
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoreduction properties of novel Z-scheme structured Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6 composites for the removal of Cr(VI)†
Bin Liu‡
 ,
Zhili Fan‡,
Wangjian Zhai,
Junfeng He,
Shaofeng Wang*,
Fuming Chen,
Yinzhen Wang,
Wei Li,
Xianhua Hou and
Qinyu He*
,
Zhili Fan‡,
Wangjian Zhai,
Junfeng He,
Shaofeng Wang*,
Fuming Chen,
Yinzhen Wang,
Wei Li,
Xianhua Hou and
Qinyu He*
Guangdong Provincial Key Laboratory of Quantum Engineering and Quantum Materials, Guangdong Provincial Engineering Technology Research Center for Quantum Precision Measurement, Guangdong Provincial Engineering Technology Research Center of Efficient Green Energy and Environmental Protection Materials, School of Physics and Telecommunication Engineering, South China Normal University, China. E-mail: gracylady@163.com
First published on 16th April 2021
Abstract
Novel Z-scheme structured Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6 (LSTBM) composites were prepared via a facile two-step solvothermal method. Several characterization techniques were employed to investigate the phases, microstructures, compositions, valence states, oxygen vacancies, surface oxygen absorption, energy band structures and lifetime of photoproduced carriers. It was found that the lifetime and transfer of the photoproduced carriers of LSTBM were better than those of Bi2MoO6 (BMO) and Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3 (LSTO). The LSTBM with a molar ratio of BMO/(LSTO + BMO) = 0.07 (denoted as LSTBM7) showed 1.9 and 3.1 times removal rates than those for BMO and LSTO, respectively. Importantly, the built-in electric field in the heterojunction of LSTBM and Ov-s, especially in Ov-s on the higher-Fermi-level side of the heterojunction, had co-played roles in prolonging the lifetime and improving the transfer of photogenerated carriers. The photoproduced e− played a dominant role in reducing Cr(VI) to Cr(III) and the produced Cr(III) tends to form Cr(OH)3 and adsorb onto the surface of the photocatalyst to decrease the nucleation energy. The possible reduction route for Cr(VI) to Cr(III) over LSTBM7 was figured out. This study implies that inducing Ov-s on the higher-Fermi-level side of the Z-scheme heterojunction is a more effective route for separating the photogenerated electrons and holes and improving the transfer of photogenerated carriers.
Introduction
With the development of industry, more and more waste water containing heavy metal ions, such as hexavalent chromium [Cr(VI)], are being discharged into rivers and soil.1 Cr(VI) is toxic and is a threat to human beings and animals.2 In order to solve this problem, numerous methods have been explored, such as adsorption,3 coagulation–flocculation technique,4 membrane separation,5 ion exchange,6 electrochemical method7 and photocatalysis.8 Due to the safety and simplicity of the process, photocatalytic technology has attracted increasing attention in removing Cr(VI).9 However, there are several shortcomings, particularly low removal efficiency, that should be addressed.To resolve the problem of low removal efficiency, many routes have been developed. Two approaches have dominantly attracted attention due to the high effectiveness. One is to improve the lifetime of photogenerated carriers. Another is to enhance the transfer efficiency of the photogenerated carriers. The routes to prolonging the lifetime of the photogenerated carriers include establishing a built-in field through heterojunction,10 introducing an electron-capturing level such as oxygen vacancy (Ov) in the band gap,11 and employing semiconductor (SM) possessing intrinsic local electric field such as perovskites.12 Among the above-mentioned strategies, constructing a heterojunction and introducing an electron-capturing level in the band gap has attracted increasing attention due to its considerable effect on prolonging the lifetime of photogenerated carriers.11,13 The constructed heterojunctions include p–n heterojunction,14 type II heterojunction15 and Z-scheme heterojunction, among which Z-scheme heterojunction is the most effective strategy to improve the lifetime of photogenerated carriers.16,21 Meanwhile, it is unanimously accepted that introducing Ov is also an effective way to separate photogenerated electron–hole pairs by capturing electrons.17–19 To enhance the transfer efficiency of the photogenerated carriers, several strategies have been developed, such as hybridizing expensive metal nanoparticles, such as Au and Ag nanoparticles, with photocatalysts, and compositing good-conductivity material such as g-C3N4 with photocatalysts.20
In this study, we designed a photocatalyst that was expected to superpose the effects of Ov and Z-scheme heterojunction on prolonging the lifetime of photogenerated carriers as well as improving the transfer of photogenerated electrons. In general, the matched energy band edge position between the two SM needs to be considered for constructing Z-scheme heterojunction. As depicted in Fig. S1,† the edge of the valence band (VB) and conduction band (CB) of SM I are higher than those of SM II. The CB of SM II is higher than the VB of SM I. After contacting, electrons would transfer from SM I to SM II. Thus, a built-in electric field with a direction from SM I to SM II will be formed. This built-in electric field would drive photogenerated electrons in the heterojunction to transfer from SM II to SM I and drive photogenerated holes in the heterojunction to transfer from SM I to SM II. Eventually, the photogenerated electron–hole pairs are separated efficiently and the lifetimes of photogenerated carriers are prolonged.
Recently, strontium titanate (SrTiO3), an ABO3-type perovskite, has received considerable attention because of its physicochemical stability, non-toxicity and interesting structure compatibility.22–25 It was reported that doping with La in SrTiO3 (LSTO) would transform it into an n-type degenerate semiconductor, which can trap extra charge carriers (electrons) generated in the conduction band bottom of n-type semiconductor,26 consequently resulting in inhibiting the recombination of photo-generated charge carriers.25 A previous study showed that La-doping in SrTiO3 resulted in a higher concentration of oxygen vacancy clustering.27 Furthermore, it has been reported that doping La at the Sr site of SrTiO3 could result in the transformation of Ti4+ (3d0 orbital) into Ti3+ (3d1 orbital), therefore the formation of Ov for a charge balance.28 The presence of Ti3+ would improve the transfer of photoinduced electrons.29 In addition, Ov-s can modulate both the coordination structures and electronic states of adsorbates via the surface-dangling bonds and localized electrons, therefore improving the photocatalytic efficiency.30,31 It has also been found that the Ti3+/Ti4+(%) ratio, therefore the Ov concentration increases with the increase in La concentration.28 On the other hand, it is well-documented that a large amount of Ov-s will be generated at the heterostructure interface. Consequently, when the components of the heterostructure had different crystal structures, it could present excellent photocatalytic properties owning to the high separation rate of photo-generated carriers induced by Ov-s and the built-in electric field.32 Based on the above discussion, we designed Z-scheme structured composites and chose LSTO to be one SM in a Z-scheme structured composite, whereas another chosen SM with energy-structure-matching in this composite was Bi2MoO6 (BMO), which has Aurivillius structure, is non-toxic, low-cost, chemically stable and visible-light responsible.33,34
In this work, a facile two-step solvothermal method was employed to prepare novel Z-scheme structured LSTO/BMO composites (LSTBM). As expected, it exhibits excellent Cr(VI) photoreduction efficiency, resulting from a longer lifetime of photogenerated carriers and improved transfer. It is found that, besides the Z-scheme structured heterojunction, the ionized Ov is one of the dominant factors effectively separates the photogenerated electrons and holes. Furthermore, the presence of Ti3+ in LSTO due to La doping leads to higher transfer ability.
Experimental
Synthesis of photocatalysts
The sample LSTO was fabricated using the following procedure. 3.40 g titanium butoxide was dissolved in 30 mL ethylene glycol solution, in which 1.69 g of Sr(NO3)2 and 0.87 g of La(NO3)3·6H2O were dissolved and magnetically stirred at 38 °C for 4 h. The obtained solution was dried at 60 °C for days and then transferred into a 45 mL aqueous solution of NaOH (5 M). The mixture was put into a Teflon-lined stainless-steel autoclave and sealed for reacting at 180 °C for 24 h. After cooling to room temperature, the sample was washed with deionized water and ethanol until the pH was 7, then dried at 60 °C for 12 h. The as-prepared powder is LSTO. For comparison, pure SrTiO3 was prepared using the above-described procedure without lanthanum source, and the as-prepared pure SrTiO3 was denoted as STO.The LSTBM composites and BMO were synthesized through a solvothermal method. In the first step, 0.05 g of Na2MoO4·2H2O and 0.2 g Bi(NO3)3·5H2O were dissolved in 15 mL ethylene glycol and magnetically stirred for 85 min (denoted as solution A). In the second step, 20 mL of absolute ethanol was added to solution A under magnetic stirring for 10 min to obtain solution B. In the third step, a certain amount of LSTO was added in 20 mL of absolute ethanol and sonicated for 30 min to obtain turbid liquid (denoted as mixture C). In the fourth step, solution B and mixture C were mixed with the molar ratios of Mo/(Mo + Ti) of 5%, 7% and 10%, and stirred for 10 min to obtain a mixture (denoted as “mixture D”). Finally, the mixture D was poured into a Teflon-lined stainless-steel autoclave and kept at 160 °C for 6 h. The product was washed with deionized water and ethanol and subsequently dried at 60 °C for 12 h. A series of LSTBM was obtained and denoted as LSTBM5, LSTBM7, and LSTBM10 for those with starting molar ratios of Mo/(Mo + Ti) of 5%, 7% and 10%, respectively.
In order to clarify the design effects of LSTBM on the photocatalytic properties, pure BMO was prepared through the identical hydrothermal procedure and parameters.
Characterization
X-ray diffraction (XRD) was carried out to analyze the phases of as-prepared samples using an X'Celerator detector (XRD, X'Pert Pro, Panalytical, Los Altos, CA) with Cu Kα radiation (λ = 1.5406 Å) in the 2θ range of 20°–80°. The microstructures of the as-prepared powders were investigated using scanning electron microscopy (SEM, ZEISS ULTRA 55). The elements in samples were tested by energy-dispersive X-ray spectrometry (EDS). The detailed microstructures and crystallographic structures were further observed using high-resolution transmission electron microscopy (HRTEM, JEM-210HR, Japan). Brunauer–Emmett–Teller (BET) specific surface area and pore size distribution were measured on JW-BK200C. The electron spin resonance spectrum (ESR) was utilized to confirm the presence of Ov by Bruker EMXPLUS. X-ray photoelectron spectroscopy (XPS) investigation was carried out to identify the surface elemental composition and chemical states of samples using a Thermo Scientific K-Alpha XPS system (Thermo Fisher Scientific, America). UV-vis diffuse reflectance (UV-vis DRS) measurements were performed on a UV-2600 spectrophotometer (Shimadzu, Japan) using BaSO4 as a reference sample. Thermal gravimetric (TG) analysis and derivative thermal gravimetric (DTG) analysis were performed on a TGA5500 instrument at a heating rate of 10 °C min−1. Photoluminescence (PL) spectra of as-prepared samples were recorded on a Hitachi F-4500 fluorescence spectrophotometer (Hitachi, Japan) to qualitatively characterize the recombination rates of photogenerated electron–hole pairs. Mott–Schottky curves (M–S) and electrochemical impedance spectroscopy (EIS) data were collected using the CHI760E Electrochemical Workstation (Shanghai Chenhua Instrument Corp., China) at room temperature with an Ag/AgCl (0.5 M Na2SO4) as the reference electrode and a Pt wire as the counter electrode. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) measurements were performed using the SPECTRO ARCOS instrument.In order to examine the absorbed O2, H2 temperature programming reduction analysis (H2-TPR) was performed using AutoChem1 II 2920. First, 120 mg catalyst was pretreated in pure He flow (30–50 mL min−1) for 1 h at 300 °C for dry pretreatment. After cooling to 50 °C, 10% H2/He gas with a flow rate of 30–50 mL min−1 was introduced for 1 h for saturation, and then He flow (50 mL min−1) to purge for 1 h to remove the weak physically adsorbed gas on the surface. The temperature of the furnace was increased at a rate of 10 °C min−1 to 700 °C and a TCD was used as the detector.
Photocatalytic activity experiments
The photocatalytic properties of all catalysts were assessed by testing the properties of photoreducing Cr(VI) in a quartz reactor irradiating by a simulated solar light source generated by a 350 W xenon lamp (Shanghai Lansheng Company, China). In these experiments, 20 mg of the catalyst was plugged into 100 mL K2Cr2O7 solution (10 mg L−1). Before irradiation, the mixed solution was stirred in the dark for 30 min to attain the adsorption–desorption equilibrium. During the photoreduction process, a 1.5 mL-mixed solution was sampled at a given time interval and centrifuged to remove the containing photocatalyst. The concentration of Cr(VI) was tested using a UV-vis spectrophotometer at 540 nm based on the diphenyl carbazide colorimetric method.35 The relationship between concentration (C) and absorbance (A) follows an empirical formula set up by our group as shown in Fig. S2.† The correlation coefficient R2 was close to 1 (R2 = 0.99996), suggesting a high fitting degree of the regression line to the observed absorbance of Cr(VI). Therefore, the concentration of Cr(VI) can be calculated from eqn (1).| C = (A − 0.00197)/0.09639 | (1) |
The removal rate of Cr(VI) at different reaction times can be calculated from eqn (2),
| Removal rate = (C0 − Ct)/C0 × 100 | (2) |
Results and discussion
Phases and microstructures
The XRD patterns of the as-prepared samples are shown in Fig. 1(a). The LSTO exhibits strong diffraction peaks at 2θ = 22.71°, 32.33°, 39.83°, 46.37°, 57.71°, 67.77° and 77.01°, corresponding to the (100), (110), (111), (200), (211), (220) and (310) faces of cubic SrTiO3 (PDF #35-0734), respectively. After the introduction of La3+, diffraction peaks of LSTO were basically close to those of STO. Fig. 1(b) shows the enlarged diffraction peak (110) of LSTO, clearly indicating that after introducing La3+, the peaks have shifted to lower angles, probably due to the substitution by a larger-ionic-radius La3+ (1.15 Å) for a smaller-ionic-radius Sr2+ (1.13 Å).25 The peaks of LSTO are more diffusive than those of STO due to the introduction of La3+. The above results indicate that La atoms were successfully introduced into the crystal structure of SrTiO3. BMO exhibits five well-defined peaks at 2θ = 28.10°, 32.38°, 46.78°, 55.18° and 58.29°, which can be indexed to the (131), (200), (202), (331) and (262) planes, respectively, of crystalline Bi2MoO6 (PDF #72-1524). After the formation of composite catalysts, there was no obvious shift of the diffraction peaks, indicating that BMO exists as a separate phase rather than being incorporated into the LSTO lattice.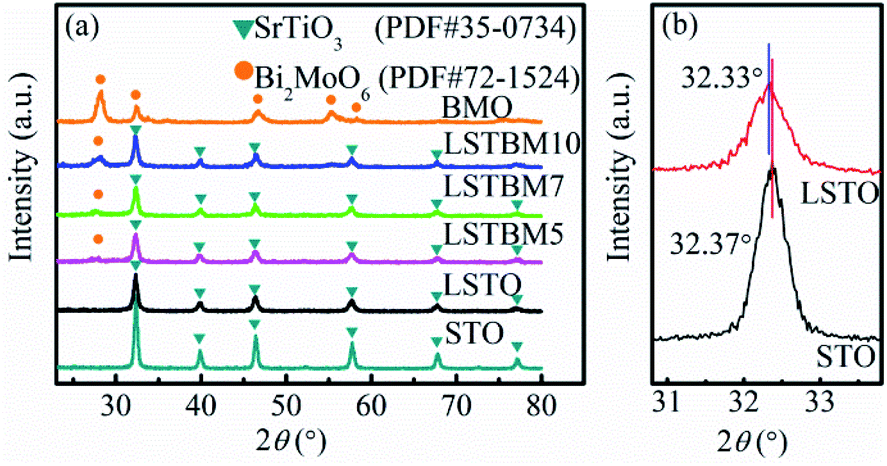 | ||
| Fig. 1 (a) XRD patterns of STO, LSTO, BMO, LSTBM5, LSTBM7 and LSTBM10; (b) the enlarged diffraction peak (110) of STO and LSTO. | ||
The morphologies and particle sizes of LSTO, BMO and LSTBM7 were investigated using SEM. As displayed in Fig. 2(a), the sample LSTO possesses an irregular spherical morphology with a diameter of 100–160 nm, the surface of which has a small island with a dimension of around 20 nm. From Fig. 2(b), it can be found that BMO particles are basically spheres with the size of 1.5–3 μm and assembled into nano-sheets. Additionally, some BMO particles were merged into larger irregular particles of the size of 6–9 μm. Fig. 2(c) is the SEM image of LSTBM7. It is obviously seen from Fig. 2(c) that there are two particle types, a larger one and a smaller one. The larger one possesses a size of ∼9 μm and the smaller one possesses a size of ∼100 nm. By comparing the sizes of BMO and LSTO in Fig. 2(a) and (b), respectively, it can be concluded that the larger ones are probably the merged BMO and the smaller ones are LSTO. A high-resolution image of LSTBM7 is shown in Fig. 2(d). It can be seen that the larger particle in Fig. 2(c) possesses nanosheet-structure, a characteristic of BMO, and the smaller particle possesses a size of 100 nm and island surface, a characteristic of LSTO. From Fig. 2(c) and (d), it can be concluded that LSTO mainly lands on the surface of BMO, and some LSTO microspheres are embedded in the layered structure of BMO to form a 3D hierarchical structure. Moreover, such a hybrid structure could increase the specific surface area and expose more active sites to participate in photocatalytic reactions. The BET surface area, pore volume and pore size of LSTO, BMO and LSTBM7 are shown in Table 1. The sample LSTBM7 possesses a BET surface area of 6.614 m2 g−1, higher than that of LSTO (5.795 m2 g−1). Notably, from Fig. 2(c), it can be seen that the contact area between LSTO and BMO is small. Fig. 2(e) is the EDS spectrum of LSTBM7. It can be confirmed that LSTBM7 contains La, Sr, Ti, Bi, Mo and O elements. Further information about LSTBM7 composites was obtained from HRTEM images as shown in Fig. 2(f). We chose two contacted particles for HRTEM investigation, in which the smaller one was located on the surface of the larger one. The smaller one possesses lattice spacing of 0.198 nm, possibly corresponding to the crystal plane of (200) of LSTO and slightly larger than that of SrTiO3 (PDF #35-0734). The larger one possesses a lattice spacing of 0.312 nm, consistent with the (131) crystal plane of BMO. The above-mentioned results imply that the heterostructure of LSTO/BMO is in LSTBM7.
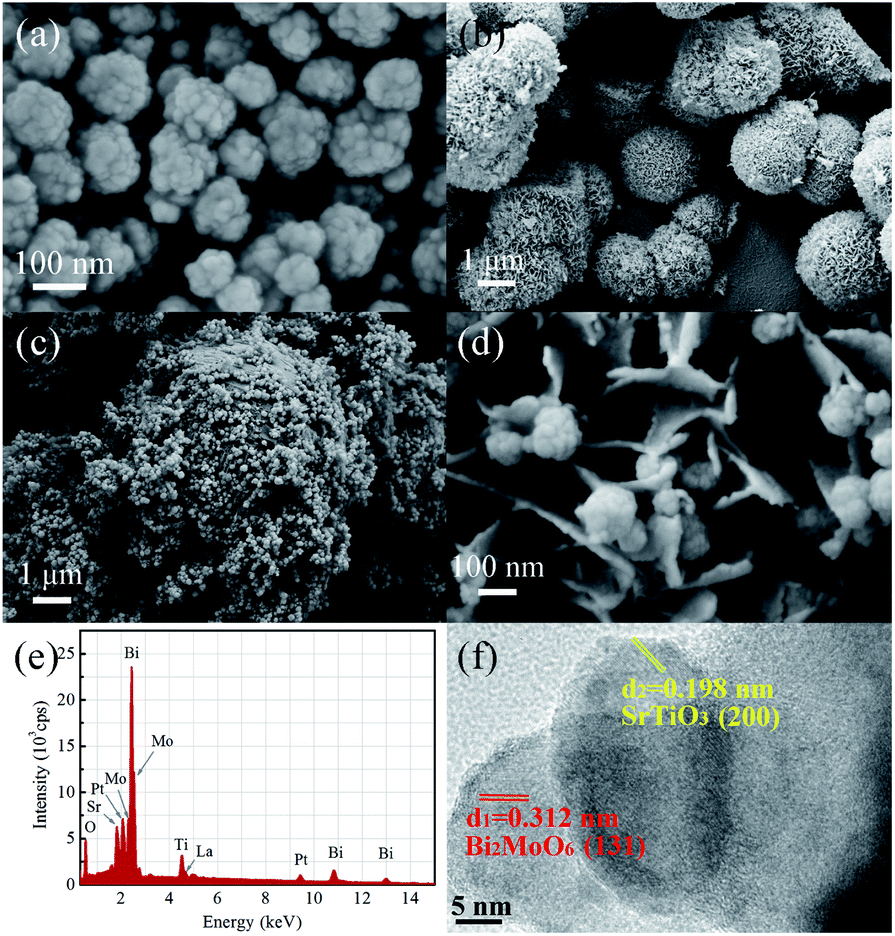 | ||
| Fig. 2 Typical SEM image of (a) LSTO, (b) BMO, (c) and (d) LSTBM7 at different magnifications; (e) EDS spectrum of LSTBM7 and (f) HRTEM images of LSTBM7. | ||
| Sample | BET area (m2 g−1) | Pore volume (m2 g−1) | Pore size (nm) |
|---|---|---|---|
| LSTO | 5.795 | 0.015 | 20.082 |
| BMO | 55.276 | 0.127 | 15.311 |
| LSTBM7 | 6.614 | 0.010 | 10.360 |
ESR and H2-TPR analysis
To confirm the existence of Ov, ESR spectra of BMO, LSTO and LSTBM7 were recorded and are shown in Fig. 3(a). It can be seen that there is a peak at g = 2.004, identified for electrons trapped on Ov-s36 in the ESR spectra of the above-said samples, indicating the presence of Ov-s in BMO, LSTO and LSTBM7. In addition, we can see that the ESR intensity of LSTBM7 was much higher than the sum of LSTO and BMO, suggesting that LSTBM7 possesses much more Ov-s than that in LSTO and BMO, probably due to lattice distortion and mismatch in the heterojunction LSTO/BMO.30 Since the molar proportion of BMO in LSTBM7 is only 7%, the above results show that a small amount of heterojunction can produce considerable Ov-s, i.e., the heterojunction is a very effective tool to produce Ov-s.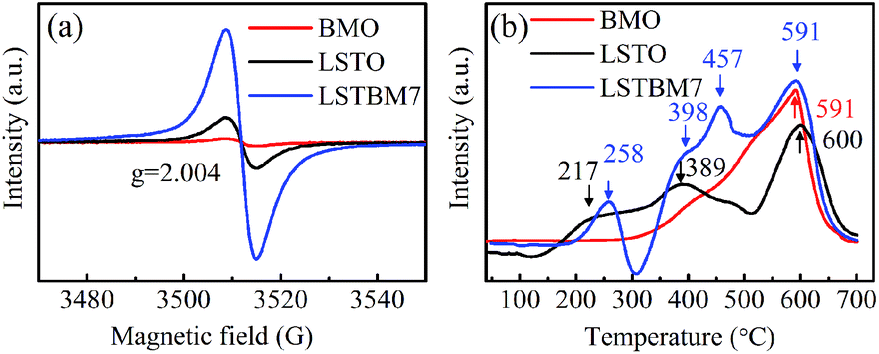 | ||
| Fig. 3 (a) ESR spectra and (b) H2-TPR profile of the BMO, LSTO and LSTBM7. | ||
H2-TPR analysis was performed to investigate the reducibility of the samples. The H2-TPR diagrams of BMO, LSTO and LSTBM7 are shown in Fig. 3(b). There is only one reduction region around 591 °C in the H2-TPR profile of BMO, indicating that there is only lattice oxygen in BMO. For LSTO, there is a wide reduction in the range of 175–500 °C (attributed to the surface lattice oxygen37) and one reduction region around 600 °C (attributed to lattice oxygen38). LSTBM7 shows four distinctive reduction regions around 258 °C, 398 °C, 457 °C and 591 °C. The first and second ones could be ascribed to the absorbed O by Ov-s and surface lattice O, respectively, whereas the reduction peak around 591 °C could be attributed to lattice oxygen. The peaks around 457 °C could be attributed to the oxygen absorbed by Ov-s in the heterojunction of LSTO/BMO, suggesting that the absorbed oxygen by Ov-s has a higher binding energy than that of the surface lattice oxygen. In the low-temperature region around 150–300 °C, LSTBM7 showed higher reduction temperature, indicating higher reducibility. At a higher reduction temperature region, the peak of LSTBM7 moved to a higher temperature, possibly owing to the presence of Ti3+, since the presence of Ti3+ ions in LSTO structure can lead to coupling between those Ti3+ ions through O ions (Ti3+–O2−–Ti4+) or superexchange interaction.28
Elemental valence state analysis
The chemical compositions and chemical states of the as-prepared samples were studied by XPS analysis. Fig. 4(a) shows the XPS survey spectra of LSTO, BMO and LSTBM7. Fig. 4(b), (c) and (d) shows the high-resolution XPS spectra of Sr 3d, Ti 2p and La 3d in LSTO and LSTBM7, respectively. Fig. 4(e) and (f) exhibit the high-resolution XPS spectra of Bi 4f and Mo 3d in BMO and LSTBM7, respectively. Fig. 4(g) shows high-resolution XPS spectra of O 1s in LSTO, BMO and LSTBM7.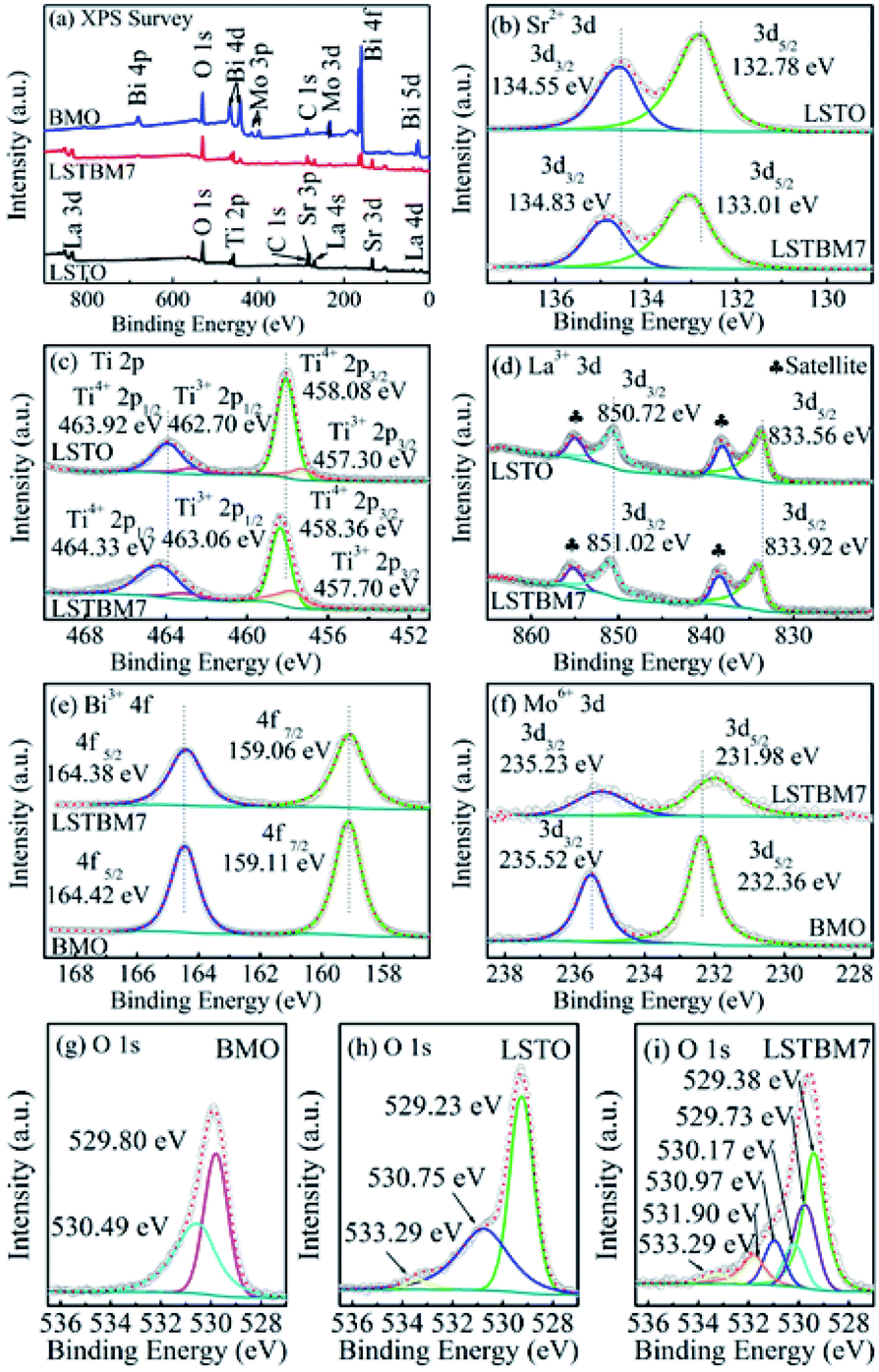 | ||
| Fig. 4 (a) XPS survey spectrum of LSTO, BMO and LSTBM7; (b)–(d) high-resolution XPS spectra of Sr 3d, Ti 2p and La 3d in LSTO and LSTBM7, respectively; (e) and (f) high-resolution XPS spectra of Bi 4f and Mo 3d in BMO and LSTBM7, respectively; (g)–(i) high-resolution XPS spectra of O 1s in BMO, LSTO and LSTBM7. | ||
From Fig. 4(a), it is easy to find that the expected coexistence of Sr, Ti, La, Bi, Mo, O, and C elements (C came from the reference sample) in LSTBM7, which is consistent with the results obtained from EDS investigations.
Fig. 4(b) shows that the Sr 3d spectra of LSTO consist of two individual peaks at 132.78 eV and 134.55 eV, which can be attributed to Sr 3d5/2 and Sr 3d3/2 of SrTiO3,25 respectively. Whereas in LSTBM7, both peaks of Sr 3d5/2 and Sr 3d3/2 shift to higher values of 133.01 eV and 134.83 eV, respectively.
Ti 2p spectra in Fig. 4(c) reveals that LSTO exhibits two peaks at 458.08 eV (ref. 39) and 463.92 eV,40 attributed to Ti4+ 2p3/2 and Ti4+ 2p1/2, respectively, in SrTiO3, while there are higher binding energies of 458.36 eV and 464.33 eV for Ti4+ 2p3/2 and Ti4+ 2p1/2 in the LSTBM7, respectively. Furthermore, the peaks located at 457.30 eV and 462.70 eV of LSTO correspond to Ti3+ 2p3/2 and Ti3+ 2p1/2,41 respectively. So, Ov-s may have been introduced in the LSTO by mixed-valence of titanium (Ti4+/Ti3+) to balance the charge of the compound. The amounts of Ti3+ and Ov in La-doped SrTiO3 were calculated from the XPS peak area, and the results are illustrated in Fig. S3 and Table S1.† We can conclude that Ti3+ and Ov amounts in La-doped SrTiO3 increased with the increase of La concentration. The results of Ti 2p spectra agree with those obtained from H2-TPR analysis.
XPS peaks of La 3d5/2 and La 3d3/2 of LSTO in Fig. 4(d) are located around 833.56 eV (ref. 42) and 850.72 eV,43 respectively. The shake-up satellite peaks of La 3d5/2 and La 3d3/2 are located at 838.11 eV and 854.90 eV.44 Apparently, LSTBM7 possesses a higher binding energy for La 3d5/2, La 3d3/2, shake-up satellite peaks of La 3d5/2 and La 3d3/2 as 833.92 eV, 851.02 eV, 838.39 eV and 855.04 eV, respectively.
Fig. 4(e) shows two strong peaks for Bi 4f5/2 and Bi 4f7/2 located at 164.42 eV and 159.11 eV, respectively, that belong to BMO.45 It is noteworthy that peaks of Bi 4f5/2 and Bi 4f7/2 in LSTBM7 composites exhibit a slight shift towards the lower binding energy compared to those in BMO.
In Fig. 4(f), two peaks at 232.36 eV and 235.52 eV are observed in the XPS spectrum of BMO. The two peaks are ascribed to Mo 3d5/2 and Mo 3d3/2,45 respectively. The peaks of Mo 3d5/2 and Mo 3d3/2 of LSTBM7 also shift to lower values as 231.98 eV and 235.23 eV, respectively.
Fig. 4(g), (h) and (i) show high-resolution XPS spectra of O 1s in BMO, LSTO and LSTBM7, respectively. Three peaks can be fitted for XPS spectra of O 1s of LSTO at 529.23 eV, 530.75 eV and 533.29 eV. The peak at 529.23 eV could be attributed to the lattice oxygen of SrTiO3, while the middle peak at 530.75 eV could be attributed to Ov in LSTO.46 The peak at 533.29 eV is usually assigned to the loosely bound oxygen on the LSTO surface.47 BMO exhibits two peaks at 529.80 eV and 530.49 eV, which are ascribed to the Bi–O and Mo–O bonds, respectively, of Bi2MoO6.45 Six peaks can be fitted at 529.73 eV, 529.38 eV, 530.17 eV, 530.97 eV, 531.90 eV and 533.29 eV. The peak at 529.38 eV can be assigned to the lattice oxygen of LSTO, the one at 530.97 eV can be attributed to Ov of LSTO, and the one at 533.29 eV is assigned to the loosely bound oxygen on the LSTO surface. The peaks at 529.73 eV and 530.17 eV can be assigned to Bi–O and Mo–O bonds, respectively, of Bi2MoO6. It is worth noting that a new peak appears at 531.90 eV in LSTMB7. This peak has also been reported in ref. 48, which was assigned to surface hydroxyl groups. In our sample LSTBM7, there are a few surface hydroxyl groups. On the other hand, the oxygen absorbed by Ov-s with higher binding energy is found in the heterojunction based on the above conclusion from H2-TPR data. We would like to attribute the peak at 531.90 eV to Ov on the LSTO side of the heterojunction LSTO/BMO. More reasons would be given below.
The above-mentioned XPS results reveal that the binding energies of Sr 3d, La 3d, Ti 2p in LSTBM7 are accordingly higher than those in LSTO, while the binding energies of Bi 4f and Mo 3d in the LSTBM7 composite are accordingly lower than those in BMO. Previous works revealed the internal field in heterojunction could result in a shift of binding energy.49,50 From the above-mentioned results, it seems that the produced internal field in the heterojunction LSTO/BMO in LSTBM7 led to a rise in binding energies on the LSTO side and reduction in binding energies on the BMO side, suggesting that the internal field direction is from LSTO to BMO.
It is well known that Ov possesses a positive charge. Therefore, Ov would tend to combine long-distance electrons. After the heterojunction LSTO/BMO is formed, a built-in electric field would form in the junction, in case of which on the LSTO side, some Ov closer to the interface LSTO/BMO would lose the combined electrons and some Ov located away from the interface would not lose the combined electrons. Consequently, the binding energy of O 1s would possess reducing value according to the order, Ov of LSTO in the junction and closer to the interface, the Ov of LSTO in the junction and located away from the interface, the other Ov of LSTO and BMO outside the junction LSTO/BMO. Thus, in the XPS spectrum of LSTBM7, the peak located at 530.97 eV could be attributed to Ov which did not lose combined electrons, while the peak located at 531.90 eV could be ascribed to Ov which lost combined electrons. The mechanism is shown in Fig. S4.† It can be expected that if the Ov was exposed to built-in field produced by low-energy electrons, the Ov is more likely to combine photogenerated electrons, therefore resulting in a longer lifetime for photogenerated carriers.
Band structure
The UV-vis DRS is commonly used to investigate the light absorption and energy band features of SM. As shown in Fig. S5(a),† LSTO presents a similar light response to the reported results.25 BMO exhibits absorption in the visible light range with an absorption edge approaching 437 nm. It can be found that composites LSTBM5, LSTBM7 and LSTBM10 possess stronger absorption within the visible light range than that of LSTO but lower than that of BMO.Based on the absorption spectra, the band gap (Eg) of SM can be calculated from eqn (3).
| αhν = A(hν − Eg)n/2 | (3) |
| Sample | Eg (eV) | Ecb (V) | Evb (V) |
|---|---|---|---|
| LSTO | 3.14 | −1.39 | 1.75 |
| BMO | 2.84 | −0.35 | 2.49 |
According to ref. 8 and 33, SrTiO3 and Bi2MoO6 are indirect and direct SMs, respectively. Based on this knowledge, the estimated-Eg-s of LSTO and BMO are 3.14 eV and 2.84 eV, respectively.
Mott–Schottky plots were used to determine the SM type, as well as the band edge positions of LSTO and BMO. Since the slopes of the fitting curves are positive, as shown in Fig. S6,† both BMO and LSTO are typical n-type SMs. For an n-type SM, the flat band potential (Efb) is located near the conduction band minimum (Ecb) and is readily obtained from the horizontal intercepts as 1/C2 equals zero. According to eqn (4),52 Ecb of LSTO and BMO are estimated to be −1.39 V and −0.35 V vs. NHE, respectively. The valence band maximum (Evb) of LSTO and BMO are calculated using eqn (5).49
| ENHE = Evs. Ag/AgCl + 0.197 | (4) |
| Evb = Eg + Ecb | (5) |
Consequently, the evaluated Evb of LSTO and BMO are 1.75 V and 2.49 V, respectively. The Eg-s and band edge positions of LSTO and BMO are summarized in Table 2.
Photoreduction ability and stability of photocatalysts
The photocatalytic activity of the as-fabricated samples was evaluated by the photoreduction of simulated wastewater containing Cr(VI). Fig. 5(a) and (b) are the dark adsorption–time curve and photoreduction rate–time curves of aqueous-phase Cr(VI) over the as-prepared samples. As shown in Fig. 5(a), it can be found that all samples had reached the dark adsorption–desorption equilibrium within 30 min. It can be seen from Fig. 5(b) that LSTBM7 has the highest photoreduction rate of 99.7% within 180 min, much higher than that of pure BMO and LSTO. The samples, LSTBM5 and LSTBM10 also display a higher removal rate than that of pure BMO and LSTO. Meanwhile, all kinetic curves are linear and the photoreduction of Cr(VI) follows the pseudo-first-order kinetic model:| −ln(Ct/C0) = kt | (6) |
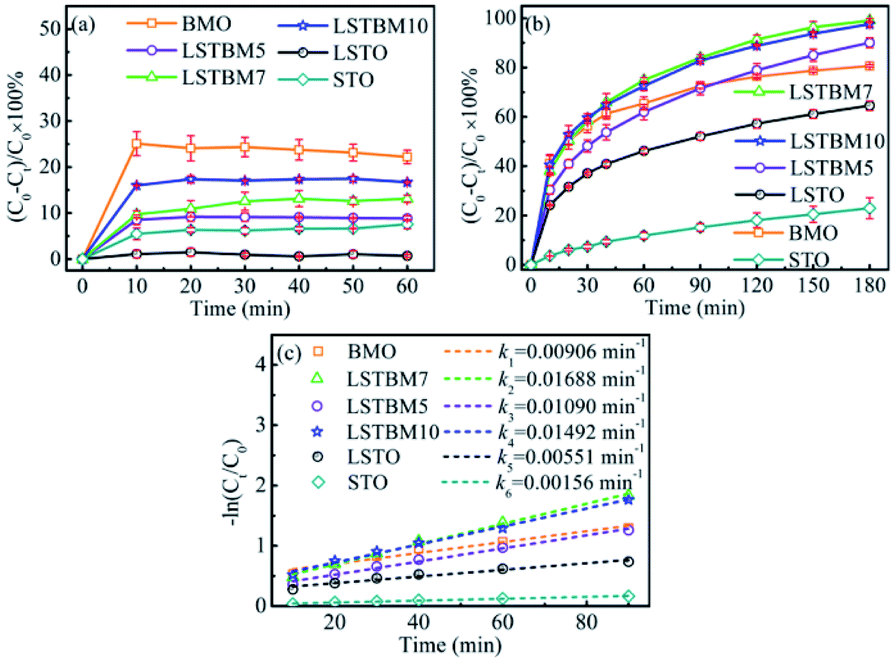 | ||
| Fig. 5 (a) Adsorption experiments of the samples in dark; (b) normal photocatalytic reduction of Cr(VI) and (c) pseudo-first-order kinetic plots for the photocatalytic reduction of Cr(VI). | ||
The ability of separation and transfer of photogenerated carriers
PL spectroscopy was utilized in this work to investigate the recombination of photo-generated electron–hole pairs in SMs.40 In general, a lower PL intensity indicates a lower recombination rate of the photo-generated electron–hole pairs. Fig. 6(a) shows the PL spectra of as-prepared samples under the excitation of 325 nm-wavelength light. It was found that the PL intensity around 370 nm is caused by the recombination of photoproduced electron–hole pairs in LSTO.53 Peaks around 400–500 nm could be ascribed to Ov in materials.53–55 Obviously, the PL intensity of LSTBM5, LSTBM7 and LSTBM10 was lower than that in LSTO, suggesting that the hybridization of LSTO with BMO can effectively impede the recombination of the photogenerated carriers.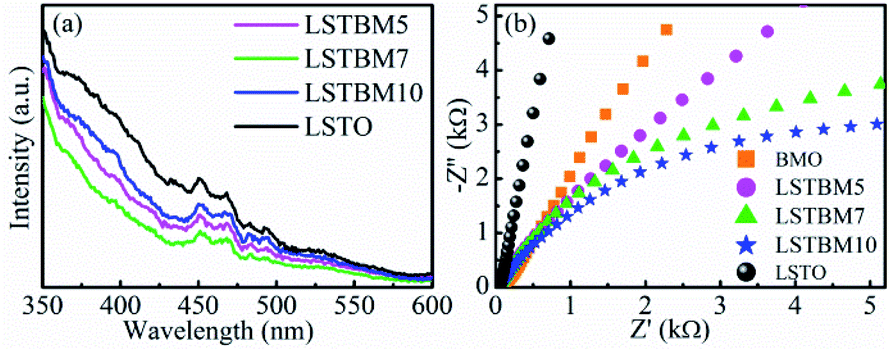 | ||
| Fig. 6 (a) PL spectra and (b) Nyquist plots of as-prepared samples. | ||
The electrochemical impedance spectroscopy (EIS) measurements can be utilized to investigate the ability of electron transfer in the as-prepared samples since a small arc radius means good electronic conductivity and efficient electron separation.56 As shown in Fig. 6(b), LSTBM5, LSTBM7 and LSTBM10 show smaller Nyquist radii than those in BMO and LSTO, suggesting that hybridization of LSTO and BMO would favor the charge transfer. The possible reason will be given in the mechanism section. The possible reason accounting for the better transfer ability of LSTBM7 could be ascribed to the higher-binding-energy Ov after more ionized-Ov was freed by the built-in field in the heterojunction.57
Possible reaction mechanism and route from Cr(VI) to Cr(III)
In order to examine the reaction route to reducing Cr(VI) to Cr(III), a series of investigations were performed to compare the compositions and phases between the fresh LSTBM7 and used LSTBM7. There are color changes (Fig. S7(a)†) that occurred during light irradiation in LSTBM7, as well as a color change in testing solution (simulated Cr(VI) wastewater at the beginning of photoreduction), accompanying a pH value change from 7.03 to 7.81 (Fig. S7(a)†). Before plugging into the simulated Cr(VI) wastewater, the color of LSTBM7 and simulated Cr(VI) wastewater were light yellow color and yellow (heavier than that of LSTBM7), respectively. However, after the process (including LSTBM7 plugging into the simulated Cr(VI) wastewater, then dark absorption for 30 min and photoreduction for 180 min) was finished, the color of LSTBM7 and the simulated Cr(VI) wastewater changed into grey-green and nearly colorless, respectively. These changes meant a decrease of Cr(VI) in the testing solution and something deposited on the surface of LSTBM7 particles after being used (the word “used” means “over which the Cr(VI) was photoreduced”). The XPS spectra shown in Fig. S7(b) and (c)† indicate that only Cr(III) species (576.96 eV and 586.90 eV) exist on the surface of the used LSTBM7 photocatalyst.58,59 Since Cr(OH)3 is a grey-green color,9 it can be concluded that it is Cr(OH)3 is deposited on the surface of LSTBM7 after the photoreduction of Cr(VI) solution. The chemical reaction of the used LSTBM7 around 472 °C, exhibited in TG and DTG curves (Fig. S7(d)†), further confirms the existence of Cr(OH)3 on the surface of the used LSTBM7 since when the temperature reaches 400, Cr(OH)3 gradually lose the OH- and completely transforms into Cr2O3 at 500 °C.60,61 In short, all of the above-mentioned investigations showed the deposition of Cr(OH)3 on the surface of the used LSTBM7. But there are no XRD peaks seen in the used LSTBM7 (Fig. S7(e)†). This result means the deposited Cr(OH)3 is amorphous.In order to further figure out the possible mechanism for photoreduction from Cr(VI) to Cr(III), the active species of LSTBM7 during photoreduction were investigated by performing the active species trapping experiments. Different scavengers, including carbon tetrachloride (CCl4), ethylenediaminetetraacetic acid disodium (EDTA-2Na), isopropanol (IPA) and nitrogen (N2) purging were employed for quenching the photogenerated electrons (e−), holes (h+), hydroxyl radicals (·OH) and superoxide radicals (·O2−) generated during the photoreduction process.62,63 Generally, the inhibition of photocatalytic activity after adding the scavenger mean the existence of corresponding active species.35,62,64,65 Fig. 7 displays the results of active species trapping experiments. Obviously, there are three active species, namely, – e−, ·O2− and ·OH – during photoreduction. The three active species play a rising role in photoreduction according to the order, ·OH < ·O2− < e−. The addition of EDTA–2Na shows an abnormal phenomenon that the photocatalytic activity was reinforced by introducing EDTA–2Na. This reinforcement was attributed to the reaction between EDTA–2Na and h+, a reaction that could reduce the recombination of photogenerated electron–hole pairs, allowing more electrons, and therefore, more ·O2−-s to participate in the reduction reaction, resulting in the rise in photocatalytic activity.62,66
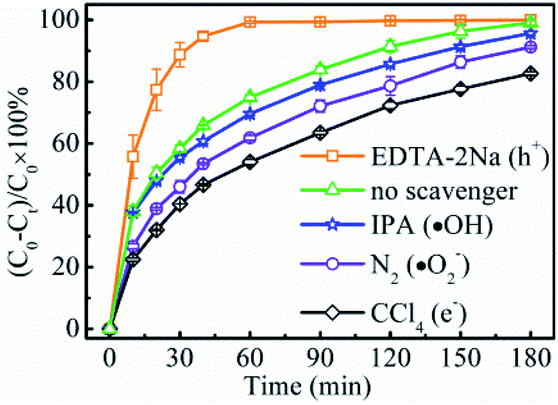 | ||
| Fig. 7 Effects of various scavengers on the photoreduction rate of Cr(VI) of LSTBM7. | ||
Based on the aforementioned results and analysis, a possible energy band structure and reaction mechanism are put forward in Fig. 8. Since Ecb and Evb of LSTO and BMO are (−1.39 V, 1.75 V), (−0.35 V, 2.49 V), respectively. LSTO and BMO could form a Z-scheme heterojunction in LSTBM7. In the case of this Z-scheme mechanism, the following inference can be obtained. When BMO and LSTO contact closely, the electrons will move from LSTO to BMO and form a built-in field in the interface with the direction of LSTO to BMO. The built-in field direction has been confirmed from the above-discussed XPS analysis. Under irradiation, as shown in Fig. 8, the electrons in the VB of LSTO and BMO would be excited and jump to their CB. Due to the existence of a built-in electric field, the photogenerated electrons of BMO would move to LSTO and tend to recombine with photogenerated holes in VB of LSTO, leaving behind photogenerated electrons in the CB of LSTO and photogenerated holes in the VB of BMO, resulting in a longer lifetime of photogenerated carriers. This inference agrees well with the PL results.
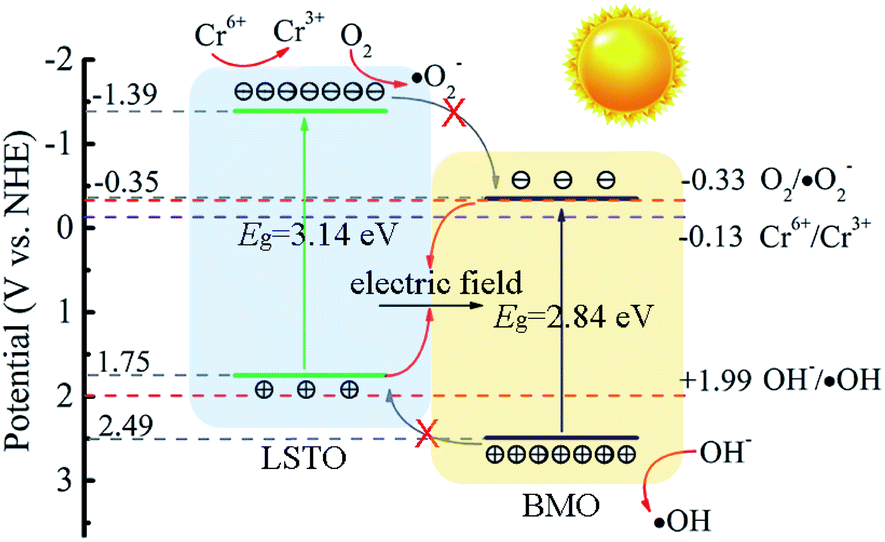 | ||
| Fig. 8 Proposed mechanism for the separation of photogenerated charge carriers in LSTBM7. | ||
A comparison of high-solution XPS between the samples after 20 min of photoreduction and 180 min of photoreduction can give detailed evidence of the photoreduction route from Cr(VI) to Cr(III) in Fig. S8.† It indicates that after 20 min of photoreduction, there appeared Cr(VI) on the surface of LSTBM7 particles, while at the end of photoreduction (180 min), only Cr(III) existed on the surface of LSTBM7, suggesting that Cr(VI) in the simulated wastewater was adsorbed on the surface of LSTBM7 first, then photo-reduced into Cr(III). In addition, during the photoreduction, the produced Cr(III) tends to form Cr(OH)3 in case of pH 7.81 and adsorb onto the surface of the photocatalyst to decrease the nucleation energy.
Based on all of the above results and analyses, a possible reaction mechanism for Cr(VI) to Cr(III) over LSTBM7 composites is proposed as follows. Actually, during the photoreduction, there was a competition between the reaction of photogenerated electrons with Cr(VI) (Cr6+/Cr3+, −0.13 V)9 and the reaction of photogenerated electrons with O2 (O2/·O2−, −0.33 V). But from the results of active species experiments, it can be seen that the photogenerated electrons tend to directly reduce Cr(VI) to Cr(III) rather than form ·O2−. As for the phenomenon that ·OH played less role in photoreduction, the previous research can explain it. According to previous research,62,67 ·OH can be attributed to the equilibrium transmitting to Cr(VI) as shown in eqn (7)–(9),
| ·O2− + H+ → ·HO2 | (7) |
| ·HO2 + ·O2− + H+ → H2O2 + O2 | (8) |
| H2O2 + Cr(III) + H+ → Cr(VI) + H2O + ·HO | (9) |
Meanwhile, because the energy difference (0.5 V) between the VB of BMO and the potential of OH−/·OH is smaller than that (1.06 V) between the CB of LSTO and the potential of O2/·O2−, the production of ·OH is smaller. That is why during photoreduction of Cr(VI) over LSTBM7, ·OH play less role than ·O2−.
All in all, the above discussion confirmed the Z-scheme mechanism in LSTBM7. The Z-scheme structured heterojunction and Ov in the junction have enhanced the lifetime of photogenerated carriers and improved the transferability of photoproduced carriers. On the basis of experimental results and the discussion above, the process for Cr(VI) photoreduction of LSTBM7 can be described as follows:
| LSTBM7 + hν → e− + h+ | (10) |
| CrO42− + 4H2O + 3e− → Cr(OH)3 + 5OH− | (11) |
| O2 + e− → ·O2− | (12) |
| CrO42− + 4H2O + 3·O2− → Cr(OH)3 + 3O2 + 5OH− | (13) |
| H2O + h+ → ·OH + H+ | (14) |
Conclusion
In summary, a novel heterogeneous Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6 composites were prepared via a facile two-step solvothermal method. The energy band structure analyses showed that the heterojunction Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6 forms a Z-scheme. The Z-scheme structured Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6 photocatalyst showed excellent photocatalytic reduction performance for Cr(VI) removal. The photocatalytic reduction of the sample LSTBM7 is 1.9, 3.1 and 10.6 times that of BMO, LSTO and STO, respectively. The dominant factors which led to improved photocatalytic efficiency are the prolonged lifetime of photoproduced carriers and improved transfer of carriers. The prolonged lifetime possibly originated from the effective separation of photogenerated electrons and holes by the build-in electric field in the heterojunction of Sr0.8La0.2(Ti1−δ4+Tiδ3+)O3/Bi2MoO6, as well as Ov which lost combined electrons in the junction. The improved transfer of photogenerated carriers could be attributed to the increased Ov which lose captured electrons in the built-in field. It was also found that e− plays a dominant role in reducing Cr(VI) to Cr(III). Meanwhile, it was found that during the photoreduction, the produced Cr(III) tends to form Cr(OH)3 in the case of pH 7.81 and adsorb onto the surface of the photocatalyst to decrease the nucleation energy.This work implies that inducing Ov-s on the higher-Fermi-level side of the Z-scheme heterojunction is an effective route to separating the photogenerated electrons and holes and improving the transfer of photogenerated carriers. Such heterostructured photocatalyst design strategy based on perovskite-related materials and bismuth molybdate might provide new insights for treating Cr-containing wastewater.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 51972122 and 51672090).Notes and references
- Z. Zhao, H. An, J. Lin, M. Feng, V. Murugadoss, T. Ding, H. Liu, Q. Shao, X. Mai, N. Wang, H. Gu, S. Angaiah and Z. Guo, Chem. Rec., 2018, 19, 873–882 CrossRef PubMed.
- C. E. Barrera-Díaz, V. Lugo-Lugo and B. Bilyeu, J. Hazard. Mater., 2012, 223–224, 1–12 CrossRef PubMed.
- C. Wan, R. Zhang, L. Wang, X. Liu, D. Bao and G. Song, Appl. Surf. Sci., 2021, 536, 147980 CrossRef CAS.
- Z. A. Zakaria and W. A. Ahmad, Water, Air, Soil Pollut., 2020, 231, 490 CrossRef CAS.
- M. Pishnamazi, S. Koushkbaghi, S. S. Hosseini, M. Darabi, A. Yousefi and M. Irani, J. Mol. Liq., 2020, 317, 113934 CrossRef CAS.
- Z. Shao, C. Huang, Q. Wu, Y. Zhao, W. Xu, Y. Liu, J. Dang and H. Hou, J. Hazard. Mater., 2019, 378, 120719 CrossRef CAS PubMed.
- J. Zhou, M. Li, W. Zhou, J. Hu, Y. Long, Y. F. Tsang and S. Zhou, Sci. Total Environ., 2020, 748, 141425 CrossRef CAS PubMed.
- Y. Yang, X.-A. Yang, D. Leng, S.-B. Wang and W.-B. Zhang, Chem. Eng. J., 2018, 335, 491–500 CrossRef CAS.
- T. Li, Y. Gao, L. Zhang, X. Xing, X. Huang, F. Li, Y. Jin and C. Hu, Appl. Catal., B, 2020, 277, 119065 CrossRef CAS.
- W. Dong, Y. Liu, G. Zeng, S. Zhang, T. Cai, J. Yuan, H. Chen, J. Gao and C. Liu, J. Colloid Interface Sci., 2018, 518, 156–164 CrossRef CAS PubMed.
- Z. Shayegan, C.-S. Lee and F. Haghighat, Chem. Eng. J., 2018, 334, 2408–2439 CrossRef CAS.
- B. Lin, A. Chaturvedi, J. Di, L. You, C. Lai, R. Duan, J. Zhou, B. Xu, Z. Chen, P. Song, J. Peng, B. Ma, H. Liu, P. Meng, G. Yang, H. Zhang, Z. Liu and F. Liu, Nano Energy, 2020, 76, 104972 CrossRef CAS.
- H. Jiang, H. Song, Z. Zhou, X. Liu and G. Meng, J. Phys. Chem. Solids, 2007, 68, 1830–1835 CrossRef CAS.
- W. Mao, L. Zhang, T. Wang, Y. Bai and Y. Guan, Front. Environ. Sci. Eng., 2021, 15, 52 CrossRef CAS.
- C. Liu, W. Fang, Y. Song, F. Li, Z. Sun and L. Xu, Mater. Sci. Semicond. Process., 2020, 120, 105276 CrossRef CAS.
- A. Raja, P. Rajasekaran, B. Vishnu, K. Selvakumar, J. Yeon Do, M. Swaminathan and M. Kang, Sep. Purif. Technol., 2020, 252, 117446 CrossRef CAS.
- R. Hailili, C. Wang and E. Lichtfouse, Appl. Catal., B, 2018, 232, 531–543 CrossRef CAS.
- D. Chen, S. Wu, J. Fang, S. Lu, G. Zhou, W. Feng, F. Yang, Y. Chen and Z. Fang, Sep. Purif. Technol., 2018, 193, 232–241 CrossRef CAS.
- S. Song, J. Wang, T. Peng, W. Fu and L. Zan, Appl. Catal., B, 2018, 228, 39–46 CrossRef CAS.
- C. Yu, P. Wang, X. Wang, F. Chen and H. Yu, J. Alloys Compd., 2019, 806, 263–271 CrossRef CAS.
- G. Liao, C. Li, X. Li and B. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef.
- H. Che, J. Chen, K. Huang, W. Hu, H. Hu, X. Liu, G. Che, C. Liu and W. Shi, J. Alloys Compd., 2016, 688, 882–890 CrossRef CAS.
- F. Xiao, J. Xu, L. Cao, S. Jiang, Q. Zhang and L. Wang, Environ. Sci. Pollut. Res., 2019, 27, 5788–5796 CrossRef PubMed.
- Y. Wu, Y. Wei, Q. Guo, H. Xu, L. Gu, F. Huang, D. Luo, Y. Huang, L. Fan and J. Wu, Sol. Energy Mater. Sol. Cells, 2018, 176, 230–238 CrossRef CAS.
- D. Yang, X. Zou, Y. Sun, Z. Tong and Z. Jiang, Front. Chem. Sci. Eng., 2018, 12, 440–449 CrossRef.
- G. Wang, P. Wang, H.-K. Luo and T. S. A. Hor, Chem.–Asian J., 2014, 9, 1854–1859 CrossRef CAS PubMed.
- S. Suthirakun, S. C. Ammal, G. Xiao, F. Chen, K. Huang, H.-C. zur Loye and A. Heyden, Solid State Ionics, 2012, 228, 37–45 CrossRef CAS.
- P. Songwattanasin, A. Karaphun, S. Phokha, S. Hunpratub, S. Maensiri, V. Amornkitbamrung and E. Swatsitang, Phys. B, 2019, 571, 213–221 CrossRef CAS.
- X. Zhou, N. Liu, T. Yokosawa, A. Osvet, M. E. Miehlich, K. Meyer, E. Spiecker and P. Schmuki, ACS Appl. Mater. Interfaces, 2018, 10, 29532–29542 CrossRef CAS PubMed.
- W. Hou, H. Xu, Y. Cai, Z. Zou, D. Li and D. Xia, Appl. Surf. Sci., 2020, 530, 147218 CrossRef CAS.
- J. L. Hao Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef PubMed.
- K. Zhao, Z. Zhang, Y. Feng, S. Lin, H. Li and X. Gao, Appl. Catal., B, 2020, 268, 118740 CrossRef CAS.
- X. Li, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, J. Hazard. Mater., 2020, 400, 123243 CrossRef CAS PubMed.
- X. Liu, S. Gu, Y. Zhao, G. Zhou and W. Li, J. Mater. Sci. Technol., 2020, 56, 45–68 CrossRef.
- R. Yuan, J. Qiu, C. Yue, C. Shen, D. Li, C. Zhu, F. Liu and A. Li, Chem. Eng. J., 2020, 401, 126020 CrossRef CAS.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- Q. Liang, X. Wu, D. Weng and H. Xu, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- C. Huang, J. Liu, P. Sun, X. Liang, C. Tang and H. Wu, Energy Sources, Part A, 2019, 43, 326–336 CrossRef.
- R. P. Vasquez, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 217–240 CrossRef CAS.
- M. Abdi, V. Mahdikhah and S. Sheibani, Opt. Mater., 2020, 102, 109803 CrossRef CAS.
- N. K. A. Hamed, M. K. Ahmad, N. H. H. Hairom, A. B. Faridah, M. H. Mamat, A. Mohamed, A. B. Suriani, N. Nafarizal, F. I. M. Fazli, S. M. Mokhtar, W. I. W. Omar and M. Shimomura, Appl. Surf. Sci., 2020, 534, 147571 CrossRef CAS.
- C. N. R. R. D. D. Sarma, J. Electron Spectrosc. Relat. Phenom., 1980, 20, 25–45 CrossRef.
- Y. Liu, S. Zhou, J. Li, Y. Wang, G. Jiang, Z. Zhao, B. Liu, X. Gong, A. Duan, J. Liu, Y. Wei and L. Zhang, Appl. Catal., B, 2015, 168–169, 125–131 CrossRef CAS.
- Y. Yu, L. Piao, J. Xia, W. Wang, J. Geng, H. Chen, X. Xing and H. Li, Mater. Chem. Phys., 2016, 182, 77–85 CrossRef CAS.
- M. Sun, P. Guo, M. Wang and F. Ren, Optik, 2019, 199, 163319 CrossRef CAS.
- Z. Meng, B. Tong, L. Shen, L. Xu, W. Chu, W. Wei, X. Xu, Y. Liu, L. Pi, S. Zhang, C. Zhou, Z. Ma, G. Meng and Z. Sheng, Ceram. Int., 2020, 46, 22140–22145 CrossRef CAS.
- H. Tan, Z. Zhao, W.-b. Zhu, E. N. Coker, B. Li, M. Zheng, W. Yu, H. Fan and Z. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 19184–19190 CrossRef CAS PubMed.
- F. Fu, H. Shen, X. Sun, W. Xue, A. Shoneye, J. Ma, L. Luo, D. Wang, J. Wang and J. Tang, Appl. Catal., B, 2019, 247, 150–162 CrossRef CAS.
- K. Wei, B. Wang, J. Hu, F. Chen, Q. Hao, G. He, Y. Wang, W. Li, J. Liu and Q. He, RSC Adv., 2019, 9, 11377–11384 RSC.
- W. Shi, M. Li, X. Huang, H. Ren, F. Guo, Y. Tang and C. Lu, Chem. Eng. J., 2020, 394, 125009 CrossRef CAS.
- X. Ren, K. Wu, Z. Qin, X. Zhao and H. Yang, J. Alloys Compd., 2019, 788, 102–109 CrossRef CAS.
- C. Chen, M. Li, Y. Jia, R. Chong, L. Xu and X. Liu, J. Colloid Interface Sci., 2020, 564, 442–453 CrossRef CAS PubMed.
- S. Li, M. Li, A. Tao, M. Song, B. Wang, J. Niu, F. Yu and Y. Wu, J. Sol-Gel Sci. Technol., 2020, 94, 288–297 CrossRef CAS.
- B. G. Anitha and L. G. Devi, Chem. Phys. Lett., 2020, 742, 137138 CrossRef CAS.
- A. Rosy and G. Kalpana, Curr. Appl. Phys., 2018, 18, 1026–1033 CrossRef.
- S. Zhang, M. Li, J. Zhao, H. Wang, X. Zhu, J. Han and X. Liu, Appl. Catal., B, 2019, 252, 24–32 CrossRef CAS.
- J. Jing, Z. Chen, Y. Bu, M. Sun, W. Zheng and W. Li, Electrochim. Acta, 2019, 304, 386–395 CrossRef CAS.
- R. Yan, D. Luo, C. Fu, Y. Wang, H. Zhang, P. Wu and W. Jiang, Sep. Purif. Technol., 2020, 234, 116130 CrossRef CAS.
- E. Desimoili, C. Malitesta, P. G. Zambonin and J. C. Riviere, Surf. Interface Anal., 1988, 13, 173–179 CrossRef.
- K. Yan, Z. Liu, Z. Li, R. Yue, F. Guo and Z. Xu, Hydrometallurgy, 2019, 186, 42–49 CrossRef CAS.
- A. S. O. Gomes, N. Yaghini, A. Martinelli and E. Ahlberg, J. Raman Spectrosc., 2017, 48, 1256–1263 CrossRef CAS.
- H. Li, C. Zhao, X. Li, H. Fu, Z. Wang and C.-C. Wang, J. Alloys Compd., 2020, 844, 156147 CrossRef CAS.
- B. Wang, K. Wei, F. Chen, Y. Wang, G. He, W. Li, J. Liu and Q. He, J. Alloys Compd., 2019, 804, 100–110 CrossRef CAS.
- P. Shao, J. Tian, F. Yang, X. Duan, S. Gao, W. Shi, X. Luo, F. Cui, S. Luo and S. Wang, Adv. Funct. Mater., 2018, 28, 1705295 CrossRef.
- P. Shao, S. Yu, X. Duan, L. Yang, H. Shi, L. Ding, J. Tian, L. Yang, X. Luo and S. Wang, Environ. Sci. Technol., 2020, 54, 8464–8472 CrossRef CAS PubMed.
- Z. Wang, M. Murugananthan and Y. Zhang, Appl. Catal., B, 2019, 248, 349–356 CrossRef CAS.
- C. Zhao, Z. Wang, X. Li, X. Yi, H. Chu, X. Chen and C.-C. Wang, Chem. Eng. J., 2020, 389, 123431 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01466h |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |