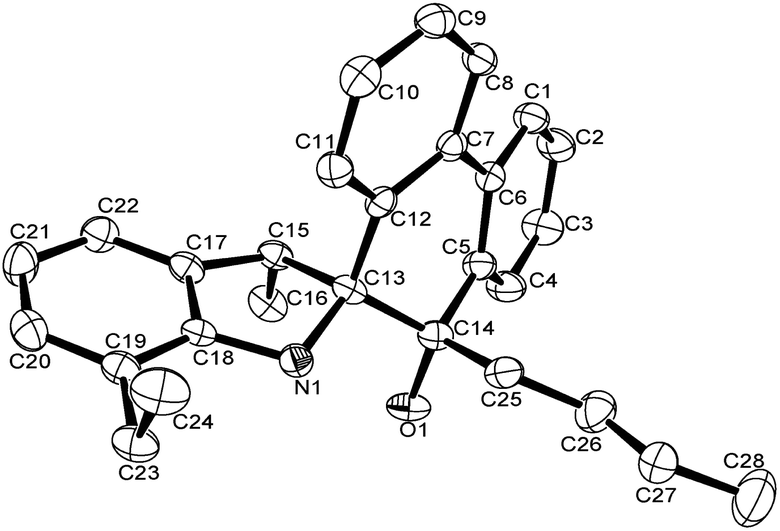
A single crystal of complex Al1 suitable for the X-ray diffraction analysis was obtained from its toluene solution. The molecular structure of complex Al1 is shown in Fig. 4, and the selected bond lengths and bond angles are collected in Table 2.
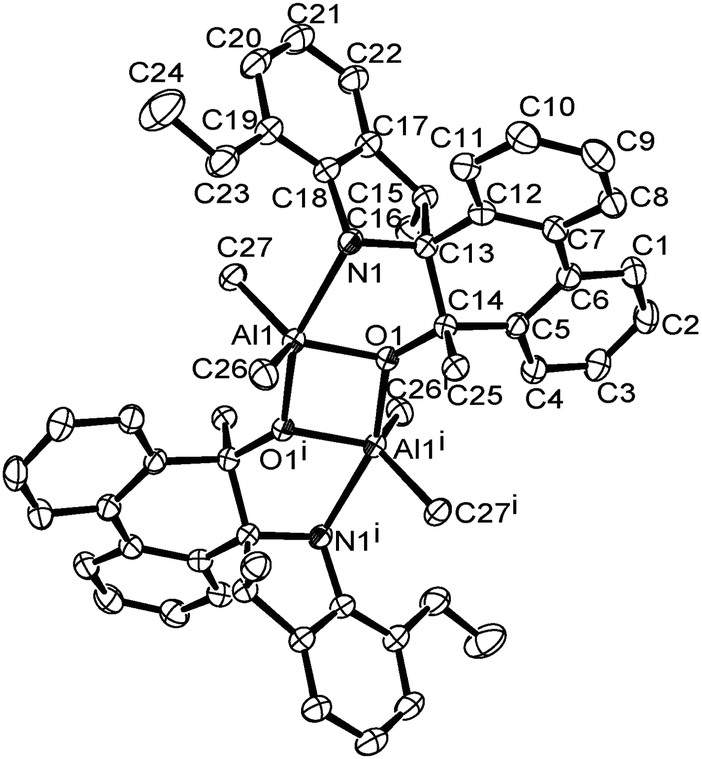 | ||
| Fig. 4 ORTEP drawing of Al1. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and free solvent molecules have been omitted for clarity. | ||
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| Al1–O1 | 1.8669 (15) | O1–Al1–O1i | 79.51 (5) |
| Al1–O1i | 1.9881 (13) | O1–Al1–N1 | 79.59 (5) |
| Al1–N1 | 2.3291 (15) | O1i–Al1–N1 | 159.10 (5) |
| Al1–C26 | 1.9960 (18) | O1–Al1–C26 | 115.68 (7) |
| Al1–C27 | 1.9813 (17) | O1–Al1–C27 | 122.84 (7) |
| O1–Al1i | 1.9882 (13) | O1i–Al1–C26 | 100.96 (6) |
| O1–C14 | 1.4504 (18) | O1i–Al1–C27 | 100.87 (7) |
| N1–C13 | 1.490 (2) | C26–Al1–C27 | 120.09 (8) |
| N1–C18 | 1.4388 (19) | N1–Al1–C26 | 88.41 (6) |
| Al1–Al1i | 2.9647 (15) | N1–Al1–C27 | 90.18 (7) |
Fig. 4 shows the complex Al1 as a dimeric form, which is symmetrically penta-coordinated by two bridged oxygen atoms (O1 and O1i), one nitrogen atom (N1) from the spiro-phenanthrene-monoketone ligand and two coordinating methyl anions from the trimethylaluminum. Each aluminum possessed a distorted trigonal bipyramidal geometry. Another methyl anion from trimethylaluminum was added to the carbonyl of the spiro-phenanthrene-monoketone ligand. Complex Al1 possessed a C2 symmetric axis through the centroid of the Al1–O1–Ali–O1i plane. However, there is a slight asymmetry in the Al2O2 four-membered ring system. The bond lengths of the Al1–O1 [1.8669 (15) Å] and Al1–N1 [2.3291 (15) Å] in Al1 are shorter than the corresponding ones in the dimethylaluminum 2-chloro-6,6-dimethylcyclopentyl-pyridin-7-oxylates [1.8735 (14) Å, 2.3500 (17) Å].15c The quadrilateral center comprises Al1, O1, Al1i and O1i, but there is no direct bonding between the two aluminum atoms with an intramolecular distance of 2.9647 (15) Å. The dihedral angle between the two phenyl rings of spiro-phenanthrene is about 27.29°, much larger than that in L2, indicating the greater distortion of the six-membered ring after coordination with Al. In addition, although N–H remained in the Al1 complex, there was no hydrogen bonding observed in Al1 due to the bulky environment around the N atom.
Ring-opening polymerization of ε-CL catalyzed by Al complexes Al1–Al6
First, complex Al1 was evaluated for the ring opening polymerization of rac-LA under various conditions, but there was no polymer product. In contrast, the ROP of ε-CL proceeded smoothly. Therefore, the ring opening polymerization of ε-CL was investigated in detail, and the polymerization results are collected in Table 3. The complex Al1 was employed to study the effect of temperature, molar ratio, and reaction time on the ROP of ε-CL. The results showed that there was no polymer observed at room temperature or 30 °C when the ROP of ε-CL was conducted at a molar ratio [CL]:[Al]:[BnOH] of [250:1:1]. When increasing the temperature from 60 °C to 90 °C (Table 3, runs 1–3), the polymer yield with 30 minutes gradually increased from 3% to 45%. Further increasing the temperature to 100 °C led to a significant increase of the polymer yield (up to 93%) (Table 3, run 4). This temperature effect on the polymerization agreed well with our previous results, in which the mononuclear species is proposed as the real active species for the ring opening polymerization of ε-CL and the ratio of the mononuclear species increased at higher temperature (verified by 27Al NMR).15b However, a higher temperature of 110 °C resulted in a quick decrease of yield (64%, run 5, Table 3). The higher monomer ratio led to a slight decrease of the polymer yield and increase of the molecular weight. Prolonging the reaction time from 10 minutes to 60 minutes led to the gradual increase of yields from 89% to almost 100%. At the same time, the molecule weights are quite similar (4.28–4.86 × 104 g mol−1), indicating the minimal effect of the reaction time. Without BnOH, Al1 still exhibited good efficiency for ROP of ε-CL with 87% polymer yield, but producing the PCL with high molecular weight (8.95 × 104 g mol−1). When two equivalents of BnOH were used, the polymer yield was kept at 85%. However, the molecular weight sharply decreased from 8.95 to 2.55 × 104 g mol−1, indicating the significant chain transfer of BnOH (runs 4, 11 and 12, Table 3).
| Run | Cat. | CL:Al:BnOH |
T/°C | t/min | Isolated yieldsb (%) | Mnc (×104) | Mncald (×104)d | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 20 μmol Al; 1.0 M ε-CL toluene solution.b Isolated yield: weight of the polymer obtained/weight of monomer used.c GPC data in THF vs. polystyrene standards, using a correcting factor of 0.56.20d Mn (calcd) = (monomer/initiator) × (conversion) × 114 + 108 (Mw of BnOH). | ||||||||
| 1 | Al1 | 250:1:1 |
60 | 30 | 3 | 1.10 | 0.10 | 1.32 |
| 2 | Al1 | 250:1:1 |
80 | 30 | 26 | 2.05 | 0.75 | 1.44 |
| 3 | Al1 | 250:1:1 |
90 | 30 | 45 | 1.86 | 1.29 | 1.67 |
| 4 | Al1 | 250:1:1 |
100 | 30 | 93 | 4.37 | 2.66 | 1.41 |
| 5 | Al1 | 250:1:1 |
110 | 30 | 64 | 3.27 | 1.83 | 1.33 |
| 6 | Al1 | 400:1:1 |
100 | 30 | 85 | 5.48 | 3.89 | 1.39 |
| 7 | Al1 | 250:1:1 |
100 | 10 | 89 | 4.71 | 2.55 | 1.34 |
| 8 | Al1 | 250:1:1 |
100 | 20 | 91 | 4.28 | 2.60 | 1.51 |
| 9 | Al1 | 250:1:1 |
100 | 40 | 96 | 4.54 | 2.75 | 1.38 |
| 10 | Al1 | 250:1:1 |
100 | 60 | >99 | 4.86 | 2.86 | 1.36 |
| 11 | Al1 | 250:1:0 |
100 | 30 | 87 | 8.95 | — | 1.89 |
| 12 | Al1 | 250:1:2 |
100 | 30 | 85 | 2.55 | 1.22 | 1.19 |
| 13 | Al5 | 250:1:0 |
100 | 30 | 81 | 12.45 | — | 1.87 |
| 14 | Al5 | 250:1:1 |
100 | 30 | 83 | 2.82 | 2.38 | 1.47 |
| 15 | Al5 | 250:1:2 |
100 | 30 | 71 | 2.70 | 1.02 | 1.24 |
| 16 | Al5 | 250:1:1 |
80 | 30 | 36 | 2.92 | 1.04 | 1.39 |
| 17 | Al5 | 250:1:1 |
90 | 30 | 67 | 4.05 | 1.92 | 1.31 |
| 18 | Al5 | 250:1:1 |
110 | 30 | 48 | 2.15 | 1.38 | 1.86 |
| 19 | Al5 | 250:1:1 |
100 | 40 | 87 | 1.99 | 2.49 | 1.70 |
| 20 | Al5 | 250:1:1 |
100 | 60 | 95 | 2.16 | 2.72 | 2.04 |
| 21 | Al5 | 400:1:1 |
100 | 30 | 31 | 1.56 | 1.42 | 1.96 |
| 22 | Al5 | 500:1:1 |
100 | 30 | 0 | — | — | — |
| 23 | Al2 | 250:1:1 |
100 | 30 | 77 | 3.34 | 2.21 | 1.31 |
| 24 | Al3 | 250:1:1 |
100 | 30 | 96 | 4.42 | 2.75 | 1.34 |
| 25 | Al4 | 250:1:1 |
100 | 30 | 85 | 4.27 | 2.43 | 1.31 |
| 26 | Al6 | 250:1:1 |
100 | 30 | 86 | 1.91 | 2.46 | 1.70 |
Considering the potential role of the long chain of the butyl group on improving the polymerization activity or molecular weight of PCL, Al5 was also investigated for ROP under the same condition. The results showed that without BnOH, the molecular weight of the polymer by Al5 was much higher than that by Al1 (run 13 vs. 11, Table 3), up to 12.4 × 104 g mol−1. When increasing the BnOH amount, the polymer yield did not change a lot. However, the molecular weight of PCL sharply decreased (runs 13–15, Table 3), also indicating the chain transfer of BnOH. In the presence of BnOH, the polymerizations were conducted at various temperatures and the highest yield was achieved at 100 °C. In prolonging the reaction time from 30 min to 60 minutes, the polymer yield gradually increased from 83% to 95%. However, upon increasing the monomer molar ratio from 250 to 400 and 500, the polymer yield dramatically decreased from 83% to 31% and 0% (runs 20–22, Table 3). It is probable that the deactivation of the complexes by impurities in the monomer account for this.
At 100 °C and with the molar ratio of [CL]:[Al] as 250 within 30 minutes, all other aluminum complexes were investigated for ROP of ε-CL. The results showed that the polymer yields by Al1–Al6 bearing the n-Bu group are generally lower than those by Al1–Al3 having the methyl group, which was demonstrated by the yield order: Al4 < Al1, Al5 < Al2, Al6 < Al3, indicating a steric effect of the bulkier substituent around the Al center. In addition, the R2 group has a small effect on the polymer yield, which was demonstrated by the same order of polymer yield: Al3 (R2 = H) > Al1 (R2 = H) > Al2 (R2 = Me); Al6 (R2 = H) > Al4 (R2 = H) > Al5 (R2 = Me). The reason is probably due to the stronger electron donor ability of methyl group.
The obtained PCL was characterized by 1H NMR and MALDI-TOF spectra (shown in Fig. 5). 1H NMR clearly showed the signal of PhCH2O (7.35, 5.11 ppm), indicating the active species of Al–OCH2Ph in the polymerization process. The MALDI-TOF spectrum of the polymer also showed that there was one major family in the product (Fig. 5), in which the interval 114.2 Da was observed between the adjacent peaks (one repeat unit of polymer), and it can be ascribed to PhCH2O + 114.1 × n (number of repeat units) + Na+, a typical character of the linear structure, which agrees well with its 1H NMR spectrum.
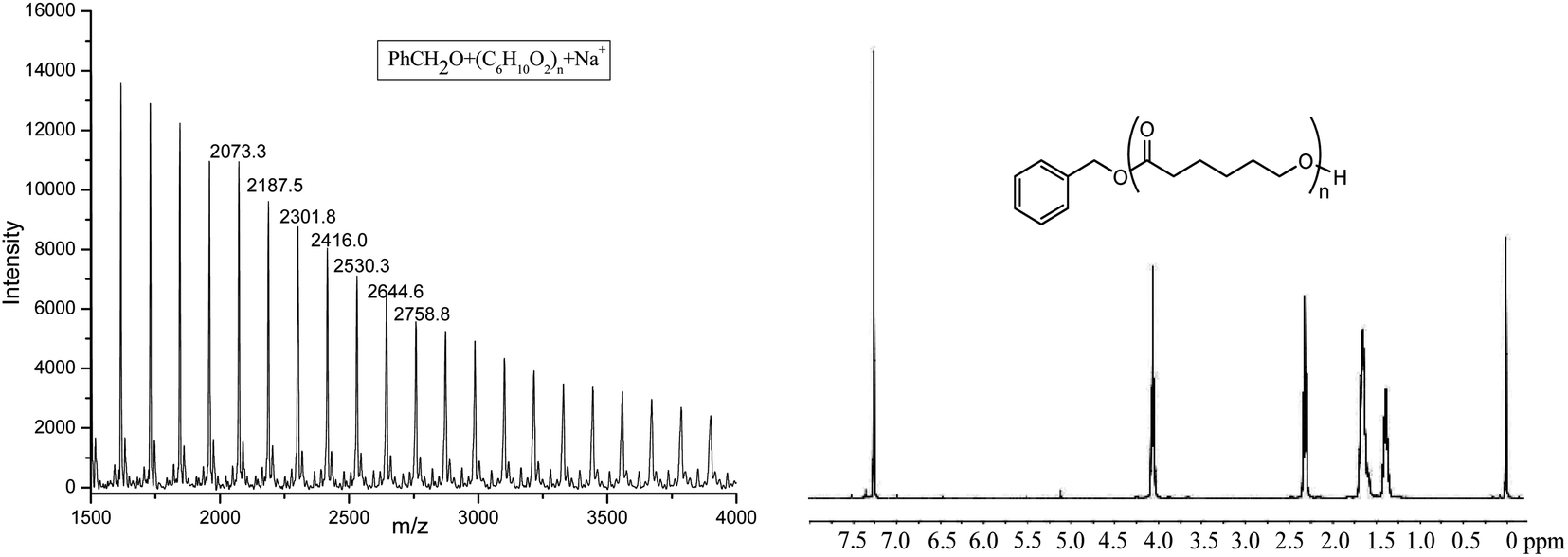 | ||
| Fig. 5 The MALDI-TOF spectrum and 1H NMR of PCL obtained by Al1 + BnOH (run 4, Table 3). | ||
Conclusion
The oxygen-bridged binuclear aluminum complexes (Al1–Al6) bearing spiro-phenanthrene-monoketone/OH derivatives (L1–L6) were prepared and fully characterized by 1H/13C NMR spectroscopy, elemental analyses and single crystal X-ray diffraction. The crystal structure of Al1 indicated an oxygen-bridged dimeric form in the solid state. All aluminum complexes demonstrated moderate to high efficiency for the ROP of ε-caprolactone (ε-CL) in the presence of benzyl alcohol (BnOH), producing the linear PCL capped with the BnO. The complexes Al3 and Al6 containing bulkier iPr substitutes at the ortho-position of arylamine can enhance the catalytic activities for the ROP of ε-CL. More importantly, the complexes Al1 and Al5 also exhibited good catalytic activities in the absence of BnOH.Experimental section
General consideration
All manipulations involving air- and moisture-sensitive compounds were performed using standard Schlenk techniques under a nitrogen atmosphere. Toluene was refluxed over sodium and distilled under nitrogen prior to use. Other reagents were purchased from Aldrich, Acros, or local suppliers. NMR spectra were recorded on a Bruker DMX 400 MHz instrument at ambient temperature using TMS as an internal standard; δ values were given in ppm and J values in Hz. 1H NMR spectra are referenced using the residual solvent peak at δ 7.26 ppm for CDCl3 and δ 7.16 ppm for C6D6. 13C NMR spectra are referenced using the residual solvent peak at 77.16 ppm for CDCl3 and δ 128.06 ppm for C6D6. Elemental analysis was carried out using a Flash EA 1112 micro-analyzer. Molecular weights and the molecular weight distribution (MWD) of polyethylene were determined by PL-GPC220 at room temperature, with tetrahydrofuran (THF) as the solvent. The compounds spiro-[7-ethyl-3-methylindoline-2,10-phenanthren-9-one] (L1)18 and spiro-[7-isopropyl-3,3-dimethylindoline-2,10-phenanthren-9-one] (L3)16c were prepared according to the reported literature.Synthesis of phenanthrene-based ligands
:1) as the eluent. It was then recrystallized in heptane to afford the product as a yellow powder (0.966 g) in 28% yield. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.97 (d, J = 8.0 Hz, 1H), 7.91–7.87 (m, 2H), 7.77–7.75 (m, 1H), 7.70–7.66 (m, 1H), 7.43–7.29 (m, 3H), 6.85 (s, 1H), 6.57 (s, 1H), 4.59 (s, 1H), 3.45–3.39 (m, 1H), 2.75–2.65 (m, 2H), 2.25 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H), 0.97 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 25 °C): δ 201.5, 146.2, 143.1, 137.6, 134.7, 131.4, 131.3, 130.0, 129.9, 129.5, 128.6, 128.1, 128.0, 127.2, 127.0, 126.5, 124.2, 123.1, 121.8, 77.6, 54.7, 24.5, 21.1, 17.8, 13.8. Anal. calcd for C25H23NO: C, 84.95; H, 6.56; N, 3.96. Found: C, 84.67; H, 6.36; N, 4.01.Synthesis of aluminum complexes Al1–Al6
General procedure for ε-caprolactone ring-opening polymerization
A typical ring-opening polymerization procedure of ε-CL in the presence of one equivalent of benzyl alcohol (Table 3, run 5) is presented as follows. The precatalyst Al1 (0.0082 g, 0.020 mmol) was dissolved in 2 mL of toluene in a Schlenk flask at room temperature. To this mixture, benzyl alcohol (0.020 mmol) in toluene solution was added and stirred at room temperature for 5 min. Then, the flask was placed in a temperature-controlled oil bath preheated at 100 °C for 5 min, and ε-CL (0.571 g, 5.0 mmol) was injected. The polymerization was terminated by the addition of glacial acetic acid (ca. 0.2 mL) into the reaction mixture until the solution was stirred for the described amount of time. The resulting viscous solution was diluted with dichloromethane, and then transferred into a beaker containing cold methanol (100 mL) with stirring. The resultant polymer was collected on filter paper, and dried under reduced pressure to give a white solid.X-ray crystallographic studies
Crystals of L2, L4, and L5 suitable for X-ray diffraction analyses were obtained from their heptane solution at room temperature overnight. Crystals of the complex Al1 suitable for the X-ray diffraction analyses were obtained from its toluene solution. X-ray studies were carried out on a Rigaku Saturn724 + CCD with graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å) at 173 (2) K. Cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least squares on F2. All hydrogen atoms were placed in calculated positions. Structure solution and refinement were performed by using the SHELXL-2014 package.21 Within the structure refinement of complex Al1, there were free solvent molecules that had no influence on the geometry of the main compounds. Therefore, the SQUEEZE option of the crystallographic program PLATON22 was used to remove these free solvents from the structure. Details of the X-ray structure determinations and refinements are provided in Table S1.† Details of the hydrogen-bonding interactions of ligands (L2, L4 and L5) are listed in Table S2.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51973005 and 21871275). Dr Yue also acknowledges the support of the Yan'an High-level Talents Special Foundation (No. 2019-33) and the Doctoral Scientific Research Startup Project of Yan’an University (No. YDBK2019-22).References
- (a) M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS; (b) I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS; (c) M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC.
- (a) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC; (b) J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC; (c) O. Santoro, X. Zhang and C. Redshaw, Catalysts, 2020, 10, 800 CrossRef CAS; (d) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- (a) Y. Wei, S. Wang and S. Zhou, Dalton Trans., 2016, 45, 4471–4485 RSC; (b) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (c) J. Lewiński and A. E. H. Wheatley, Top. Organomet. Chem., 2013, 41, 1–58 Search PubMed.
- P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965–1975 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le. Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- (a) Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS; (b) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS PubMed; (c) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed; (d) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS PubMed; (e) X. Pang, R. Duan, X. Li and X. Chen, Polym. Chem., 2014, 5, 3894–3900 RSC; (f) X. Pang, R. Duan, X. Li, C. Hu, X. Wang and X. Chen, Macromolecules, 2018, 51, 906–913 CrossRef CAS; (g) P. Wang, X. Hao, J. Cheng, J. Chao and X. Chen, Dalton Trans., 2016, 45, 9088–9096 RSC; (h) L. Qin, Y. Zhang, J. Chao, J. Cheng and X. Chen, Dalton Trans., 2019, 48, 12315–12325 RSC.
- C.-L. Lee, Y.-F. Lin, M.-T. Jiang, W.-Y. Lu, J. K. Vandavasi, L.-F. Wang, Y.-C. Lai, M. Y. Chiang and H.-Y. Chen, Organometallics, 2017, 36, 1936–1945 CrossRef CAS.
- (a) S. Gesslbauer, R. Savela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 9, 7912–7920 CrossRef CAS; (b) S. Gesslbauer, H. Cheek, A. J. P. White and C. Romain, Dalton Trans., 2018, 47, 10410–10414 RSC.
- F.-J. Lai, L.-L. Chiu, C.-L. Lee, W.-Y. Lu, Y.-C. Lai, S. Ding, H.-Y. Chen and K.-H. Wu, Polymer, 2019, 182, 121812 CrossRef.
- (a) J. A. Francis, S. G. Bott and A. R. Barron, J. Chem. Soc., Dalton Trans., 1998, 3305–3310 RSC; (b) J. A. Francis, S. G. Bott and A. R. Barron, J. Organomet. Chem., 2000, 597, 29–37 CrossRef CAS.
- J. C. Gálvez-Ruiz, H. Nöth and A. Flores-Parra, Inorg. Chem., 2003, 42, 7569–7578 CrossRef PubMed.
- (a) S. Yoon, S. H. Kim, J. Heo and Y. Kim, Inorg. Chem. Commun., 2013, 29, 157–159 CrossRef CAS; (b) S. H. Kim, S. Y. Han, J. H. Kim, Y. Y. Kang, J. Lee and Y. Kim, Eur. J. Inorg. Chem., 2015, 2323–2329 CrossRef CAS.
- M. Li, M. Chen and C. L. Chen, Polymer, 2015, 64, 234–239 CrossRef CAS.
- W.-H. Sun, M. Shen, W. J. Zhang, W. Huang, S. F. Liu and C. Redshaw, Dalton Trans., 2011, 40, 2645–2653 RSC.
- (a) Q. Zhang, W. Zhang, N. M. Rajendran, T. Liang and W.-H. Sun, Dalton Trans., 2017, 46, 7833–7843 RSC; (b) Q. Zhang, W. Zhang, G. A. Solan, T. Liang and W.-H. Sun, Polymers, 2018, 10, 764–782 CrossRef; (c) D. Zhu, L. Guo, W. Zhang, X. Hu, K. Nomura, A. Vignesh, X. Hao, Q. Zhang and W.-H. Sun, Dalton Trans., 2019, 48, 4157–4167 RSC.
- (a) D. N. Nicolaides, K. E. Litinas, G. K. Papageorgiou and J. Stephanidou-Stephanatou, J. Heterocycl. Chem., 1991, 28, 139–143 CrossRef CAS; (b) G. A. Abakumov, V. K. Cherkasov, N. O. Druzhkov, Y. A. Kurskii, G. K. Fukin, L. G. Abakumova and T. N. Kocherova, Synth. Commun., 2006, 36, 3241–3247 CrossRef CAS; (c) L. D. Li, C. S. B. Gomes, P. T. Gomes, L. F. Veiros and S. Y. Kim, ARKIVOC, 2009, 95–111 Search PubMed; (d) V. K. Cherkasov, N. O. Druzhkov, T. N. Kocherova, A. S. Shavyrin and G. K. Fukin, Tetrahedron, 2012, 68, 1422–1426 CrossRef CAS; (e) C. Dong, D. A. Dickie, W. A. Maio and T. A. Manz, ACS Omega, 2018, 3, 16858–16865 CrossRef CAS PubMed; (f) S. Roy, B. Sarkar, D. Bubrin, M. Niemeyer, S. Zalis, G. K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 15230–15231 CrossRef CAS PubMed; (g) L. Li, M. Jeon and S. Y. Kim, J. Mol. Catal. A: Chem., 2009, 303, 110–116 CrossRef CAS; (h) S. Sa, M. Jeon and S. Y. Kim, J. Mol. Catal. A: Chem., 2014, 393, 263–271 CrossRef CAS; (i) A. Anjum, S. Paswan, M. Kumar, H. Ali and R. K. Dubey, Asian J. Chem., 2019, 31, 943–950 CAS.
- B. Gao, X. Luo, W. Gao, L. Huang, S.-M. Gao, X. Liu, Q. Wu and Y. Mu, Dalton Trans., 2012, 41, 2755–2763 RSC.
- Q. Mahmood, E. Yue, W. Zhang, G. A. Solan, T. Liang and W.-H. Sun, Org. Chem. Front., 2016, 3, 1668–1679 RSC.
- N. O. Druzhkov, G. G. Kazakov, A. S. Shavyrin, E. V. Baranov, E. N. Egorova, A. V. Piskunov and G. A. Abakumov, Inorg. Chem. Commun., 2018, 90, 92–96 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of crystallography and hydrogen bonding interactions of ligands. CCDC 2063065 (L2), 2063064 (L4), 2063180 (L5) and 2063066 (Al1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01288f |
| This journal is © The Royal Society of Chemistry 2021 |