
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow do the doping concentrations of N and B in graphene modify the water adsorption?
Thi Tan Pham†
ab,
Thanh Ngoc Pham†ab,
Viorel Chihaiac,
Quang Anh Vuab,
Thuat T. Trinh d,
Trung Thanh Phame,
Le Van Thang*ab and
Do Ngoc Son*ab
d,
Trung Thanh Phame,
Le Van Thang*ab and
Do Ngoc Son*ab
aHo Chi Minh City University of Technology, 268 Ly Thuong Kiet Street, Ward 14, District 10, Ho Chi Minh City, Vietnam. E-mail: dnson@hcmut.edu.vn; vanthang@hcmut.edu.vn
bVietnam National University Ho Chi Minh City, Quarter 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
cInstitute of Physical Chemistry “Ilie Murgulescu” of the Romanian Academy, Splaiul Independentei 202, Sector 6, 060021 Bucharest, Romania
dDepartment of Civil and Environmental Engineering, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway
eNamur Institute of Structured Matter (NISM), Department of Physics, University of Namur, 61 Rue de Bruxelles, B-5000 Namur, Belgium
First published on 1st June 2021
Abstract
Understanding the interaction of water and graphene is crucial for various applications such as water purification, desalination, and electrocatalysis. Experimental and theoretical studies have already investigated water adsorption on N- and B-doped graphene. However, there are no reports available that elucidate the influences of the N and B doping content in graphene on the microscopic geometrical structure and the electronic properties of the adsorbed water. Thus, this work is devoted to solving this problem using self-consistent van der Waals density functional theory calculations. The N and B doping contents of 0.0, 3.1, 6.3, and 9.4% were considered. The results showed that the binding energy of water increases almost linearly as a function of doping content at all concentrations for N-doped graphene but below 6.3% for B-doped graphene. In the linear range, the binding energy increases by approximately 30 meV for each increment of the doping ratio. Analyses of the geometric and electronic structures explained the enhancement of the water–graphene interaction with the variation in doping percentage.
1. Introduction
The interface between water and graphene plays an important role in diverse applications such as wetting, water purification, desalination, and electrocatalysis owing to its frictionless properties.1,2 The water–graphene interface has been intensively investigated by experimental and computational methods.2–10 Experimentally, the interface can be studied by measuring static water contact angles, providing macroscopic understanding, and different values of the water contact angles were reported for water droplets on graphene.1–4 The discrepancy in the reported data originates from several factors, i.e., the number of graphene layers, the underlying substrate, and the effects of doping. Hong et al. showed that the supporting substrate and physical doping by applying a voltage can unambiguously affect the hydrophilicity of graphene, which was later confirmed by Ashraf et al.3,4 Both studies claimed that the origin of the physical doping-induced wettability of graphene is the shift of the Dirac point away from the Fermi level of graphene. To provide an atomistic understanding, density functional theory (DFT) calculations have been widely employed to investigate the adsorption of water on graphene.5–10 Nevertheless, the different computational parameters result in a discrepancy in the binding energy (Eb) of the water monomer on graphene, which is mainly due to lack of treatment of the van der Waals interactions. By using the conventional GGA-PBE method, the Eb is 30 meV, whereas the vdW-DF2C09x result6 is 80 meV, which is more consistent with the random-phase approximation (98 meV) and the diffusion Monte Carlo method (100 meV).8 The variance between the GGA-PBE and vdW inclusive methods is about 50 meV; however, Brandenburg et al. found that a change in Eb of H2O on graphene of 50 meV leads to the graphene surface changing from hydrophobic to hydrophilic.8 This highlights the importance of using van der Waals inclusive DFT methods to study the water–graphene interaction.7,8 To elucidate the wetting dynamics of water on graphene, classical molecular dynamics simulations were employed.11,12 Andrews et al. pointed out that at rather low coverage, water molecules tend to spread out and form water layers on graphene, whereas water nanodroplets are formed when more water molecules are exposed.11 Similarly, Akaishi et al. also found that water forms a double layer covering the entire graphene surface, followed by the formation of three-dimensional water clusters. Their results indicated that the pristine graphene itself exhibits hydrophilicity and the stabilization of the water double layer on graphene leads to the hydrophobicity observed in experiments.12 The predictions of two previous theoretical studies have been proved by recent experiments on the water–graphite interface.13 Wang et al. experimentally observed a monolayer of water grown on the graphite surface using STM imaging.13 By using DFT calculations, they also found that all water molecules within this monolayer form a strong hydrogen bonding network, giving rise to the stabilization of this water monolayer.Compared to the wealth of information concerning water on pristine graphene, the physical properties of water on nitrogen (N) and boron (B)-doped graphene are not fully understood yet.14,15 N-doped graphene quantum dots showed hydrophilicity (hydrophobicity) with mono-layered (multi-layered) structures.16 Graphene doped with boron has been synthesized and used in various applications.17 Dai et al. theoretically investigated the adsorption of water and gases on single site X-doped graphene (X = B, N, Al, and S) using the GGA-PBE method, and N and B doping enhanced the adsorption strength of water.18 Yang et al. studied water/fluorinated graphene using DFT calculations with the vdW-DF functional and found that fluorine doping could increase the Eb of the adsorbed water and change the water dipole moment.19 Recently, Xu et al. investigated water on hydrogenated pyridinic N-doped, graphitic, and pyrrolic N-doped graphene using the GGA-PBE approximation with Grimme’s D2 dispersion correction.20 It was found that both the Eb and the orientation of the water molecules are significantly altered upon N doping. Cardozo-Mata et al. also studied water dimers on the N terminated vacancies of graphene and found that the interaction mainly originates from hydrogen bonding between water H and N.21 All of the previous reports considered water adsorption on N- and B-doped graphene at a specific doping content and on the N terminated graphene edges and defects; however, the influences of the N and B doping content on the water–substrate interaction remain unknown. Therefore, this work addresses this issue by systematically considering the water adsorption on graphene with N and B doping at the graphitic sites and low concentrations of 0.0, 3.1, 6.3, and 9.4%. Using DFT calculations with the rev-vdW-DF2 functional, we elucidated a microscopic picture of the structural–electronic property relationships for water–graphene systems. There are several types of doping in graphene. The graphitic type refers to replacing the C atoms in graphene with dopant atoms like N and B, which retains the honeycomb structure of perfect graphene, and the pyridinic and pyrrolic types indicate doping accompanied by the creation of vacancies and edges. The graphitic type of doping22–25 was considered in the current work because it exhibits higher stability than the pyridinic and pyrrolic types26,27 and also has potential applications in fuel cells, Li-ion batteries, photocatalysis, and electrochemical sensing.28 Clarification of the hydrophilicity and hydrophobicity of water on N,B-doped graphene with variation of the doping content is out of scope. Also, we only focused on doping with single components of N and B without mixing of these elements.
2. Computational details
All our DFT calculations were performed within the periodic supercell approach using the Quantum ESPRESSO package.29 Core electrons are represented by Vanderbilt’s ultrasoft pseudopotentials30 with the GBRV library,31 whereas the valence states were expanded in plane waves with cut-off energies of 50 and 600 Ry for wave functions and augmented charge densities, respectively. We used the rev-vdW-DF2 exchange–correlation functional,32 which has been shown to give reasonable accuracy for water on carbon materials,33 as well as other systems.34–37 All atoms are allowed to relax until the force acting on them is less than 0.02 eV Å−1. A Γ-centered 6 × 6 × 1 k-point mesh is used for structural optimizations. To accurately calculate the density of states (DOS), a Γ-centered 16 × 16 × 1 k-point mesh is employed. To estimate the vacuum level and thus the work function accurately, we employed dipole moment correction to eliminate the spurious electric field in the surface normal direction.38 This correction is used only for the calculation of the work function, and it is omitted during structural optimization.The pristine and doped graphene films were modeled by using a unit cell of lateral (4 × 4) periodicity composed of 32 C atoms with a vacuum of 18 Å in the normal surface direction. To evaluate the stability of graphene doped with N and B, the formation energy is calculated by
| Ef = Egra − NX × μX, | (1) |
The binding energy of water on pristine and doped graphene is estimated by
| Eb = (Ewater + Egra) − Ewater/gra, | (2) |
| Δρ = ρwater/gra − ρwater − ρgra. | (3) |
Here, ρwater/gra, ρwater, and ρgra are the charge densities of water adsorbed on graphene and of the fragments, i.e., water and graphene in their adsorbed atomic geometries.
3. Results and discussion
3.1. N- and B-doped graphene
Various positions of N and B substitutions for C atoms are shown in Fig. 1. The dopant contents in the graphene sheet are ca. 3.1, 6.3, and 9.4% for single, double, and triple site replacement, respectively, and are comparable with the N content in experimentally reported N-doped graphene.22,39 Formation energies are estimated and summarized in Table 1.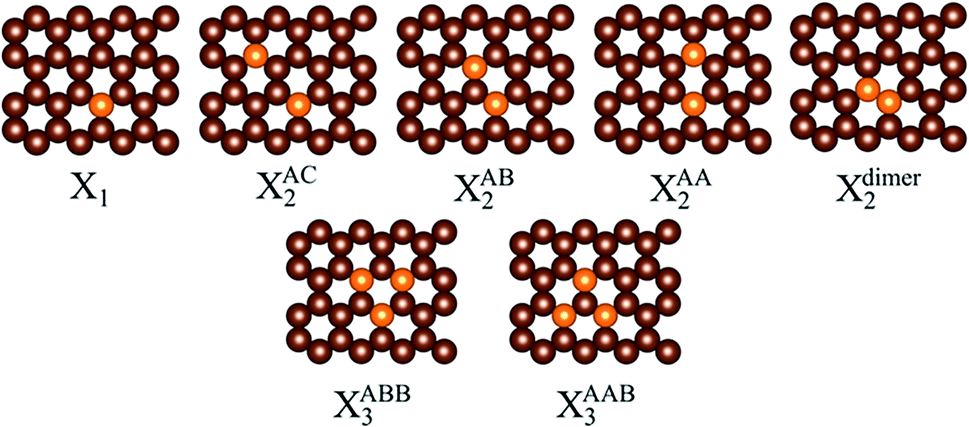 | ||
| Fig. 1 Atomic configurations of single, double, and triple substitutions with dopant X (X = N, B) in graphene. The positions for the double substitutions are AC, AB, and AA, whereas those for triple substitutions are ABB and AAB. A periodic 4 × 4 supercell of graphene is used. Color scheme: carbon (brown); dopant (orange). Single, double, and triple site doping are denoted by X1, X2, and X3, respectively. | ||
| N doping | B doping | ||||||
|---|---|---|---|---|---|---|---|
| Structure | Ef/eV | εF − εD/eV | ϕ/eV | Structure | Ef/eV | εF − εD/eV | ϕ/eV |
| N1 | 0.885 | 0.82 | 3.74 | B1 | 1.131 | −0.87 | 5.15 |
| NAC2 | 1.994 | 1.24 | 3.48 | BAC2 | 2.659 | −1.38 | 5.43 |
| NAB2 | 2.187 | 1.17 | 3.55 | BAB2 | 2.788 | −1.19 | 5.36 |
| NAA2 | 1.817 | 1.03 | 3.72 | BAA2 | 2.362 | −1.14 | 5.15 |
| Ndimer2 | 2.815 | 1.32 | 3.63 | Bdimer2 | 3.564 | −1.19 | 5.20 |
| NABB3 | 3.414 | 1.12 | 3.61 | BABB3 | 4.071 | −1.30 | 5.31 |
| NAAB3 | 3.566 | 1.22 | 3.59 | BAAB3 | 4.243 | −1.32 | 5.37 |
| Graphene | 4.47 | ||||||
We found that all formation energies are positive, and thus the formation of doped graphene is thermodynamically unfavorable with respect to gas-phase N2, and bulk boron. It should be noted that the calculated positive formation energies are not abnormal, as discussed earlier by Okamoto.40 Experimentally, N- and B-doped graphene are usually synthesized by the chemical vapor deposition (CVD) method in which the N and B sources are not gas-phase N2 and bulk boron. For example, double substitution of N in graphene can be achieved by CVD with NH3 and CH4 as precursors.22–26 In addition, the atomic configuration of N- and B-doped graphene can be tuned by using different synthesis methods and a less stable configuration can be obtained.22–27,40,41 As shown in Table 1, the Fermi level shifts relative to the Dirac point calculated by εF − εD are positive and negative for N- and B-doped graphene, respectively, indicating that N and B doping correspond to n- and p-type doping, respectively. The formation energies of both types of doped graphene increase in the order X1 < XAA2 < XAC2 < XAB2 < Xdimer2 < XABB3 < XAAB3 (X = N, B), implying that single substitution is preferable in comparison to double and triple substitution. For double site doping by N atoms, the calculated formation energy and εF − εD of NAA2 are the lowest, in good agreement with other theoretical studies.22,24,40 We found that the formation energies of doped graphene correlate with εF − εD; namely the lower perturbation of the π-electrons of graphene results in a more stable configuration, and the observation that higher concentration of dopant results in higher formation energy agrees with the literature.42–45 Experimentally, the NAB2 configuration was found to be present in more than 80% of N doping sites.22 In contrast, the calculated formation energy of the NAB2 configuration is greater than that of NAA2 by 0.37 eV, indicating that NAB2 is less stable than NAA2. Therefore, the positions of the N dopants in graphene strongly depend on the experimental synthesis conditions.22 For B-doped graphene, the B1 and BAA2 configurations are found experimentally.17,25 The BAA2 configuration is synthesized by doping two B atoms into polyaromatic hydrocarbon precursors.25 B-doped graphene can also be formed by incorporating B atoms within the graphene vacancies.46 For triple site doping, we consider two doping configurations, namely XABB3 and XAAB3, where the dopants alternatively replace the C atoms in graphene. These configurations are similar to the C3N4 family, which has proved to have excellent catalytic activity for several important reactions.47 Here, we don’t consider the doping configurations with large distances between the dopant atoms because these configurations cause effects similar to single-site doping.48
The work functions of pristine and doped graphene are also listed in Table 1. The work function of pristine graphene is about 4.47 eV which is in good agreement with other theoretical studies (4.38 eV) and experimental values (4.45–4.5 eV).49 The N- and B-doped systems exhibit lower and higher work functions than pristine graphene and also the BCN monolayer, respectively.44,49 The different work functions have different effects on the adsorption configuration of water on the substrates, as shown in the below section.
3.2. Water adsorption on graphene substrates
As a reference for the doped systems, the adsorption of water on a pristine graphene substrate was studied. Three stable configurations of water adsorbed on pristine graphene, namely 1-leg, 2-leg, and O-down, were obtained after the geometric optimization (see Fig. 2). The binding energies (Eb), the structural parameters, and the work functions for these three adsorption configurations are summarized in Table 2. We found that 1-leg and 2-leg are more favorable than O-down, implying that the water molecule prefers making H bonds to the pristine graphene surface, and 1-leg is the most favorable structure. Also, the rev-vdW-DF2 functional provided a binding energy for water in good agreement with the results obtained by the diffusion Monte Carlo method (100 meV)8 and the projector augmented wave method.50 The 1-leg and 2-leg configurations also have a longer vertical bond distance to the surface and a wider dipole moment angle compared to the O-down structure. In addition, the work function variation is positive for 1-leg and 2-leg, while it is negative for the O-down structure.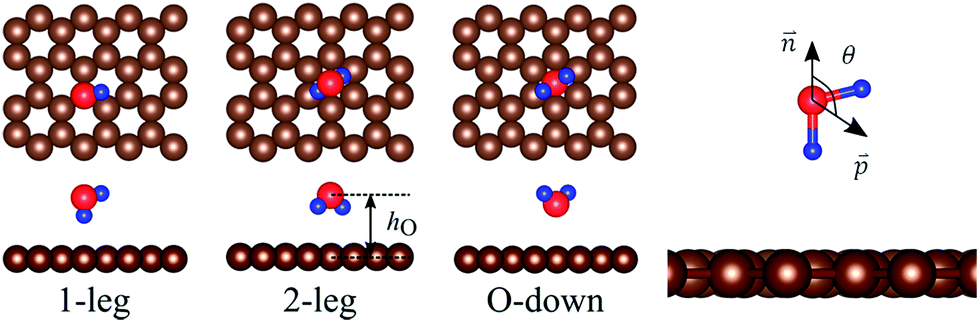 | ||
Fig. 2 Adsorption configurations of a water monomer on pristine graphene. A periodic 4 × 4 supercell of graphene was used. Color scheme: carbon (brown), oxygen (red), and hydrogen (blue). The angle (θ) is between the water dipole moment 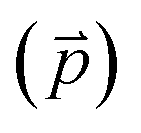 and the surface normal and the surface normal 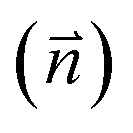 . . | ||
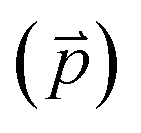 and the surface normal
and the surface normal 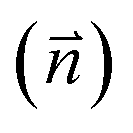 on pristine graphene. The work function (ϕ) and work function change (Δϕ) relative to the clean surface are also included
on pristine graphene. The work function (ϕ) and work function change (Δϕ) relative to the clean surface are also included
For the adsorption of H2O on N- and B-doped graphene, many possible configurations of water were considered. After performing the DFT optimization, the O-down and 1-leg configurations were found on the N- and B-doped substrates, respectively. For each substrate, the most favorable adsorption sites are presented in Fig. 3, and the binding energies are listed in Table 3. Notably, the 2-leg configuration was not found on the N- and B-doped substrates. For single site doping with N, the preferential position of water is on top of the N atom. However, for double and triple site doping with N and all cases of B doping, the water adsorbs stably near the doping atoms. Furthermore, the N-doped substrates retain their planar structure upon water adsorption, while the structure of BAAB3 becomes corrugated, leading to a much higher binding energy. Therefore, we predict that a further increase in the concentration may cause the doped substrates to become unstable towards the adsorption of water.
 | ||
| Fig. 3 Adsorption configurations of a water molecule on doped graphene. A periodic 4 × 4 supercell of graphene was used. Color scheme: carbon (brown), nitrogen (green), boron (orange), oxygen (red), and hydrogen (blue). | ||
| N doping | B doping | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure | Eb /meV | hO/Å | θ/° | ϕ/eV | Δϕ/eV | Structure | Eb /meV | hO/Å | θ/° | ϕ/eV | Δϕ/eV |
| a GGA-PBE results in ref. 18.b PBE + Grimme D2 results in ref. 20.c The vertical bond distance between the O of H2O and the topmost atom of the substrate. | |||||||||||
| H2O/N1 | 154 | 2.97 | 30 | 3.10 | −0.65 | H2O/B1 | 148 | 3.30 | 106 | 5.52 | 0.37 |
| 60a | 40b | ||||||||||
| 140b | 180b | ||||||||||
| H2O/NAC2 | 163 | 3.01 | 42 | 3.25 | −0.23 | H2O/BAC2 | 152 | 3.26 | 105 | 5.84 | 0.41 |
| H2O/NAB2 | 187 | 2.86 | 12 | 2.90 | −0.65 | H2O/BAB2 | 182 | 3.10 | 92 | 5.75 | 0.39 |
| H2O/NAA2 | 160 | 2.89 | 30 | 3.04 | −0.68 | H2O/BAA2 | 144 | 3.24 | 107 | 5.41 | 0.26 |
| H2O/NABB3 | 194 | 2.80 | 23 | 2.79 | −0.74 | H2O/BABB3 | 170 | 3.28 | 119 | 5.77 | 0.05 |
| H2O/NAAB3 | 208 | 2.85 | 14 | 2.77 | −0.81 | H2O/BAAB3 | 280 | 2.72c | 89 | 5.51 | 0.13 |
The binding energies of the water molecule on single site N- and B-doped graphene obtained by the rev-vdW-DF2 functional in the present work are significantly improved compared to those obtained by the GGA-PBE method.18 Also, they are in good agreement with those obtained by Grimme’s D2 method with van der Waals treatment.20 Single site N and B doping increase the Eb by 39 and 33 meV, respectively, relative to the most favorable configuration (1-leg) on pristine graphene. The order of preference for the adsorption of water on double site N- and B-doped graphene was found to be AB > AC > AA. Doping in the AB positions provides the best substrate for improving the binding energy of H2O. Noticeably, the AC and AA structures have a higher scattering level of the doping atoms in comparison to the AB structure, and the binding energy of the water molecule on the AC and AA substrates approaches that for single site doping. Therefore, a significant increase in the binding energy can be achieved if the doping atoms are located close together. Also, double site doping with N and B further enhanced the binding energy of H2O by 33 and 34 meV upon comparing NAB2 and BAB2 to N1 and B1, respectively. Notably, triple site N doping (NAAB3) enhances the binding energy of H2O by 21 meV compared to the best double site doping (structure NAB2). In contrast, the BAAB3 substrate increases the binding energy by 98 meV compared to BAB2. Notably, the BAAB3 structure is deformed by water adsorption. Fig. 4 shows that the binding energy increases almost linearly as a function of doping content at all considered concentrations for N-doped graphene but below 6.3% for B-doped graphene. We estimated that each increase in N and B doping concentration in the linear range could increase the binding energy of H2O by around 30 meV, indicating that the increase in doping content enhances the water–graphene interaction.
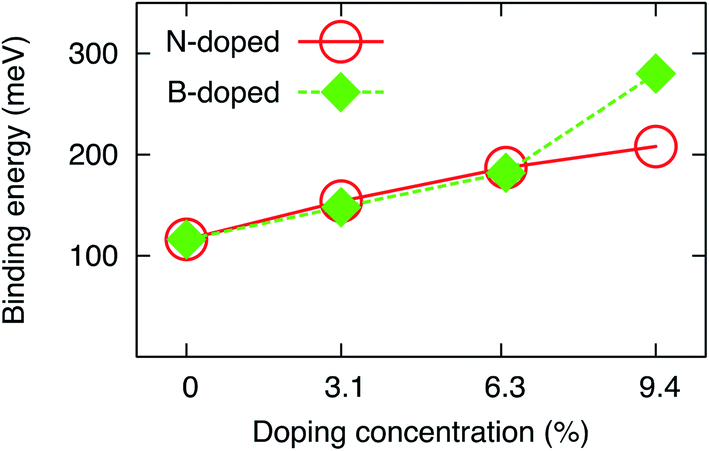 | ||
| Fig. 4 The binding energy of the water molecule versus N and B doping content. | ||
The vertical bond distance from the oxygen atom to the surface decreases from 3.38 > 2.97 > 2.86 ≈ 2.85 Å (3.38 > 3.30 > 3.10 > 2.72 Å) for pristine graphene, N1, NAB2, and NAAB3 (pristine graphene, B1, BAB2, and BAAB3), respectively. This trend in the bond distance was found to correlate with the increase in the binding energy. The angle θ of the dipole moment relative to the surface normal is acute and obtuse for the O-down and 1-leg configurations of the water molecule on the N-doped and B-doped substrates, respectively, which have the dipole moment pointing away from and towards the surface, in that order. In addition, the increase in the binding energy was found to closely correlate with the reduction of the dipole moment angle, i.e., 128 > 30 > 12 ≈ 14° (128 > 106 > 92 > 89°) for H2O on pristine graphene, N1, NAB2, and NAAB3 (pristine graphene, B1, BAB2, and BAAB3), respectively. This result implies that the water molecule rotates to minimize the dipole moment angle and stabilize its adsorption on the substrates upon the increase in doping content. The angle change is useful for experiments on static water contact angles.1–4 Furthermore, the work function decreases and increases relative to the clean surface upon the adsorption of water on N-doped and B-doped graphene, respectively. Also, the work function change Δϕ is negative and positive for water on N- and B-doped graphene, respectively, which correlates with the corresponding O-down and 1-leg configurations. The literature showed that the higher the work function, the higher the vacuum level of the substrate should be,51,52 and therefore, the longer the distance from the surface of the substrate over which the intermolecular forces can take effect on the water molecule. Thus, pristine graphene and B-doped graphene with their high work functions can adsorb water at longer vertical bonding distances (hO), and hence there is a higher probability of finding the most stable configuration to be the 1-leg structure. Whilst N-doped graphene with its lower work function could only offer the O-down configuration as the most favorable structure, which is located at a shorter vertical bond length. In short, the higher work function of B-doped graphene provides a more extended vacuum space from the substrate surface, allowing the water molecule to orient its structure to form a H bond with the surface, while the lower work function of N-doped graphene only allows the O-down configuration of water to stabilize. It should be noted that the average vertical distance from the water oxygen atom to the surface is shorter for N-doped graphene than B-doped graphene.
In reality, experiments have investigated the adsorption of water monomers and small water clusters via STM13,53 and AFM techniques.54 Therefore, we also expect that experiments will be set up to confirm our findings on the correlation of the geometrical and physical parameters upon variation of the doping concentration in the considered graphene systems for water adsorption. Our results demonstrated that the increase in doping concentration enhances the water–substrate interaction. Thus, the wettability of graphene is controllable, which is useful for many applications such as electrocatalysis1,2 and water condensation.3 For instance, the dissociation of H2O in the water-splitting reaction becomes easier if the water–graphene interaction increases.
3.3. Electronic structure analysis
Elucidation of the charge rearrangement, the point charges of atoms, and the atomic orbital projected density of states can help us understand the underlying physics determining the interactions of water with the substrates. Fig. 5 shows that the interface regions of O-down H2O/graphene, H2O/N1, H2O/NAB2, and H2O/NAAB3 present charge depletion, while those of 1-leg H2O/graphene, H2O/B1, H2O/BAB2, and H2O/BAAB3 exhibit charge accumulation. The quantitative charges of each species are given in Table 4. The hydrogen and oxygen atoms of the water always lose and gain charge, respectively. However, the water molecule retains its neutral charge for O-down H2O/graphene and gains or loses charge for the others. Therefore, the substrate doesn’t exchange charge with the water for O-down/graphene, but it exchanges charge with the water for the other structures. The charge exchange is ignorable for the N-doped substrates, but it is significant for the 1-leg H2O/graphene and the B-doped systems. In addition, the charge exchange for 1-leg H2O/graphene is higher than that for O-down H2O/graphene, which explains why the binding energy is higher for the former than the latter. Furthermore, the N atoms accumulate charge while the B atoms donate charge, which is in good agreement with the n-type and p-type nature of N and B doping, respectively. Finally, the obtained binding energies of the water molecule on the different substrates of below 280 meV and the small charge exchange between H2O and the substrates indicate that the water molecule is physisorbed on the surfaces.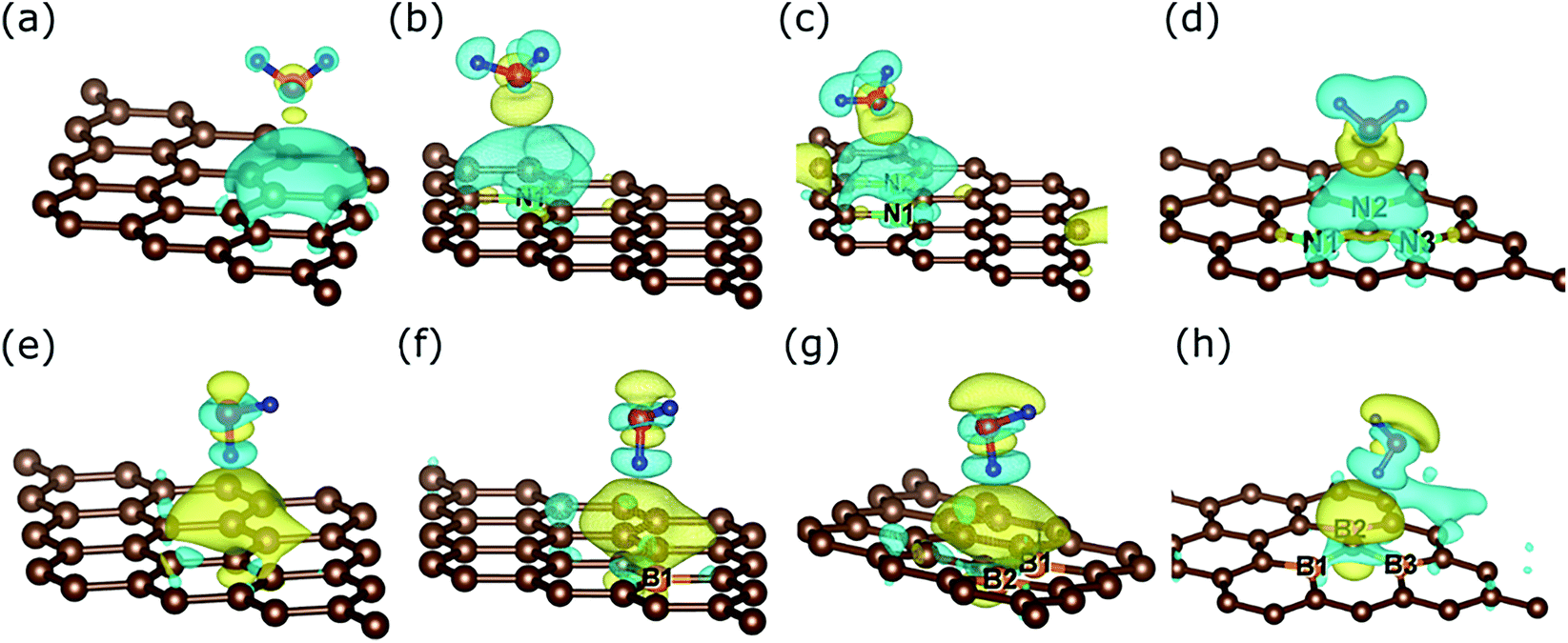 | ||
| Fig. 5 Charge density difference upon the adsorption of water on the substrate: (a) O-down H2O/pristine graphene, (b) H2O/N1, (c) H2O/NAB2, (d) H2O/NAAB3, (e) 1-leg H2O/pristine graphene, (f) H2O/B1, (g) H2O/BAB2, and (h) H2O/BAAB3. Isosurfaces are plotted at 0.0003 e− Bohr−3. Yellow and cyan indicate charge accumulation and depletion, respectively. Color scheme: carbon (brown), nitrogen (green), boron (orange), oxygen (red), and hydrogen (blue). | ||
| H2O/substrate | O-down H2O/graphene | H2O/N1 | H2O/NAB2 | H2O/NAAB3 | 1-leg H2O/graphene | H2O/B1 | H2O/BAB2 | H2O/BAAB3 |
|---|---|---|---|---|---|---|---|---|
| 2H | −1.259 | −1.170 | −1.618 | −1.243 | −1.169 | −1.245 | −1.231 | −1.275 |
| O | 1.259 | 1.175 | 1.624 | 1.234 | 1.183 | 1.265 | 1.253 | 1.306 |
| H2O | 0.000 | 0.005 | 0.006 | −0.009 | 0.014 | 0.020 | 0.022 | 0.031 |
| C atoms | 0.000 | −1.115 | −5.248 | −8.365 | −0.014 | 2.980 | 5.978 | 8.969 |
| N (B) atoms | 1.110 | 5.242 | 8.375 | −3.000 | −6.000 | −9.000 | ||
| Substrate | 0.000 | −0.005 | - 0.006 | 0.009 | −0.014 | −0.020 | −0.022 | −0.031 |
Generally, the interaction between the adsorbate and the substrate is created by the attraction between the unoccupied states of the adsorbate and the occupied states of the substrate, or the occupied states of the adsorbate and the unoccupied states of the substrate.55 The atomic orbital projected density of states in Fig. 6 shows that an increase in N doping content shifts the C pz and N p orbitals downward relative to O-down/graphene to increase the occupation at the valence band maximum (VBM). Simultaneously, the lowest unoccupied s orbital (LUMO) of the water also shifts downward towards the Fermi level. Therefore, the energy difference between the LUMO of the water and the VBM decreases in the order: 2.3 > 1.9 > 1.7 > 1.5 eV for the O-down H2O/graphene, H2O/N1, H2O/NAB2, and H2O/NAAB3 structures, respectively. Contrastingly, an increase in B doping concentration shifts the C pz and B p orbitals upward towards the Fermi level to increase the unoccupied states at the conduction band minimum (CBM) of the substrate. Simultaneously, the highest occupied peak of the water p orbital (HOMO) moves upward to reduce the energy difference between the HOMO and the CBM to 2.6 > 1.9 > 1.5 ≈ 1.5 eV for the 1-leg H2O/graphene, H2O/B1, H2O/BAB2, and H2O/BAAB3 configurations, respectively. The reduction of the energy differences between the LUMO of water and the VBM of the substrate, and the HOMO of water and the CBM of the substrate was found to correlate with the enhancement of the water binding energy. Furthermore, the position of the p orbital peak of the 1-leg configuration is also closer to the Fermi level than that of the O-down configuration. Therefore, the interaction of the water molecule with the substrate is stronger for the 1-leg structure than the O-down structure.
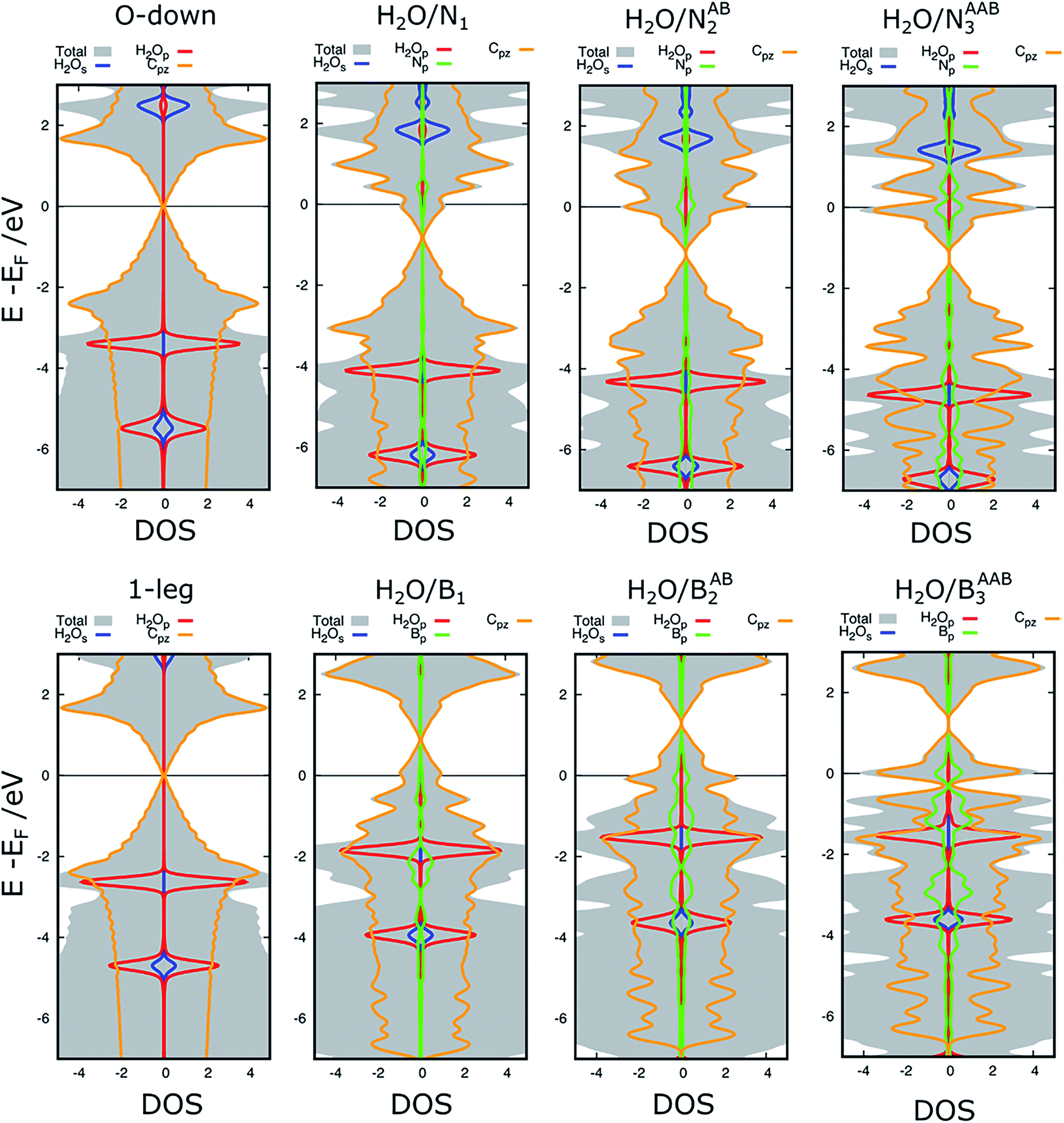 | ||
| Fig. 6 Atomic orbital projected density of states of water in the O-down and 1-leg configurations on pristine graphene and on N- and B-doped graphene. | ||
4. Conclusions
Density functional theory calculations with the rev-vdW-DF2 functional have been used to explore the effects of N and B doping and also the doping concentration in graphene on water adsorption. The most favorable adsorption configurations of water are O-down and 1-leg for the N and B dopants, respectively. The increase in doping content enhances the binding energy of water. The increase in doping concentration enriches the occupied states at the VBM for N doping and the unoccupied states at the CBM for B doping, which originates from the corresponding contributions of the C pz and N p orbitals and the C pz and B p orbitals, respectively. Simultaneously, with increasing doping content, the unoccupied s orbital (LUMO) of water shifts closer to the VBM of N-doped graphene, while the occupied p orbital (HOMO) of water moves closer to the CBM of B-doped graphene. This shift reduces the energy gap, favors the water–graphene interaction, and hence improves the water binding energy. Furthermore, the water molecule was found to rotate its structure to minimize the dipole moment angle relative to the surface normal upon increasing the doping content.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by Vietnam National University Ho Chi Minh City under grant number C2020-20-39.References
- D. Parobek and H. Liu, Wettability of graphene, 2D Mater., 2015, 2, 032001 CrossRef.
- C. Melios, C. E. Giusca, V. Panchal and O. Kazakova, Water on graphene: review of recent progress, 2D Mater., 2018, 5, 022001 CrossRef.
- G. Hong, Y. Han, T. M. Schutzius, Y. Wang, Y. Pan, M. Hu, J. Jie, C. S. Sharma, U. Müller and D. Poulikakos, On the Mechanism of Hydrophilicity of Graphene, Nano Lett., 2016, 16, 4447 CrossRef CAS PubMed.
- A. Ashraf, Y. Wu, M. C. Wang, K. Yong, T. Sun, Y. Jing, R. T. Haasch, N. R. Aluru and S. Nam, Doping-Induced Tunable Wettability and Adhesion of Graphene, Nano Lett., 2016, 16, 4708 CrossRef CAS PubMed.
- O. Leenaerts, B. Partoens and F. M. Peeters, Water on graphene: hydrophobicity and dipole moment using density functional theory, Phys. Rev. B, 2009, 79, 235440 CrossRef.
- I. Hamada, Adsorption of water on graphene: a van der Waals density functional study, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195436 CrossRef.
- A. O. Ajala, V. Voora, N. Mardirossian, F. Furche and F. Paesani, Assessment of Density Functional Theory in Predicting Interaction Energies between Water and Polycyclic Aromatic Hydrocarbons: from Water on Benzene to Water on Graphene, J. Chem. Theory Comput., 2019, 15, 2359 CrossRef CAS PubMed.
- J. G. Brandenburg, A. Zen, M. Fitzner, B. Ramberger, G. Kresse, T. Tsatsoulis, A. Grüneis, A. Michaelides and D. Alfè, Physisorption of Water on Graphene: Subchemical Accuracy from Many-Body Electronic Structure Methods, J. Phys. Chem. Lett., 2019, 10, 358 CrossRef PubMed.
- T. O. Wehling, A. I. Lichtenstein and M. I. Katsnelson, First-principles studies of water adsorption on graphene: the role of the substrate, Appl. Phys. Lett., 2008, 93, 202110 CrossRef.
- G. Revilla-López and N. López, A unified study for water adsorption on metals: meaningful models from structural motifs, Phys. Chem. Chem. Phys., 2014, 16, 22426 RSC.
- J. E. Andrews, S. Sinha, P. W. Chung and S. Das, Wetting dynamics of a water nanodrop on graphene, Phys. Chem. Chem. Phys., 2016, 18, 23482 RSC.
- A. Akaishi, T. Yonemaru and J. Nakamura, Formation of Water Layers on Graphene Surfaces, ACS Omega, 2017, 2, 2184 CrossRef CAS PubMed.
- Y.-R. Wang, J.-Y. Xu, C.-K. Ma, M.-X. Shi, Y.-B. Tu, K. S. S. Meng and J.-Z. Wang, Ice II-like Monolayer Ice Grown on Graphite Surface, J. Phys. Chem. C, 2019, 123, 20297–20303 CrossRef CAS.
- J. Fujiki and K. Yogo, Water adsorption on nitrogen-doped carbons for adsorption heat pump/desiccant cooling: Experimental and density functional theory calculation studies, Appl. Surf. Sci., 2019, 492, 776 CrossRef CAS.
- K. V. Kumar, K. Preuss, Z. X. Guo and M. M. Titirici, Understanding the Hydrophilicity and Water Adsorption Behavior of Nanoporous Nitrogen-Doped Carbons, J. Phys. Chem. C, 2016, 120, 18167 CrossRef CAS.
- N.-J. Kuo, Y.-S. Chen, C.-W. Wu, C.-Y. Huang, Y.-H. Chan and I. P. Chen, One-Pot Synthesis of Hydrophilic and Hydrophobic N-Doped Graphene Quantum Dots via Exfoliating and Disintegrating Graphite Flakes, Sci. Rep., 2016, 6, 30426 CrossRef CAS PubMed.
- S. Agnoli and M. Favaro, Doping graphene with boron: a review of synthesis methods, physicochemical characterization, and emerging applications, J. Mater. Chem. A, 2016, 4, 5002 RSC.
- J. Dai, J. Yuan and P. Giannozzi, Gas adsorption on graphene doped with B, N, Al, and S: a theoretical study, Appl. Phys. Lett., 2009, 95, 232105 CrossRef.
- Y. Yang, F. Liu and Y. Kawazoe, Adsorptionof water on fluorinated graphene, J. Phys. Chem. Solids, 2019, 124, 54 CrossRef CAS.
- A. Xu, L. Shi, L. Zeng and T. S. Zhao, First-principle investigations of nitrogen-, boron-, phosphorus-doped graphite electrodes for vanadium redox flow batteries, Electrochim. Acta, 2019, 300, 389 CrossRef CAS.
- V. A. Cardozo–Mata, J. A. Pescador–Rojas, A. Hernández–Hernández, L. A. Hernández–Hernández, A. Miralrio, F. J. M. Farías, E. Vallejo–Castañeda and E. Rangel, Chemical interaction between nitrogen-doped graphene defects and a copper (111) surface: Effects on water molecule adsorption, Appl. Surf. Sci., 2020, 502, 144149 CrossRef.
- R. Lv, Q. Li, A. R. Botello-Méndez, T. Hayashi, B. Wang, A. Berkdemir, Q. Hao, A. L. Elías, R. Cruz-Silva, H. R. Gutiérrez, Y. A. Kim, H. Muramatsu, J. Zhu, M. Endo, H. Terrones, J.-C. Charlier, M. Pan and M. Terrones, Nitrogen-doped graphene: beyond single substitution and enhanced molecular sensing, Sci. Rep., 2012, 2, 586 CrossRef PubMed.
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Toward N-Doped Graphene via Solvothermal Synthesis, Chem. Mater., 2011, 23, 1188 CrossRef CAS.
- Y.-X. Yu, Can all nitrogen-doped defects improve the performance of graphene anode materials for lithium-ion batteries?, Phys. Chem. Chem. Phys., 2013, 15, 16819 RSC.
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, A Boron-Containing PAH as a Substructure of Boron-Doped Graphene, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed.
- C. Ma, Q. Liao, H. Sun, S. Lei, Y. Zheng, R. Yin, A. Zhao, Q. Li and B. Wang, Tuning the Doping Types in Graphene Sheets by N Monoelement, Nano Lett., 2018, 18, 386 CrossRef CAS PubMed.
- P. Błoński, J. Tuček, Z. Sofer, V. Mazánek, M. Petr, M. Pumera, M. Otyepka and R. Zbořil, Doping with Graphitic Nitrogen Triggers Ferromagnetism in Graphene, J. Am. Chem. Soc., 2017, 139, 3171 CrossRef PubMed.
- H. Wang, T. Maiyalagan and X. Wang, Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications, ACS Catal., 2012, 2, 781 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef PubMed.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Pseudopotentials for high-throughput DFT calculations, Comput. Mater. Sci., 2014, 81, 446 CrossRef CAS.
- I. Hamada, van der Waals density functional made accurate, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- J. G. Brandenburg, A. Zen, D. Alfè and A. Michaelides, Interaction between water and carbon nanostructures: how good are current density functional approximations?, J. Chem. Phys., 2019, 151, 164702 CrossRef PubMed.
- T. N. Pham, M. Sugiyama, F. Muttaqien, S. E. M. Putra, K. Inagaki, D. N. Son, Y. Hamamoto, I. Hamada and Y. Morikawa, Hydrogen Bond-Induced Nitric Oxide Dissociation on Cu(110), J. Phys. Chem. C, 2018, 122, 11814 CrossRef CAS.
- T. N. Pham, Y. Hamamoto, K. Inagaki, D. N. Son, I. Hamada and Y. Morikawa, Insight into Trimeric Formation of Nitric Oxide on Cu(111): A Density Functional Theory Study, J. Phys. Chem. C, 2020, 124, 2968 CrossRef CAS.
- N. T. X. Huynh, V. Chihaia and D. N. Son, Hydrogen storage in MIL-88 series, J. Mater. Sci., 2019, 54, 3994–4010 CrossRef.
- D. N. Son, T. T. T. Huong and V. Chihaia, Simultaneous adsorption of SO2 and CO2 in an Ni(bdc)(ted)0.5 metal–organic framework, RSC Adv., 2018, 8, 38648 RSC.
- L. Bengtsson, Dipole correction for surface supercell calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301 CrossRef CAS.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Efficient Synthesis of Heteroatom (N or S)-Doped Graphene Based on Ultrathin Graphene Oxide–Porous Silica Sheets for Oxygen Reduction Reactions, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS.
- Y. Okamoto, First-principles molecular dynamics simulation of O2 reduction on nitrogen-doped carbon, Appl. Surf. Sci., 2009, 256, 335 CrossRef CAS.
- M. Telychko, P. Mutombo, P. Merino, P. Hapala, M. Ondráček, F. C. Bocquet, J. Sforzini, O. Stetsovych, M. Vondráček, P. Jelínek and M. Švec, Electronic and Chemical Properties of Donor, Acceptor Centers in Graphene, ACS Nano, 2015, 9, 9180 CrossRef CAS PubMed.
- S. Thomas and M. A. Zaeem, A new planar BCN lateral heterostructure with outstanding strength and defect-mediated superior semiconducting to metallic properties, Phys. Chem. Chem. Phys., 2020, 22, 22066 RSC.
- S. Beniwal, J. Hooper, D. P. Miller, P. S. Costa, G. Chen, S.-Y. Liu, P. A. Dowben, E. C. H. Sykes, E. Zurek and A. Enders, Graphene-like Boron–Carbon–Nitrogen Monolayers, ACS Nano, 2017, 11, 2486 CrossRef CAS PubMed.
- S. Thomas, M. S. Manju, K. M. Ajith, S. U. Lee and M. Asle Zaeem, Strain-induced work function in h-BN and BCN monolayers, Phys. E, 2020, 123, 114180 CrossRef CAS.
- S. Thomas and S. U. Lee, Atomistic insights into the anisotropic mechanical properties and role of ripples on the thermal expansion of h-BCN monolayers, RSC Adv., 2019, 9, 1238 RSC.
- R. Lv, G. Chen, Q. Li, A. McCreary, A. Botello-Méndez, S. V. Morozov, L. Liang, X. Declerck, N. Perea-López, D. A. Cullen, S. Feng, A. L. Elías, R. Cruz-Silva, K. Fujisawa, M. Endo, F. Kang, J.-C. Charlier, V. Meunier, M. Pan, A. R. Harutyunyan, K. S. Novoselov and M. Terrones, Ultrasensitive gas detection of large-area boron-doped graphene, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14527 CrossRef CAS PubMed.
- W. J Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability?, Chem. Rev., 2016, 116, 7159 CrossRef PubMed.
- T. M. Dieb, Z. Hou and K. Tsuda, Structure prediction of boron-doped graphene by machine learning, J. Chem. Phys., 2018, 148, 241716 CrossRef PubMed.
- M. Legesse, F. El Mellouhi, E. T. Bentria, M. E. Madjet, T. S. Fisher, S. Kais and F. H. Alharbi, Reduced work function of graphene by metal adatoms, Appl. Surf. Sci., 2017, 394, 98 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- D. N. Son, B. T. Cong and H. Kasai, Hydronium Adsorption on OOH Precovered Pt(111) Surface: Effects of Electrode Potential, J. Nanosci. Nanotechnol., 2011, 11, 2983–2989 CrossRef CAS PubMed.
- D. N. Son, P. V. Cao, T. T. T. Hanh, V. Chihaia and M. P. Pham-Ho, Influences of Electrode Potential on Mechanism of Oxygen Reduction Reaction on Pd-Skin/Pd3Fe(111) Electrocatalyst: Insights from DFT-Based Calculations, Electrocatalysis, 2018, 9, 10–21 CrossRef CAS.
- C. L. Muhich, J. Y. Westcott, T. C. Morris, A. W. Weimer and C. B. Musgrave, The effect of N and B doping on graphene and the adsorption and migration behavior of Pt atoms, J. Phys. Chem. C, 2013, 117, 10523 CrossRef CAS.
- T. Kumagai, M. Kaizu, H. Okuyama, S. Hatta, T. Aruga, I. Hamada and Y. Morikawa, Direct Observation of Hydrogen-Bond Exchange within a Single Water Dimer, Phys. Rev. Lett., 2008, 100, 166101 CrossRef CAS PubMed.
- O. K. Le, V. Chihaia, M. P. Pham-Ho and D. N. Son, Electronic and optical properties of monolayer MoS2 under the influence of polyethyleneimine adsorption and pressure, RSC Adv., 2020, 10, 4201–4210 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |